

Not long ago, the blood vessel’s adventitial layer was considered nondescript connective tissue comprised of fibroblasts and perivascular nerves. Several observations over the past twenty years have changed this misconception. The overexpression of endothelial nitric oxide synthase in adventitial fibroblasts rescued agonist-mediated vasodilation in arteries without endothelium and reactive oxygen species generated in the adventitial layer inactivated endothelium-derived nitric oxide.1, 2 These data provided direct evidence that substances released from the adventitia could modify vascular function. The adventitia is now recognized as the most complex layer of the vessel wall, comprised not only of fibroblasts and nerves but of dynamic interactions between inflammatory cells, microvessels, lymphatics, and distinct subpopulations of progenitor cells.
Fibroblasts are the most abundant cell in the adventitia and operate as early sensors of environmental stress. Their response to activation is diverse and includes proliferation, secretion of cytokines and growth factors, modification of the extracellular matrix, and expansion of the vasa vasorum.3 Through their reciprocal communication with other cells, adventitial fibroblasts coordinate the blood vessel’s response to injury and stress. Furthermore, the fibroblast displays extraordinary plasticity and can acquire a smooth muscle phenotype (myofibroblast) and participate in vessel remodeling or undergo epigenetic alterations to a proinflammatory phenotype and recruit monocytes and macrophages.4
Nonetheless, the molecular mechanisms responsible for fibroblasts’ signaling in vascular disease are poorly understood because specific labeling and genetic manipulation of these cells have been challenging. In this issue of Arteriosclerosis, Thrombosis, and Vascular Biology, Harrison et al. utilize a novel fibroblast-specific knockout mouse to assess the fibroblast’s role in angiotensin (Ang) II-induced hypertension and vascular remodeling.5
Ang II infusion in mice is a common model for studying cellular mechanisms of hypertension and vascular smooth muscle cell growth in cardiovascular disease. Multiple studies have contributed to our understanding of the complex intracellular signaling of Ang II.6 Reactive oxygen species derived from activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidases function as a second messenger to mediate Ang II signaling in vascular cells. Ang II increases the expression and activation of the Nox1 NADPH oxidase in smooth muscle cells and Nox2 in fibroblasts, endothelial cells, and myeloid cells. Harrison et al. employed an inducible Cre recombinase under the control of a fibroblast-specific regulatory enhancer element of the type I collagen α2 gene (Col1α2-CreERT) to generate fibroblast specific deletion of Nox2 in mice. With this model, the authors examined the role of fibroblast Nox2 in Ang II’s vascular effects.
The study’s first key finding was that deficiency of fibroblast Nox2 mitigated Ang II-induced hypertension without an effect on basal blood pressure. Prior studies have shown mixed results regarding the role of Nox2 in blood pressure regulation. The global Nox2 knockout mouse demonstrated a lower basal systolic blood pressure and a similar increase in pressure with Ang II infusion compared to control mice.7 Most comparable to the current study, the overexpression of a Nox2 inhibitor in adventitial fibroblasts did not affect basal blood pressure or Ang II-induced hypertension.8 The research group responsible for the current study has previously demonstrated that myeloid Nox2 regulated basal blood pressure, whereas deficiency of endothelial Nox2 reduced Ang II-induced hypertension.9 Of note, the resting blood pressure in humans with nonfunctional Nox2 was similar to normal subjects, despite evidence of increased nitric oxide levels.10 The primary mechanism by which Nox2-derived reactive oxygen species affects Ang II-mediated effects on blood pressure remains unclear and likely involves inactivation of nitric oxide, altered calcium handling, and increased vascular compliance, depending on the cell type examined.
Harrison et al. ‘s second major finding was that deficiency of fibroblast Nox2 prevented Ang II-dependent vascular remodeling. Infusion of Ang II increased the size of the medial layer in large (aorta), medium (carotid), and small (myocardial septal) arteries, in part by increasing smooth muscle cell proliferation. The observation that fibroblast Nox2 is necessary for Ang II-mediated medial hyperplasia suggests that fibroblasts activate smooth muscle cell growth via a paracrine mechanism. In contrast to the disparate reported effects on blood pressure, published data is consistent with the conclusion that adventitial Nox2 mediates Ang II-induced hyperplasia.7–9
The third major finding of Harrison et al. is the most interesting. The authors performed a proteomic analysis of conditioned media collected from cells treated with Ang II and identified 121 factors secreted by fibroblasts but not smooth muscle cells. Additional analysis identified growth differentiation factor 6 (GDF6), a member of the transforming growth factor β superfamily, as a regulator of smooth muscle cell growth. Ang II infusion increased GDF6 expression in the aorta of control mice, and this effect was blunted in mice deficient in fibroblast Nox2. Experiments in vitro showed that treatment with GDF6 increased smooth muscle cell proliferation, whereas conditioned media from Ang II treated fibroblasts after knockdown of GDF6 failed to induce smooth muscle cell growth.
The report by Harrison et al. is the first to demonstrate a role for GDF6 in mammalian vascular remodeling and provides a new mechanism of how adventitial fibroblasts regulate medial hyperplasia. However, the study offers no new information on how fibroblast Nox2 mediates Ang II-induced hypertension. The authors suggest that since the effect of fibroblast Nox2 on Ang II- hypertension was temporally associated with the thickening of the medial layer, the two may be related. However, prior data suggested independence between blood pressure and medial hyperplasia following Ang II infusion.7 There is no direct evidence that GDF6 signaling contributed to Ang II hypertension via vascular remodeling or other mechanisms. Additional studies will be needed to understand the cellular mechanisms by which GDF6 and other fibroblast-derived factors signal to endothelial and smooth muscle cells in the vessel wall. For example, the specific receptors transducing GDF6 activity remain unclear. Of note, it was recently shown in ATVB that GDF6 maintains endothelial barrier dysfunction by modulating the vascular endothelial growth factor (VEGF) receptor.11 It is also interesting that, like Nox2, the Nox1 NADPH oxidase has been implicated in Ang II-induced hypertension and medial hyperplasia.12 These results can be reconciled with the present study if the signaling of GDF6 to smooth muscle cells involves activation of Nox1, as reported with other growth factors.13
A fibroblast-specific Nox2 knockout mouse is a valuable tool for exploring the vascular response to injury. To confirm the specificity of the Col1α2-CreERT model, the authors showed no overlap between tdTomato labeled fibroblasts and the endothelial cell marker VE-cadherin. This observation contrasts with a recent report that used the Col1α2-CreERT model to study the mesenchymal-endothelial transition of cardiac fibroblasts and found that more than one-third of tdTomato labeled fibroblasts expressed endothelial markers 14 days after Ang II infusion.14 Mesenchymal-endothelial transition also altered the expression of paracrine factors by fibroblasts. Future studies using the Col1α2-CreERT model to study fibroblasts will need to consider these findings when interpreting their results. Given the phenotypic and functional heterogeneity of fibroblasts in vivo, single-cell transcriptomic data will complement cell-specific knockout models to strengthen mechanistic insights.
In summary, Harrison et al. identify GDF6 as a Nox2-dependent mitogen secreted by adventitial fibroblasts in response to Ang II and essential for medial hyperplasia. It will be interesting to learn the redox-dependent mechanism for GDF6 expression and whether fibroblast-derived GDF6 has a role in other vascular diseases.
Figure. Outside-in-signaling by the adventitia.
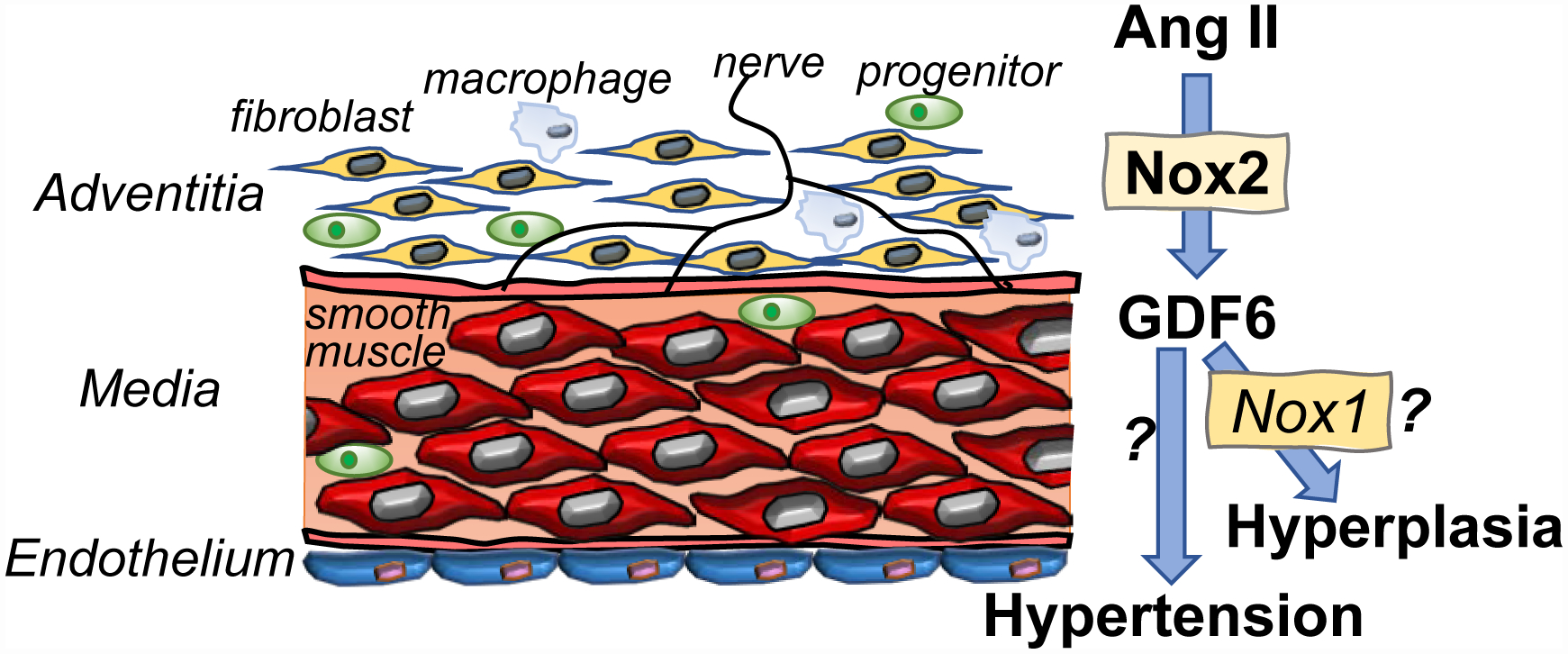
Nox2-dependent secretion of GDF6 by adventitial fibroblasts mediate the vascular response to Ang II. The activation of smooth muscle cell Nox1 by GDF6 is speculative.
b). Sources of Funding
BMS is supported by the National Institutes of Health (T32HL007101). FJM is supported in part by Merit Reviews (BX005450 and BX001729) from the United States Department of Veterans Affairs Biomedical Laboratory R&D Service, the National Institutes of Health (R01EB028798), National Science Foundation (ECCS1936793), and the American Heart Association (19TPA3490002).
Footnotes
Disclosures
None.
References
- 1.Tsutsui M, Chen AFY, O’Brien T, Crotty TB and Katusic ZS. Adventitial expression of recombinant eNOS gene restores NO production in arteries without endothelium. Arteriosclerosis, Thrombosis & Vascular Biology. 1998;18:1231–1241. [DOI] [PubMed] [Google Scholar]
- 2.Di Wang H, Hope S, Du Y, Quinn MT, Cayatte A, Pagano PJ and Cohen RA. Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension. 1999;33:1225–1232. [DOI] [PubMed] [Google Scholar]
- 3.Stenmark KR, Yeager ME, El Kasmi KC, Nozik-Grayck E, Gerasimovskaya EV, Li M, Riddle SR and Frid MG. The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol. 2013;75:23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li M, Riddle SR, Frid MG, El Kasmi KC, McKinsey TA, Sokol RJ, Strassheim D, Meyrick B, Yeager ME, Flockton AR, McKeon BA, Lemon DD, Horn TR, Anwar A, Barajas C and Stenmark KR. Emergence of fibroblasts with a proinflammatory epigenetically altered phenotype in severe hypoxic pulmonary hypertension. J Immunol. 2011;187:2711–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harrison CB, Trevelin SC, Richards DA, Santos CX, Sawyer G, Markovinovic A, Zhang X, Zhang M, Brewer AC and Yin X. Fibroblast Nox2 (NADPH Oxidase-2) Regulates ANG II (Angiotensin II)–Induced Vascular Remodeling and Hypertension via Paracrine Signaling to Vascular Smooth Muscle Cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2020:ATVBAHA. 120.315322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, Scalia R and Eguchi S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol Rev. 2018;98:1627–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ and Cohen RA. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. CircRes. 2001;88:947–953. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Ormsby A, Oja-Tebbe N and Pagano PJ. Gene transfer of NAD(P)H oxidase inhibitor to the vascular adventitia attenuates medial smooth muscle hypertrophy. Circ Res. 2004;95:587–594. [DOI] [PubMed] [Google Scholar]
- 9.Sag CM, Schnelle M and Shah AM. Response by Sag et al to Letter Regarding Article, “Distinct Regulatory Effects of Myeloid Cell and Endothelial Cell NAPDH Oxidase 2 on Blood Pressure”. Circulation. 2017;136:2090–2091. [DOI] [PubMed] [Google Scholar]
- 10.Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, Martino S, Gambineri E, Soresina AR, Pignatelli P, Martino F, Basili S and Loffredo L. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation. 2009;120:1616–1622. [DOI] [PubMed] [Google Scholar]
- 11.Krispin S, Stratman AN, Melick CH, Stan RV, Malinverno M, Gleklen J, Castranova D, Dejana E and Weinstein BM. Growth Differentiation Factor 6 Promotes Vascular Stability by Restraining Vascular Endothelial Growth Factor Signaling. Arterioscler Thromb Vasc Biol. 2018;38:353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F and Krause K-H. Decreased blood pressure in NOX1-deficient mice. FEBS Letters. 2006;580:497–504. [DOI] [PubMed] [Google Scholar]
- 13.Miller FJ Jr., Chu X, Stanic B, Tian X, Sharma RV, Davisson RL and Lamb FS. A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid Redox Signal. 2010;12:583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong W, Li R, Yang H, Lu Y, Zhou L, Sun L, Wang D and Duan J. Mesenchymal-endothelial transition-derived cells as a potential new regulatory target for cardiac hypertrophy. Scientific reports. 2020;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]