

Abstract
To mimic a hypothetical pathway for protein evolution, we previously developed a design strategy (Metal-Templated Interface Redesign), in which a monomeric protein (cytochrome cb562) was tailored for metal-mediated self-assembly, followed by the re-design of the resulting oligomers for enhanced stability and metal-based functions. Here we show that a single hydrophobic mutation on the cytochrome cb562 surface can drastically alter the outcome of metal-directed oligomerization to yield a new trimeric architecture, (TriCyt1)3, featuring an unusual hexa-histidine coordination motif. Through computational and rational redesign, this nascent trimer is converted into second and third-generation variants (TriCyt2)3 and (TriCyt3)3 with increased structural stability and preorganization for metal coordination. The three TriCyt variants combined furnish a unique design platform to a) provide tunable coupling between protein quaternary structure and metal coordination, b) enable the construction of metal/pH-switchable protein oligomerization motifs, and c) generate a robust metal coordination site that can accommodate all mid-to-late first-row transition metal ions with high affinity, including Mn(II) with nanomolar dissociation constants, rivaling those of the strongest Mn(II)-binding protein, calprotectin.
Keywords: Bioinorganic Chemistry, EPR spectroscopy, Metalloproteins, Protein design, Protein structures, Supramolecular Chemistry
Graphical Abstract

A metal-template-based protein design strategy is used to generate a series of trimeric protein assemblies with chemically tunable self-assembly properties and a rare hexa-histidine coordination motif that can accommodate all mid-to-late first-row transition metal ions with high affinity, including Mn(II) with nanomolar dissociation constants.
Metalloproteins perform countless biological functions despite the fact that they co-opt barely more than a handful of transition metal ions.[1] Underlying this functional diversity is a complex interplay between metal coordination/reactivity and protein structure/dynamics.[2] Although the metal-protein interplay can often be understood through detailed, top-down studies of natural metalloproteins,[3] it remains considerably more challenging to build this interplay from scratch in the form of new metalloproteins.[4] Starting with pioneering studies in the 1990’s,[5] there have been notable successes in the de novo design of functional metalloproteins,[6] which are predominantly based on four-helix bundle and α-helical coiled-coiled motifs with readily parametrizable structures.[7] On the one hand, the fact that diverse bioinorganic functions can be obtained only with a limited set of structural motifs illustrates the versatility of the de novo design approach. On the other hand, it also highlights the challenge of – and the need for – devising alternative strategies and designing new protein architectures for building bioinorganic complexity in a bottom-up fashion.
It has been hypothesized that some modern metalloproteins may have emerged through the metal-nucleated oligomerization of small peptides or protein domains, followed by the evolution of the resulting assemblies into stable, functional architectures.[8] Based on this hypothetical trajectory, we previously developed a protein design strategy termed Metal-Templated Interface Redesign (MeTIR),[9] primarily using a monomeric, four-helix bundle protein (cytochrome cb562) as a building block.[10] First, we installed two bis-His motifs (H59/H63, H73/H77) on the Helix3 surface of cyt cb562 to enable metal coordination.[11] The resulting construct, MBPC1, assembled into different oligomeric states depending on the coordination preferences of nucleating metal ions (NiII-trimer; CuII-dimer; ZnII-tetramer) (Figure S1).[11–12] Given the extensive protein-protein interfaces in the D2 symmetric Zn4:MBPC14 tetramer, this assembly was chosen as a platform for MeTIR.[9a] Zn4:MBPC14 was elaborated through rational redesign and directed-evolution to build functional architectures that selectively bound metal ions,[13] displayed allostery,[14] and performed catalytic reactions in vivo.[15] Yet, despite the functional versatility of the Zn4:MBPC14 progeny, they are inherently biased by the metal-templating strategy toward ZnII coordination chemistry. Moreover, because of their D2 symmetry, they possess at least four copies of each metal center of interest, complicating the examination and modification of the individual metal centers.
Here we aimed to direct the self-assembly of MBPC1 toward more pre-organized architectures in lower oligomerization states, possibly with fewer metal centers. It is well-established that surface-exposed hydrophobic residues can effectively induce protein-protein interactions and aggregation.[16] Thus, we incorporated a Trp residue (W70) onto the MBPC1 Helix3 surface between the H59/H63 and H73/H77 motifs to enable the formation of a hydrophobic core upon metal-mediated oligomerization (Figure 1a and S1). We also mutated the negatively charged Asp66 sidechain on the same surface to Asn to avoid repulsive electrostatic interactions during self-assembly.
Figure 1.
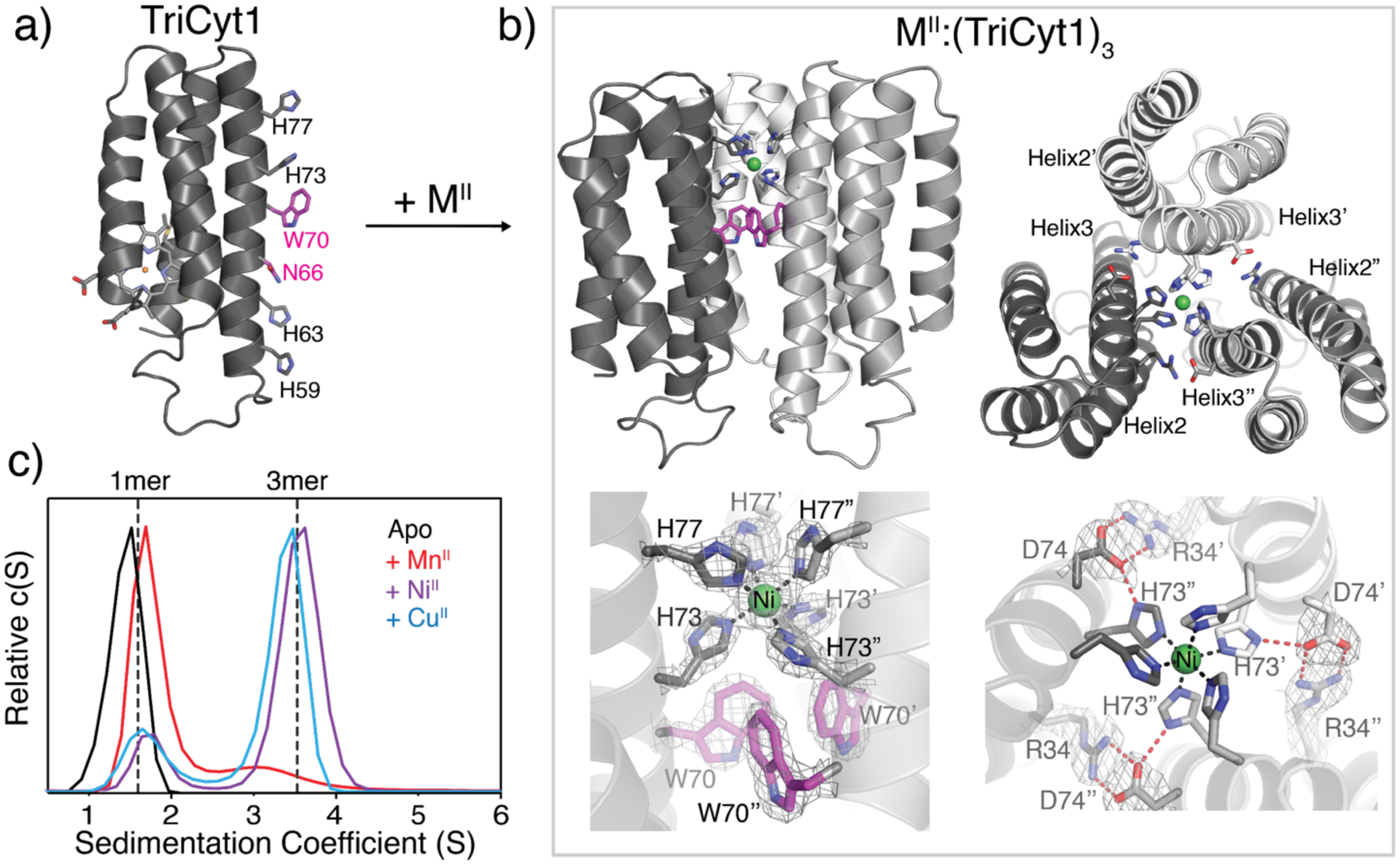
A) Structural model of TriCyt1 monomer. B) Crystal structure of the NiII:(TriCyt1)3 trimer (PDB ID: 6WZA), which is essentially identical to that of CuII:(TriCyt1)3 (Figure S2, PDB ID: 6X8X). The upper panels show the side- and top-views of the trimer and the bottom panels depict the primary and secondary coordination environments that comprise the “core motif”. The 2Fo–Fc maps (grey mesh) are contoured at 1σ. C) Sedimentation velocity (SV) profiles of TriCyt1 (30 μM monomer) in the absence and presence of 10 μM MnCl2, NiCl2, and CuCl2 (see Figure S4 for a complete set).
The metal-dependent assembly of the resulting variant, TriCyt1, was first screened by crystallization in the presence of one equivalent (3 mM) of all mid-to-late first-row transition metal ions (MnII to ZnII). Regardless of the metal ion identity, we observed crystals with hexagonal morphologies, suggesting that they shared an underlying protein arrangement with three-fold symmetry. We obtained 2.5-Å resolution crystal structures of the NiII- and CuII-TriCyt1 complexes (Figure 1b and S2). These structures revealed isostructural trigonal (P321) lattices, formed by trimeric TriCyt1 substructures containing a single NiII or CuII ion coordinated in a near-octahedral geometry by three pairs of H73/H77 residues. The trimeric substructures feature a closepacked, parallel arrangement of TriCyt1 monomers. Near the center is a hub of T-stacked W70 sidechains that non-covalently buttress the metal-coordinating H73 residues (Figure 1b and S3). Additionally, there are three pairs of intermonomer, salt-bridging interactions between R34 and D74 residues that surround the metal coordination site and the tris-W70 hub. The D74 carboxylates are further H-bonded to the δ-N’s of H73 imidazoles from the same monomer, thus completing an extensive network of interactions surrounding the metal coordination site (Figure 1b). Interestingly, H59/H63 pairs are not coordinated by metal ions.
Collectively, these structural details suggested that the crystallographically observed trimeric TriCyt1 structure possesses a high degree of preorganization, which allows it to accommodate only a single metal ion within the same His6 coordination motif independently of the metal identity. Indeed, analytical ultracentrifugation (AUC) and size-exclusion chromatography (SEC) experiments showed that TriCyt1 was monomeric in solution, but exclusively formed trimers upon addition of one equiv. of MnII, FeII, CoII, NiII, CuII and ZnII (Figure 1c and S4–S5). The yield of trimer formation in solution roughly followed the Irving-Williams series,[1c, 17] ranging from 12% for MnII to ~95% for CoII (Figure 1c and S4).
Metal coordination by a His6 motif is exceedingly rare in bioinorganic chemistry. The only well-established biological example is found in the immune protein calprotectin,[18] which is involved in the sequestration of metal ions (particularly MnII) to limit microbial growth and boasts one of the highest MnII affinities among natural proteins.[19] Given the rarity of the His6 motif, in addition to the challenge of generating stable Mn coordination sites in proteins, we asked whether the TriCyt1 structure could be subjected to MeTIR to design progressively more stable trimers, which can subsequently bind MnII (and other divalent ions) with high affinity. The MII:(TriCyt1)3 trimer presents extensive intermonomer interactions (>3000 Å2), which are dominated by a central interface formed by Helices3 of the monomers and peripheral interfaces between neighboring Helices2 and 3 (Figure 1b). We first undertook a computational redesign of these interfaces, whereby the “core motif” (His6 site + tris-W70 hub + R34/D74 salt-bridges) and the protein backbone positions were maintained. Through an iterative process involving sidechain and rotamer optimization with Rosetta and visual inspection, we generated the second-generation variant, TriCyt2, which includes six additional surface mutations on TriCyt1. AUC and SEC experiments showed that TriCyt2 was stable and monomeric in solution but now trimerized in near-quantitative yield upon binding MnII (as well as the other tested metal ions) (Figure 2a and S4–5). Notably, this represents an 8-fold improvement in MnII-induced oligomerization.
Figure 2.
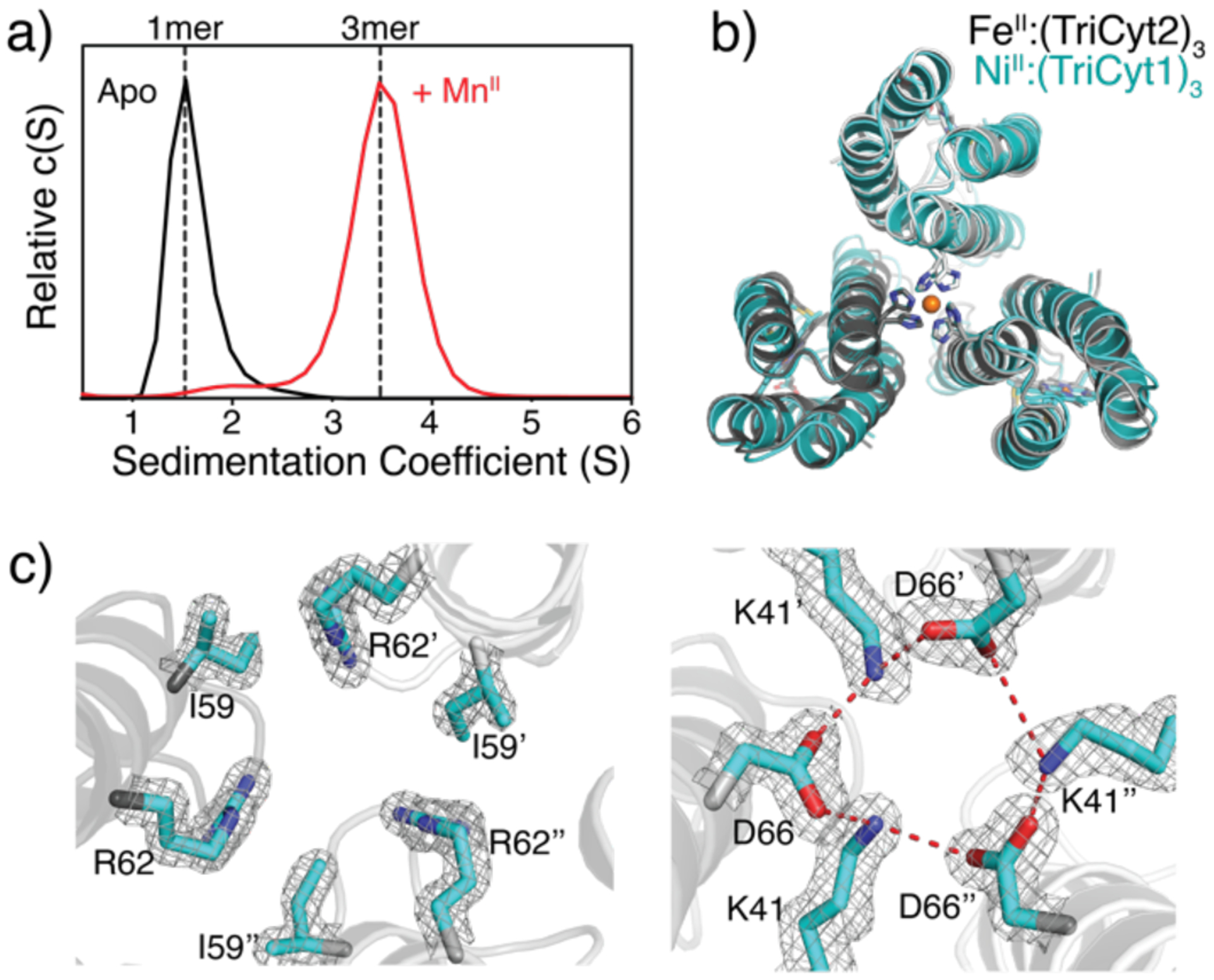
A) SV profiles of TriCyt2 (30 μM monomer) in the absence and presence of 10 μM MnCl2. B) Structural overlay of FeII:(TriCyt2)3 (grey, PDB ID: 6WZ0) and NiII:(TriCyt1)3 (cyan, PDB ID: 6WZA). C) Hydrophobic packing (left) and H-bonding (right) interactions at the core interface of FeII:(TriCyt2)3. The 2Fo–Fc maps (grey mesh) are contoured at 1σ.
These observations indicate that interface redesign was successful and furnished a unique protein construct that can conditionally assemble into trimers in the presence of all relevant first-row transition metal ions. We determined the 1.7-Å-resolution crystal structure of FeII-bound TriCyt2 complex, which is nearly isostructural with MII:(TriCyt1)3 complexes (rmsd = 0.86 Å over all 318 α-C’s) (Figure 2b). As designed, the H59I, H63V, V69L mutations contribute to hydrophobic packing in the Helix3 central core while eliminating the non-coordinating H59/H63 motif, whereas the Q41K and N66D substitutions generate a closed network of H-bonding interactions in the same core (Figure 2c). Additionally, the D54A mutation eliminates the potential repulsive interactions between the Asp54 chains, which causes a slight compaction of the trimer near the 50’s loops (Figure S6). In combination, the six designed mutations yield an increase in sidechain packing in the TriCyt2 trimer interior (buried surface area or BSA = 3440 Å2) compared to TriCyt1 (BSA = 3050 Å2) (Figure S7), consistent with increased trimer stability. Notably, TriCyt2 forms a trimer in the crystal lattice even in the absence of metal ions, revealing an essentially identical structure (rmsd = 0.37 Å) to the FeII:(TriCyt2)3 complex that includes a pre-organized His6 site (Figure S8).
In the next stage of redesign, we sought to stabilize the peripheral interfaces between Helices2 and 3 from neighboring monomers to obtain a metal-independent trimer in solution. Although the peripheral interfaces are wider and less packed compared to the central interface, they appeared amenable to engineering complementary electrostatic interactions. Accordingly, we incorporated three Lys (T31K, A35K, N80K) and two Glu (I67E, Q71E) residues into TriCyt2. The resulting third-generation variant, TriCyt3, indeed formed trimers even in the absence of metal ions (Figure 3a). Consistent with their electrostatic stabilization, TriCyt3 trimers reversibly dissociate into monomers upon lowering the solution pH to <4 (likely due to protonation of Glu/Asp residues), even in the presence of tightly binding metal ions such as CuII (Figure S9), thus providing a pH-switchable protein assembly platform. We determined the TriCyt3 trimer structure in complex with MnII, CoII, NiII and CuII ions (resolutions ranging from 1.8 to 2.2 Å), which showed little deviation from the TriCyt2 trimers (overall rmsd = 0.37 Å) (Figure 3b and S10). The redesigned peripheral interfaces exhibit increased electrostatic complementarity, owing largely to a network of H-bonding/electrostatic interactions involving the Lys and Glu residues (Figure 3b and Figure S11). An examination of metal coordination in the four MII:(TriCyt3)3 structures point to a stable His6 site that can accommodate all tested metal ions in near-octahedral geometries (Figures 3c and S12). Of particular note is the unusual CuII-His6 coordination, which has–to the best of our knowledge–not been previously observed in a protein scaffold and highlights the ability of the TriCyt3 scaffold to enforce a hexacoordinate geometry. Electron paramagnetic resonance (EPR) spectra of Mn-, Co- and Cu-TriCyt3 complexes are all consistent with metal centers in +2 oxidation states (Figures 4a and S16). Despite the enforcement of His6 binding by the TriCyt3 scaffold, there appears to be some flexibility in metal coordination, as evidenced by a) the relatively high temperature factors of the H77 residues in all structures, b) the observation of both Λ and Δ isomers for the CoII-His6 species (Figure S12), and c) varying extents of deviation of the coordination bond angles from perfect octahedral geometry among different metal centers (Figure S13).
Figure 3.
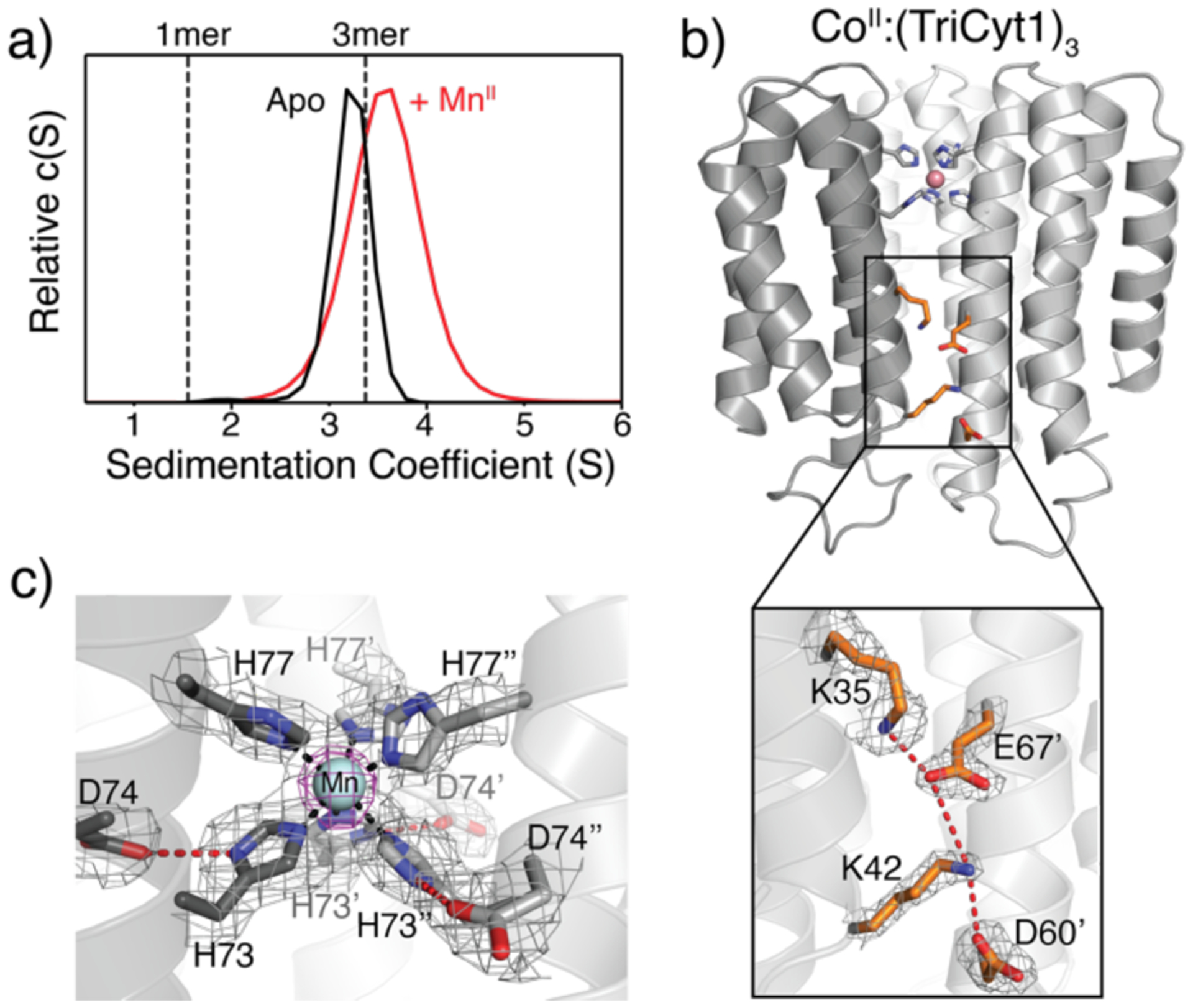
A) SV profiles of TriCyt3 in the absence and presence of MnCl2. B) Crystal structure of CoII:(TriCyt3)3 (PDB ID: 6WZ2), highlighting engineered H-bonding/electrostatic interactions in peripheral interfaces. C) His6-MnII coordination environment in MnII:(TriCyt3)3 (PDB ID: 6WZ1). The 2Fo–Fc (grey) and MnII-anomalous difference (purple) maps are contoured at 1σ and 5σ, respectively.
Figure 4.

A) X-band EPR spectrum of MnII:(TriCyt3)3. B) MnII-binding isotherm for competitive binding titration of TriCyt3 in the presence of Mag-Fura-2. C) Dissociation constants for MII:(TriCyt3)3 complexes determined by competition titrations (see also Figures S14 and S15, and Tables S7 and S8). Metal was added in 2.5 μM increments from 2.5 mM metal chloride stock solutions. The standard errors shown are the standard errors of the fits as calculated by the data fitting program DynaFit (see Supporting Materials and Methods section of the SI).
Having uncoupled protein oligomerization from metal binding, we measured the metal binding affinities of (TriCyt3)3 via competitive titrations, using Mag-Fura-2 and Fura-2 as chelating indicators.[13c, 20] All titrations were consistent with one MII/one trimer stoichiometry, yielding dissociation constants (Kd) ranging from 50 nM for MnII to <1 pM for CuII (Figures 4b, 4c and S14–15). The MnII affinity is noteworthy as it approximates the lowest Kd’s reported for the Mn-His6 center of calprotectin (which range from low nM to low μM)[19] and is >1000-fold lower than that for the Mn-regulatory protein, MntR (Kd = 50–160 μM)[21] and >14-fold lower than that for a designed protein with the highest reported MnII-binding affinity (Kd = 700 nM).[22] Despite the apparent crystallographic disorder in H77 positions, the X-band EPR spectrum of MnII:(TriCyt3)3 (Figure 4a) is very similar to that of the His6-MnII site in calprotectin.[18a] The zero-field splitting of MnII:(TriCyt3)3 (300 MHz; E/D = 0.30) is in fact lower than that of MnII-calprotectin (485 MHz; E/D = 0.30), consistent with a highly symmetrical coordination environment.
In summary, we have reported here the metal-templated design of a series of trimeric protein assemblies (TriCyt1–3), which a) provide tunable coupling between protein quaternary structure and metal coordination, b) furnish metal/pH-switchable protein oligomerization motifs, and c) enable the construction of a robust coordination site for all mid-to-late first-row transition metal ions, including the highest MnII affinities achieved in an artificial protein. From a practical standpoint, the TriCyt platform offers important advantages for the bottom-up design of functional metalloproteins. Owing to its construction from cytochrome cb562 monomers (rather than peptide chains), it is stable and structurally tractable. At the same time, it enables the metal centers to be built in extensive, evolutionarily-naïve interfaces that can be liberally modified to tune the metal-protein interplay and protein oligomerization without affecting protein stability (which stands in contrast to de novo designed α-helical metalloproteins).[7] This could, among other possibilities, enable systematic investigations of the redox properties of the unusual Cu-His6 (and likely also Fe-His6) coordination motif, as well as the design of coordinatively unsaturated metal centers with potential chemical reactivities. From an evolutionary standpoint, our findings illustrate that even a single mutation on a protein’s surface (e.g., W70) can divert protein self-assembly into drastically different pathways, in turn leading to the emergence of new structural motifs with nascent functional sites in protein-protein interfaces.
Supplementary Material
Acknowledgements
We thank R. Subramanian and T. Choi for helpful discussions. This work was funded by the NSF (CHE1607145), by the NIH (a CBI traineeship to A.K. through T32GM112584 and R01GM138884 to F.A.T), by NASA (80NSSC18M0093; ENIGMA: Evolution of Nanomachines in Geospheres and Microbial Ancestors (NASA Astrobiology Institute Cycle 8)). Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource at the Stanford Linear Accelerator Center and the Advanced Light Source at the Lawrence Berkeley National Laboratory, which are supported by the DOE, Office of Science, Office of Basic Energy Sciences under contracts DE-AC02-76SF00515 and DE-AC02-05CH11231, respectively. Coordinate and structure factor files for the crystal structures have been deposited into the Protein Data Bank (www.rcsb.org) with the following accession codes: 6WZA (NiII:TriCyt13), 6X8X (CuII:TriCyt13), 6WYU (TriCyt23), 6WZ0 (FeII:TriCyt23), 6WZ1 (MnII:TriCyt33), 6WZ2 (CoII:TriCyt33, Λ isomer), 6X7E (CoII:TriCyt33, Δ isomer), 6WZC (NiII:TriCyt33), and 6WZ3 (CuII:TriCyt33).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Waldron KJ, Rutherford JC, Ford D, Robinson NJ, Nature 2009, 460, 823–830; [DOI] [PubMed] [Google Scholar]; b) Williams RJP, J. Chem. Soc., Dalton Trans 1991, 539‒546; [Google Scholar]; c) Frausto da Silva JJR, Williams RJP, The biological chemistry of the elements, Oxford University Press, Oxford, 2001. [Google Scholar]
- [2].a) Cook SA, Hill EA, Borovik AS, Biochemistry 2015, 54, 4167–4180; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Churchfield Lewis A., George A, Tezcan FA, Essays in Biochem 2017, 61, 245–258; [DOI] [PubMed] [Google Scholar]; c) Nastri F, D’Alonzo D, Leone L, Zambrano G, Pavone V, Lombardi A, Trends in Biochem. Sci 2019. [DOI] [PubMed] [Google Scholar]
- [3].a) Bertini I, Gray HB, Stiefel EI, Valentine JS, Biological Inorganic Chemistry, Structure & Reactivity, University Science Books, Sausalito, 2007; [Google Scholar]; b) Lippard S, Berg J, Principles of Bioinorganic Chemistry, University Science Books, Mill Valley, 1994; [Google Scholar]; c) Holm RH, Kennepohl P, Solomon EI, Chem. Rev 1996, 96, 2239–2314; [DOI] [PubMed] [Google Scholar]; d) Holm RH, Solomon EI, 2014, 114, 3367–3368. [DOI] [PubMed] [Google Scholar]
- [4].a) Lu Y, Yeung N, Sieracki N, Marshall NM, Nature 2009, 460, 855–862; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yu F, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL, Chem. Rev 2014, 114, 3495–3578; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schwizer F, Okamoto Y, Heinisch T, Gu Y, Pellizzoni MM, Lebrun V, Reuter R, Köhler V, Lewis JC, Ward TR, Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- [5].a) Handel TM, Williams SA, Degrado WF, Science 1993, 261, 879–885; [DOI] [PubMed] [Google Scholar]; b) DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A, Annu. Rev. Biochem 1999, 68, 779–819. [DOI] [PubMed] [Google Scholar]
- [6].a) Faiella M, Andreozzi C, de Rosales RTM, Pavone V, Maglio O, Nastri F, DeGrado WF, Lombardi A, Nat. Chem. Biol 2009, 5, 882–884; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reig AJ, Pires MM, Snyder RA, Wu Y, Jo H, Kulp DW, Butch SE, Calhoun JR, Szyperski T, Solomon EI, DeGrado WF, Nat. Chem 2012, 4, 900; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Joh NH, Wang T, Bhate MP, Acharya R, Wu Y, Grabe M, Hong M, Grigoryan G, DeGrado WF, Science 2014, 346, 1520–1524; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ghirlanda G, Osyczka A, Liu WX, Antolovich M, Smith KM, Dutton PL, Wand AJ, DeGrado WF, J. Am. Chem. Soc 2004, 126, 8141–8147; [DOI] [PubMed] [Google Scholar]; e) Roy A, Sommer DJ, Schmitz RA, Brown CL, Gust D, Astashkin A, Ghirlanda G, J. Am. Chem. Soc 2014, 136, 17343–17349; [DOI] [PubMed] [Google Scholar]; f) Nanda V, Rosenblatt MM, Osyczka A, Kono H, Getahun Z, Dutton PL, Saven JG, DeGrado WF, J. Am. Chem. Soc 2005, 127, 5804–5805; [DOI] [PubMed] [Google Scholar]; g) Mutter AC, Tyryshkin AM, Campbell IJ, Poudel S, Bennett GN, Silberg JJ, Nanda V, Falkowski PG, Proc. Natl. Acad. Sci. USA 2019, 116, 14557–14562; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Shifman JM, Gibney BR, Sharp RE, Dutton PL, Biochemistry 2000, 39, 14813–14821; [DOI] [PubMed] [Google Scholar]; i) Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, DeGrado WF, Dutton PL, Nature 1994, 368, 425–432; [DOI] [PubMed] [Google Scholar]; j) Koder RL, Anderson JLR, Solomon LA, Reddy KS, Moser CC, Dutton PL, Nature 2009, 458, 305–309; [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Bender GM, Lehmann A, Zou H, Cheng H, Fry HC, Engel D, Therien MJ, Blasie JK, Roder H, Saven JG, DeGrado WF, J. Am. Chem. Soc 2007, 129, 10732–10740; [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Zastrow ML, Pecoraro VL, Biochemistry 2014, 53, 957–978; [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Matzapetakis M, Pecoraro VL, J. Am. Chem. Soc 2005, 127, 18229–18233; [DOI] [PubMed] [Google Scholar]; n) Tegoni M, Yu F, Bersellini M, Penner-Hahn JE, Pecoraro VL, Proc. Natl. Acad. Sci. USA 2012, 109, 21234–21239; [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Tolbert AE, Ervin CS, Ruckthong L, Paul TJ, Jayasinghe-Arachchige VM, Neupane KP, Stuckey JA, Prabhakar R, Pecoraro VL, Nat. Chem 2020, 12, 405–411; [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Zhang S-Q, Chino M, Liu L, Tang Y, Hu X, DeGrado WF, Lombardi A, J. Am. Chem. Soc 2018, 140, 1294–1304; [DOI] [PMC free article] [PubMed] [Google Scholar]; q) Nanda V, Senn S, Pike DH, Rodriguez-Granillo A, Hansen WA, Khare SD, Noy D, Biochim. Biophys. Acta A 2016, 1857, 531–538; [DOI] [PMC free article] [PubMed] [Google Scholar]; r) Ulas G, Lemmin T, Wu Y, Gassner GT, DeGrado WF, Nat. Chem 2016, 8, 354–359; [DOI] [PMC free article] [PubMed] [Google Scholar]; s) Berwick MR, Lewis DJ, Jones AW, Parslow RA, Dafforn TR, Cooper HJ, Wilkie J, Pikramenou Z, Britton MM, Peacock AFA, J. Am. Chem. Soc 2014, 136, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Lombardi A, Pirro F, Maglio O, Chino M, DeGrado WF, Acc. Chem. Res 2019, 52, 1148–1159; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tebo AG, Pecoraro VL, Curr. Opin. Chem. Biol 2015, 25, 65–70; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chino M, Leone L, Maglio O, D’Alonzo D, Pirro F, Pavone V, Nastri F, Lombardi A, Angew. Chem., Int. Ed 2017, 56, 15580–15583; [DOI] [PubMed] [Google Scholar]; d) Stenner R, Steventon JW, Seddon A, Anderson JLR, Proc. Natl. Acad. Sci. U.S.A, 2020, 117, 1419–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Eck RV, Dayhoff MO, Science 1966, 152, 363–366; [DOI] [PubMed] [Google Scholar]; b) Armstrong RN, Biochemistry 2000, 39, 13625–13632; [DOI] [PubMed] [Google Scholar]; c) Bergdoll M, Eltis LD, Cameron AD, Dumas P, Bolin JT, Prot. Sci 1998, 7, 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Salgado EN, Ambroggio XI, Brodin JD, Lewis RA, Kuhlman B, Tezcan FA, Proc. Natl. Acad. Sci. USA 2010, 107, 1827–1832; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Salgado EN, Radford RJ, Tezcan FA, Acc. Chem. Res 2010, 43, 661–672; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Churchfield LA, Tezcan FA, Acc. Chem. Res 2019, 52, 345–355. [DOI] [PubMed] [Google Scholar]
- [10].Faraone-Mennella J, Tezcan FA, Gray HB, Winkler JR, Biochemistry 2006, 45, 10504–10511. [DOI] [PubMed] [Google Scholar]
- [11].Salgado EN, Faraone-Mennella J, Tezcan FA, J. Am. Chem. Soc 2007, 129, 13374–13375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Salgado EN, Lewis RA, Faraone-Mennella J, Tezcan FA, J. Am. Chem. Soc 2008, 130, 6082–6084; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Salgado EN, Lewis RA, Mossin S, Rheingold AL, Tezcan FA, Inorg. Chem 2009, 48, 2726–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Medina-Morales A, Perez A, Brodin JD, Tezcan FA, J. Am. Chem. Soc 2013, 135, 12013–12022; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Salgado EN, Brodin JD, To MM, Tezcan FA, Inorg. Chem 2011, 50, 6323–6329; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Brodin JD, Medina-Morales A, Ni T, Salgado EN, Ambroggio XI, Tezcan FA, J. Am. Chem. Soc 2010, 132, 8610–8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Churchfield LA, Medina-Morales A, Brodin JD, Perez A, Tezcan FA, J. Am. Chem. Soc 2016, 138, 13163–13166; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Churchfield LA, Alberstein RG, Williamson LM, Tezcan FA, J. Am. Chem. Soc 2018, 140, 10043–10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Song WJ, Tezcan FA, Science 2014, 346, 1525–1528; [DOI] [PubMed] [Google Scholar]; b) Song WJ, Yu J, Tezcan FA, J. Am. Chem. Soc 2017, 139, 16772–16779. [DOI] [PubMed] [Google Scholar]
- [16].a) Fink AL, Fold. Des 1998, 3, R9–R23; [DOI] [PubMed] [Google Scholar]; b) Garcia-Seisdedos H, Empereur-Mot C, Elad N, Levy ED, Nature 2017, 548, 244–247. [DOI] [PubMed] [Google Scholar]
- [17].Irving H, Williams RJP, Nature 1948, 162, 746–747. [Google Scholar]
- [18].a) Gagnon DM, Brophy MB, Bowman SEJ, Stich TA, Drennan CL, Britt RD, Nolan EM, J. Am. Chem. Soc 2015, 137, 3004–3016; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hayden JA, Brophy MB, Cunden LS, Nolan EM, J. Am. Chem. Soc 2013, 135, 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zygiel EM, Nolan EM, Annu. Rev. Biochem 2018, 87, 621–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Grynkiewicz G, Poenie M, Tsien RY, J. Biol. Chem 1985, 260, 3440–3450. [PubMed] [Google Scholar]
- [21].Golynskiy MV, Gunderson WA, Hendrich MP, Cohen SM, Biochemistry 2006, 45, 15359–15372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rittle J, Field MJ, Green MT, Tezcan FA, Nat. Chem 2019, 11, 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.