

Abstract
Aims
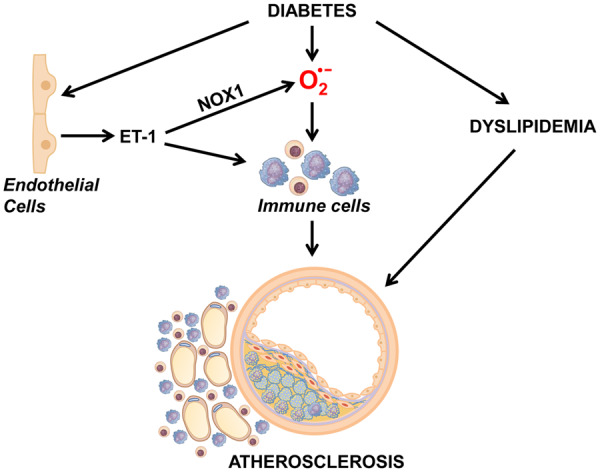
NADPH oxidase (NOX) 1 but not NOX4-dependent oxidative stress plays a role in diabetic vascular disease, including atherosclerosis. Endothelin (ET)-1 has been implicated in diabetes-induced vascular complications. We showed that crossing mice overexpressing human ET-1 selectively in endothelium (eET-1) with apolipoprotein E knockout (Apoe−/−) mice enhanced high-fat diet-induced atherosclerosis in part by increasing oxidative stress. We tested the hypothesis that ET-1 overexpression in the endothelium would worsen atherosclerosis in type 1 diabetes through a mechanism involving NOX1 but not NOX4.
Methods and results
Six-week-old male Apoe−/− and eET-1/Apoe−/− mice with or without Nox1 (Nox1−/y) or Nox4 knockout (Nox4−/−) were injected intraperitoneally with either vehicle or streptozotocin (55 mg/kg/day) for 5 days to induce type 1 diabetes and were studied 14 weeks later. ET-1 overexpression increased 2.5-fold and five-fold the atherosclerotic lesion area in the aortic sinus and arch of diabetic Apoe−/− mice, respectively. Deletion of Nox1 reduced aortic arch plaque size by 60%; in contrast, Nox4 knockout increased lesion size by 1.5-fold. ET-1 overexpression decreased aortic sinus and arch plaque alpha smooth muscle cell content by ∼35% and ∼50%, respectively, which was blunted by Nox1 but not Nox4 knockout. Reactive oxygen species production was increased two-fold in aortic arch perivascular fat of diabetic eET-1/Apoe−/− and eET-1/Apoe−/−/Nox4−/− mice but not eET-1/Apoe−/−/Nox1y/− mice. ET-1 overexpression enhanced monocyte/macrophage and CD3+ T-cell infiltration ∼2.7-fold in the aortic arch perivascular fat of diabetic Apoe−/− mice. Both Nox1 and Nox4 knockout blunted CD3+ T-cell infiltration whereas only Nox1 knockout prevented the monocyte/macrophage infiltration in diabetic eET-1/Apoe−/− mice.
Conclusion
Endothelium ET-1 overexpression enhances the progression of atherosclerosis in type 1 diabetes, perivascular oxidative stress, and inflammation through NOX1.
Keywords: Atherosclerosis, Diabetes, Endothelin-1, NADPH oxidases
Graphical Abstract
Translational perspective
We demonstrate that endothelial cell-restricted human ET-1 overexpression worsens atherosclerosis in diabetes and causes perivascular oxidative stress and inflammation, extending our previous findings to a model of type 1 diabetes mellitus. We also show that these effects are mediated through NOX1 and that atherosclerosis is worsened by loss of NOX4, providing evidence that NADPH oxidase isoforms play differential roles in plaque progression. These results offer new approaches to prevent the progression of atherosclerosis in diabetes, which is associated with significantly increased risk of cardiovascular events.
1. Introduction
The major cause of mortality and disability in patients with diabetes mellitus is vascular disease. This includes macrovascular complications such as atherosclerosis and microvascular complications such as diabetic nephropathy.1 Although the mechanism linking diabetes and vascular disease is complex, overproduction of reactive oxygen species (ROS) seems to play an important role.2 The major source of ROS in the vasculature is the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase family of enzymes. Four isoforms of NADPH oxidase (NOX) have been described in the vasculature and each is characterized by its catalytic subunit: NOX1, NOX2, NOX4, and NOX5. Unlike NOX2, NOX1, and NOX4 do not participate in pathogen clearance and may therefore represent safer pharmacological targets in the prevention of diabetes-related vascular disease. NOX5 is not present in rodents.3
Several groups have investigated the role of NOX isoforms in diabetic cardiovascular disease. Using the murine model of streptozotocin (STZ)-induced type 1 diabetes, Youn et al.4 found that diabetes-associated endothelial dysfunction, endothelial nitric oxide synthase uncoupling, and vascular ROS production was blunted in NOX1-deficient mice. More recently, deletion of Nox1 has been shown to markedly reduce aortic atherosclerosis, ROS production, and inflammation in diabetes induced with STZ in apolipoprotein E knockout (Apoe−/−) mice.5 In contrast to NOX1, the role of NOX4 in vascular disease is more controversial. Deletion of Nox4 in rodent models of diet-induced atherosclerosis caused endothelial dysfunction and/or worsened atherosclerosis in the aorta.6–8 Deletion of Nox4 increased aortic atherosclerosis after 20 weeks of diabetes mellitus,9 whereas it had no effect on plaque formation after 10 weeks.5 Yet, deletion of Nox4 prevented diabetic nephropathy in STZ-treated Apoe−/− mice10 and flagellin-induced atherosclerosis in Apoe−/− mice fed a high-fat diet (HFD).11
Endothelin (ET)-1 is a proinflammatory and potent vasoconstrictor peptide produced largely by endothelial cells.12 Plasma ET-1 levels are increased in patients with diabetes mellitus13–15 and correlate with severity of atherosclerosis in humans.16 In the vasculature, ET-1 stimulates proliferation, fibrosis, and inflammation through pro-oxidant pathways.17 ET-1 was shown to induce expression of NOX2 in human umbilical vein endothelial cells18 and NOX1, NOX2, and NOX4 in rat mesenteric artery smooth muscle cells (SMCs).19 Transgenic mice with endothelium-restricted human ET-1 overexpression (eET-1) present mesenteric artery endothelial dysfunction and vascular remodelling that are associated with increased NOX activity and gp91Phox (NOX2) protein levels.20 Likewise, ET-1 overexpression worsened mesenteric artery endothelial dysfunction and oxidative stress in STZ-treated mice.21 ET type A (ETA) receptor blockade with avosentan reduced aortic atherosclerosis and nephropathy in diabetic STZ-treated Apoe−/− mice.22 Furthermore, human ET-1 overexpression worsened HFD-induced atherosclerosis and was associated with increased oxidative stress in Apoe−/− mice.23 However, it remains unknown whether human ET-1 overexpression enhances atherosclerosis in diabetes through NOX1 but not NOX4.
We hypothesized that endothelium-restricted ET-1 overexpression would worsen atherosclerosis in diabetes through a mechanism involving NOX1 but not NOX4. To test this hypothesis, we first crossed eET-1 with Apoe−/− mice and evaluated whether ET-1 overexpression accelerated the progression of atherosclerosis in STZ-induced diabetes. Thereafter, we evaluated whether this enhancement of atherosclerosis would be prevented in eET-1/Apoe−/− deficient in Nox1 but not Nox4. We also evaluated aortic perivascular oxidative stress and inflammation, as these processes are known to contribute to vascular damage and atherosclerosis.
2. Methods
Expanded Methods are provided in the Supplementary material online.
2.1 Experimental design
The study was approved by the Animal Care Committee of the Lady Davis Institute for Medical Research and McGill University, followed recommendations of the Canadian Council for Animal Care and was in agreement with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. C57BL/6 transgenic mice overexpressing the human ET-1 (eET-1) driven by the Tie2 promoter conferring endothelial-specific expression were described previously.24 C57BL/6 Apoe−/− mice were obtained from Jackson Laboratory (B6.129P2-Apoetm1Unc/J, Bar Harbor, ME, USA). Apoe−/−/Nox1−/y and Apoe−/−/Nox4−/− mice were generated by backcrossing Nox1−/y and Nox4−/− mice with Apoe−/−/Nox4−/− mice (Animal Resources Centre, Canning Vale, WA, Australia) at the Baker IDI Heart & Diabetes Research Institute (Melbourne, Australia).5Nox1−/y and Nox4−/− mice have been described previously.25,26 The eET-1/Apoe−/− mice were generated at the Lady Davis Institute for Medical Research (Montreal, QC, Canada) by crossing eET-1 mice with Apoe−/− mice obtained from Jackson Laboratories (Bar Harbor, ME, USA). eET-1/Apoe−/−/Nox1−/y and eET-1/Apoe−/−/Nox4−/− mice were generated at the Lady Davis Institute for Medical Research by crossing eET-1/Apoe−/− mice with Apoe−/−/Nox1−/y and Apoe−/−/Nox4−/− mice, respectively.
Six-week-old male Apoe−/−, Apoe−/−/Nox1−/y, Apoe−/−/Nox4−/−, eET-1/Apoe−/−, eET-1/Apoe−/−/Nox1−/y, and eET-1/Apoe−/−/Nox4−/− mice were injected IP with either vehicle (sodium citrate buffer) or with STZ (55 mg/kg/day) for five consecutive days to induce type 1 diabetes mellitus.5 Ten days after the last injection of STZ, only mice presenting a blood glucose level ≥15 mmol/L after 4–6 h of fasting time were considered diabetic and included in the study. Mice were studied 14 weeks after the last IP injection of either vehicle or STZ. At the end of the protocol, blood glucose level was evaluated after 4–6 h of fasting. The following day, mice were euthanized by anaesthesia followed by blood and tissue collection as follows. Mice were weighed and anaesthetized with 3% isoflurane mixed with O2 at 1 L/min and the depth of anaesthesia was confirmed by rear foot squeezing. Blood was collected by cardiac puncture for plasma ET-1 determination and measurement of plasma cholesterol, high-density lipoprotein (HDL) and triglycerides. Tissues were harvested in ice-cold phosphate buffered saline and weighed, and tibia length was determined. The base of the heart (which comprises the aortic sinus) and the aortic arch with perivascular adipose tissue (PVAT) were dissected and embedded in VWR Clear Frozen Section Compound (VWR international, Edmonton, AL, Canada) for characterization of atherosclerotic plaque, ROS production, alpha-smooth muscle actin (α-SMA) expression, collagen content, buried fibrous caps, and monocyte/macrophage and CD3+ T-cell infiltration. The remaining tissues were frozen in liquid nitrogen and stored at −80°C until used.
2.2 Data analysis
Results are presented as means ± SEM. Data were compared with one-way analysis of variance (ANOVA) followed by a Student–Newman–Keuls post hoc test. In absence of normal distribution, a Kruskal–Wallis one-way ANOVA followed by a Dunn’s multiple comparison test was used to compare data. P < 0.05 was considered statistically significant.
3. Results
3.1 Fasting glycaemia, plasma ET-1, and lipaemia
STZ treatment induced type 1 diabetes efficiently in all the groups. Fasting glycaemia was increased three-fold in diabetic Apoe−/− mice, which was unaffected by ET-1 overexpression, Nox1 or Nox4 knockout (Supplementary material online, Figure S1). eET-1/Apoe−/− mice presented an eight-fold higher plasma ET-1 level compared to Apoe−/− mice (Supplementary material online, Figure S1). Plasma ET-1 was unaffected by diabetes and Nox1 or Nox4 knockout in Apoe−/− mice. Plasma ET-1 tended to be decreased in diabetic eET-1/Apoe−/− mice, but remained five-fold higher, which was unaffected by Nox1 or Nox4 knockout. Plasma cholesterol levels were increased to a similar extent in diabetic Apoe−/− and eET-1/Apoe−/− mice, which was unaffected by Nox1 or Nox4 knockout (Supplementary material online, Figure S2). Diabetes tended to increase plasma HDL levels in Apoe−/− but not eET-1/Apoe−/− mice, which was unaffected by Nox1 or Nox4 knockout. Plasma triglycerides were increased three-fold in diabetic Apoe−/− mice and 1.7-fold in diabetic eET-1/Apoe−/− mice, which was blunted by Nox1 and Nox4 knockout in diabetic Apoe−/− mice and tended to be decreased in diabetic eET-1/Apoe−/− mice (Supplementary material online, Figure S2).
3.2 ET-1 overexpression worsened atherosclerosis in diabetes through NOX1
Atherosclerotic lesions were characterized in 8 µm cryosections of aortic sinus and aortic arch obtained at 90-µm intervals. In the aortic sinus, diabetes increased plaque size 4.4-fold in Apoe−/− mice, which was unaffected by Nox1 or Nox4 deletion (Figure 1A and B). ET-1 overexpression further enhanced plaque size 2.5-fold, which tended to be reduced by Nox1 but not Nox4 knockout. In the aortic arch, no plaques were detected in vehicle-treated Apoe−/− mice whereas they were present in diabetic Apoe−/− mice (Figure 1C and D). Nox1 but not Nox4 knockout tended to decrease diabetes-induced atherosclerosis in Apoe−/− mice. ET-1 overexpression augmented lesion size five-fold in diabetic Apoe−/− mice, which was reduced 60% by Nox1 knockout and was further increased 1.5-fold by Nox4 knockout.
Figure 1.

Endothelial cell-restricted endothelin-1 overexpression worsened atherosclerosis in type 1 diabetes through NOX1. Atherosclerotic lesion areas were determined by oil red O staining in aortic sinus (A, B) and arch (C, D) of Apoe−/−, eET-1/Apoe−/−, eET-1/Apoe−/−/Nox1−/y, and eET-1/Apoe−/−/Nox4−/− mice 14 weeks after intraperitoneal injections with either vehicle (Veh) or streptozotocin (STZ). Representative images of oil red O-stained and Mayer’s haematoxylin-counterstained aortic sinus (A) and arch (C) cryosections are shown. Lesion sizes were calculated as mean μm2 of four sections obtained at 90-μm intervals of aortic sinus or arch. Data are presented as means ± SEM, n = 5–9. Data were analysed using one-way ANOVA followed by a Student–Newman–Keuls post hoc test. *P < 0.05 and **P < 0.001 vs. non-diabetic Apoe−/−, †P < 0.01 and ††P < 0.001 vs. diabetic Apoe−/−, ‡P < 0.01 and ‡‡P < 0.001 vs. non-diabetic eET-1/Apoe−/−, §P < 0.05 and §§P < 0.001 vs. diabetic eET-1/Apoe−/−.
3.3 ET-1 overexpression decreased plaque α-SMA content through NOX1
α-SMA expression, a marker of plaque stability, was assessed in aortic sinus and arch lesions. In the aortic sinus, ET-1 overexpression decreased α-SMA content per lesion area by ∼35%, which was blunted by Nox1 but not Nox4 knockout (Supplementary material online, Figure S4). Nox1 knockout also tended to exhibit increased α-SMA plaque content in diabetic Apoe knockout mice. In the aortic arch, ET-1 overexpression decreased α-SMA content per lesion area by ∼50%, which was also blunted by Nox1 but not Nox4 knockout (Figure 2). Aortic sinus and arch plaque collagen content was unaffected by ET-1 overexpression, or lack of Nox4 (Supplementary material online, Figures S5 and S6). However, mice deficient in Nox1 tended to present more plaque collagen content in the aortic sinus but not the aortic arch. Buried fibrous caps that have been proposed as evidence of prior plaque rupture, tended to be more numerous in aortic sinus and arch plaques, and were unaffected by Nox1 nor Nox4 knockout.
Figure 2.
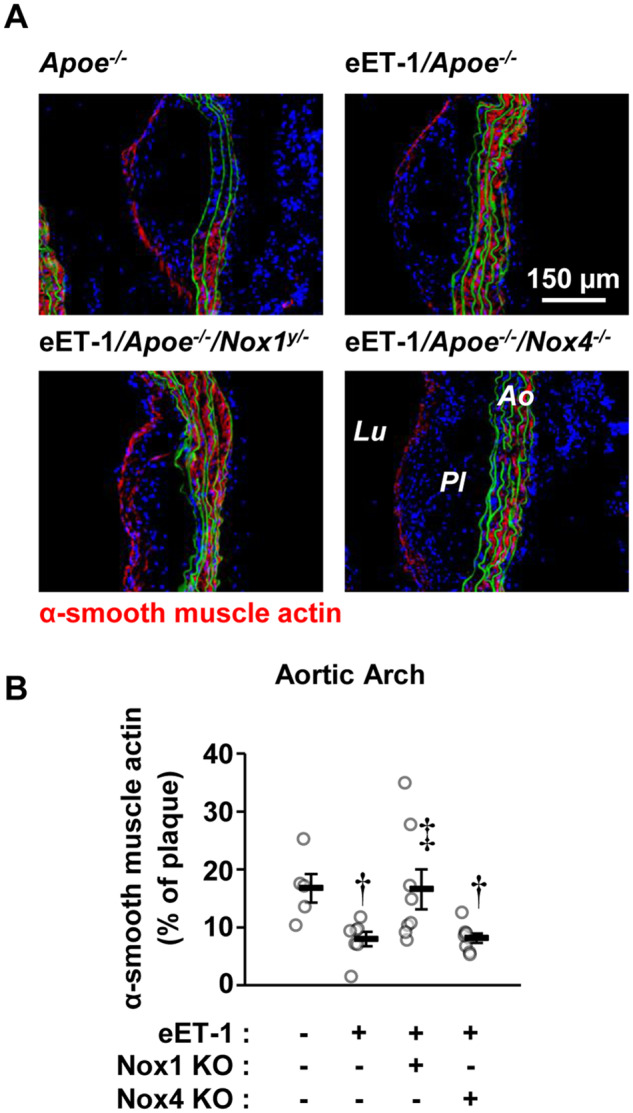
Endothelial cell-restricted endothelin-1 overexpression (eET-1) decreased alpha-smooth actin content in atherosclerotic plaques via NOX1. Alpha-smooth muscle actin content (A and B) was determined by immunofluorescence in aortic arch plaques of Apoe−/−, eET-1/Apoe−/−, eET-1/Apoe−/−/Nox1−/y, and eET-1/Apoe−/−/Nox4−/− mice 14 weeks after intraperitoneal injections with either vehicle or streptozotocin. Representative fluorescence images of α-smooth muscle actin (red) are shown in A. Green and blue represent elastin autofluorescence and 4′,6-diamidino-2-phenylindole fluorescence, respectively. Ao, aorta; Lu, lumen; Pl, plaque. Data are presented as means ± SEM, n = 5–8. Data were analysed using one-way ANOVA followed by a Student–Newman–Keuls post hoc test. †P < 0.05 vs. diabetic Apoe−/−, ‡P < 0.05 vs. diabetic eET-1/Apoe−/−.
3.4 ET-1 overexpression produced oxidative stress and PVAT immune cell infiltration through NOX1 or NOX4
Oxidative stress assessed by dihydroethydium (DHE) fluorescence in the aortic arch media and plaque was similar between all groups (Supplementary material online, Figure S3). ET-1 overexpression increased ROS production two-fold in aortic PVAT of diabetic Apoe−/− mice, which was blunted by Nox1 but not Nox4 knockout (Figure 3). Nox1, Nox2, and Nox4 mRNA expressions were examined by reverse transcription-quantitative PCR in two highly vascularized tissues, the renal cortex and lung. Since the number of samples per group was less than 5 in some groups, the data were only analysed qualitatively. The results showed that the mRNA expression of Nox1 and Nox2 but not Nox4 was increased in renal cortex and lung of diabetic Apoe−/− compared to non-diabetic Apoe−/− mice (Supplementary material online, Figure S8). The mRNA expression of Nox1 and Nox2 was not further increased by ET-1 overexpression, and Nox4 mRNA expression was unaltered by high ET-1 levels in diabetic Apoe−/− mice. Nox1 and Nox4 mRNAs were undetectable in Nox1−/y and Nox4−/− mice, respectively.
Figure 3.
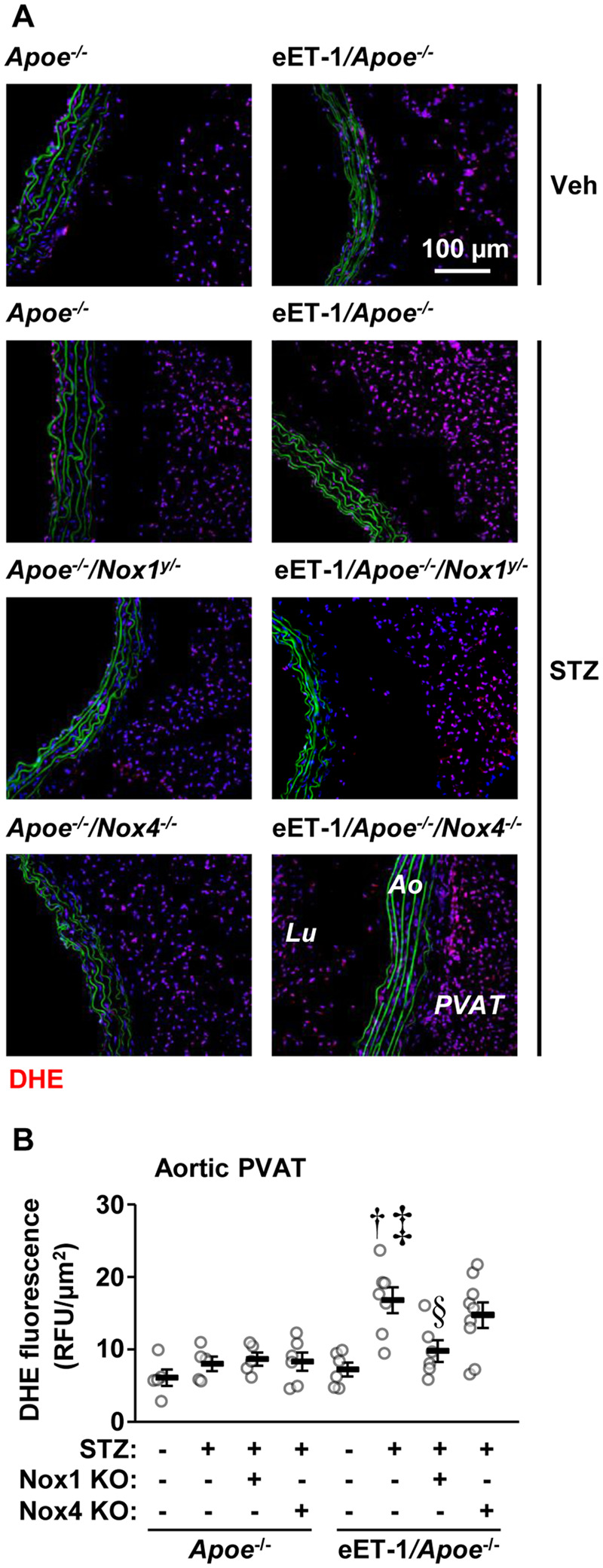
Endothelial cell-restricted endothelin-1 overexpression (eET-1) produced perivascular adipose tissue oxidative stress through NOX1. Reactive oxygen species generation was determined by dihydroethidium (DHE) staining in aortic arch perivascular adipose tissue of Apoe−/−, eET-1/Apoe−/−, eET-1/Apoe−/−/Nox1−/y, and eET-1/Apoe−/−/Nox4−/− mice 14 weeks after intraperitoneal injections with either vehicle (Veh) or streptozotocin (STZ). Representative images of DHE-stained sections are shown in A. Red, green, and blue represent DHE fluorescence, elastin autofluorescence, and 4′,6-diamidino-2-phenylindole fluorescence, respectively. Ao, aorta; Lu, lumen; PVAT, perivascular adipose tissue; RFU, relative fluorescent units. Data are presented as means ± SEM, n = 5–9. Data were analysed using one-way ANOVA followed by a Student–Newman–Keuls post hoc test. †P < 0.001 vs. diabetic Apoe−/−, ‡P < 0.01 vs. non-diabetic eET-1/Apoe−/−, §P < 0.05 vs. diabetic eET-1/Apoe−/−.
ET-1 overexpression decreased by 35% monocyte/macrophage infiltration per lesion area in aortic sinus but not in aortic arch of Apoe−/− mice (Supplementary material online, Figure S7). Aortic sinus and arch plaque monocyte/macrophage infiltration was unaffected by Nox1 or Nox4 knockout. ET-1 overexpression increased 2.7-fold aortic PVAT monocyte/macrophage infiltration in diabetic Apoe−/− mice, which was prevented by Nox1 but not Nox4 knockout (Figure 4A and B). Diabetes tended to increase aortic PVAT CD3+ T-cell infiltration in Apoe−/− and Apoe−/−/Nox4−/− mice but not in Apoe−/−/Nox1−/y mice (Figure 4C and D). ET-1 overexpression increased CD3+ T-cell infiltration 2.6-fold in diabetic Apoe−/− mice, which was abrogated by Nox1 knockout and reduced by Nox4 knockout (Figure 4).
Figure 4.

Endothelial cell-restricted endothelin (eET)-1 overexpression caused perivascular adipose tissue immune cell infiltration through NOX1 or NOX4. Infiltration of monocytes/macrophages (A and C) and CD3+ T cells (B and D) was determined by immunofluorescence in aortic arch perivascular adipose tissue of Apoe−/−, eET-1/Apoe−/−, eET-1/Apoe−/−/Nox1−/y, and eET-1/Apoe−/−/Nox4−/− mice 14 weeks after intraperitoneal injections with either vehicle (Veh) or streptozotocin (STZ). Representative images of MOMA-2 (monocyte/macrophages, red) and CD3 (red) immunofluorescence are shown in A and B, respectively. Green and blue represent elastin autofluorescence and 4′,6-diamidino-2-phenylindole (DAPI) fluorescence, respectively. Ao, aorta; Lu, lumen; PVAT, perivascular adipose tissue. Data are presented as means ± SEM, n = 5–8. Data were analysed using one-way ANOVA followed by a Student–Newman–Keuls post hoc test. †P < 0.05 vs. diabetic Apoe−/−, ‡P < 0.01 vs. non-diabetic eET-1/Apoe−/−, §P < 0.05 and §§P < 0.01 vs. diabetic eET-1/Apoe−/−.
4. Discussion
It was previously demonstrated that the ET system plays a role in diabetes mellitus-induced development of aortic atherosclerotic plaques, oxidative stress, and inflammation in Apoe−/− mice, since they were reduced by ETA receptor blockade with avosentan.22 The present study demonstrates that endothelial human ET-1 overexpression worsens atherosclerosis, perivascular oxidative stress, and inflammation in diabetes, extending our previous findings to a model of type 1 diabetes mellitus.23 It was also shown using gene deletion that two sources of ROS, NOX1 and NOX4, producing respectively superoxide and hydrogen peroxide, have opposite action, promoting and counteracting atherosclerotic plaque development and inflammation in diabetic mellitus.5,9 In this study, we show that ET-1 overexpression induced an enhancement of atherosclerosis in type 1 diabetes that is mediated through NOX1 and worsened by the loss of NOX4, providing additional evidence that increased NOX isoforms play differential roles in atherosclerotic plaque progression.
It is well-established that diabetes mellitus increases the risk of developing atherosclerosis and other vascular complications.27 Increased ET-1 expression may play a role in diabetes-associated atherosclerosis. Plasma ET-1 levels are increased in patients with diabetes13–15 and correlate with severity of atherosclerosis in humans16 and inhibition of the ETA receptors with avosentan reduced aortic atherosclerosis in diabetic Apoe−/− mice.22 To investigate the role of ET-1 overexpression in atherosclerosis in diabetes, we crossed eET-1 transgenic mice with atherosclerosis-prone Apoe−/− mice and induced type 1 diabetes by administration of STZ. Consistent with our previous findings, eET-1/Apoe−/− mice presented an eight-fold higher plasma ET-1 level compared to Apoe−/− mice.23 Plasma ET-1 levels in eET-1/Apoe−/− mice were not statistically altered by diabetes induction, Nox1 or Nox4 knockout, suggesting that alterations in plasma ET-1 are unlikely to explain the enhanced atherosclerosis. Fasting blood glucose and total cholesterol levels were also increased to a similar extent in all diabetic mice at the end of the study groups.
In the current study, we found that endothelial ET-1 overexpression worsened atherosclerosis in diabetic mice in both the aortic arch and sinus. These findings extend our previous results showing that increased ET-1 worsens atherosclerosis caused by HFD and demonstrate that ET-1’s modulation of atherogenesis is not limited to a single aetiology.23 This is particularly important for application in humans, where the causes of atherosclerosis are often heterogeneous and multifactorial. However, ET-1 overexpression alone is not sufficient to consistently promote atherosclerosis, as non-diabetic eET-1/Apoe−/− mice presented increased plaque size in the aortic sinus but not the arch. This suggests that increased ET-1 accelerates the progression of atherosclerosis in predisposed conditions such as diabetes and hypercholesterolaemia and it is therefore likely that patients with such conditions would benefit most from ET-1 blockade.
Increased ROS generation has been implicated in the development and progression of vascular complications in diabetes.2 In this study, we observed that ET-1 overexpression induced enhanced progression of atherosclerosis in diabetic mice that was associated with an increase in ROS generation in aortic arch PVAT. We made a similar observation in HFD-induced atherosclerosis.23 The increase in ROS generation could be a direct consequence of ET-1 overexpression. ET-1 was shown to induce NOX activity or ROS generation in vitro in vascular endothelial18 and SMCs19,28,29 and ex vivo in carotid arteries30 and aortic rings.31 One study has shown that ET-1 inhibits NOX activity in aortic endothelial cells.32 Interestingly, ET-1 was revealed to mediate the action of angiotensin II in aortic SMCs. Six-hour angiotensin II stimulation-induced NOX activity and NOX1 and NOX4 protein expression were prevented by ET receptor blockers.28 Furthermore, it has been reported that ET-1 increases the mRNA expression of Nox2 (identified as gp91Phox in the publication) in endothelial cells18 and protein levels of NOX1, NOX2 and NOX4 in SMCs.19 In the latter study, the investigators demonstrated that knockdown of Nox1 or Nox2 with small interfering RNA prevented activation of NOX by 4-hour ET-1 stimulation.19 However, it has not been determined whether Nox4 knockdown affects ET-1 response. We previously showed that NOX activity and NOX2 protein levels were increased in mesenteric arteries of 10-week-old male eET-1 mice overexpressing ET-1 in endothelial cells.20 Furthermore, we have observed that 14-week diabetes-induced ROS generation in mesenteric arteries was enhanced by ET-1 overexpression.21 However, it has never been determined whether NOX1 or NOX4 plays a role in endothelial ET-1 overexpression-induced ROS generation and atherosclerosis in diabetes. NOX5 is expressed in human vascular cells and is not expressed in rodents. ET-1 has been shown to increase NOX5 protein levels in human aortic SMCs and microvascular endothelial cells.33,34 It would be interesting to determine whether NOX5 plays a role in atherosclerosis in humans with diabetes mellitus.
Gray et al.9 have previously shown that increased ROS generation in diabetic mice was associated with increased Nox1 and Nox2 mRNA expression levels in aorta. Increased ET-1 may have augmented ROS generation in diabetic mice by enhancing the increase in Nox1 and Nox2 mRNA expression levels. Qualitative investigation of two highly vascularized tissues, renal cortex and lungs, revealed that diabetes-induced elevation in Nox1 and Nox2 mRNA expression levels was unaffected by ET-1 overexpression. Furthermore, Nox4 mRNA expression was unaltered by diabetes or high ET-1 levels. This study demonstrated that NOX1 is required for the enhancement of atherosclerosis and immune cell infiltration caused by high ET-1 expression, but the mechanisms remain unclear.
Several studies have reported a role for NOX1 in the development of atherosclerosis in Apoe−/− mice.5,35,36 In our study, Nox1 knockout blunted the atherosclerosis enhancement caused by ET-1 overexpression in the aortic arch which, to our knowledge, is the first time that ET-1 has been shown to mediate its proatherogenic actions through NOX1. In the aortic sinus of diabetic eET-1/Apoe−/− mice, Nox1 knockout tended to reduce plaque size although this did not achieve statistical significance. It is possible that Nox1 knockout only slows plaque progression and that the absence of significant difference in the aortic sinus reflects the fact that plaques are already near-maximal in size. Indeed, animals were studied 14 weeks after induction of diabetes and plaques were larger in the aortic sinus than in the aortic arch. In the current study, Nox4 knockout further worsened aortic arch atherosclerosis in diabetic eET-1/Apoe−/− mice but not Apoe−/− mice and had no effect in the aortic sinus. Several studies using Apoe−/− or Ldlr−/− mice fed a HFD have reported that Nox4 knockout enhances HFD-induced atherosclerosis.6–8 On the other hand, studies using STZ-treated Apoe−/−/Nox4−/− mice have found no differences 10 weeks after induction of diabetes5 and enhanced atherosclerosis after 20 weeks.9 Our findings therefore seem consistent with the notion that NOX4 has an atheroprotective role, at least in certain vascular territories. The qualitative difference of actions between NOX1 and NOX4 is best explained by the type of ROS they produce, with NOX1-derived superoxide being deleterious to vascular health and NOX4-derived hydrogen peroxide being beneficial.37
Plaque stability, which depends on the thickness of the fibrous cap and the degree of cap inflammation, is particularly important in human atherosclerosis as it determines which plaques will rupture and ultimately cause myocardial infarction or stroke. While this is true in humans, traditional mouse models of atherosclerosis are particularly resistant to extensive plaque destabilization and have only been reported by a few investigators to exhibit spontaneous plaque rupture and secondary thrombosis.38–41 Less severe plaque injury such as endothelial denudation and plaque fissure without necrotic core exposure still do occur in mice, and terms such as plaque disruption have been suggested to describe these in order to distinguish them from overt plaque rupture.42 Nevertheless, features of plaque instability found in human atherosclerosis have widely been investigated in rodents,43,44 as they may still provide insights into the biology of plaque remodelling. We found in this study that ET-1 overexpression decreased α-SMA content, a marker of SMCs, which are a component of the fibrous cap,45 which has been suggested by some investigators to indicate previous silent plaque disruption.43,46 Noteworthy, it has been shown in humans that coronary plaque ET-1 immunoreactivity is greater in patients with unstable angina,47 who have decreased plaque SMCs due to apoptosis than in those with stable angina.48 Interestingly, the change in α-SMA content but not in buried fibrous caps was blunted by Nox1 but not Nox4 knockout in both the aortic sinus and arch. Other studies have also implicated oxidative stress in plaque instability in humans. Indeed, ROS production and NOX subunit expression are both high in the shoulder region of plaques,49 and their levels are higher in patients with unstable angina compared to stable angina independently of the degree of vessel stenosis.50 It is therefore possible that ET-1 modulated SMC loss by stimulating NOX1-dependent superoxide production and triggering redox-sensitive pathways such as those involving nuclear factor κB and activating protein-1. However, whether ET-1 overexpression plays a role in atherosclerotic plaque instability and rupture needs to be demonstrated using an appropriate model of plaque rupture.
In the current study, we found that ET-1 overexpression increased ROS production in the aortic PVAT of diabetic Apoe−/− mice through NOX1. Although previous studies using the non-selective NOX inhibitor apocynin have implicated a role for NOX in ET-1-induced vascular damage,20,30 our findings show for the first time that ET-1 produces oxidative stress through the specific NOX1 isoform. We did not find that Nox4 knockout reduced ROS production, although this may have been a result of concomitant up-regulation of NOX1 and/or NOX2. Indeed, Gray et al. showed a reduction in aortic hydrogen peroxide but not total ROS generation in diabetic Apoe−/−/Nox4−/− mice, the latter ascribed to increased expression of NOX1, NOX2 and p47phox in these mice.9 Increase in other sources of ROS such as xanthine oxidase in peroxisomes, and NADH dehydrogenase and ubiquinone cytochrome c reductase in mitochondria may also explain the lack of effect of Nox4 deficiency on ROS generation.
Interestingly, ROS production in aortic arch plaque was similar between groups and is therefore unlikely to have contributed to the enhanced atherosclerosis seen in diabetic eET-1/Apoe−/− mice. Instead, it seems that NOX1-dependent PVAT oxidative stress is playing a more important role here. There is accumulating evidence that PVAT modulates the development of vascular disease through so-called ‘outside-in’ mechanisms. Transplant of proinflammatory visceral PVAT to the mid-perivascular area of the common carotid arteries, a site usually devoid of atherosclerosis, impairs vascular relaxation and causes atherosclerotic lesions to develop in that area.51 In obese mice, PVAT-derived ROS production and p67phox expression are increased and removal of such pro-oxidant PVAT restores aortic endothelium-dependent relaxation.52 Thus, it is possible that abluminal secretion of endothelium-derived ET-1 causes PVAT oxidative stress, which creates a pro-inflammatory milieu that in turn promotes endothelial dysfunction and atherosclerosis.
It is widely accepted that inflammatory processes play an important role in the pathogenesis of atherosclerosis.53,54 These include innate responses involving monocytes/macrophages55 and adaptive immunity involving T cells.56 In the present study, we found that ET-1 overexpression increased aortic PVAT infiltration of both monocyte/macrophage and CD3+ T cells in diabetic Apoe−/− mice. This observation is consistent with our previous findings of enhanced PVAT inflammation in eET-1/Apoe−/− mice fed a HFD.23 Studies in Apoe−/− mice have reported that Nox1 knockout reduces diabetes-induced aortic macrophage infiltration5 and HFD-induced atherosclerotic plaque macrophage infiltration.35 Here, Nox1 knockout blunted ET-1-induced monocyte/macrophage and T-cell infiltration but did not statistically reduce inflammation in Apoe−/− mice. This suggests that NOX1 participates in ET-1-induced immune cell recruitment through activation by pro-inflammatory stimuli such as chemokines and cytokines. It is also interesting to note that enhanced inflammation occurred only in aortic PVAT where ROS production was also increased, possibly because one induces the other or because of reciprocal induction.
In contrast to NOX1, the role of NOX4 in inflammation is less clear. Studies in diabetic Apoe−/−/Nox4−/− mice have reported increased aortic macrophage infiltration 20 weeks after induction of diabetes9 but no differences 10 weeks post-induction.5 Inducible deletion of Nox4 increased plaque macrophage infiltration,8 whereas endothelial overexpression of NOX4 had no effect on it.6 Here, we found that Nox4 knockout blunted ET-1-induced T-cell infiltration but not macrophage infiltration. It is unclear to what extent aortic T-cell infiltration contributes to the progression of aortic atherosclerosis seen in diabetic eET-1/Apoe−/− mice, since T-cell infiltration was prevented by both Nox1 and Nox4 deletion.
4.1 Limitations
A limitation of the present study is that DHE staining did not reveal changes in ROS generation in the aortic wall of diabetic Apoe−/− and eET-1/Apoe−/− mice and in PVAT of diabetic Apoe−/− mice. The reasons for this are unclear, since this technique has been previously demonstrated to detect changes in ROS generation in aorta and PVAT of HFD fed Apoe−/− and eET-1/Apoe−/− mice.23 It is likely that many factors in our experimental design [environment (microbiota), genetic models used, method of ROS detection and operators] may have contributed to the differences between our findings and others. Another significant limitation is that only one method of ROS generation, DHE staining, was used. Although DHE staining can accurately measure ROS generation as noted by a scientific statement from the American Heart Association,57 it is far from being perfect, and there is a need to validate ROS generation by other methods. An additional limitation is that plaque rupture is largely absent in the atherosclerotic Apoe knockout mouse model. Nevertheless, this model presents features of plaque instability found in human atherosclerosis, which may still provide insights into the biology of plaque remodelling. There is a need for a murine model with plaque rupture. An important limitation is that while we found that endothelial cell-restricted ET-1 overexpression worsens atherosclerosis and immune cell infiltration in vivo through NOX1, we were unable to determine the mechanism whereby ET-1 interacts with NOX1. Our qualitative data suggest that Nox1 mRNA is not up-regulated by ET-1 overexpression, but more in-depth future studies are required to clarify the mechanisms.
5. Conclusion
Taken together, our results demonstrate for the first time that endothelium-restricted ET-1 overexpression worsens atherosclerosis in type 1 diabetes through NOX1. Enhanced perivascular oxidative stress and/or inflammation may be responsible for this effect, as they represent key steps in atherogenesis and were also blunted by deletion of Nox1. The finding that Nox4 knockout further augmented atherosclerotic burden supports the hypothesis that NOX1 and NOX4 have somewhat opposing roles within the vasculature. Clinically, this study provides a rationale for using ET receptor antagonists to slow down the progression of atherosclerosis in predisposed patients such as those with diabetes mellitus. Our results also suggest that ET receptor blockade may improve plaque stability in humans and therefore reduce the risk of major cardiovascular events such as myocardial infarction in diabetic subjects.
Data availability
The data underlying this article are available in the article and in its online supplemental material.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
We are grateful to Adriana Cristina Ene, Guillem Colell Dinarès, and Isabelle Miguel for excellent technical support.
Conflict of interest: none declared.
Funding
This work was supported by the Canadian Institutes of Health Research (CIHR) (123465) and CIHR First Pilot Foundation (143348), a Canada Research Chair (CRC) on Hypertension and Vascular Research by the CRC Government of Canada/CIHR Program, and by the Canada Fund for Innovation, all to E.L.S.; and by fellowships to N.I.-K. [Société Québécoise d’hypertension artérielle (SQHA) and ‘Fonds de recherché du Québec en Santé’], O.B. (SQHA), S.O. (CIHR Canada Graduate Scholarship-Master’s scholarship), and M.T. and S.C.C. (Science without Borders of the National Council for Scientific and Technological Development of Brazil).
Time for primary review: 37 days
References
- 1. Paneni F, Beckman JA, Creager MA, Cosentino F.. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I. Eur Heart J 2013;34:2436–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gao L, Mann GE.. Vascular NAD(P)H oxidase activation in diabetes: a double-edged sword in redox signalling. Cardiovasc Res 2009;82:9–20. [DOI] [PubMed] [Google Scholar]
- 3. Montezano AC, De Lucca Camargo L, Persson P, Rios FJ, Harvey AP, Anagnostopoulou A, Palacios R, Gandara ACP, Alves-Lopes R, Neves KB, Dulak-Lis M, Holterman CE, de Oliveira PL, Graham D, Kennedy C, Touyz RM.. NADPH oxidase 5 is a pro-contractile Nox isoform and a point of cross-talk for calcium and redox signaling-implications in vascular function. J Am Heart Assoc 2018;7:e009388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Youn JY, Gao L, Cai H.. The p47phox- and NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin-induced murine model of diabetes. Diabetologia 2012;55:2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JP, Touyz RM, Wingler K, Cooper ME, Schmidt HH, Jandeleit-Dahm KA.. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013;127:1888–1902. [DOI] [PubMed] [Google Scholar]
- 6. Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, Sugamura K, Fitzgibbons T, Keaney JF Jr.. Endothelial NADPH oxidase 4 protects ApoE-/- mice from atherosclerotic lesions. Free Radic Biol Med 2015;89:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, Deussen A, Koch E, Morawietz H.. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J 2016;37:1753–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schürmann C, Rezende F, Kruse C, Yasar Y, Löwe O, Fork C, van de Sluis B, Bremer R, Weissmann N, Shah AM, Jo H, Brandes RP, Schröder K.. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J 2015;36:3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, Calkin AC, Biessen EA, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA.. Reactive oxygen species can provide atheroprotection via NOX4-dependent inhibition of inflammation and vascular remodeling. Arterioscler Thromb Vasc Biol 2016;36:295–307. [DOI] [PubMed] [Google Scholar]
- 10. Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA.. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. JASN 2014;25:1237–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim J, Seo M, Kim SK, Bae YS.. Flagellin-induced NADPH oxidase 4 activation is involved in atherosclerosis. Sci Rep 2016;6:25437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T.. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988;332:411–415. [DOI] [PubMed] [Google Scholar]
- 13. Schneider JG, Tilly N, Hierl T, Sommer U, Hamann A, Dugi K, Leidig-Bruckner G, Kasperk C.. Elevated plasma endothelin-1 levels in diabetes mellitus. Am J Hypertens 2002;15:967–972. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi K, Ghatei MA, Lam H-C, O'Halloran DJ, Bloom SR.. Elevated plasma endothelin in patients with diabetes mellitus. Diabetologia 1990;33:306–310. [DOI] [PubMed] [Google Scholar]
- 15. Collier A, Leach JP, McLellan A, Jardine A, Morton JJ, Small M.. Plasma endothelinlike immunoreactivity levels in IDDM patients with microalbuminuria. Diabetes Care 1992;15:1038–1040. [DOI] [PubMed] [Google Scholar]
- 16. Lerman A, Edwards BS, Hallett JW, Heublein DM, Sandberg SM, Burnett JC Jr.. Circulating and tissue endothelin immunoreactivity in advanced atherosclerosis. N Engl J Med 1991;325:997–1001. [DOI] [PubMed] [Google Scholar]
- 17. Schiffrin EL. Vascular endothelin in hypertension. Vascul Pharmacol 2005;43:19–29. [DOI] [PubMed] [Google Scholar]
- 18. Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ, Morawietz H.. Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun 2000;269:713–717. [DOI] [PubMed] [Google Scholar]
- 19. Briones AM, Tabet F, Callera GE, Montezano AC, Yogi A, He Y, Quinn MT, Salaices M, Touyz RM.. Differential regulation of Nox1, Nox2 and Nox4 in vascular smooth muscle cells from WKY and SHR. J Am Soc Hypertens 2011;5:137–153. [DOI] [PubMed] [Google Scholar]
- 20. Amiri F, Virdis A, Neves MF, Iglarz M, Seidah NG, Touyz RM, Reudelhuber TL, Schiffrin EL.. Endothelium-restricted overexpression of human endothelin-1 causes vascular remodeling and endothelial dysfunction. Circulation 2004;110:2233–2240. [DOI] [PubMed] [Google Scholar]
- 21. Idris-Khodja N, Ouerd S, Mian MOR, Gornitsky J, Barhoumi T, Paradis P, Schiffrin EL.. Endothelin-1 overexpression exaggerates diabetes-induced endothelial dysfunction by altering oxidative stress. Am J Hypertens 2016;29:1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watson AM, Li J, Schumacher C, de Gasparo M, Feng B, Thomas MC, Allen TJ, Cooper ME, Jandeleit-Dahm KA.. The endothelin receptor antagonist avosentan ameliorates nephropathy and atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 2010;53:192–203. [DOI] [PubMed] [Google Scholar]
- 23. Li MW, Mian MO, Barhoumi T, Rehman A, Mann K, Paradis P, Schiffrin EL.. Endothelin-1 overexpression exacerbates atherosclerosis and induces aortic aneurysms in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 2013;33:2306–2315. [DOI] [PubMed] [Google Scholar]
- 24. Amiri F, Paradis P, Reudelhuber TL, Schiffrin EL.. Vascular inflammation in absence of blood pressure elevation in transgenic murine model overexpressing endothelin-1 in endothelial cells. J Hypertens 2008;26:1102–1109. [DOI] [PubMed] [Google Scholar]
- 25. Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, Krause KH.. Decreased blood pressure in NOX1-deficient mice. FEBS Lett 2006;580:497–504. [DOI] [PubMed] [Google Scholar]
- 26. Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH.. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol 2010;8:e1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kannel WB, McGee DL.. Diabetes and cardiovascular disease. The Framingham study. JAMA 1979;241:2035–2038. [DOI] [PubMed] [Google Scholar]
- 28. Laplante MA, Wu R, Moreau P, de Champlain J.. Endothelin mediates superoxide production in angiotensin II-induced hypertension in rats. Free Radic Biol Med 2005;38:589–596. [DOI] [PubMed] [Google Scholar]
- 29. Wedgwood S, McMullan DM, Bekker JM, Fineman JR, Black SM.. Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy. Circ Res 2001;89:357–364. [DOI] [PubMed] [Google Scholar]
- 30. Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, Chen AF.. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation 2003;107:1053–1058. [DOI] [PubMed] [Google Scholar]
- 31. Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS.. Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric-oxide synthase in the rat aorta. J Pharmacol Exp Ther 2005;315:1058–1064. [DOI] [PubMed] [Google Scholar]
- 32. Dammanahalli JK, Sun Z.. Endothelin (ET)-1 inhibits nicotinamide adenine dinucleotide phosphate oxidase activity in human abdominal aortic endothelial cells: a novel function of ETB1 receptors. Endocrinology 2008;149:4979–4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Montezano AC, Burger D, Paravicini TM, Chignalia AZ, Yusuf H, Almasri M, He Y, Callera GE, He G, Krause KH, Lambeth D, Quinn MT, Touyz RM.. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ Res 2010;106:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pandey D, Patel A, Patel V, Chen F, Qian J, Wang Y, Barman SA, Venema RC, Stepp DW, Rudic RD, Fulton DJ.. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol 2012;302:H1919–H1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ Jr.. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011;216:321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sobey CG, Judkins CP, Rivera J, Lewis CV, Diep H, Lee HW, Kemp-Harper BK, Broughton BR, Selemidis S, Gaspari TA, Samuel CS, Drummond GR.. NOX1 deficiency in apolipoprotein E-knockout mice is associated with elevated plasma lipids and enhanced atherosclerosis. Free Radic Res 2015;49:186–198. [DOI] [PubMed] [Google Scholar]
- 37. Schroder K, Zhang M, Benkhoff S, Mieth A, Pliquett R, Kosowski J, Kruse C, Luedike P, Michaelis UR, Weissmann N, Dimmeler S, Shah AM, Brandes RP.. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ Res 2012;110:1217–1225. [DOI] [PubMed] [Google Scholar]
- 38. Calara F, Silvestre M, Casanada F, Yuan N, Napoli C, Palinski W.. Spontaneous plaque rupture and secondary thrombosis in apolipoprotein E-deficient and LDL receptor-deficient mice. J Pathol 2001;195:257–263. [DOI] [PubMed] [Google Scholar]
- 39. Johnson J, Carson K, Williams H, Karanam S, Newby A, Angelini G, George S, Jackson C.. Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation 2005;111:1422–1430. [DOI] [PubMed] [Google Scholar]
- 40. Johnson JL, Jackson CL.. Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis 2001;154:399–406. [DOI] [PubMed] [Google Scholar]
- 41. Williams H, Johnson JL, Carson KG, Jackson CL.. Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 2002;22:788–792. [DOI] [PubMed] [Google Scholar]
- 42. Schwartz SM, Galis ZS, Rosenfeld ME, Falk E.. Plaque rupture in humans and mice. Arterioscler Thromb Vasc Biol 2007;27:705–713. [DOI] [PubMed] [Google Scholar]
- 43. Jackson CL, Bennett MR, Biessen EA, Johnson JL, Krams R.. Assessment of unstable atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2007;27:714–720. [DOI] [PubMed] [Google Scholar]
- 44. Shai SY, Sukhanov S, Higashi Y, Vaughn C, Kelly J, Delafontaine P.. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe-/- mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arterioscler Thromb Vasc Biol 2010;30:1916–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. van der Wal AC, Becker AE.. Atherosclerotic plaque rupture–pathologic basis of plaque stability and instability. Cardiovasc Res 1999;41:334–344. [DOI] [PubMed] [Google Scholar]
- 46. Ouweneel AB, Verwilligen RAF, Van Eck M.. Vulnerable plaque and vulnerable blood: two critical factors for spontaneous atherothrombosis in mouse models. Atherosclerosis 2019;284:160–164. [DOI] [PubMed] [Google Scholar]
- 47. Zeiher AM, Goebel H, SchäChinger V, Ihling C.. Tissue endothelin-1 immunoreactivity in the active coronary atherosclerotic plaque. A clue to the mechanism of increased vasoreactivity of the culprit lesion in unstable angina. Circulation 1995;91:941–947. [DOI] [PubMed] [Google Scholar]
- 48. Bauriedel G, Hutter R, Welsch U, Bach R, Sievert H, Luderitz B.. Role of smooth muscle cell death in advanced coronary primary lesions: implications for plaque instability. Cardiovasc Res 1999;41:480–488. [DOI] [PubMed] [Google Scholar]
- 49. Sorescu D, Weiss D, Lassègue B, Clempus RE, Szöcs K, Sorescu GP, Valppu L, Quinn MT, Lambeth JD, Vega JD, Taylor WR, Griendling KK, Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002;105:1429–1435. [DOI] [PubMed] [Google Scholar]
- 50. Azumi H, Inoue N, Ohashi Y, Terashima M, Mori T, Fujita H, Awano K, Kobayashi K, Maeda K, Hata K, Shinke T, Kobayashi S, Hirata K, Kawashima S, Itabe H, Hayashi Y, Imajoh-Ohmi S, Itoh H, Yokoyama M.. Superoxide generation in directional coronary atherectomy specimens of patients with angina pectoris: important role of NAD(P)H oxidase. Arterioscler Thromb Vasc Biol 2002;22:1838–1844. [DOI] [PubMed] [Google Scholar]
- 51. Ohman MK, Luo W, Wang H, Guo C, Abdallah W, Russo HM, Eitzman DT.. Perivascular visceral adipose tissue induces atherosclerosis in apolipoprotein E deficient mice. Atherosclerosis 2011;219:33–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ketonen J, Shi J, Martonen E, Mervaala E.. Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet-induced obese C57Bl/6 mice. Circ J 2010;74:1479–1487. [DOI] [PubMed] [Google Scholar]
- 53. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 54. Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol 2012;32:2045–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, Merval R, Proudfoot A, Tedgui A, Mallat Z.. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008;117:1649–1657. [DOI] [PubMed] [Google Scholar]
- 56. Zhou X, Nicoletti A, Elhage R, Hansson GK.. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation 2000;102:2919–2922. [DOI] [PubMed] [Google Scholar]
- 57. Griendling KK, Touyz RM, Zweier JL, Dikalov S, Chilian W, Chen YR, Harrison DG, Bhatnagar A; American Heart Association Council on Basic Cardiovascular Sciences. Measurement of reactive oxygen species, reactive nitrogen species, and redox-dependent signaling in the cardiovascular system: a scientific statement from the American Heart Association. Circ Res 2016;119:e39–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplemental material.