

Abstract
A large population in the world has been infected by COVID-19. Understanding the mechanisms of Severe Acute Respiratory Syndrome CoronaVirus 2 (SARS-CoV-2) is important for the management and treatment of COVID-19. When it comes to the infection process, one of the most important proteins in SARS-CoV-2 is the spike (S) protein, which is able to bind to human Angiotensin-Converting Enzyme 2 (ACE2) and initializes the entry of the host cell. In this study, we implemented multiscale computational approaches to study the electrostatic features of the interfaces of the SARS-CoV-2 S protein receptor binding domain and ACE2. The simulations and analyses were performed on high-performance computing resources in the Texas Advanced Computing Center. Our study identified key residues on SARS-CoV-2, which can be used as targets for future drug design. The results shed light on future drug design and therapeutic targets for COVID-19.
The number of confirmed cases of Coronavirus Disease 2019 (COVID-19) is increasing dramatically1 due to the fast spread of SARS-CoV-2. The large coronavirus family includes hundreds of viruses that usually do not pose a threat to human health. SARS-CoV-2 is the seventh member of those coronaviruses that infect the human body. Of these, four (HCoV-229E, HCoV-OC43, HCoV-NL63, HKU1)2 cause mild to moderate symptoms, while the other three can cause serious, even fatal diseases. SARS coronavirus (SARS-CoV) broke out in 2002 and caused Severe Acute Respiratory Syndrome (SARS). MERS coronavirus (MERS-CoV) found in 2012 caused Middle East Respiratory Syndrome (MERS), which continues to cause sporadic and localized outbreaks. The animal-to-human and human-to-human transmissions of these two fatal viruses caused the outbreak of the 2003 SARS pandemic and the following 2012 MERS pandemic, which resulted in over 8000 and 2400 reported cases, and killed 774 and 858 people, respectively. SARS-CoV-2 is the third known death-leading coronavirus that appeared in 2019, has caused a global pandemic, and compared to SARS and MERS, COVID-19 spreads much faster and infects a larger population globally. Many studies have been made on SARS-CoV-2 to develop COVID-19 diagnostics, therapeutics, and vaccines. Meanwhile, a great number of fundamental studies have also been focused on revealing the mechanisms of SARS-CoV-2 and other coronaviruses to deeply understand them and assist the treatment and drug design for COVID-19.
Coronaviridae is a family of enveloped, positive-sense, single-stranded RNA viruses that infects a wide range of hosts, including amphibians, birds, and mammals.3 The spherical viruses are typically decorated with spike (S) proteins, making the whole virus the shape of solar corona. The structure of a coronavirus mainly contains several types of proteins, including: the nucleocapsid (N) proteins which are inside the envelope of the coronavirus, the membrane (M) proteins and the envelope (E) proteins which form the virus envelope, and the spike (S) proteins which are trimmers on the envelope of the virus. The spike (S) proteins initialize an infection by binding to the human Angiotensin-Converting Enzyme 2 (ACE2). Then, SARS-CoV-2 enters the host cell and reproduces more viruses, which are released later to infect more cells (see Figure 1). Inhibiting the interactions between S proteins and ACE2 can block the infections, which is a direction for therapeutic drug design. Therefore, many of studies have been focused on the interactions of coronaviruses’ S proteins and human ACE2.
Figure 1.

Process of SARS-CoV-2 infecting host cells. (A) SARS-CoV-2 S protein binds to ACE2 of host cell, and enters the host cell after binding. (B) SARS-CoV-2 utilizes the host cell as a factory to reproduce more SARS-CoV-2 and release them to infect more cells.
Computational approaches are successfully used to study protein–protein interactions.4–6 Such approaches can also be used to study the protein–protein interactions of viruses. Computations of viruses are challenging due to the complexity of the viruses. Two categories of computational approaches have been implemented to study the viruses: atomic simulations, which study a whole virus or part of a virus at the atomic level, and coarse-grained simulations, which treat a virus at the amino acid level. Some research efforts are focused on determining the structures of viruses, others reveal fundamental mechanisms for viruses, such as viral capsid assembly7 and viral capsid electrostatic features.8 Due to the fast growth of supercomputer performance and rapid developments of computational algorithms, computational approaches play significant roles in virus studies.
This article studies the fundamental mechanisms of SARS-CoV-2 S protein binding to ACE2.
This article studies the fundamental mechanisms of SARS-CoV-2 S protein binding to ACE2. Based on the structure of the SARS-CoV-2 S protein and ACE2 (see Figure 2), several computational approaches were utilized to perform calculations and simulations. These approaches can be widely utilized to study more viruses. The electrostatic features were analyzed based on the distribution of the charged residues. The hydrogen bonds and salt bridges are studied after molecular dynamic (MD) simulations. Key residues, which significantly contribute to binding energy, are also identified in this work. Previous studies demonstrated that the interactions between ACE2 and SARS-CoV-2 is more temperature-sensitive than ACE2 and SARS-CoV. The binding affinity of ACE2 and SARS-CoV-2 was also calculated.9 In this article, more detailed binding mechanisms, such as the key residues, salt bridges, and hydrogen bonds are revealed. Such details provide more specific information for future drug design. The findings in this article will pave the way for other research related to drug design and treatments for COVID-19 and other coronavirus-caused diseases.
Figure 2.

Structure of S protein homotrimer and ACE2. Positive and negative residues on a monomer of S protein are colored in blue and red, respectively. (A) ACE2 is colored in gray. The S protein trimer contains three identical monomers, colored in cyan, green, and orange. One monomer (cyan)'s RBD flips out to bind with the ACE2. The S protein is composed by S1 and S2 subunits. (B) Single S protein monomer structure. The orange circle shows the RBD and the black circle marks the flexible hinge connecting RBD and other part of the S protein.
METHODS
Structure Preparation
The genetic sequence of SARS-CoV-2 was obtained from Genebank,10 which was from the early patients in December 2019. The SARS-CoV-2 with the ACE2 complex structure was modeled by the Swiss-Model web server (https://swissmodel.expasy.org/) based on the template of the complex structures of SARS-CoV and ACE2 (pdb ID: 6ACG, https://www.rcsb.org/structure/6ACG). To study their electrostatic interactions, we mainly focus on the receptor binding domain (RBD) of the S protein and the binding domain of ACE2.
Electrostatic Calculations Using DelPhi
In order to study the electrostatic features, DelPhi11 was utilized to calculate the electrostatic potential for the S protein RBD and ACE2 binding domain. In the framework of continuum electrostatics, DelPhi calculates the electrostatic potential 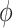 (in systems comprised of biological macromolecules and water in the presence of mobile ions) by solving the Poisson–Boltzmann equation (PBE)
(in systems comprised of biological macromolecules and water in the presence of mobile ions) by solving the Poisson–Boltzmann equation (PBE)
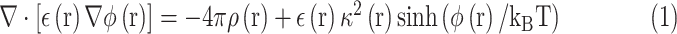
|
where 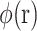 is the electrostatic potential,
is the electrostatic potential, 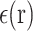 is the dielectric distribution,
is the dielectric distribution, 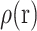 is the charge density based on the atomic structures,
is the charge density based on the atomic structures, 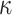 is the Debye–Huckel parameter,
is the Debye–Huckel parameter, 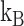 is the Boltzmann constant, and
is the Boltzmann constant, and 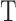 is the temperature. Due to the irregular shape of macromolecules, DelPhi uses a finite difference method to solve the PBE.
is the temperature. Due to the irregular shape of macromolecules, DelPhi uses a finite difference method to solve the PBE.
The electrostatic potential of the SARS-CoV-2 S protein with ACE2 was calculated by DelPhi. The calculated electrostatic potential on the surface was visualized with Chimera (see Figure 3). In order to visualize electric field lines between SARS-CoV-2 and ACE2, the visual molecular dynamics (VMD: https://www.ks.uiuc.edu/Research/vmd/) software package (see Figure 3) was implemented based on the electrostatic potential map from DelPhi calculations. The color scale range was set from –1.0 to 1.0 kT/Å.
Figure 3.

Electrostatic surface and electric field lines of SARS-CoV-2 S protein and ACE2 binding domain. (A) Electrostatic surface of S protein and ACE2. (B) Binding interface of ACE2. (C) Binding interface of S protein RBD. (D) Electric field lines of S protein RBD and ACE2 binding domain. (E) A closeup of electric field lines at the binding interfaces. The residues forming salt bridges are highlighted.
MD Simulations SARS-CoV-2 RBDs
To simulate the dynamic interactions between S proteins and ACE2 protein, 20-ns MD simulations were carried out using NAMD on GPUs using Lonestar5 at the Texas Advanced Computing Center (TACC https://www.tacc.utexas.edu/). A 2000-step minimization was performed for each simulation, followed by a 51000-step simulation to relax the complex structures to avoid the clashes at the binding interface. During the MD simulations, the temperature was set to be 300K, and the pressure was set as standard using the Langevin dynamics. For PME, which is set for full-system periodic electrostatics, the grid size is (86, 88, 132) as x, y, and z values, respectively. After the first 0.05 ns of the simulation, atoms that were not involved in binding domains were constrained within a margin of 5.0 Å of their natural movement maximum length value, since the binding forces were strong enough for proteins to bond completely after 51000 steps, which is not suitable for finding salt bridges. In order to get a more accurate result of the simulation, data of the last 10 ns of simulations were used for data analysis.
To analyze the binding interaction between S protein and ACE2 by finding the key residues, the salt bridges that were formed within the distance of 4 Å were extracted from the last 2000 frames of simulations, and for hydrogen bonds, the cutoff was 3.2 Å. The several top-strongest salt bridges in each binding domain were determined by calculating their formation frequency (occupancy in Figure 4) during MD simulation. The simulation is shown in Movie 1.
Figure 4.

H-bonds between SARS-CoV-2 RBD and ACE2 binding domain. (A) Numbers of H-bonds at different time stamps from 10 to 20 ns, the average value is marked as red line with the value of 11.30. (B) Occupancy of H-bonds ordered by decreasing values of occupancy, and the residues are labeled in y-axis with the SARS-Cov-2 in the left and ACE2 in the right. (C) List of hydrogen bonds that are ranked by their electrostatic energy values.
Binding Energy Calculations Using MM/PBSA
For the binding energy calculations of the S protein and ACE2, the widely used MM/PBSA method was involved in calculating the average binding energy from the last 2000 frames, which were the frames of the last 10 ns trajectory of the MD simulation (see Tables 1 and 2).
Table 1. Binding Energy of SARS-CoV-2 S Protein RBDs Binding With ACE2 Binding Domain. SD Stands for Standard Deviation.
| Energy terms | Magnitude (kcal/mol) | |
|---|---|---|
| Mean | SD | |
| Polar solvation energy | 630.18 | 11.7 |
| Nonpolar solvation energy | –18.41 | 0.41 |
| Van der Waals energy | –128.53 | 8.43 |
| Coulombic energy | –656.69 | 16.04 |
| Electrostatic energy | –26.51 | 10.48 |
Table 2. Binding Energy of Individual Hydrogen Bonds Between SARS-CoV-2 S Protein RBD and ACE2 Binding Domain, With the Decreasing Order of the Electrostatic Energy Mean Values.
| H-Bonds | Electrostatic energy (kcal/mol) | Hydrogen bonds Occupancy | |
|---|---|---|---|
| Mean | **SD | (%) | |
| *LYS417-ASP30 | –6.97 | 1.57 | 39.64 |
| *GLU484-LYS31 | –5.13 | 1.53 | 55.12 |
| *LYS444-GLU56 | –5.02 | 1.67 | 45.7 |
| *ARG403-GLU37 | –2.21 | 2.4 | 13.09 |
| TYR473-ASP30 | –2.14 | 2.28 | 44.83 |
| THR500-ASP355 | –2.07 | 1.52 | 67.46 |
| TYR495-GLU37 | –2.07 | 1.65 | 46.5 |
| ASN487-TYR83 | –2.04 | 1.57 | 50.22 |
| TYR453-HSD34 | –1.53 | 1.29 | 41.73 |
| TYR449-ASN64 | –1.04 | 1.38 | 27.52 |
| PHE490-LYS31 | –0.67 | 1.12 | 40.08 |
| TYR505-ALA386 | –0.6 | 1.45 | 21.08 |
| THR478-GLN24 | –0.39 | 1.46 | 13.86 |
| GLN506-GLN325 | –0.37 | 0.87 | 22.8 |
| SER477-GLN24 | –0.36 | 2.13 | 35.29 |
| GLN498-ASP38 | –0.35 | 0.94 | 43.96 |
| TYR505-ALA387 | –0.26 | 1.73 | 28.57 |
| SER477-THR20 | -0.25 | 1.12 | 19.51 |
| ASN440-GLU329 | –0.22 | 0.92 | 23.05 |
| PHE486-GLN76 | –0.13 | 0.75 | 40.43 |
| GLY447-GLN42 | –0.06 | 0.72 | 38.64 |
| LYS444-GLN60 | 0.64 | 1.58 | 43.36 |
| GLY446-GLU56 | 0.67 | 0.75 | 37.66 |
| GLN493-GLU35 | 1.04 | 1.06 | 44.31 |
| GLY496-ASP38 | 1.79 | 1.07 | 67.76 |
*Salt bridges at the binding interface.
**SD stands for Standard Deviation.
For each pair of the hydrogen bond, residue ID on the left side is from SARS-CoV-2 S protein RBD and the right side is from ACE2 binding domain.
In the MM/PBSA calculation method, Δ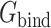 is calculated as
is calculated as
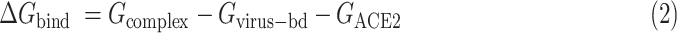
|
where 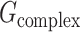 is the total energy of complex of S protein and ACE2 pair;
is the total energy of complex of S protein and ACE2 pair; 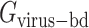 and
and 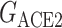 are the total energies of the two individual proteins, and virus-bd stands for the binding domain of SARS-CoV-2 to ACE2. The total energy is calculated as
are the total energies of the two individual proteins, and virus-bd stands for the binding domain of SARS-CoV-2 to ACE2. The total energy is calculated as

|
where 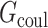 is the Coulombic energy,
is the Coulombic energy, 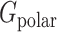 is the polar part of the solvation energy,
is the polar part of the solvation energy, 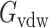 is the Van der Waals energy, and
is the Van der Waals energy, and 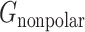 is the nonpolar part of the solvation energy. The Coulombic and polar electrostatic energies were calculated by DelPhi. The Van der Waals binding energy was calculated using NAMD. The nonpolar term of solvation energy was calculated via the solvent accessible surface area method
is the nonpolar part of the solvation energy. The Coulombic and polar electrostatic energies were calculated by DelPhi. The Van der Waals binding energy was calculated using NAMD. The nonpolar term of solvation energy was calculated via the solvent accessible surface area method

|
where γ = 0.0054 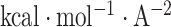 , b = 0.92
, b = 0.92 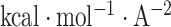 ,12 and SA denotes the solvent accessible surface area, which is calculated using the Naccess2.1.1 program (http://www.bioinf.manchester.ac.uk/naccess/).
,12 and SA denotes the solvent accessible surface area, which is calculated using the Naccess2.1.1 program (http://www.bioinf.manchester.ac.uk/naccess/).
RESULTS AND DISCUSSIONS
The structure of SARS-CoV-2 S protein binding with human ACE2 is shown in Figure 2. The S protein is a homotrimer that contains three identical monomers, as shown in cyan, orange, and green. ACE2 is shown in gray. The main function of the S1 subunit is to bind its RBD with ACE2, while the S2 subunit mainly focuses on the cell membranes' fusion between the virus and host cell. The structure shows that the RBD from the S protein monomer (cyan) flips out and binds with the ACE2, while the other two monomers’ RBDs are hidden in the S1 subunit. This configuration change of RBD is essential for the S protein binding to ACE2. The hinge structure responsible for the configuration change is shown in the black circle in Figure 2(B).
The electrostatic features are important for the protein–protein binding process. Therefore, we indicated the positive and negative residues in blue and red, respectively, on one of the S protein monomers [see Figure 2(A) and (B)]. Most of the charged residues are distributed on the surface of the S protein. At the binding interface of the RBD, the dominant charge is positive, which provides attractive force to ACE2, as the net charge of the ACE2 binding interface is negative. Details are shown in electrostatic surfaces and electric field lines in the following sections.
Electrostatic Surfaces
Electrostatic features are important for protein–protein interactions and drug design. We calculated the electrostatic potential on the surface of the SARS-CoV-2 S protein and ACE2. The binding interface of ACE2 shows significant negative electrostatic potential, while the binding interface of SARS-CoV-2 S protein RBD shows dominantly positive potential (see Figure 3). Therefore, the S protein RBD is attracted by the ACE2 due to their opposite net charges at their binding interfaces. Such electrostatic binding force is common in other strong protein–protein interactions, which provides long-range interactions.
Since SARS-Cov-2 attracts hACE2 to bind, the binding energy is one of the essential terms that can show how much contribution each different residue makes in the binding process.
To further illustrate the electrostatic interactions, electric field lines between the SARS-CoV-2 S protein and ACE2 are calculated and shown in Figures 3(D) and (E). Dense electric field lines indicate strong electrostatic forces. From the electric field line distribution, it is clear that there are four groups of dense field lines between the S protein RBD and ACE2. The sources of the four groups of field lines are all charged residues, which are labeled in Figure 3(E). Those residues are further proved to be the salt-bridge residues at the binding interfaces of S protein RBD and ACE2, as shown in Figure 3(E).
Binding Energy
Since SARS-Cov-2 attracts hACE2 to bind, the binding energy is one of the essential terms that can show how much contribution each different residue makes in the binding process. The binding energy of the SARS-CoV-2 S protein RBD and ACE2 binding domain are calculated using the MM/PBSA method. The results of the binding energy are shown in Table 1. More detailed calculations and analyses of salt bridges and hydrogen bonds are shown in Table 2. In Table 1, the nonpolar solvation energy is the energy of excluding the water molecules when the protein complex immerses in water. The Van der Waals energy decreases significantly when the S protein RBD is away from ACE2. The nonpolar solvation energy and Van der Waals binding energy are nonspecific to amino acids. These two energy terms mainly depend on the binding surface area. Normally, a larger binding surface results in greater nonpolar solvation energy and Van der Waals binding energy values. In this article, we mainly focus on the long-range electrostatic binding energy, which is the sum of the Coulombic energy and polar solvation energy. These two energy terms strongly depend on the distribution of amino acids, especially the charged amino acids. For the SARS-CoV-2 S protein RBD and ACE2, the electrostatic binding energy is –26.51 kcal/mol. To further examine which residues significantly contribute to the electrostatic binding energy, we performed the analyses on hydrogen bonds and salt bridges.
Hydrogen Bonds
The drug targets usually are the residues that form hydrogen bonds or salt bridges. Therefore, studying hydrogen bonds and salt bridges at the binding interfaces is crucial for COVID19. Hydrogen bonds and salt bridges at the interfaces of S protein and ACE2 are calculated based on the MD simulations and are analyzed using the VMD H-bond tool. The average number of H-bonds is 11.30, shown as the red lines in Figure 4(A). From Figure 4(B), the H-bonds with over 10% occupancy are presented with a decreasing order of occupancy. Among the 25 pairs of hydrogen bonds, four salt bridges are included and marked with asterisks. The occupancy of each hydrogen bond pair is shown in Table 2 and visualized in Figure 4(B). The highest occupancy of the salt bridge is 67.76%, from the S protein GLY469 and ACE2 ASP38. Another hydrogen bonds' list is shown in Figure 4(C), which is ranked by the electrostatic binding energy contributed by each pair of hydrogen bond. The top four pairs are LYS417-ASP30, GLU484-LYS31, LYS444-GLU56, ARG403-GLU37 (for each pair, the residue id on the left side is from the SARS-CoV-2 S protein and that on the right side is from ACE2). By default, the VMD Salt Bridge tool identifies a salt-bridge when the distance between a positive residue and a negative residue is less than 4Å, no matter if these two residues form hydrogen bond or not. It is notable that the top four hydrogen bonds are also salt bridges. We therefore can conclude that the S protein is attracted to ACE2, and there are a total of 25 residue pairs between the interfaces that contribute significantly to the electrostatic binding energy. The top four residue pairs are all salt bridges. The average distance between each pair of residues in these top salt bridges is calculated: LYS417-ASP30, 2.5Å; GLU484-LYS31, 4.4Å; LYS444-GLU56, 2.8Å; ARG403-GLU37, 2.6 Å. The sum of the electrostatic binding energies of the four salt bridges is –19.33 kcal/mol, which takes 73% of the total electrostatic binding energy. Thus, the salt-bridge involved residues are the most important residues that can be considered as future drug targets. The structures of the four salt bridges at the interfaces are illustrated in Figure 5.
Figure 5.

Structural demonstration of key residues in salt bridges. SARS-CoV-2 S protein RBD (yellow) with human ACE2 binding domain (gray). Key residues are marked with its amino acid types, sequence numbers, and charges, where blue stands for positively charged amino acid and red stands for the negative ones. Four pairs of salt bridges are found by VMD. All the structures in this figure are rendered by Chimera.
CONCLUSION
COVID-19 infects a large population in the world. This fatal disease is caused by SARS-CoV-2, a novel coronavirus. Understanding the mechanisms of SARS-CoV-2 is critically important, not only for ongoing COVID-19 but also for the upcoming challenges from other disease-causing coronaviruses in the future. A deep understanding of the mechanisms of this virus is critical to developing treatments and vaccines for SARS-CoV-2, and to be better prepared for potential future novel viruses.
Among the types of proteins in coronaviruses, S protein plays a significant role of attaching the virus to the host cell. Blocking the interactions between S protein and human ACE2 may result in effective therapeutic targets for COVID-19. This study implemented several computational approaches to study the fundamental mechanisms of COVID-19 S protein binding with human ACE2. The electrostatic features of COVID-19 S protein and ACE2 are analyzed according to the charge distribution on their structures. Hydrogen bonds and salt bridges on the interfaces are studied based on MD simulations. Residues involved in salt bridges are identified as key residues that stabilize the interactions of S protein and ACE2 complex structure. Blocking these key residues may inhibits the function of S protein, thus preventing the infection of SARS-CoV-2. This study provides potential targets for the drug design of COVID-19. The methods implemented in this research can be widely used to study other viruses.
ACKNOWLEDGMENTS
The calculations and analyses were performed at the Texas Advanced Computing Center. This work was supported in part by the National Institutes of Health under Grant SC1GM132043-01; in part by the National Institutes on Minority Health and Health Disparities, a component of the NIH under Grant 5U54MD007592.
Biographies
Yixin Xie received the B.S. degree in computer science from Southern Polytechnic State University, Marietta, GA, USA, in 2014 and the M.A. degree in media arts from the University of Technology, Sydney, Ultimo NSW, Australia, in 2016. She is currently working toward the Ph.D. degree in computational science with the University of Texas at El Paso, El Paso, TX, USA. Her research is focused on utilizing comprehensive computational approaches to study molecular motors and disease-causing viruses including SARS-CoV-2. Contact her at yxie4@miners.utep.edu.
Dan Du received the B.S. degree in electronic information from Central China Normal University, Wuhan, China, in 2007, the M.S. degree in physics from Huazhong University of Science and Technology,Wuhan,China, in 2011, and she received the M.S. degree in computer engineering from Clemson University, Clemson, SC, USA, in 2016. She is currently working toward the Ph.D. degree in computational science with the University of Texas at El Paso, El Paso, TX, USA. Her research interests include DNA/RNA secondary structure predictions and mechanisms of viral infections such as SARS-CoV-2. Contact her at ddu2@miners.utep.edu.
Chitra B. Karki received the B.S. degree in physics from Prithivi Narayan Campus, Tribhuvan University, Nepal, in 2011, the M.S. degree in physics from the University Campus, Tribhuvan University, Pokhara, Nepal, in 2013, and the M.S. degree in physics in 2019 from the University of Texas at El Paso, where he is currently working toward the graduate degree with the Computational Science Department. His research interest is in understanding different disease related proteins using various computational approaches. Contact him at cbkarki@miners.utep.edu.
Wenhan Guo received the M.S. degree in public health from Zhengzhou University, Zhengzhou, China, in 2018, and is currently working toward the master's degree with the Computational Science Department, University of Texas at El Paso, El Paso, TX, USA. Her research is mainly focused on the study of the structure of virus and scientific software development. Contact her at wguo1@miners.utep.edu.
Alan E Lopez-Hernandez received the B.S. degree in physics from University of Texas at El Paso in 2020, and is currently working toward the master's degree with the Computational Science Department, University of Texas at El Paso, El Paso, TX, USA. His research is mainly focused on using computational approaches to study virus biophysical properties. Contact him at aelopezhern@miners.utep.edu.
Shengjie Sun received the B.S. degree in physics from Central South University, Changsha, China, in 2016, and is currently working toward the Ph.D. degree with the Computational Science Department, University of Texas at El Paso, El Paso, TX, USA. His research interests are in the study of the interaction between myosin and actin and scientific software development. Contact him at ssun1@miners.utep.edu.
Brenda Y Juarez is currently working toward the bachelor's degree in physics with a pre-med concentration with the University of Texas at El Paso, El Paso, TX, USA. She wants to further her studies and pursue the M.D./Ph.D. degrees with a Ph.D. concentration on biophysics. Contact her at byjuarez@miners.utep.edu.
Haotian Li's main research includes biomolecular structure prediction and related method development. He received the B.S. degree in physics and the Ph.D. degrees in physics from the Huazhong University of Science and Technology, Wuhan, China, in 2011 and 2019, respectively. Contact her at 767265653@qq.com.
Jun Wang received the B.S. degree in physics the Ph.D. degree in physics major from the Huazhong University of Science and Technology, Wuhan, China, in 2009 and 2017, respectively. His main research focuses on developing programs to solve computational biology problems. Contact him at junwangwx@gmail.com.
Lin Li is currently an Assistant Professor with the Physics Department, University of Texas at El Paso, El Paso, TX, USA. His research is mainly focused on developing and applying novel computational approaches to study health related biology problems. He received the B.S. and Ph.D. degrees in physics from the Huazhong University of Science and Technology, Wuhan, China, in 2005 and 2011, respectively. He worked as a Postdoctoral Researcher, Research Associate, and then Research Assistant Professor with Clemson University from 2011 to 2017. Since 2017, he has been working at the University of Texas at El Paso as an Assistant Professor. He has authored or coauthored more than 40 peer reviewed scientific papers in the area of computational biophysics. His research bridges physics, computing, and biology. He is the corresponding author of this article. Contact him at lli5@utep.edu.
Funding Statement
This work was supported in part by the National Institutes of Health under Grant SC1GM132043-01; in part by the National Institutes on Minority Health and Health Disparities, a component of the NIH under Grant 5U54MD007592.
REFERENCES
- 1.World Health Organization, Coronavirus Disease 2019 (COVID-19): Situation Report, 85, 2020.
- 2.Fehr A. R. and Perlman S., “Coronaviruses: An overview of their replication and pathogenesis,” in Coronaviruses. Berlin, Germany: Springer, 2015, pp. 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li G., et al. , “Coronavirus infections and immune responses,” J. Med. Virol., vol. 92, no. 4, pp. 424–432, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L., Wang L., and Alexov E., “On the energy components governing molecular recognition in the framework of continuum approaches,” Frontiers Mol. Biosci., vol. 2, 2015, Art. no. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L., Alper J., and Alexov E., “Cytoplasmic dynein binding, run length, and velocity are guided by long-range electrostatic interactions,” Sci. Rep., vol. 6, 2016, Art. no. 31523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L., Alper J., and Alexov E., “Multiscale method for modeling binding phenomena involving large objects: Application to kinesin motor domains motion along microtubules,” Scientific Rep., vol. 6, 2016, Art. no. 23249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xian Y., et al. , “The roles of electrostatic interactions in capsid assembly mechanisms of giant viruses,” Int. J. Mol. Sci., vol. 20, no. 8, 2019, Art. no. 1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li C., et al. , “Highly efficient and exact method for parallelization of grid-based algorithms and its implementation in DelPhi,” J. Comput. Chem., vol. 33, no. 24, pp. 1960–1966, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He J., et al. , “Molecular mechanism of evolution and human infection with sars-cov-2,” Viruses, vol. 12, no. 4, 2020, Art. no. 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu F., et al. , “A new coronavirus associated with human respiratory disease in China,” Nature, vol. 579, no. 7798, pp. 265–269, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L., et al. , “DelPhi: A comprehensive suite for DelPhi software and associated resources,” BMC Biophysics, vol. 5, no. 1, 2012, Art. no. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sitkoff D., Sharp K.A., and Honig B., “Accurate calculation of hydration free energies using macroscopic solvent models,” J. Phys. Chem., vol. 98, no. 7, pp. 1978–1988, 1994. [Google Scholar]