

Abstract
Aims
Hypertension is a major risk factor for cardiovascular diseases. However, vascular remodelling, a hallmark of hypertension, has not been systematically characterized yet. We described systematic vascular remodelling, especially the artery type- and cell type-specific changes, in hypertension using spontaneously hypertensive rats (SHRs).
Methods and results
Single-cell RNA sequencing was used to depict the cell atlas of mesenteric artery (MA) and aortic artery (AA) from SHRs. More than 20 000 cells were included in the analysis. The number of immune cells more than doubled in aortic aorta in SHRs compared to Wistar Kyoto controls, whereas an expansion of MA mesenchymal stromal cells (MSCs) was observed in SHRs. Comparison of corresponding artery types and cell types identified in integrated datasets unravels dysregulated genes specific for artery types and cell types. Intersection of dysregulated genes with curated gene sets including cytokines, growth factors, extracellular matrix (ECM), receptors, etc. revealed vascular remodelling events involving cell–cell interaction and ECM re-organization. Particularly, AA remodelling encompasses upregulated cytokine genes in smooth muscle cells, endothelial cells, and especially MSCs, whereas in MA, change of genes involving the contractile machinery and downregulation of ECM-related genes were more prominent. Macrophages and T cells within the aorta demonstrated significant dysregulation of cellular interaction with vascular cells.
Conclusion
Our findings provide the first cell landscape of resistant and conductive arteries in hypertensive animal models. Moreover, it also offers a systematic characterization of the dysregulated gene profiles with unbiased, artery type-specific and cell type-specific manners during hypertensive vascular remodelling.
Keywords: Single-cell RNA sequencing, Cell atlas, Cellular communication, Hypertension
Graphical Abstract

1. Introduction
Hypertension is a leading risk factor for a panel of cardiovascular diseases worldwide including ischaemic heart disease and stroke, contributing to more than 10 million deaths and more than 212 million disability life years in 2015 alone. 1 Current anti-hypertensive treatment strategies aim to limit the overactivated sympathetic nervous system and renin–angiotensin–aldosterone system in hypertension. However, limited efficacy was still seen in ∼10–30% hypertensive patients who are not responsive even to combined use of current anti-hypertensive medications, thus necessitating further mechanistic studies of hypertension. 2 Along with the heart and kidney, blood vessels, which encompass both conductive and resistant arteries, are among the key organs that operate in concert to determine blood pressure. However, approaches to define vascular remodelling during hypertension at the transcriptomic level rely mainly on flow-sorted cells or in vitro cultured cells and subsequent microarray analysis. Limitations lie in the biased selection of markers for flow sorting or the changes incurred to cells by in vitro cell culture. As a result, although endothelial dysfunction and smooth muscle cell (SMC)-centred vascular changes in hypertension have been described, 3 there has been a lack of systematic depiction of the remodelling process. With single-cell RNA sequencing (scRNA-seq), unbiased identification of cell types and systematic transcriptomic analysis are made possible.
Additionally, mesenchyme and immune cells within the vasculature have not been sufficiently characterized in hypertension. As to adventitial mesenchyme cells, they are gradually recognized as important in atherosclerosis and pulmonary hypertension, whereas their role in vascular remodelling in primary hypertension has been elusive. 4,5 For immune cells, as in atherosclerosis, a wealth of evidence supports the involvement of circulating immune cells in hypertension. 6 However, the phenotype of immune cells residing in the artery and their functional interaction with other vascular cell types under hypertension remain significant yet unexamined issues. Adding to the complexity of hypertensive vascular remodelling are the heterogeneity of major vascular cell types and their dynamic cellular communication status, which are only possible to be studied with recent use of scRNA-seq. 7,8
ScRNA-seq offers an unparalleled opportunity to systematically resolve the cellular diversity and dynamic cellular communication changes. Here, we performed scRNA-seq of mesenteric artery (MA) and aortic artery (AA), which represent resistant artery and conductive artery, respectively, from spontaneously hypertensive rats (SHRs) and their healthy controls Wistar Kyoto (WKY) rats to decipher the artery-specific and vascular cell type-specific changes in hypertension, discover potentially important cell sub-clusters, and unravel the altered cell–cell interactions.
2. Methods
2.1 Ethics statement
All animal procedures conformed to Guide for Care and Use of Laboratory Animals published by US National Institute of Health (8th edition, 2011) and were approved by the Institutional Animal Care and Treatment Committee of Southwest Medical University, China.
2.2 scRNA-seq of artery cells with 10× chromium
We performed scRNA-seq of MA and AA cells from WKY/N and SHR/N rats (male, 16–18 weeks old, 7–8 rats in each group). Only male rats were included to avoid the sex influence on experiment results. Rats were anaesthetized with intraperitoneal injection of 50 mg/kg sodium pentobarbital. The adequacy of anaesthesia throughout the procedure was confirmed by the lack of reflex response to toe stretch. Direct blood pressure was measured and recorded with a heparinized fluid-filled catheter that was inserted into the isolated femoral artery. While still being under 50 mg/kg intraperitoneal sodium pentobarbital anaesthesia, the rats were sacrificed by exsanguination via transcardial perfusion with 50 mL phosphate buffered saline (PBS) and subsequent heart excision. All branches of superior and inferior MAs (excluding the superior and inferior MAs themselves) and AA (thoracic artery and abdominal artery; aortic arch is not included) were collected and perivascular tissues were carefully cleaned. Pooled tissue from each group and artery type, respectively, was obtained. As described before, 8 the arteries were digested with 2 mg/mL collagenase I (Invitrogen; 17018-029) and 2 mg/mL dispase II (Sigma; D4693) in Hank’s balanced salt solution (with calcium and magnesium) at 37°C for 30 min. Single nucleated live cells were sorted into PBS with 0.04% bovine serum albumin and then loaded to 10× Genomics Chromium chip. Standard 10× Chromium Single Cell 3′ v2 (10× Genomics) protocols were followed for scRNA-seq for reverse transcription and library preparation. For details, see the Supplementary material online.
2.3 Quality control and pre-clustering of scRNA-seq data
The library was demultiplexed and aligned to the rat genome with CellRanger (version 3.0.2; 10× Genomics) pipeline. Separate datasets were merged with cellranger aggr pipeline (default parameters) that equalizes sequencing depth between samples. R package Seurat (version 3.0.2) was used for cell filtration, data normalization, dataset integration, dimension reduction, and cell clustering and cluster visualization with default parameters unless otherwise specified. 9 Bells expressing <500 or >3500 genes were filtered out to exclude non-cell or cell aggregates and cells with >15% mitochondrial gene percentage were also filtered out to exclude cells at a compromised state. For details, see the Supplementary material online.
2.4 Cell type annotation and sub-clustering
According to classic major vascular cell type marker expression, the clusters were assigned to major vascular cell types and immune cells. For sub-clustering, gene count of cells from the same major cell type were retrieved and integrated using 30 dimensions. Cell type-specific and sub-cluster-specific markers were found with FindAllMarkers function in Seurat with arguments avg_logFC >0.41, min.pct >0.1 and only.pos = TRUE. P-value <0.01 determined by Wilcoxon rank-sum tests was used to further filter for significantly enriched genes. Avg_logFC at cut-off value 0.41 was chosen to gate for genes with an average fold change of 1.5-fold. Differentially expressed genes between indicated cell types or sub-clusters were found with FindMarkers function in Seurat with similar conditions as FindAllMarkers function. For details, see the Supplementary material online.
2.5 Curation of gene lists
Cytokine and chemokine lists are manually curated on the basis of UniProt protein knowledgebase 10 keywords searches. Genes related to hypertension-associated single nucleotide polymorphism (SNP) sites, rat transcription factors (TFs), surface proteins, and extracellular matrix (ECM) are from published studies. 11–15 The Genenames resources were used for curation of ligand and ion channels lists. 16 Ligand–receptor interaction pair lists were generated according to published study. 17 Genes causal for blood pressure change in SMC-specific knock-out mice and genes contributing to monogenic hypertension are curated according to publications. 3,18,19 The curated lists were included in the Supplementary material online, Table S1.
2.6 Ligand–receptor cellular communication analysis
Ligand–receptor pair analysis was performed as described 8 using lists from published data. 17 Briefly, using normalized data, the interaction strength of a specific ligand–receptor pair between ligand cell type and receptor cells was the sum of multiplied expression level of ligand gene and receptor gene between each cell in the ligand cell type and each cell in the receptor cell type divided by cell number in each cell type to adjust for the influence of cell number. Changes of ligand–receptor communication strength were represented by interaction strength-weigted fold changes between SHR cell type and corresponding WKY control. R package pheatmap was used for visualization.
2.7 Immunofluorescent staining
MA or AA was snap frozen in liquid nitrogen and stored in −80°C. Sections were fixed with acetone and then blocked for 1 h with 10% donkey serum in PBS with 0.1% Tween-20. For en face immunofluorescent staining, aorta adventitia peeled off the aorta or whole MAs were processed as previously published (for details, see the Supplementary material online). 8
2.8 Statistical analysis
Blood pressure and plasma level of glucose and lipid from WKY rat and SHR were represented as mean ± standard error of mean. P-value <0.05 determined by unpaired Student’s t-test was considered statistically significant.
3. Results
3.1 Landscape of major cell types in hypertensive MA and AA
ScRNA-seq has been used to characterize the vascular landscape in atherosclerosis, but a single-cell level description of vascular changes in hypertension is lacking. To bridge the knowledge gap, we performed single-cell transcriptomic profiling of 16–18 weeks old SHR-MA and SHR-AA with their counterparts in WKY rats as controls (Figure 1A). SHR is the most commonly used rat model for human essential hypertension which has a genetic background and develops elevated blood pressure spontaneously with a normal diet. 20 Blood pressure was directly measured with a heparinized fluid-filled catheter which was inserted into the isolated femoral artery. High blood pressure was detected in 16–18 weeks old SHRs confirming the establishment of hypertension model and no significant dysregulation of glucose or lipid levels was observed (Supplementary material online, Figure S1A). Histological difference of MA and AA properties between SHR and WKY rats was not observed (Supplementary material online, Figure S1B). After sequencing of flow-sorted single nucleated live cells (Supplementary material online, Figure S1C), datasets from different groups were aggregated to control for comparable sequencing depth. After quality control, 6631, 5652, 5138, and 4462 cells with similar median number of genes were retained in WKY-MA, SHR-MA, WKY-AA, and SHR-AA datasets, respectively (Supplementary material online, Figure S1D and E). The unbiased clusters were annotated to major vascular cell types according to known marker genes: SMCs (Myh11 and Cnn1; clusters 1, 2, 4, 6, and 7), endothelial cells (ECs; Cdh5 and Vwf; clusters 8 and 13), mesenchymal stromal cells (MSCs; Gpx3, and Dcn; clusters 3, 5, 10, and 11), immune cells (Ptprc; clusters 9, 12, and 15), and neurons (Plp1) ( Figure 1B and Supplementary material online, Figure S2A and B). Similar distribution of gene numbers was observed in each major cell type (Supplementary material online, Figure S1F). Regardless of rat strain, MA displayed a higher percentage of SMCs compared with AA, whereas the percentages of MSCs and immune cells in AA were higher than those in MA ( Figure 1C). SHR-MA MSC percentage demonstrated a more than two-fold increase in comparison with that of WKY-MA MSC and a similar increase was observed in the AA immune cell percentage in SHRs ( Figure 1D).
Figure 1.

Transcriptomic atlas of WKY and SHR MA and AAs. (A) Experimental design and analysis pipeline. (B) Integrated and split view of the scRNA-seq datasets from WKY and SHR AA and MA. Cells were from pooled MA or AA samples of eight WKY rats or seven SHRs. Cell number: WKY-MA, n = 6631 cells from 8 WKY rats; SHR-MA, n = 5652 cells from 7 SHRs; WKY-AA, n = 5138 cells from 8 WKY rats; SHR-AA, n = 4462 cells from 7 SHRs. (C) Percentage of major cell types in each dataset. (D) Percentage shift of major cell types in AA and MA in SHRs compared with WKY rats. AA, aortic artery; EC, endothelial cell; MA, mesenteric artery; MSC, mesenchymal stromal cell; SHR, spontaneously hypertensive rats; SMC, smooth muscle cell; WKY, Wistar Kyoto.
3.2 Cell type-specific and artery-specific genes of rat arteries
Knowledge about SMCs, ECs, and MSCs from MA is lacking since separation of each vascular cell type is difficult due to the small MA diameter and a lack of established surface markers for SMCs and MSCs. The overlap of EC and MSC markers with immune cells ( Figure 2A) suggests their potential expression of inflammation-related genes. Then, we intersected the cell type-specific genes with receptors and ligands, through which the vascular cells might act on and respond to each other (Supplementary material online, Figure S3A). For cytokines, it is notable that MSCs and immune cells both demonstrated expression of Ccl2 and Cxcl1 ( Figure 2B). Cd36, a marker found important in insulin resistance of hypertensive rats 21 is expressed in both ECs and immune cells (Supplementary material online, Figure S3A). Different vascular cell types showed distinct expression of superoxide dismutases, with Sod3 and Sod2 highly expressed in MSCs and immune cells, respectively ( Figure 2B). For ion channels, overlapped expression between multiple cell types was not frequent, with only P2rx4 and Kcnn4 shared between ECs and immune cells ( Figure 2B).
Figure 2.

Cell type- and artery type-specific genes of WKY rat AA and MA. (A) Venn diagram of cell type-specific genes (average fold change > 1.5, P-value < 0.01 determined by Wilcoxon rank-sum tests). Yellow highlighted Venn diagram parts g, i, j, and n demonstrate the overlap of EC, MSC, and immune cell type-specific genes. (B) Dot plot of genes in indicated Venn diagram parts of (A) in selected gene classifications (cytokines, ROS-related enzymes, and ion channels). (C) Venn diagram of cell type-specific genes that belong to ECM classification. Highlighted Venn diagram part c showed the number of ECM genes exclusively expressed in MSCs. (D) Average expression heatmap of top five genes in Venn diagram parts e–o of (C). (E) Average expression heatmap of top three SNP genes in Venn diagram parts a–d and all SNP genes from Venn diagram part e–o from (A). (F and G) Venn diagram of genes expressed higher in WKY-MA SMCs, ECs, and MSCs in comparison with WKY-AA analogue (F) or genes expressed higher genes in WKY-AA SMCs, ECs, and MSCs in comparison with WKY-MA analogue (G). Cells were from pooled MA or AA samples of eight WKY rats. Cell number: MA SMC, n = 6012 cells; AA SMC, n = 2362 cells; MA EC, n = 302 cells; AA EC, n = 142 cells; MA MSC, n = 308 cells; AA MSC, n = 2291 cells. Dot plot of selected genes from the central Venn diagram part which were expressed higher in all three cell types. AA, aortic artery; EC, endothelial cell; ECM, extracellular matrix; MA, mesenteric artery; MSC, mesenchymal stromal cell; SMC, smooth muscle cell; SNP, single nucleotide polymorphism.
Not surprisingly, MSCs were the major source for ECM ( Figure 2C, Venn diagram part c), especially for collagens and proteoglycans (Supplementary material online, Figure S3B). ECs and MSCs displayed the largest overlap of ECM genes including Fn1 and Fbln2 ( Figure 2C, Venn diagram part g and Figure 2D). Noticeably, Mmp2, a major contributor of vascular remodelling under hypertension 22 and its inhibitor Timp2 were both expressed mainly in MSCs (Supplementary material online, Figure S3B). The number of genes involved in essential hypertension-related SNP was similar across cell types (Supplementary material online, Figure S3C). Various SNP genes were cell type-specific: SMCs (Tagln and Myh11), ECs (Aqp1 and Tie1), MSCs (Ogn and Col15a1), and immune cells (Zfat and Rarres1) ( Figure 2E). Interestingly, the expression of hypertension-related SNP Nfkbia, an inhibitor of Nfkb, is shared between ECs, MSCs, and immune cells ( Figure 2E). A significant proportion of the SMC and EC cell type-specific genes (46.1% and 39.4%, respectively) were differentially expressed between MA and AA, and this number (23.11%) is much lower in MSCs (Supplementary material online, Figure S4). More than 10 TFs including Klf and Fosb were found higher in all MA non-immune cells suggesting that these TFs might contribute to the difference between MA and AA ( Figure 2F). Less genes were found higher in AA across non-immune cell types, among them were ion channel gene (Aqp1) and ECMs (Ctgf and Lum) ( Figure 2G).
3.3 Artery-specific changes of SMCs in hypertension
Hypertensive vascular remodelling is dependent on the vessel type. This is demonstrated by the uniquely dysregulated genes of MA and AA SMCs under hypertension ( Figure 3A). More genes are downregulated in SHRs compared to WKY control ( Figure 3A), among which a significant proportion (50.0%) were expressed higher in WKY-MA than WKY-AA (Supplementary material online, Figure S5A). In AA SMCs, more than 50% of the upregulated genes in SHR-AA SMCs were expressed lower in WKY-AA than WKY-MA and more than 50% of the downregulated genes in SHR-AA SMCs were expressed higher in WKY-AA than WKY-MA (Supplementary material online, Figure S5A). This suggests a potential shrinking of difference between AA and MA SMCs under hypertension.
Figure 3.

Artery-specific transcriptomic changes and sub-cluster alterations in SMCs during hypertension. (A) Venn diagram of dysregulated genes (average fold change > 1.5, P-value < 0.01) in MA and AA SMCs from SHRs with WKY counterparts as controls. SMC cell number: WKY-MA, n = 6012 cells from 8 WKY rats; SHR-MA, n = 4528 cells from 7 SHRs; WKY-AA, n = 2362 cells from 8 WKY rats; SHR-AA, n = 1255 cells from 7 SHRs. Character before the underscore indicates corresponding Venn diagram part. (B) Volcano plot of differentially expressed genes (average log fold change > 0.41, average fold change > 1.5, P value < 0.01 determined by Wilcoxon rank-sum tests) in SHR-MA SMCs in comparison with WKY counterparts. SMC cell number: WKY-MA, n = 6012 cells from 8 WKY rats; SHR-MA, n = 4528 cells from 7 SHRs. (C) Feature plot showing the expression of PCP4 in MA of WKY rat and SHR. (D) Immunostaining of PCP4 and ACTA2 in MA of WKY rat and SHR. White scale bar, 50 μm; and yellow scale bar, 10 μm. (E) Average expression heatmap of selected dysregulated genes from corresponding Venn diagram parts of (A). (F) Dot plot of selected ECM genes from corresponding Venn diagram parts of (A). (G) TSNE plot showing the distribution of sub-clusters in all SMCs and in each dataset. (H) Percentage of each sub-cluster in each dataset. (I) Sub-cluster percentage fold difference in WKY rat MA and AA. (J) Violin plot showing the expression of indicated markers in each sub-cluster. (K) EC sub-cluster percentage fold difference in SHR MA and AA in comparison with WKY rat counterpart. (L and M) Violin plot showing the expression of markers in indicated sub-cluster and GOMF analysis of sub-cluster-specific genes. AA, aortic artery; GOMF, gene ontology molecular function; MA, mesenteric artery; SHR, spontaneously hypertensive rats; SMC, smooth muscle cell; tSNE, t-distributed stochastic neighbour embedding; WKY, Wistar Kyoto.
Resistant arteries and conductive arteries undergo different vascular remodelling, to which vascular SMCs contribute through modulation of its contractile function, secretory function, ECM dysregulation, etc. 3 In hypertension, a hypercontractile state of SMCs was reported. 23 Consistently, Pcp4, which modulates the binding of calcium to calmodulin, 24 was among the top 10 upregulated genes in SHR-MA SMCs compared to WKY-MA ( Figure 3B and C and Supplementary material online, Figure S5B). The upregulation was confirmed at the protein level ( Figure 3D). Pcp4 upregulation was not observed in SHR-AA SMCs (Supplementary material online, Figure S5C–E). Additionally, Sphk1, which was important in angiotensin II-induced intracellular calcium elevation and subsequent smooth muscle contraction, was also upregulated in SHR-MA only (Figure 3E). The exclusive upregulation of Pcp4 and Sphk1 in SHR-MA SMCs but not SHR-AA SMCs suggests a potential hypercontractile state of SHR-MA SMCs. However, in both in MA and AA, downregulation of multiple genes involved in SMC contraction including Itpr1, Flna, and Mylk was observed (Figure 3E). These genes were found causal of hypertension in SMC-specific gene knout-out models. 3 Concomitantly, SMC markers Myh11 and Srf were also downregulated in both MA and AA SMCs under hypertension, accompanied by upregulation of TF Klf4 which promotes SMC phenotypic switch towards secretory/migratory phenotype ( Figure 3F). Despite the upregulation of Klf4 in both MA and AA SMCs under hypertension, SHR-AA SMCs exclusively secrete more proinflammatory cytokines (Cxcl1, Il6, and Ccl2), vascular calcification-promoting factor Bmp2, and fibrotic Tgfb3 ( Figure 3E).
For ECM remodelling, SHR-AA SMCs exhibited decrease of genes that encode proteins which constitute elastin fibres (Fbln5 and Fbn1) and increase of collagen (Col1a1) ( Figure 3F). However, in SHR-MA SMCs, although downregulation of Eln (encoding tropoelastin) that also constitutes elastin fibres was observed, multiple collagens (Col4a1, Col4a5, Col18a1, and Col12a1) demonstrated significant downregulation ( Figure 3F and Supplementary material online, Figure S5F). Enzymes which promote degradation of ECMs decreased in both MA and AA (MA: Cstb, Adamts1, and Adamts15; AA: Serpine1) ( Figure 3F). Less production (except Col1a1 in SHR-AA SMCs) and less degradation of ECM implicated a decreased ECM turn-over rate in SHRs. Full list of dysregulated ECM genes was presented as well as genes from classifications including receptors, TFs, and SNPs (Supplementary material online, Figure S5G).
Currently, there are limited studies about SMC heterogeneity. ScRNA-seq offers an opportunity to examine this issue. After dataset integration, 9 SMC sub-clusters were found, among which the percentage of sub-cluster 7 is significantly higher in MA and sub-clusters 3 and 8 were mainly found in AA (Figure 3G–I). Marker genes for sub-cluster 7 include Hmgcs2, which is a ketogenic enzyme involving in regulation of glucose level, and Bcr, which could be activated by angiotensin II, 25 whereas sub-clusters 3 and 8 expressed ECM genes Fbln5 and Lum, respectively ( Figure 3J and Supplementary material online, Figure S6A). Notably, multiple genes that inhibit (Mgp26 ) or promote (Sparc27 and Bgn28 ) vascular calcification were expressed higher in sub-clusters 3 and 8 (Supplementary material online, Figure S6B). During hypertension, the percentage of sub-cluster 6 doubled in MA ( Figure 3K). Markers for sub-cluster 6 include heat shock proteins Hspb1 and Hspa1b which were involved in hypertension ( Figure 3L). 29,30 Consistently, gene ontology term heat shock protein binding was enriched in sub-cluster 6 ( Figure 3L). For AA SMCs, sub-clusters 2 and 5 cells were enriched in SHRs ( Figure 3K). Marker genes for sub-cluster 5 include proinflammatory genes Nfkbia, Nfkb2, and Il6, which resulted in enriched gene ontology term cytokine activity in sub-cluster 5 ( Figure 3M).
3.4 Artery-specific changes in ECs under hypertension
Although endothelial dysfunction has been an established mechanism for hypertension, artery type-dependent changes in ECs were not studied. In AA ECs, significantly more genes were upregulated than downregulated, whereas a reverse trend was observed in MA ECs (Supplementary material online, Figure S7A). A total of 46.8% of the downregulated genes in SHR-MA EC and 57.3% of the upregulated genes in SHR-AA EC in comparison with WKY counterparts were among the genes expressed higher in MA ECs than AA ECs (Supplementary material online, Figure S7B and C), suggesting a decreased difference between MA and AA ECs in SHRs.
Hypertensive vascular changes encompass a complex network including inflammation, ECM, reactive oxygen species (ROS), and vasoregulation. 31 To examine these aspects, we assigned the dysregulated gene classifications including cytokines, ECM, enzymes, and receptors (Supplementary material online, Figure S7D). For inflammation-related genes, SHR-AA ECs displayed upregulation of proinflammatory Cxcl1, Il6, Cxcl2, Icam1, and Bmp4, which were increased in angiotensin II-induced hypertension model, whereas downregulation of proinflammatory Cxcl1, Ccl7, Cxcl10, and Vcam1 was observed in SHR-MA ECs in comparison with WKY control ( Figure 4A). Notably, Cxcl1 and Il6 were shown above to be increased in SHR-AA SMCs ( Figure 3E). For ECM genes, Sulf1, which desulphates heparan sulphate proteoglycans, and multiple glycoproteins (Vwf, Fn1, Igfbp4, and Lamb4) were decreased in SHR-MA ECs ( Figure 4B). SHR-AA ECs demonstrated downregulation of metalloproteinase inhibitor Timp4, and upregulation of Adamts4, which could be inhibited by Timp4. Col3a1 and Fgl2 were upregulated in SHR-AA ECs ( Figure 4B). Overall, SHR-MA ECs demonstrated decreased production of ECM, whereas the decreased degradation and increased production of ECM were implicated in SHR-AA ECs.
Figure 4.

Artery-specific transcriptomic changes and sub-cluster alterations in ECs of SHRs. (A and B) Dot plot showing the expression of selected significantly differentially expressed genes (P < 0.01 determined by Wilcoxon rank-sum tests) in WKY or SHR ECs from MA or AA. EC cell number: WKY-MA, n = 302 cells from 8 WKY rats; SHR-MA, n = 475 cells from 7 SHRs; WKY-AA, n = 142 cells from 8 WKY rats; SHR-AA, n = 173 cells from 7 SHRs. (C and D) Violin plot showing the expression of selected differentially expressed (P < 0.01 determined by Wilcoxon rank-sum tests) genes in WKY or SHR ECs from MA or AA. EC cell number: WKY-MA, n = 302 cells from 8 WKY rats; SHR-MA, n = 475 cells from 7 SHRs; WKY-AA, n = 142 cells from 8 WKY rats; SHR-AA, n = 173 cells from 7 SHRs. (E) TSNE plot showing the distribution of sub-clusters in all ECs and in each dataset. (F) Percentage of each sub-cluster in each dataset. (G) EC sub-cluster percentage fold difference in SHRs with WKY rats as control in MA and AA. (H) Percentage of cells at different stages of cell cycle in each sub-cluster. (I) Heatmap showing EC sub-cluster identity and the top 10 EC sub-cluster genes (by average fold change). (J) Dot plot showing indicated genes upregulated in SHRs in EC sub-clusters 1–3 of MA and AA compared to WKY controls. (K) Volcano plot of differentially expressed genes (average log fold change > 0.41, average fold change > 1.5, P-value < 0.01) in AA EC4 sub-cluster (lymphatic ECs) from SHRs compared to WKY rats. EC sub-cluster 4 cell number: WKY-AA, n = 11 cells; SHR-AA, n = 13 cells. (L) Immunostaining of LYVE1 and SOX18 in WKY and SHR-AA adventitia. Scale bar, 50 μm. AA, aortic artery; EC, endothelial cell; MA, mesenteric artery; SHR, spontaneously hypertensive rats; SMC, smooth muscle cell; WKY, Wistar Kyoto.
For ROS-related genes, protective responses that might result in decreased ROS were observed in both MA and AA ECs. Lox, which could generate ROS, 32 was downregulated in SHR-MA ECs, and Nfe2l2, encoding anti-oxidant TF NRF2, and its downstream gene Hmox1 (encoding haem oxygenase-1) were upregulated in SHR-AA ECs ( Figure 4C). For vasoregulation, reduced Ptgs2 (encoding COX2) in SHR-MA ECs may lead to reversed endothelial-dependent vasoconstriction 33 ( Figure 4D) in comparison with WKY-MA ECs. For SHR-AA ECs, Ednrb (encoding endothelin receptor type B), which mediates vasodilation when expressed on ECs, 34 was increased. However, Adrb2 (encoding β2-adrenergic receptor), the gene delivery of which mitigated high blood pressure in SHRs, 35 was downregulated in SHR-AA ECs ( Figure 4D). Thus, in SHR-MA ECs, mainly protective changes with potentially less vasoconstriction were shown. For SHR-AA ECs, mixed responses that may lead to vasodilation (Ednrb increase) and vasoconstriction (Adrb2 decrease) were observed. Notably, Cd36, which was revealed by genetics analysis to be related with hypertension, 21 was upregulated in both SHR-MA and SHR-AA ECs compared to WKY control (Supplementary material online, Figure S7E).
ECs were a heterogeneous population. 36 Consistently, six EC sub-clusters were discovered ( Figure 4E). Compared to WKY-MA ECs, significantly more WKY-AA ECs were in sub-cluster 6 ( Figure 4F), which showed high expression of lipid processing genes Lpl and Cd36 ( Figure 4I and Supplementary material online, Figure S8A). This sub-cluster was also observed in AA in other studies. 37 Under hypertension, cell number of sub-cluster 3 expanded in both SHR-MA and SHR-AA ECs ( Figure 4G). A higher percentage of S-phase cells was in EC sub-cluster 3 compared to other sub-clusters ( Figure 4H). Interestingly, endothelial progenitor marker Procr38 was enriched in EC sub-cluster 3, whereas progenitor markers Cd34 and Mki67 were not enriched in this sub-cluster (Supplementary material online, Figure S8A and B). Notably, marker genes for EC sub-clusters 1–3 displayed some overlap ( Figure 4I and Supplementary material online, Figures S8A and S9A). Enriched gene ontology term for EC sub-clusters 1–3 also showed overlap, and sub-cluster 3 exhibited exclusively enriched term ‘the role of GTSE1 in G2/M progression after G2 checkpoint’ (Supplementary material online, Figure S9B). During hypertension, multiple genes including Eno1 and Cd36 were upregulated in both SHR-MA and SHR-AA in EC sub-clusters 1–3 ( Figure 4J). For EC sub-cluster 4, marker genes (Lyve1 and Pdpn) and enriched gene ontology term ‘lymph vessel development’ support its identity of lymphatic ECs ( Figure 4I and Supplementary material online, Figure S10A). Its presence in MA and AA was confirmed by immunostaining (Supplementary material online, Figure S10B). In lymphatic ECs, Sox18, a TF crucial in lymphatics development, 39 was significantly upregulated in SHR-AA compared to WKY control ( Figure 4K and L).
3.5 Artery-specific changes in MSCs under hypertension
As suggested above, MSCs are an important source of inflammatory factors and ECM. In SHRs, MSC genes were dysregulated depending on the artery type ( Figure 5A). Cxcl1, Il6, Ccl2, Ccl7, and Cxcl10 were exclusively upregulated in SHR-AA MSCs in comparison with WKY counterparts ( Figure 5B and Supplementary material online, Figure S11A and B). The former four inflammatory cytokines were demonstrated above upregulated in SHR-AA SMCs. Notably, anti-inflammatory glycoprotein Tnfaip640 was also upregulated only in SHR-AA MSCs ( Figure 5B). Increase of osteo-inductive Bmp2 in SHR-AA MSCs and osteogenic marker Alpl in SHR-MA MSCs suggested enhanced vascular calcification in both artery types in hypertension ( Figure 5C and Supplementary material online, Figure S11A). For ECM, more genes were dysregulated in SHR-MA MSCs than SHR-AA MSCs (Supplementary material online, Figure S11B). Decrease of serine protease inhibitors (Serpine1 and Serpine2) in both SHR-MA and SHR-AA MSCs suggest less ECM degradation. More production of Col8a1, which is related to hypertension-associated SNP, was also observed in both SHR-MA and SHR-AA MSCs (Supplementary material online, Figure S11C). Like in SHR-AA SMCs, Col1a1 is upregulated in SHR-AA MSCs but downregulated in SHR-MA MSCs (Supplementary material online, Figure S11C). The ECM remodelling in SHR-AA MSCs may a result of increased Tgfb1 ( Figure 5D). In SHR-MA MSCs, however, both genes that promote (Ctgf and receptor Eng) and inhibit (Cilp41 and TF Nr4a142 ) transforming growth factor beta (TGFb) signalling were upregulated ( Figure 5D).
Figure 5.

Artery-specific transcriptomic changes and sub-cluster alterations in MSCs of SHRs. (A) Venn diagram of dysregulated genes (average fold change > 1.5, P-value < 0.01 determined by Wilcoxon rank-sum tests) in MA and AA MSCs in SHRs compared to WKY counterparts. MSC cell number: WKY-MA, n = 308 cells from 8 WKY rats; SHR-MA, n = 617 cells from 7 SHRs; WKY-AA, n = 2290 cells from 8 WKY rats; SHR-AA, n = 2312 cells from 7 SHRs. (B) Dot plot of inflammation-related genes that are significantly changed in SHR-AA MSCs in comparison with WKY-AA counterparts. Character before the gene symbol indicates Venn diagram part from (A). (C) Violin plot of Bmp2 and Alpl in indicated cells. MSC cell number: WKY-MA, n = 308 cells from 8 WKY rats; SHR-MA, n = 617 cells from 7 SHRs; WKY-AA, n = 2290 cells from 8 WKY rats; SHR-AA, n = 2312 cells from 7 SHRs. (D) Dot plot of fibrosis-related genes that are significantly changed (average fold change > 1.5, P-value < 0.01 determined by Wilcoxon rank-sum tests) in SHR-AA MSCs in comparison with WKY-AA counterparts. Character before the gene symbol indicates Venn diagram part from (A). (E) TSNE plot showing the distribution of sub-clusters in all MSCs and in each dataset. (F) Percentage of each sub-cluster in indicated dataset. (G) Sub-cluster percentage fold difference in WKY-MA and WKY-AA. (H) Sub-cluster percentage fold difference in SHR-MA and SHR-AA with WKY counterparts as control. (I) Dot plot showing expression of the top five genes (by average fold change) in each sub-cluster. Triangles indicate genes that are mentioned in the main text. AA, aortic artery; MA, mesenteric artery; MSC, mesenchymal stromal cell; SHR, spontaneously hypertensive rats; TGFb, transforming growth factor beta; WKY, Wistar Kyoto.
Artery MSCs have long established to be a heterogeneous population. Sub-clustering analysis of MSCs revealed 10 sub-clusters, of which sub-cluster 8 expressing apolipoproteins Apod and Apoe was found significantly enriched in MA and sub-clusters 5 and 9 expressing genes (Cytl143 and Sfrp4, 44 respectively) important for bone homeostasis were enriched in AA ( Figure 5E–G and I). Under hypertension, sub-cluster 1 expressing Alpl, a vascular calcification marker, displayed increased cell number in both SHR-MA and SHR-AA in comparison with WKY analogues ( Figure 5H and I). Importantly, Col8a1, which was increased in both SHR-MA and SHR-AA MSCs compared to WKY controls, was specific for sub-cluster 1 ( Figure 5I and Supplementary material online, Figure S12A). However, sub-cluster 2 that expressed multiple proinflammatory genes (Ccl2, Cxcl1, and Esm1) 8,45 demonstrated a reduced cell number percentage in SHR-MA but increased percentage in SHR-AA MSCs ( Figure 5H and I and Supplementary material online, Figure S12A). Trend of increased cell number in SHR-MA and reduced cell number in SHR-AA was observed for MSC sub-clusters 3–5 ( Figure 5H). Chad, a cartilage matrix protein, is a marker for sub-cluster 3 ( Figure 5I). Sub-cluster 4 expressed vessel dilator peptide Adm ( Figure 5I). In AA, the cell number of sub-cluster 6 expressing Abca8a, which is important for cholesterol efflux and Lrp1, which is crucial for lipid homeostasis, increased most significantly under hypertension ( Figure 5H and I). Acta2, which is a myofibroblast marker, was seen in sub-cluster 7 ( Figure 5I). Sub-cluster 10 expressing cardioprotective Gdf1546 displayed a decreased percentage of cell number in SHR-MA compared to WKY control ( Figure 5H and I). Gene ontology terms regulation of cell growth (sub-cluster 1), response of tumour necrosis factor (sub-cluster 2), ossification (sub-cluster 3), and actin-filament organization (sub-cluster 7) were seen in different sub-clusters, suggestive of their unique functions (Supplementary material online, Figure S12B).
3.6 Similar gene dysregulation across non-immune vascular cell types
After examining cell type-specific changes, commonly dysregulated genes across SMCs, ECs, and MSCs were investigated to help shed light on general vascular remodelling events that are not specific for one cell type. Eno1, a glycolytic enzyme that involves metabolic reprogramming in pulmonary hypertension, 47 and LOC306079, with unknown function, were upregulated across SMCs, ECs, and MSCs in both SHR-MA and SHR-AA (Supplementary material online, Figure S13). Moreover, chemokines Cxcl1 and Il6, were upregulated across the non-immune vascular cells in SHR-AA compared to WKY analogue (Supplementary material online, Figure S13).
3.7 Immune cell diversity in hypertensive artery
Accumulating evidence supports the participation of immune cells in hypertension. 6 However, immune cell landscape in hypertensive artery has not been described yet. ScRNA-seq revealed eight immune sub-clusters in AA, among which the four macrophage sub-clusters expressing Cd14 accounted for more than 70% (Figure 6A–C and Supplementary material online, Figure S14A). The four macrophage sub-clusters were annotated Pf4+ M2 macrophages (Pf4 and Cd163), inflammatory macrophages (Cd74 and Il1b), Fcna+ resident macrophages (F13a1 and Fcna), and Fn1+ M2 macrophages (Fn1 and Mmp12), respectively ( Figure 6C and Supplementary material online, Figure S14A). Sub-cluster 5 expressed Cd7 and major histocompatibility complex Class II antigen RT1-Bb, but lacks the expression of classical dendritic cell (DC) marker Itgam (Cd11b), and thus defined as DC-like ( Figure 6C and Supplementary material online, Figure S14B). Staining of CD7 confirmed the presence of DC-like cells in AA adventitia (Supplementary material online, Figure S14C and D). Sub-cluster 6 might be a mixture of immune cells, as demonstrated by its high expression of both Fcgr3a, a marker for myeloid lineages and Mal, a marker for T cells ( Figure 6C). Sub-cluster 7, expressing T cell-specific TF Gata3 and surface marker Cd3e, was a T cell cluster and demonstrated the highest enrichment of cell number in SHR-AA compared to WKY control ( Figure 6C–E). Cytotoxic enzyme Gzmk was also highly expressed in sub-cluster 7 T cells (Supplementary material online, Figure S14B and C). Immunostaining of CD3D and GZMK confirmed the presence of T cells in the AA (Figure 6F and Supplementary material online, Figure S14D). Expression of Mcemp1 and S100a9 defined sub-cluster 8 as monocytes ( Figure 6C and Supplementary material online, Figure S14C), the presence of which in AA was confirmed by MCEMP1 immunostaining (Supplementary material online, Figure S14D).
Figure 6.

Immune cell heterogeneity and responses in hypertension. (A) TSNE plot showing the distribution of sub-clusters in all immune cells and in each dataset. (B) Percentage of each immune sub-cluster in each dataset. (C) Heatmap of top 10 significantly changed genes (by average log fold change) in each immune cell sub-cluster. (D) Sub-cluster percentage fold difference in WKY mesenteric artery and AA. (E) Feature plot showing the expression of Gata3 and Cd3d in AA from WKY rat and SHR. (F) Immunostaining of CD3D in AA intima and adventitia. Scale bar, 10 μm. (G) Volcano plot of differentially expressed genes (average log fold change > 0.41, average fold change > 1.5, P-value < 0.01) in macrophages (immune sub-clusters 1–4) in WKY-AA and SHR-AA datasets. Total cell number of macrophages: WKY-AA, n = 243 cells from 8 WKY rats; SHR-AA, n = 525 cells from 7 SHRs. (H) Feature plot showing the expression of Cd14 and Lilrb3a in WKY-AA and SHR-AA. (I) Immunostaining of LILRB3 and CD14 in WKY and SHR aortic adventitia. Scale bar, 10 μm. AA, aortic artery; DC-like, dendritic cell-like; Mφ, macrophages; SHR, spontaneously hypertensive rats; WKY, Wistar Kyoto.
Apart from cell number expansion of sub-cluster 7 T cells in SHR-AA compared to WKY control, enrichment of sub-cluster 2 Il1b+ inflammatory macrophages was also observed, supporting the involvement of macrophages and T cells in hypertension ( Figure 6D). Interestingly, the accumulation of T cells was mainly observed in the aortic endothelium but not aortic adventitia ( Figure 6F). In addition to the cell number expansion, gene dysregulation was observed as well in hypertension. Lilrb3, which is an inhibitory leucocyte immunoglobulin-like receptor, was upregulated across all macrophage sub-clusters ( Figure 6G and H). This downregulation was confirmed by immunostaining ( Figure 6I).
3.8 Cellular communication changes in hypertension
Upregulation of numerous proinflammatory genes and growth factors were observed in SHR cells compared to WKY control. Interaction of Eng with multiple growth factors from TGF signalling pathway (Tgfb1, Tgfb3, and Bmp2) was enhanced in both SHR-AA and SHR-MA cells in comparison with WKY controls ( Figure 7A). However, upregulated interaction of EC-secreted Hbegf with SMC and MSC Cd9 and Cd44 was only seen in SHR-AA cells ( Figure 7A). Additionally, cell surface integrins could interact with ECM and provide a connection of cells with ECM environment. MSCs, as the major source of ECM, demonstrated enhanced interaction of Cyr61, Ctgf, and Col8a1 with integrins in both SHR-AA and SHR-MA cells compared to WKY cells. However, enhanced interaction of Col1a1 and Col3a1 with Itgb1 was only seen in SHR-AA cells (Supplementary material online, Figure S15).
Figure 7.
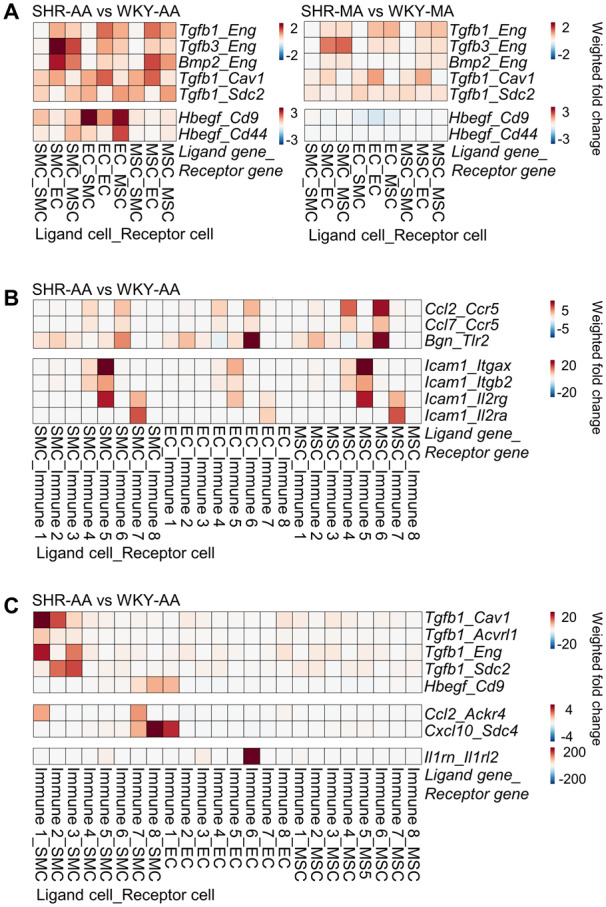
Changes of cell–cell interactions in hypertension. (A) Changes of cell–cell interaction pairs involving growth factors among non-immune vascular cells of SHR-AA and SHR-MA with WKY counterparts as control. (B) Changes of cell–cell interaction pairs involving proinflammatory molecules with non-immune cells as broadcasting cells in SHR-AA with WKY counterparts as control. (C) Changes of cell–cell interaction pairs involving growth factors and proinflammatory molecules with immune cells as broadcasting cells in SHR-AA with WKY counterparts as control. Cells before the underscore contribute to the interaction pair as ligand cells, and cells after are receptor cells. AA, aortic artery; EC, endothelial cell; MA, mesenteric artery; MSC, mesenchymal stromal cell; SHR, spontaneously hypertensive rats; SMC, smooth muscle cell; TGFb, transforming growth factor beta; WKY, Wistar Kyoto.
Apart from growth factor broadcasting, proinflammatory molecules secreted by non-immune vascular cells interact with different immune sub-clusters. Enhanced interaction of Ccl2 and Ccl7 with Ccr5 expressed by immune sub-cluster 4 (Fn1+ M2 macrophages) and immune sub-cluster 6 (undefined) was observed ( Figure 7B). For proinflammatory Icam1, upregulated interaction with immune sub-cluster 5 (DC-like cells; Icam1_Itgax, Icam1_Itgb2, Icam1_Il2rg) and immune sub-cluster 7 (T cells; Icam1_Il2rg, Icam1_Il2ra) was observed ( Figure 7B).
Immune cells also secrete growth factors and inflammatory cytokines to interact with non-immune vascular cells. In SHR-AA cells, enhanced interaction of Tgfb1 secreted by macrophages immune sub-clusters 1–3 (Pf4+ M2 macrophages, inflammatory macrophages, and Fcna+ resident macrophages) through different receptors on SMC was observed in SHR-AA dataset compared to WKY control ( Figure 7C). Ccl2_Ackr4 interaction was strengthened between immune sub-cluster 1 (Pf4+ M2 macrophages) and immune sub-cluster 7 (T cells) with SMCs. Notably, significantly upregulated interaction of Il1rn_Il1rl2 between immune sub-cluster 6 (undefined) and EC was seen, and the significance necessitates further study ( Figure 7C).
4. Discussion
Aided by scRNA-seq of 21 883 cells across WKY and SHR MA and AA, cell type- and artery type-specific vascular remodelling at the transcriptomic level in hypertensive rats was systematically characterized. We provided characteristic changes of four major vascular cell types (SMCs, ECs, MSCs, and immune cells) in hypertension and presented heterogeneous sub-clusters of these major cell types, with some potentially involved in hypertension. Furthermore, dynamic cellular communication under hypertension was described.
Conductive or resistant arteries undergo outward or inward remodelling under hypertension. Previous efforts mainly focused on the SMCs when examining the mechanism responsible for differential response of artery types. 3 Additionally, ECs used in experiments for hypertension studies were normally derived from the aorta due to its ease of isolation. Role of MSCs and immune cells were insufficiently recognized as well. Enrichment of MSCs in MA and immune cells in AA underscores their potential involvement in hypertension.
Adding another layer of complexity to hypertensive vascular remodelling, heterogeneous vascular cell types have demonstrated sub-cluster cell number expansion or decrease. Particularly, Procr+ ECs (sub-cluster 3) have demonstrated an increased cell number in both MA and AA. This is contradictory to previous finding of reduced endothelial progenitors in human essential hypertension. 48 However, controversy still looms in the markers for endothelial progenitors and further studies are in need to confirm this observation. Moreover, in immune sub-clusters, an increased number of Il1b+ inflammatory macrophages and T cells were demonstrated, which necessitate further studies to pinpoint their role in hypertensive vascular remodelling.
Close examination of computationally identified vascular cell types reveals dysregulated genes highly specific for artery types and cell types. Intersection of the dysregulated genes with curated gene sets revealed the upregulation of cytokines in AA and downregulation of ECM genes in the MA. The curation of gene set according to published studies offers implications to aspects that are established to be crucial for the molecular level identification of vascular remodelling events, although it to some extent narrowed our interpretation to these categories. However, all datasets are made public with R shiny interactive web app, which enables interrogation of potentially interesting genes. Interestingly, multiple cytokines were demonstrated to be exclusively upregulated in non-immune AA cells—SMCs (Cxcl1, Il6, and Ccl2), ECs (Cxcl1, Il6, and Cxcl2), MSCs (Cxcl1, Il6, Ccl2, and Ccl7)—which suggested that in addition to the hallmark enrichment of immune cells in hypertensive AA, inflammatory cytokines also may contribute to the remodelling process.
Surprisingly, although vascular non-immune cells are highly specialized in the functions, global responses across different cell types were uncovered. For example, Eno1, which was involved in glycolysis, was upregulated in all SMCs, ECs, and MSCs in both MA and AA. This suggested a stress response mechanism through metabolic reprogramming from oxidative phosphorylation towards glycolysis. Interference of the metabolic reprogramming 47 might reverse the global response and thus reduce blood pressure. Moreover, upregulation of Cxcl1 and Il6 was shared by aortic SMCs, ECs, and MSCs, although its basal level was highest in MSCs. Targeting the global responses of vascular remodelling suggest potentially enhanced effects.
Through the dysregulated growth factors and cytokines (Ccl2, Ccl7, etc.), vascular non-immune cells and immune cells communicate with each other in a paracrine or autocrine manner. Consistent with traditional theory, growth factors from TGFb superfamily participate in hypertensive vascular remodelling. Apart from enhanced interaction of multiple TGFb superfamily growth factors secreted by SMC and MSC with non-immune vascular cells, it is interesting to note that AA macrophages are also an important source of TGFb superfamily growth factors and may have an effect on SMCs. Similarly, Ccl2 could be secreted by MSCs to interact with immune sub-cluster 4 (Fn1+ macrophages) through Ccr5, and it could also be secreted by immune sub-cluster 7 (T cells) to interact with SMCs through Ackr4. During hypertension, paired ligand–receptor interactions were dysregulated and different sources of the same dysregulated ligand might hold different significance in hypertensive vascular remodelling. To conclude, our study revealed several potential hypertension-specific ligand–receptor interactions.
Limitations of the study lie in the following aspects. First, we mainly presented descriptive results in the study. As the main purpose of the study was to serve as a resource for the characterization of cell type- and artery type-specific changes in SHRs, only comparison of SHR vascular cells and WKY counterparts was included in the study. Building on the present results, future work might include functional studies of potentially interesting molecules or sub-clusters revealed by the vascular landscape in hypertension. Moreover, to facilitate query or reference of our dataset, all data were submitted to an interactive web app that does not require computing background of scientists. Second, SHRs serve as an animal model for primary hypertension in human in which lifestyle and genetic background interplay plays a pivotal role. Although extensive studies have focused on characterizing the underlying genetic background of SHRs, it still remains a hurdle to accurately distinguish if the transcriptomic differences observed in our study is caused by the genetic background or high blood pressure. In our study, we also could not distinguish the causal changes from the downstream effects. However, curated gene classifications including SNP and TF which could serve as drivers for transcriptomic changes of downstream genes were examined and may shed light in these aspects. Third, although immune cell phenotypic diversity and their cellular communication with vascular cell types were inspected in our study, the relatively small number of immune cells limits the effort to examine their heterogeneity in detail. For example, sub-clustering analysis of T cells, which were established to have a pivotal role in the development of hypertension, was not possible. Sequencing of flow-sorted cells targeting only interesting populations discovered in our study might be a future solution. However, our study is one essential building block towards this direction. Finally, only male rats were included in our study, which might be different from the female. To figure out the difference in gene expression profiling in different sexes, future experiments would be needed.
To sum up, vascular remodelling is a vital part in hypertension. Systematic characterization of the cell type-specific and artery type unique transcriptomic changes during hypertensive vascular remodelling is described in our study, and the data are deposited into an interactive web app for easy interrogation. These findings support a hypothesis of environment–cell interaction for the development of hypertension. During elevation of blood pressure, resident vascular cells produce chemokines/cytokines in response to mechanical stimuli, which attracts immune cells to the vessel wall.
Data availability
The scRNA-seq datasets for WKY and SHR AA and MA are available on Gene Expression Omnibus (GSE149777). Interactive web app hosting the datasets for easier queries of gene expression or comparison between indicated conditions was built with R shiny (https://github.com/WenduoGu/young_SHR_rat_artery).
Conflict of interest: none declared.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the National Natural Science Foundation of China [31972909 and 31830039], a Joint-Fellowship from the Luzhou-Southwest Medical University [2018-8], International Cooperative Initiatives from Science and Technology Department [Sichuan Science and Technology Program-2019YFH0174], Sichuan Province, China, and the British Heart Foundation, UK [RG/14/6/31144].
Translational perspective
Hypertension is a major risk factor for cardiovascular diseases. However, as a hallmark of hypertension, vascular remodelling, especially the artery type- and cell type-specific changes, has not been systematically characterized yet. In the present study, we provide the first cell landscape of resistant and conductive arteries in hypertensive animal models, in which an increase in Procr+ endothelial cells and immune cells in the vessel wall was significant. These findings may be pivotal in developing therapeutics for hypertension that could target not only single molecules but also paired cellular interactions. Thus, the findings could be interesting for both scientists and clinicians.
Supplementary Material
References
- 1. Forouzanfar MH, Liu P, Roth GA, Ng M, Biryukov S, Marczak L, Alexander L, Estep K, Hassen Abate K, Akinyemiju TF, Ali R, Alvis-Guzman N, Azzopardi P, Banerjee A, Barnighausen T, Basu A, Bekele T, Bennett DA, Biadgilign S, Catala-Lopez F, Feigin VL, Fernandes JC, Fischer F, Gebru AA, Gona P, Gupta R, Hankey GJ, Jonas JB, Judd SE, Khang YH, Khosravi A, Kim YJ, Kimokoti RW, Kokubo Y, Kolte D, Lopez A, Lotufo PA, Malekzadeh R, Melaku YA, Mensah GA, Misganaw A, Mokdad AH, Moran AE, Nawaz H, Neal B, Ngalesoni FN, Ohkubo T, Pourmalek F, Rafay A, Rai RK, Rojas-Rueda D, Sampson UK, Santos IS, Sawhney M, Schutte AE, Sepanlou SG, Shifa GT, Shiue I, Tedla BA, Thrift AG, Tonelli M, Truelsen T, Tsilimparis N, Ukwaja KN, Uthman OA, Vasankari T, Venketasubramanian N, Vlassov VV, Vos T, Westerman R, Yan LL, Yano Y, Yonemoto N, Zaki ME, Murray CJ.. Global burden of hypertension and systolic blood pressure of at least 110 to 115 mm Hg, 1990-2015. JAMA 2017;317:165–182. [DOI] [PubMed] [Google Scholar]
- 2. Cai A, Calhoun DA.. Resistant hypertension: an update of experimental and clinical findings. Hypertension 2017;70:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown IAM, Diederich L, Good ME, DeLalio LJ, Murphy SA, Cortese-Krott MM, Hall JL, Le TH, Isakson BE.. Vascular smooth muscle remodeling in conductive and resistance arteries in hypertension. Arterioscler Thromb Vasc Biol 2018;38:1969–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu Y, Zhang Z, Torsney E, Afzal AR, Davison F, Metzler B, Xu Q.. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J Clin Invest 2004;113:1258–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li M, Riddle S, Zhang H, D’Alessandro A, Flockton A, Serkova NJ, Hansen KC, Moldovan R, McKeon BA, Frid M, Kumar S, Li H, Liu H, Caanovas A, Medrano JF, Thomas MG, Iloska D, Plecita-Hlavata L, Jezek P, Pullamsetti S, Fini MA, El Kasmi KC, Zhang Q, Stenmark KR.. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor C-terminal binding protein-1. Circulation 2016;134:1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Drummond GR, Vinh A, Guzik TJ, Sobey CG.. Immune mechanisms of hypertension. Nat Rev Immunol 2019;19:517–532. [DOI] [PubMed] [Google Scholar]
- 7. Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, Hamers AAJ, Cochain C, Vafadarnejad E, Saliba AE, Zernecke A, Pramod AB, Ghosh AK, Anto Michel N, Hoppe N, Hilgendorf I, Zirlik A, Hedrick CC, Ley K, Wolf D.. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res 2018;122:1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gu W, Ni Z, Tan YQ, Deng J, Zhang SJ, Lv ZC, Wang XJ, Chen T, Zhang Z, Hu Y, Jing ZC, Xu Q.. Adventitial cell atlas of wt (wild type) and ApoE (apolipoprotein E)-deficient mice defined by single-cell RNA sequencing. Arterioscler Thromb Vasc Biol 2019;39:1055–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R.. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, Ng FL, Evangelou M, Witkowska K, Tzanis E, Hellwege JN, Giri A, Velez Edwards DR, Sun YV, Cho K, Gaziano JM, Wilson PWF, Tsao PS, Kovesdy CP, Esko T, Magi R, Milani L, Almgren P, Boutin T, Debette S, Ding J, Giulianini F, Holliday EG, Jackson AU, Li-Gao R, Lin WY, Luan J, Mangino M, Oldmeadow C, Prins BP, Qian Y, Sargurupremraj M, Shah N, Surendran P, Theriault S, Verweij N, Willems SM, Zhao JH, Amouyel P, Connell J, de Mutsert R, Doney ASF, Farrall M, Menni C, Morris AD, Noordam R, Pare G, Poulter NR, Shields DC, Stanton A, Thom S, Abecasis G, Amin N, Arking DE, Ayers KL, Barbieri CM, Batini C, Bis JC, Blake T, Bochud M, Boehnke M, Boerwinkle E, Boomsma DI, Bottinger EP, Braund PS, Brumat M, Campbell A, Campbell H, Chakravarti A, Chambers JC, Chauhan G, Ciullo M, Cocca M, Collins F, Cordell HJ, Davies G, de Borst MH, de Geus EJ, Deary IJ, Deelen J, Del Greco MF, Demirkale CY, Dorr M, Ehret GB, Elosua R, Enroth S, Erzurumluoglu AM, Ferreira T, Franberg M, Franco OH, Gandin I, Gasparini P, Giedraitis V, Gieger C, Girotto G, Goel A, Gow AJ, Gudnason V, Guo X, Gyllensten U, Hamsten A, Harris TB, Harris SE, Hartman CA, Havulinna AS, Hicks AA, Hofer E, Hofman A, Hottenga JJ, Huffman JE, Hwang SJ, Ingelsson E, James A, Jansen R, Jarvelin MR, Joehanes R, Johansson A, Johnson AD, Joshi PK, Jousilahti P, Jukema JW, Jula A, Kahonen M, Kathiresan S, Keavney BD, Khaw KT, Knekt P, Knight J, Kolcic I, Kooner JS, Koskinen S, Kristiansson K, Kutalik Z, Laan M, Larson M, Launer LJ, Lehne B, Lehtimaki T, Liewald DCM, Lin L, Lind L, Lindgren CM, Liu Y, Loos R, Lopez LM, Lu Y, Lyytikainen LP, Mahajan A, Mamasoula C, Marrugat J, Marten J, Milaneschi Y, Morgan A, Morris AP, Morrison AC, Munson PJ, Nalls MA, Nandakumar P, Nelson CP, Niiranen T, Nolte IM, Nutile T, Oldehinkel AJ, Oostra BA, O’Reilly PF, Org E, Padmanabhan S, Palmas W, Palotie A, Pattie A, Penninx B, Perola M, Peters A, Polasek O, Pramstaller PP, Nguyen QT, Raitakari OT, Ren M, Rettig R, Rice K, Ridker PM, Ried JS, Riese H, Ripatti S, Robino A, Rose LM, Rotter JI, Rudan I, Ruggiero D, Saba Y, Sala CF, Salomaa V, Samani NJ, Sarin AP, Schmidt R, Schmidt H, Shrine N, Siscovick D, Smith AV, Snieder H, Sober S, Sorice R, Starr JM, Stott DJ, Strachan DP, Strawbridge RJ, Sundstrom J, Swertz MA, Taylor KD, Teumer A, Tobin MD, Tomaszewski M, Toniolo D, Traglia M, Trompet S, Tuomilehto J, Tzourio C, Uitterlinden AG, Vaez A, van der Most PJ, van Duijn CM, Vergnaud AC, Verwoert GC, Vitart V, Volker U, Vollenweider P, Vuckovic D, Watkins H, Wild SH, Willemsen G, Wilson JF, Wright AF, Yao J, Zemunik T, Zhang W, Attia JR, Butterworth AS, Chasman DI, Conen D, Cucca F, Danesh J, Hayward C, Howson JMM, Laakso M, Lakatta EG, Langenberg C, Melander O, Mook-Kanamori DO, Palmer CNA, Risch L, Scott RA, Scott RJ, Sever P, Spector TD, van der Harst P, Wareham NJ, Zeggini E, Levy D, Munroe PB, Newton-Cheh C, Brown MJ, Metspalu A, Hung AM, O’Donnell CJ, Edwards TL, Psaty BM, Tzoulaki I, Barnes MR, Wain LV, Elliott P, Caulfield MJ; Million Veteran Program. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet 2018;50:1412–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu H, Miao YR, Jia LH, Yu QY, Zhang Q, Guo AY.. AnimalTFDB 3.0: a comprehensive resource for annotation and prediction of animal transcription factors. Nucleic Acids Res 2019;47:D33–D38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bausch-Fluck D, Goldmann U, Müller S, van Oostrum M, Müller M, Schubert OT, Wollscheid B.. The in silico human surfaceome. Proc Natl Acad Sci USA 2018;115:E10988–E10997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Naba A, Clauser KR, Hoersch S, Liu H, Carr SA, Hynes RO.. The matrisome: in silico definition and in vivo characterization by proteomics of normal and tumor extracellular matrices. Mol Cell Proteomics 2012;11:M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, Gray AJG, Bruce L, Alexander SPH, Anderton S, Bryant C, Davenport AP, Doerig C, Fabbro D, Levi-Schaffer F, Spedding M, Davies JA; NC-IUPHAR. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2018;46:D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA.. Genenames.org: the HGNC and VGNC resources in 2017. Nucleic Acids Res 2017;45:D619–D625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Lizio M, Satagopam VP, Itoh M, Kawaji H, Carninci P, Rost B, Forrest A.. A draft network of ligand–receptor-mediated multicellular signalling in human. Nat Commun 2015;6:7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Padmanabhan S, Joe B.. Towards precision medicine for hypertension: a review of genomic, epigenomic, and microbiomic effects on blood pressure in experimental rat models and humans. Physiol Rev 2017;97:1469–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Munroe PB, Barnes MR, Caulfield MJ.. Advances in blood pressure genomics. Circ Res 2013;112:1365–1379. [DOI] [PubMed] [Google Scholar]
- 20. Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C, Coffman TM; American Heart Association Council on Hypertension and Council on Clinical Cardiology. Animal models of hypertension: a scientific statement from the American Heart Association. Hypertension 2019;73:e87–e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pravenec M, Landa V, Zidek V, Musilova A, Kren V, Kazdova L, Aitman TJ, Glazier AM, Ibrahimi A, Abumrad NA, Qi N, Wang J-M, St. Lezin EM, Kurtz TW.. Transgenic rescue of defective Cd36 ameliorates insulin resistance in spontaneously hypertensive rats. Nat Genet 2001;27:156–158. [DOI] [PubMed] [Google Scholar]
- 22. Wang M, Kim SH, Monticone RE, Lakatta EG.. Matrix metalloproteinases promote arterial remodeling in aging, hypertension, and atherosclerosis. Hypertension 2015;65:698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Touyz RM, Alves-Lopes R, Rios FJ, Camargo LL, Anagnostopoulou A, Arner A, Montezano AC.. Vascular smooth muscle contraction in hypertension. Cardiovasc Res 2018;114:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang X, Putkey JA.. PEP-19 modulates calcium binding to calmodulin by electrostatic steering. Nat Commun 2016;7:13583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexis JD, Wang N, Che W, Lerner-Marmarosh N, Sahni A, Korshunov VA, Zou Y, Ding B, Yan C, Berk BC, Abe J.. Bcr kinase activation by angiotensin II inhibits peroxisome-proliferator-activated receptor gamma transcriptional activity in vascular smooth muscle cells. Circ Res 2009;104:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yao Y, Bennett BJ, Wang X, Rosenfeld ME, Giachelli C, Lusis AJ, Bostrom KI.. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ Res 2010;107:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ciceri P, Elli F, Cappelletti L, Tosi D, Savi F, Bulfamante G, Cozzolino M.. Osteonectin (SPARC) expression in vascular calcification: in vitro and ex vivo studies. Calcif Tissue Int 2016;99:472–480. [DOI] [PubMed] [Google Scholar]
- 28. Song R, Fullerton DA, Ao L, Zheng D, Zhao KS, Meng X.. BMP-2 and TGF-beta1 mediate biglycan-induced pro-osteogenic reprogramming in aortic valve interstitial cells. J Mol Med 2015;93:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pulakazhi Venu VK, Saifeddine M, Mihara K, El-Daly M, Belke D, Dean JLE, O’Brien ER, Hirota SA, Hollenberg MD.. Heat shock protein-27 and sex-selective regulation of muscarinic and proteinase-activated receptor 2-mediated vasodilatation: differential sensitivity to endothelial NOS inhibition. Br J Pharmacol 2018;175:2063–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodriguez-Iturbe B, Lanaspa MA, Johnson RJ.. The role of autoimmune reactivity induced by heat shock protein 70 in the pathogenesis of essential hypertension. Br J Pharmacol 2019;176:1829–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Oparil S, Acelajado MC, Bakris GL, Berlowitz DR, Cifkova R, Dominiczak AF, Grassi G, Jordan J, Poulter NR, Rodgers A, Whelton PK.. Hypertension. Nat Rev Dis Primers 2018;4:18014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martínez-Revelles S, García-Redondo AB, Avendaño MS, Varona S, Palao T, Orriols M, Roque FR, Fortuño A, Touyz RM, Martínez-González J, Salaices M, Rodríguez C, Briones AM.. Lysyl oxidase induces vascular oxidative stress and contributes to arterial stiffness and abnormal elastin structure in hypertension: role of p38MAPK. Antioxid Redox Signal 2017;27:379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Widlansky ME, Price DT, Gokce N, Eberhardt RT, Duffy SJ, Holbrook M, Maxwell C, Palmisano J, Keaney JF Jr, Morrow JD, Vita JA.. Short- and long-term COX-2 inhibition reverses endothelial dysfunction in patients with hypertension. Hypertension 2003;42:310–315. [DOI] [PubMed] [Google Scholar]
- 34. Kawanabe Y, Nauli SM.. Endothelin. Cell Mol Life Sci 2011;68:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Iaccarino G, Cipolletta E, Fiorillo A, Annecchiarico M, Ciccarelli M, Cimini V, Koch WJ, Trimarco B.. Beta(2)-adrenergic receptor gene delivery to the endothelium corrects impaired adrenergic vasorelaxation in hypertension. Circulation 2002;106:349–355. [DOI] [PubMed] [Google Scholar]
- 36. Lukowski SW, Patel J, Andersen SB, Sim SL, Wong HY, Tay J, Winkler I, Powell JE, Khosrotehrani K.. Single-cell transcriptional profiling of aortic endothelium identifies a hierarchy from endovascular progenitors to differentiated cells. Cell Rep 2019;27:2748–2758.e2743. [DOI] [PubMed] [Google Scholar]
- 37. Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM.. Single-cell analysis of the normal mouse aorta reveals functionally distinct endothelial cell populations. Circulation 2019;140:147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu QC, Song W, Wang D, Zeng YA.. Identification of blood vascular endothelial stem cells by the expression of protein C receptor. Cell Res 2016;26:1079–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. François M, Caprini A, Hosking B, Orsenigo F, Wilhelm D, Browne C, Paavonen K, Karnezis T, Shayan R, Downes M, Davidson T, Tutt D, Cheah KSE, Stacker SA, Muscat GEO, Achen MG, Dejana E, Koopman P.. Sox18 induces development of the lymphatic vasculature in mice. Nature 2008;456:643–647. [DOI] [PubMed] [Google Scholar]
- 40. Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, Delafontaine P, Prockop DJ.. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009;5:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang CL, Zhao Q, Liang H, Qiao X, Wang JY, Wu D, Wu LL, Li L.. Cartilage intermediate layer protein-1 alleviates pressure overload-induced cardiac fibrosis via interfering TGF-beta1 signaling. J Mol Cell Cardiol 2018;116:135–144. [DOI] [PubMed] [Google Scholar]
- 42. Palumbo-Zerr K, Zerr P, Distler A, Fliehr J, Mancuso R, Huang J, Mielenz D, Tomcik M, Furnrohr BG, Scholtysek C, Dees C, Beyer C, Kronke G, Metzger D, Distler O, Schett G, Distler JH.. Orphan nuclear receptor NR4A1 regulates transforming growth factor-beta signaling and fibrosis. Nat Med 2015;21:150–158. [DOI] [PubMed] [Google Scholar]
- 43. Shin Y, Won Y, Yang J-I, Chun J-S.. CYTL1 regulates bone homeostasis in mice by modulating osteogenesis of mesenchymal stem cells and osteoclastogenesis of bone marrow-derived macrophages. Cell Death Dis 2019;10:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Haraguchi R, Kitazawa R, Mori K, Tachibana R, Kiyonari H, Imai Y, Abe T, Kitazawa S.. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci Rep 2016;6:25198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Balta S, Mikhailidis DP, Demirkol S, Ozturk C, Kurtoglu E, Demir M, Celik T, Turker T, Iyisoy A.. Endocan—a novel inflammatory indicator in newly diagnosed patients with hypertension: a pilot study. Angiology 2014;65:773–777. [DOI] [PubMed] [Google Scholar]
- 46. Ago T, Sadoshima J.. GDF15, a cardioprotective TGF-beta superfamily protein. Circ Res 2006;98:294–297. [DOI] [PubMed] [Google Scholar]
- 47. Dai J, Zhou Q, Chen J, Rexius-Hall ML, Rehman J, Zhou G.. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat Commun 2018;9:3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oliveras A, Soler MJ, Martínez-Estrada OM, Vázquez S, Marco-Feliu D, Vila JS, Vilaró S, Lloveras J.. Endothelial progenitor cells are reduced in refractory hypertension. J Hum Hypertens 2008;22:183–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq datasets for WKY and SHR AA and MA are available on Gene Expression Omnibus (GSE149777). Interactive web app hosting the datasets for easier queries of gene expression or comparison between indicated conditions was built with R shiny (https://github.com/WenduoGu/young_SHR_rat_artery).
Conflict of interest: none declared.