

Abstract
Ricolinostat is the first orally available, selective inhibitor of histone deacetylase 6 (HDAC6), currently under evaluation in clinical trials in patients with various malignancies. It is likely that the inevitable emergence of resistance to ricolinostat is likely to reduce its clinical effectiveness in cancer patients. In this study, we investigated the potential impact of multidrug resistance-linked ATP-binding cassette (ABC) transporters ABCB1 and ABCG2 on the efficacy of ricolinostat, which may present a major hurdle to its development as an anticancer drug in the future. We demonstrated that the overexpression of ABCB1 or ABCG2 reduces the intracellular accumulation of ricolinostat, resulting in reduced efficacy of ricolinostat to inhibit the activity of HDAC6 in cancer cells. Moreover, the efficacy of ricolinostat can be fully restored by inhibiting the drug efflux function of ABCB1 and ABCG2 in drug-resistant cancer cells. In conclusion, our results provide some insights into the basis for the development of resistance to ricolinostat and suggest that co-administration of ricolinostat with a modulator of ABCB1 or ABCG2 could overcome ricolinostat resistance in human cancer cells, which may be relevant to its use in the clinic.
Keywords: ABCB1, ABCG2, Multidrug resistance, HDAC6, Ricolinostat
1. Introduction
Histone deacetylases (HDACs) are histone-modifying enzymes that remove acetyl groups from core histone proteins, causing DNA condensation and preventing gene transcription [1]. The expression and activity of HDAC isoenzymes are higher in many tumor types, making the HDACs attractive targets for drug discovery [2]. Ricolinostat (ACY-1215) is a novel and orally available small molecule compound that inhibits α-tubulin deacetylation mediated by class IIb histone deacetylase 6 (HDAC6) [3,4]. More importantly, it is the first safe and well tolerated, isoform-selective HDAC inhibitor that has progressed to clinical trials, both as a single agent and in combination therapy [5,6]. Ricolinostat is currently under evaluation in phase I and II clinical trials in patients with multiple myeloma (ClinicalTrials.gov number NCT01323751, NCT01997840, NCT01583283, NCT02189343), relapsed or refractory lymphoid malignancies (NCT02091063), relapsed chronic lymphocytic leukemia (NCT02787369), unresectable or metastatic breast cancer (NCT02632071) and for the treatment of gynecological cancer (NCT02661815). Despite the current success of ricolinostat, the inevitable emergence of resistance to ricolinostat can present a significant therapeutic challenge and limit its clinical use in cancer patients.
The emergence of multidrug resistance (MDR) to conventional and molecularly targeted therapeutic drugs remains a major challenge in cancer treatment. The MDR phenotype is often attributed to the overexpression of one of the ATP-binding cassette (ABC) drug transporters that actively effluxes chemotherapeutic agents from cancer cells and leads to relapse and eventual death of cancer patients [7,8]. ABCB1 (MDR1; P-glycoprotein) and ABCG2 (BCRP; MXR) are two of the most well-characterized MDR-linked ABC transporters known for utilizing energy derived from ATP hydrolysis to efflux and confer resistance to a large variety of chemotherapeutic agents in cancer cells [9–12]. High expression of ABCB1 and/or ABCG2 is known to be associated with the development of MDR in multiple myeloma cells [13–18], poor clinical outcome in patients with relapsed multiple myeloma [19], acute lymphocytic leukemia (ALL), acute myelogenous leukemia (AML) [20–22], or chronic lymphocytic leukemia (CLL) [23], and in patients with metastatic breast cancer [24]. Making the matter worse, cancer cells may initially be susceptible to conventional anticancer drugs, but acquire a multidrug resistant phenotype during chemotherapy before switching to targeted therapy. For instance, studies have shown that high expression of ABCB1 or ABCG2 is often up-regulated by chemotherapeutic drugs in most multiple myeloma (MM) patients [15,16,18]. Moreover, ABCB1 and ABCG2 are highly expressed at barrier sites such as the gastrointestinal tract, the blood-brain barrier, liver and kidney, which have great clinical implications on the absorption, distribution, metabolism and elimination of substrate drugs in patients [9,25].
Here, we investigate the pharmacological impact of the drug efflux transporters ABCB1 and ABCG2 on the efficacy of ricolinostat in cancer cell lines. Our data show that ricolinostat is a substrate for ABCB1 and ABCG2. The overexpression of ABCB1 or ABCG2 in cancer cells resulted in reduced intracellular concentration of ricolinostat and subsequently reduced ability of ricolinostat to inhibit the activity of HDAC6. Our results indicate that the overexpression of ABCB1 or ABCG2 represents a novel mechanism for ricolinostat resistance in human cancer cells and that drug combination therapy may be required to overcome this clinical problem in the future.
2. Materials and methods
2.1. Chemicals
Fetal calf serum (FCS), phosphate-buffered saline (PBS), Dulbecco’s Modified Eagle’s medium (DMEM), RPMI medium, trypsin-EDTA, penicillin and streptomycin were purchased from Gibco, Invitrogen (CA, USA). Ricolinostat (ACY-1215) was purchased from Selleckchem (Houston, TX, USA). Annexin V: FITC Apoptosis Detection Kit was purchased from BD Pharmingen (San Diego, CA, USA). Tools Cell Counting (CCk-8) Kit was purchased from Biotools Co., Ltd (Taipei, Taiwan). MTT dye, doxorubicin, mitoxantrone and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless stated otherwise.
2.2. Cell lines and culture conditions
The KB-3–1 human epidermal carcinoma cell line and its ABCB1-overexpressing MDR variant KB-V-1 cell line, the MCF-7 human breast carcinoma cell line and its ABCG2-overexpressing MDR variants MCF7-FLV1000 and MCF7-AdVp3000 cell lines, pcDNA3.1-HEK293, the ABCB1-transfected HEK293 (MDR19-HEK293) and the ABCG2-trans-fected EHK293 (R482-HEK293) cell lines, were maintained in DMEM. The S1 human colon carcinoma cell line and its ABCG2-overexpressing MDR variant S1-M1–80 cell line, the NIH3T3mouse embryonic fibro-blast cell line and its human ABCB1-transfected NIH3T3-G185 cell line, the OVCAR-8 human ovarian carcinoma cell line and its ABCB1-over-expressing MDR variant NCI-ADR-RES cell line were maintained in RPMI-1640. All cell lines were cultured in medium supplemented with 10% FCS, 2 mM L-glutamine and 100 units of penicillin/streptomycin/mL. KB-V-1, NCI-ADR-RES, S1-M1–80, MCF7-FLV1000 and NIH3T3-G185 cell lines were cultured in media containing 1 μg/mL vinblastine [26], 0.85 μM doxorubicin, 1 µg/mL flavopiridol, 5 μg/mL verapamil and 3 μg/mL doxorubicin [27], 80 μM of mitoxantrone or 60 ng/mL colchicine [28], respectively. HEK293 and HEK293 transfected lines were maintained in 2 mg/mL G418, as described previously [29]. All cell lines were maintained at 37 °C in 5% CO2 humidified air and placed in drug-free medium 7 days prior to assay.
2.3. Immunoblotting
Antibodies anti-acetylated-α-tubulin (#T6793, Sigma-Aldrich, St. Louis, MO, USA) at 1:100000, C219 (#517310, Merck Millipore, Burlington, Massachusetts, USA) at 1:3000, BXP-21 (#ab3380, Abcam, Cambridge, MA, USA) at 1:15000, anti-HDAC6 (#7558, Cell Signaling Technology, Danvers, MA, USA) at 1:1000 and anti-α-tubulin (#T6199, Sigma-Aldrich, St. Louis, MO, USA) at 1:100000, were used to detect acetylated tubulin, ABCB1, ABCG2, HDAC6 and tubulin as positive control for Western blotting. The secondary antibodies used were the Horseradish peroxidase-conjugated goat anti-mouse IgG (1:10000) and anti-rabbit IgG. Signals were detected as described previously [30].
2.4. Cytotoxicity assay
MTT and CCK-8 assays were performed to determine the cytotoxicity of ricolinostat and other anticancer agents in cell lines as described previously by Ishiyama et al. [31]. Briefly, cells were plated into 96-well plates at a density of 5000 cells per well in culture medium at 37 °C for 24 h before treating cells with a drug of interest for an additional 72 h and subsequently developed as described previously [30]. For the MDR reversal assays, 1 µM of tariquidar or Ko143 was added to the cytotoxicity assay in combination with ricolinostat, and the extent of reversal was determined based on the calculated fold-reversal (FR) values as described previously [30].
2.5. Apoptosis assay
The annexin V–FITC and propidium iodide (PI) staining method was performed according to the method described by Anderson et al. [32] to determine the percentage of apoptotic cells induced by ricolinostat alone or in combination with an inhibitor of ABCB1 or ABCG2. Briefly, cells were treated with ricolinostat or combination of ricolinostat and tariquidar or Ko143 for 48 h before harvested by a series of washing, centrifugation and resuspended in FACS buffer containing 1.25 µg/mL annexin V–FITC (PharMingen) and 0.1 mg/mL PI and incubated for 15 min at room temperature. The labeled cells (10000 per sample) were analyzed by FACScan (BD Biosciences) using the CellQuest software (Becton-Dickinson). PS-positive and PI-negative cells (lower right dot-plot quadrant) were considered as apoptotic, whereas PS-positive and PI-positive cells (upper right dot-plot quadrant) were considered as either necrotic or late apoptotic [32].
2.6. ATPase assay
The effect of ricolinostat on vanadate (Vi)-sensitive ATPase activity of ABCB1 or ABCG2 in membrane vesicles of High-Five cells expressing ABCB1 or ABCG2 was recorded by endpoint Pi assay as described previously [33].
2.7. Ricolinostat accumulation assay and HPLC-MS/MS analysis
The intracellular accumulation of ricolinostat was performed as described previously [34], and quantified by HPLC-MS/MS based assay according to the method described by Cihalova et al. [35] and Shen et al. [36] with slight modification. Briefly, 2 × 106 cells were treated with 10 μM of ricolinostat in the presence or absence of 10 μM of tariquidar or Ko143 at 37 °C for 60 min, washed twice with cold PBS, harvested, and resuspended in 3 × volume of methanol and stored at −80 °C. After thawing, lysates with methanol extraction were spun down by 14000 rpm for 30 min at 4 °C. Supernatants were dried with speed vacuum, redissolved in 50% methanol/H2O and 0.1% formic acid, and analyzed using Selected Reaction Monitoring (SRM). Cell contents were analyzed using multiplexed HPLC-SRM/MS on a Waters BEH column (1 × 100 mm, particle size 1.7 μm, pore size 130 Å) in ACQUITY ultra-performance liquid chromatography (UPLC) system coupled with HCT ultra (Bruker Daltonik GmbH, Bremen, Germany). Mobile system A: 0.1% formic acid in water; B: acetonitrile with 0.1% formic acid. A flow rate of 60 µL/min with a linear gradient was set as follows: 0 min, 10% B; 2 min, 10% B; 9.5 min, 60% B; 10 min, 99% B; 13 min, 99% B; 13.1 min, 10% B; 14 min, 10% B. Ricolinostat data was acquired by SRM mode; isolation 434.5 with peak width 4 amu, followed by smart fragmentation ramping from 0.3 to 2 V. One blank run was inserted between sample injections. MS2 fragment 274.56 was selected for quantitation and the peak area was detected and integrated using software package DataAnalysis 4.2. (Bruker Corporation). The standard response curves were generated with cell lysate extracts as background, combined with eight different concentrations of ricolinostat. The response curves were 1:4 serial dilutions, and ricolinostat ranging from 50 pmol/μL to 15 fmol/μL. For higher specificity, peak area of MS2 fragment (274.56) was selected and calculated as response curves.
2.8. Docking of ricolinostat in the drug-binding pocket of human ABCB1 and ABCG2
The three dimensional structure of ABCB1 was predicted using an automated protein homology-modeling server SWISS-MODEL. The amino acid sequence of the protein was submitted to SWISS-MODEL server and templates were searched with BLAST and HHBlits against SWISS-MODEL template library. For each identified template, the template’s quality was predicted from features of the target-template alignment. The templates with the highest quality were then selected and built based on the target-template alignment using ProMod3 [37–39]. The energy was minimized for ABCB1 homology modeled structure and ABCG2 protein structure (PDB:5NJG) [40] using Acclerys Discovery Studio 4.0. Ligand preparation and docking was performed by the CDOCKER module of the same software.
2.9. Quantification and statistical analysis
Data are presented as mean ± standard error of the mean (SEM), and the IC50 values are mean ± standard deviation (SD) calculated from at least three independent experiments. Curve plotting and statistical analysis were performed with KaleidaGraph (Reading, PA, USA) and GraphPad Prism (La Jolla, CA, USA) software. The improvement in fit was analyzed by two-sided Student’s t-test and labeled “statistically significant” if the probability, p, was less than 0.05.
3. Results
3.1. Ricolinostat is less effective at inhibiting the deacetylase activity of histone deacetylase 6 (HDAC6) and inducing apoptosis in human cancer cells overexpressing either ABCB1 or ABCG2
Considering that ricolinostat selectively targets HDAC6, we examined its ability to inhibit the activity of HDAC6 in human cancer cells by monitoring the acetylation level of α-tubulin (Ac-tub), a known substrate for HDAC6, in human KB-3–1 epidermal carcinoma cells and its ABCB1-overexpressing KB-V-1 multidrug resistant variant, as well as in human S1 colon carcinoma cells and its ABCG2-overexpressing S1-M1–80 multidrug resistant variant. HDAC6 is responsible for the deacetylation of non-histone substrates such as α-tubulin in the cytoplasm and therefore, inhibition of HDAC6 increases α-tubulin acetylation [41]. As shown in Fig. 1, ricolinostat induced hyperacetylation of α-tubulin in a concentration-dependent manner in all the cell lines tested, indicating that HDAC6 is inhibited by ricolinostat. However, we noticed that the inhibition of the tubulin deacetylase HDAC6 by ricolinostat was not as effective in multidrug resistant KB-V-1 and S1-M1–80 cells as compared to their respective drug-sensitive parental KB-3–1 and S1 cells (Fig. 1A and B, upper panels). Of note, SAHA is a known HDAC inhibitor and was used here as a control. The concentration of ricolinostat required to achieve 50% inhibition of the HDAC6 activity in KB-3–1, KB-V-1, S1 and S1-M1–80 was calculated at approximately 0.8 µM, 4.5 µM, 0.7 µM and 42 µM, respectively, which was determined as a percentage of increased hyperacetylation of α-tubulin in cells treated with SAHA (Fig. 1A and B, lower panels). Although Sirt2 is also a major deacetylase for tubulin deacetylation, in the present study we are not exploring how ricolinostat affects the deacetylation of tubulin. In addition, Santo et al., reported that ricolinostat has minimal activity against Sirt1 and Sirt2 [42]. For these reasons, we did not again test effect of ricolinostat on Sirt2 levels in our cell lines, and focused to provide a proof of principle in this work that the function of ABCB1 and ABCG2 reduces the efficacy of this drug, using the level of tubulin deacetylation as an indication.
Fig. 1.
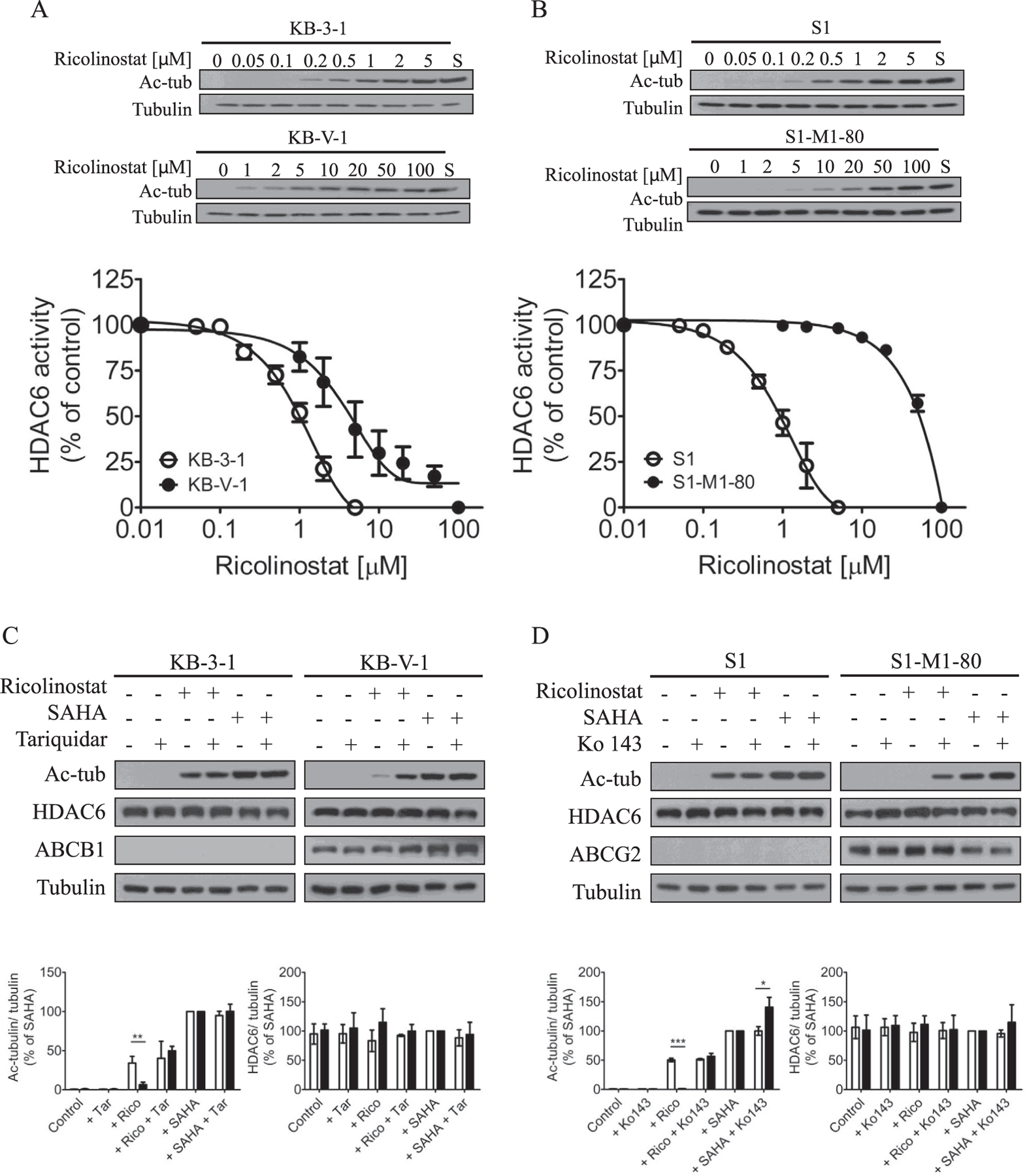
Ricolinostat inhibits deacetylation of α-tubulin. Concentration-dependent inhibition of deacetylated α-tubulin (Ac-tub) by ricolinostat in (A) drug-sensitive human KB-3–1 epidermal cancer cells (open circles) and its ABCB1-overexpressing variant KB-V-1 cells (filled circles), as well as in (B) drug-sensitive human S1 colon cancer cells (open circles) and its ABCG2-overexpressing variant S1-M1–80 cells (filled circles). Western blot analysis (upper panels) of the indicated proteins in drug-sensitive and MDR cells treated with DMSO (lane 1) or increasing concentrations of ricolinostat (lane 2–8) or 20 µM of a known HDAC inhibitor SAHA (S) as a positive control (lane 9) for 24 h at 37 °C. The inhibition of HDAC6 activity by ricolinostat was calculated as percentage inhibition of SAHA (lower panels) measured by the relative amount of Ac-tub in cell lysates from these cells, and the IC50 values of ricolinostat were determined from the dose-response curves. The expression of Ac-tub, HDAC6, ABCB1 or ABCG2 (upper panels) was determined in (C) KB-3–1 and KB-V-1 cells, (D) S1 and S1-M1–80 cells treated with DMSO (control), 2 µM of ricolinostat or 20 µM of SAHA in the presence or absence of 1 µM of tariquidar or Ko143 alone or in combination for 24 h before immunoblotting with the indicated antibodies. Quantification of Western blots (lower panels) showing the relative level of Ac-tub and HDAC6 in KB-3–1 (C, open bars) and KB-V-1 (C, closed bars) cells, S1 (D, open bars) and S1-M1–80 (D, closed bars) cells. Representative Western blots of three independent experiments are shown. Quantifications of Western blots are presented as mean ± S.E.M calculated from more than three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001, versus the same treatment in parental cells.
Next, to determine the impact of ABCB1 or ABCG2 on the efficacy of ricolinostat, we examined the effect of tariquidar or Ko143, a reference inhibitor of ABCB1 or ABCG2, on ricolinostat induced α-tubulin hyperacetylation. Cells were treated with DMSO (control), 2 µM of ricolinostat, 20 µM of SAHA, 1 µM of tariquidar (Fig. 1C) or Ko143 (Fig. 1D) alone or in combination as indicated. In the absence of ricolinostat or SAHA, HDAC6 potently deacetylates α-tubulin in all cell lines (Fig. 1C and D, lane 1 and 2). We discovered that in the presence of tariquidar, α-tubulin hyperacetylation induced by ricolinostat increased significantly in KB-V-1 cells to a comparable level as in KB-3–1 cells (Fig. 1C). Similarly, Ko143 increased ricolinostat-induced α-tubulin hyperacetylation in S1-M1–80 cells to a comparable level as in S1 cells (Fig. 1D). Interestingly, Ko143 also increased the activity of SAHA in S1-M1–80 cells. Of note, the level of HDAC6 remained unchanged in all cell lines treated with tariquidar and Ko143. Our data suggest that the overexpression of ABCB1 or ABCG2 in cancer cells can lead to reduced efficacy of ricolinostat.
Knowing that ricolinostat has a reduced effect on tubulin deacetylase activity of HDAC6 (Fig. 1) in cancer cells overexpressing ABCB1 or ABCG2, we next examined ricolinostat-induced apoptosis in human KB and S1 cancer cell lines. Exposing drug-sensitive KB-3–1 and S1 cancer cells to 20 μM of ricolinostat for 48 h markedly increased the percentage of apoptotic cells from a basal level of approximately 7% to 41%, and from 3% to 25%, respectively (Fig. 2A and B, upper panels). In contrast, 20 μM of ricolinostat resulted in only a marginal increase in apoptosis in KB-V-1 and S1-M1–80 cancer cells (Fig. 2A and B, lower panels). Additionally, we found that the apoptotic cell population in ricolinostat-treated KB-V-1 and S1-M1–80 cells can be restored by 1 μM of tariquidar and Ko143, significantly increased the percentage of apoptotic cells to approximately 60% and 25%, respectively. Of note, tariquidar and Ko143 alone had no significant effect on the level of apoptosis or cytotoxicity in all of the tested cell lines. Considering that ricolinostat at 2 μM was able to completely inhibit acetylated α-tubulin in drug-sensitive cancer cells, we wanted to determine whether ricolinostat is able to induce apoptosis in MDR cancer cells even at a very high concentration. Therefore, a significantly higher concentration of ricolinostat was used in these experiments. These results are in agreement with data shown in Fig. 1.
Fig. 2.
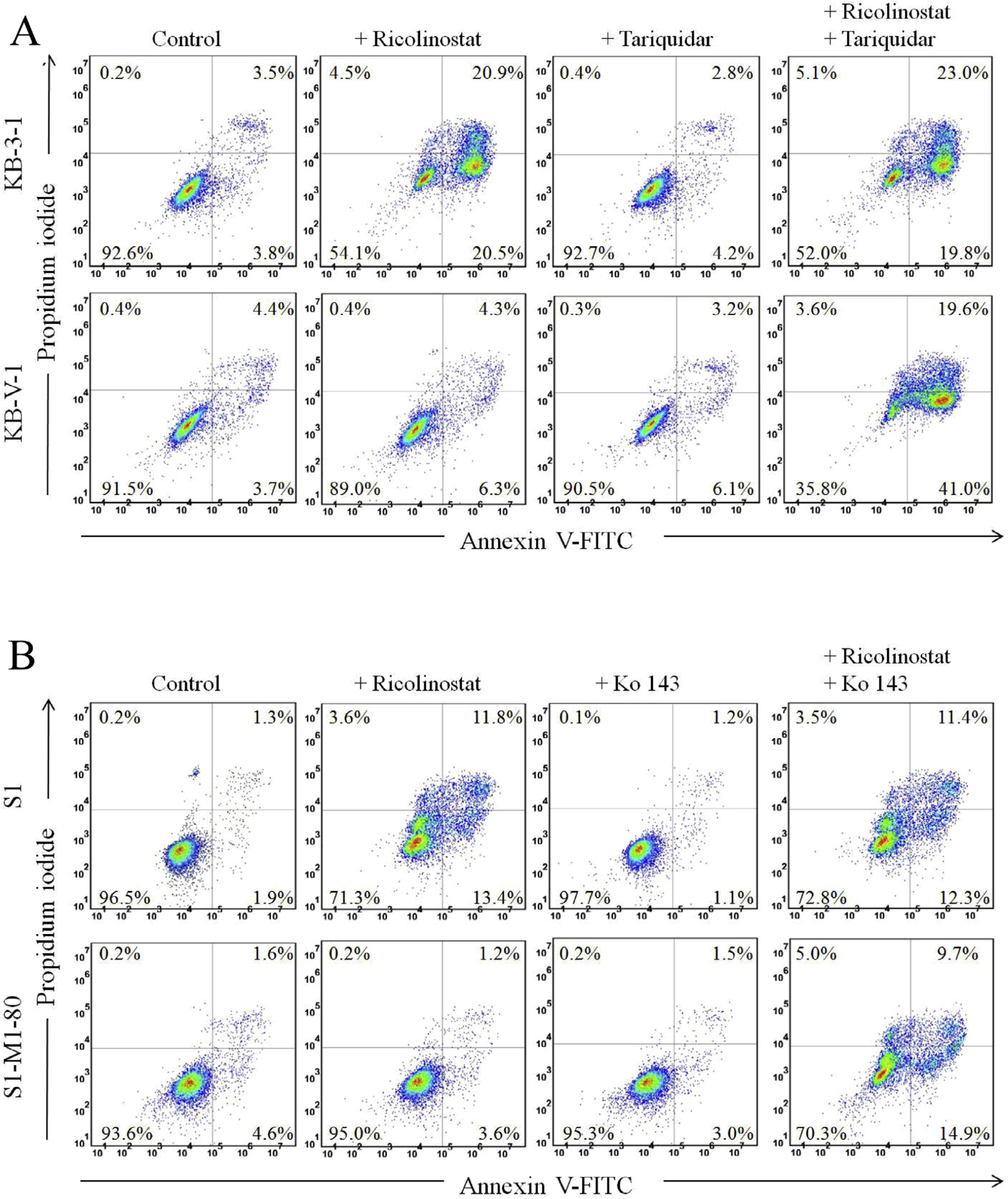
Ricolinostat induces apoptosis in human cancer cells. (A) KB-3–1 and KB-V-1 cells, as well as (B) S1 and S1-M1–80 cells were treated with DMSO (control), 20 µM of ricolinostat, 1 µM of tariquidar or 1 µM of Ko143 alone or in combination as indicated for 48 h before cells were processed and analyzed. Annexin V-FITC and PI staining method was used to detect apoptotic cells, which were quantified by flow cytometry as described previously [65]. Representative histograms are shown, and quantified values are mean values from three independent experiments.
3.2. The cytotoxicity of ricolinostat is reduced in cells overexpressing ABCB1 or ABCG2
Next, we examined cytotoxicity of ricolinostat in KB-3–1, KB-V-1, S1 and S1-M1–80 cells. We found that KB-V-1 cells (Fig. 3A, filled circles) and S1-M1–80 cells (Fig. 3B, filled circles) were significantly less sensitive to ricolinostat treatment compared to their drug-sensitive KB-3–1 and S1 parental cells (Fig. 3A and B, open circles). To confirm our observation, we tested the cytotoxicity of ricolinostat in multiple drug-sensitive and drug-resistant cell lines overexpressing either ABCB1 or ABCG2, as well as in HEK293 cells, HEK293 cells transfected with human ABCB1 (MDR19-HEK293) and HEK293 cells transfected with human ABCG2 (R482-HEK293). The IC50 values and the resistance factor (RF) values of ricolinostat in these cell lines are summarized in Table 1. The RF values were calculated by dividing the IC50 value of ricolinostat in drug-resistant variant by the IC50 value of ricolinostat in the respective parental line, representing the degree of cellular resistance to ricolinostat caused by the overexpression of ABCB1 or ABCG2. We found that all the ABCB1- and ABCG2-overexpressing variants we have tested were consistently less sensitive to ricolinostat treatment. ABCB1-overexpressing KB-V-1 and human NCI-ADR-RES ovarian cancer cells, as well as ABCB1-transfected NIH3T3-G185 cells were significantly less sensitive to ricolinostat as compared to ABCB1-negative parental cells, with RF values ranging from 6 to 10. Similarly, ABCG2-overexpressing S1-M1–80, human MCF7-FLV1000 and MCF7-AdVp3000 breast cancer cells were resistant to ricolinostat compared to that of respective ABCG2-negative parental cells, with RF values ranging from 6 to 23 (Table 1). As shown in Fig. 3C, MDR19-HEK293 cells (filled circles) and R48-HEK293 cells (open squares) were also resistant to ricolinostat, as compared to parental HEK293 cells (open circles), indicating that the expression of ABCB1 or ABCG2 reduced the cytotoxicity of ricolinostat. More importantly, we completely restored the sensitivity of ABCB1- and ABCG2-overexpressing cells to ricolinostat with 1 µM of tariquidar and Ko143 (Table 2). The fold reversal (FR) values were obtained by dividing IC50 values of cells treated with ricolinostat in the absence of tariquidar or Ko143 by IC50 values of cells treated with ricolinostat in the presence of tariquidar or Ko143. These results further demonstrate that the sensitivity to ricolinostat was dependent on the expression of ABCB1 and ABCG2 transporters whether the cells were exposed to anticancer drugs or transfected with a given gene (in the absence of anticancer drug) and other pleiotropic effects associated with the MDR phenomenon had no role.
Fig. 3.
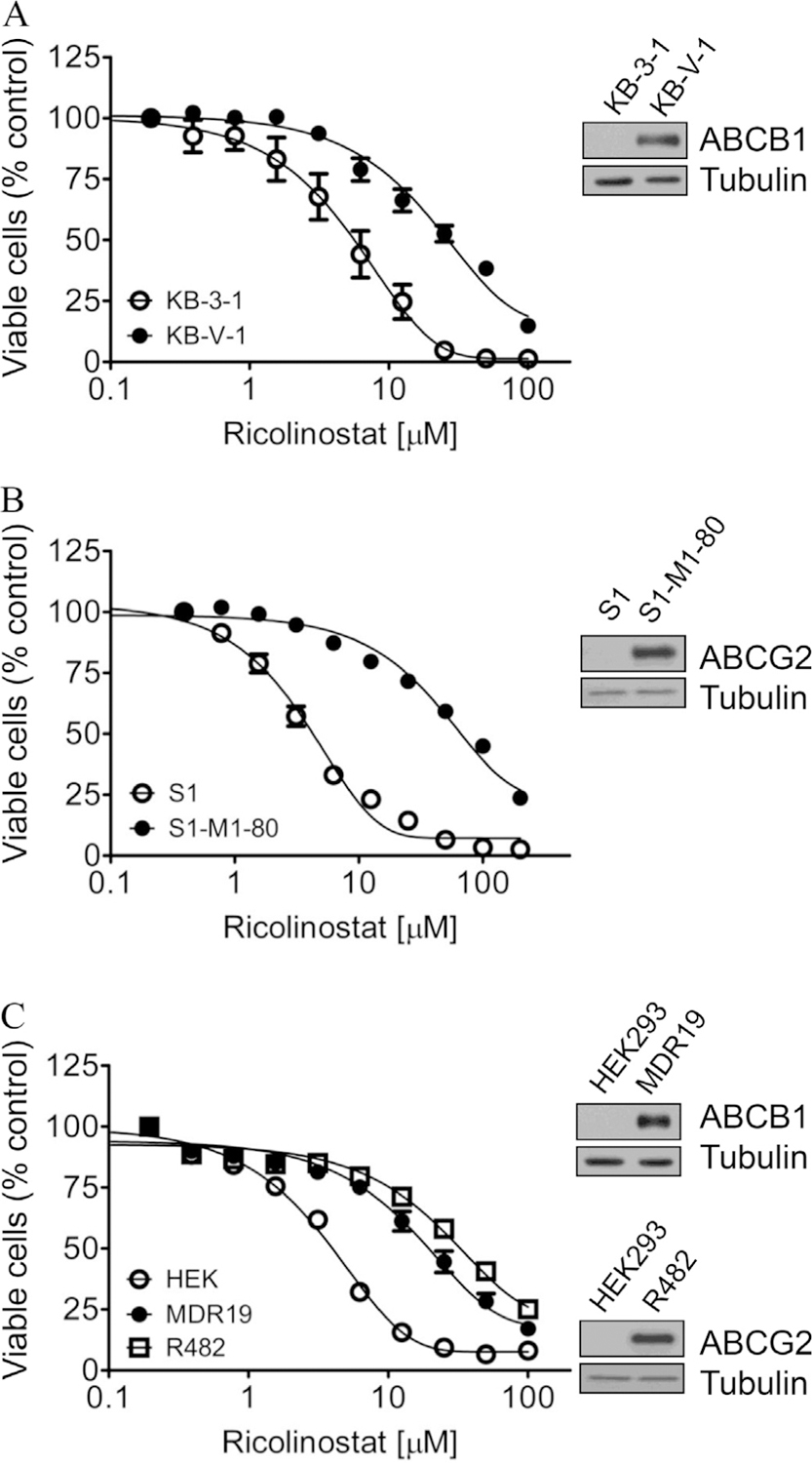
Dose-response curves for drug-sensitive versus drug-resistant cells overexpressing either ABCB1 or ABCG2 treated with ricolinostat. The cytotoxicity of (A) parental KB-3–1 cells (open circles) and ABCB1-overexpressing KB-V-1 cells (filled circles); (B) parental S1 cell line (open circles) and ABCG2-overexpressing S1-M1–80 cells (filled circles); as well as (C) parental pcDNA-HEK293 cells (open circles), ABCB1-transfected MDR19-HEK293 (filled circles) and ABCG2-transfected R482-HEK293 (open squares) cells, was determined over a period of 72 h as described previously [65]. The representative immunoblots of ABCB1, ABCG2 and tubulin as loading control in drug-sensitive and MDR cells are shown (inset). Points, mean values from at least three independent experiments; bars, SEM.
Table 1.
Cytotoxicity of ricolinostat in human cancer cell lines overexpressing ABCB1 or ABCG2.
| Cell line | Type | Transporter expressed | IC50 (μM)† | R.F‡ |
|---|---|---|---|---|
| KB-3–1 | epidermal | – | 4.28 ± 0.71 | 1.0 |
| KB-V-1 | epidermal | ABCB1 | 24.18 ± 2.05*** | 5.6 |
| OVCAR-8 | ovarian | – | 6.02 ± 0.91 | 1.0 |
| NCI-ADR-RES | ovarian | ABCB1 | 59.78 ± 7.98*** | 9.9 |
| S1 | colon | – | 3.06 ± 0.21 | 1.0 |
| S1-M1–80 | colon | ABCG2 | 69.01 ± 5.79*** | 22.6 |
| MCF7 | breast | – | 16.22 ± 3.58 | 1.0 |
| MCF7-FLV1000 | breast | ABCG2 | > 100 | > 6 |
| MCF7-AdVp3000 | breast | ABCG2 | > 100 | > 6 |
| NIH3T3 | – | – | 3.46 ± 0.70 | 1.0 |
| NIH3T3-G185 | – | ABCB1 | 32.57 ± 2.33*** | 9.4 |
| pcDNA-HEK293 | – | – | 3.40 ± 0.50 | 1.0 |
| MDR19-HEK293 | – | ABCB1 | 22.35 ± 2.05*** | 6.6 |
| R482-HEK293 | – | ABCG2 | 40.25 ± 4.36*** | 11.8 |
Abbreviation: RF, resistance factor.
IC50 values are mean ± SD calculated from dose-response curves obtained from three independent experiments using cytotoxicity assay as described in Materials and methods.
RF values were calculated by dividing IC50 values of ABC transporter overexpressing cells by IC50 values of respective parental cells.
P < 0.05;
P < 0.01;
P < 0.001.
Table 2.
The effect of tariquidar and Ko143 on the cytotoxicity of ricolinostat in cells overexpressing ABCB1 or ABCG2.
| Cell line | Mean IC50 ± SD [μM]† and (FR‡) |
||
|---|---|---|---|
| Ricolinostat | Ricolinostat + tariquidar | Ricolinostat + Ko143 | |
| KB-3–1 | 4.28 ± 0.71 (1.0) | 6.94 ± 1.83 (0.6) | N.D |
| KB-V-1 | 24.18 ± 2.05 (1.0) | 3.90 ± 1.09*** (6.2) | N.D |
| S1 | 3.06 ± 0.21 (1.0) | N.D | 3.02 ± 0.30 (1.0) |
| S1-M1–80 | 69.01 ± 5.79 (1.0) | N.D | 2.21 ± 0.18*** (31.2) |
| pcDNA-HEK293 | 3.40 ± 0.50 (1.0) | 2.05 ± 0.30* (1.7) | 2.19 ± 0.26* (1.6) |
| MDR19-HEK293 | 22.35 ± 2.05 (1.0) | 3.36 ± 0.44*** (6.7) | N.D |
| R482-HEK293 | 40.25 ± 4.36 (1.0) | N.D | 7.52 ± 1.01*** (5.4) |
Abbreviation: N.D, not determined. FR, fold reversal.
IC50 values are mean ± SD in the presence and absence of 1 μM tariquidar or 1 μM Ko143. The IC50 values were calculated from dose-response curves obtained from three independent experiments.
FR values were obtained by dividing IC50 values of cells treated with ricolinostat in the absence of tariquidar or Ko143 by IC50 values of cells treated with ricolinostat in the presence of tariquidar or Ko143.
P < 0.05;
P < 0.01;
P < 0.001.
The most likely cause for the reduced efficacy (Figs. 1 and 2) and cytotoxicity (Fig. 3) of ricolinostat in ABCB1- and ABCG2-overexpressing cells is the reduced intracellular accumulation of ricolinostat due to efflux of this compound-mediated by ABCB1 and ABCG2. Therefore, we directly measured the intracellular levels of ricolinostat (Fig. 4A) in HEK293, MDR19-HEK293 and R482-HEK293 cells in the presence or absence of tariquidar or Ko143 as described in Materials and methods. As shown in Fig. 4B, the level of ricolinostat accumulation in MDR19-HEK293 and R482-HEK293 cells was considerably lower than in parental HEK293 cells, which can be significantly restored by tariquidar and Ko143, respectively.
Fig. 4.
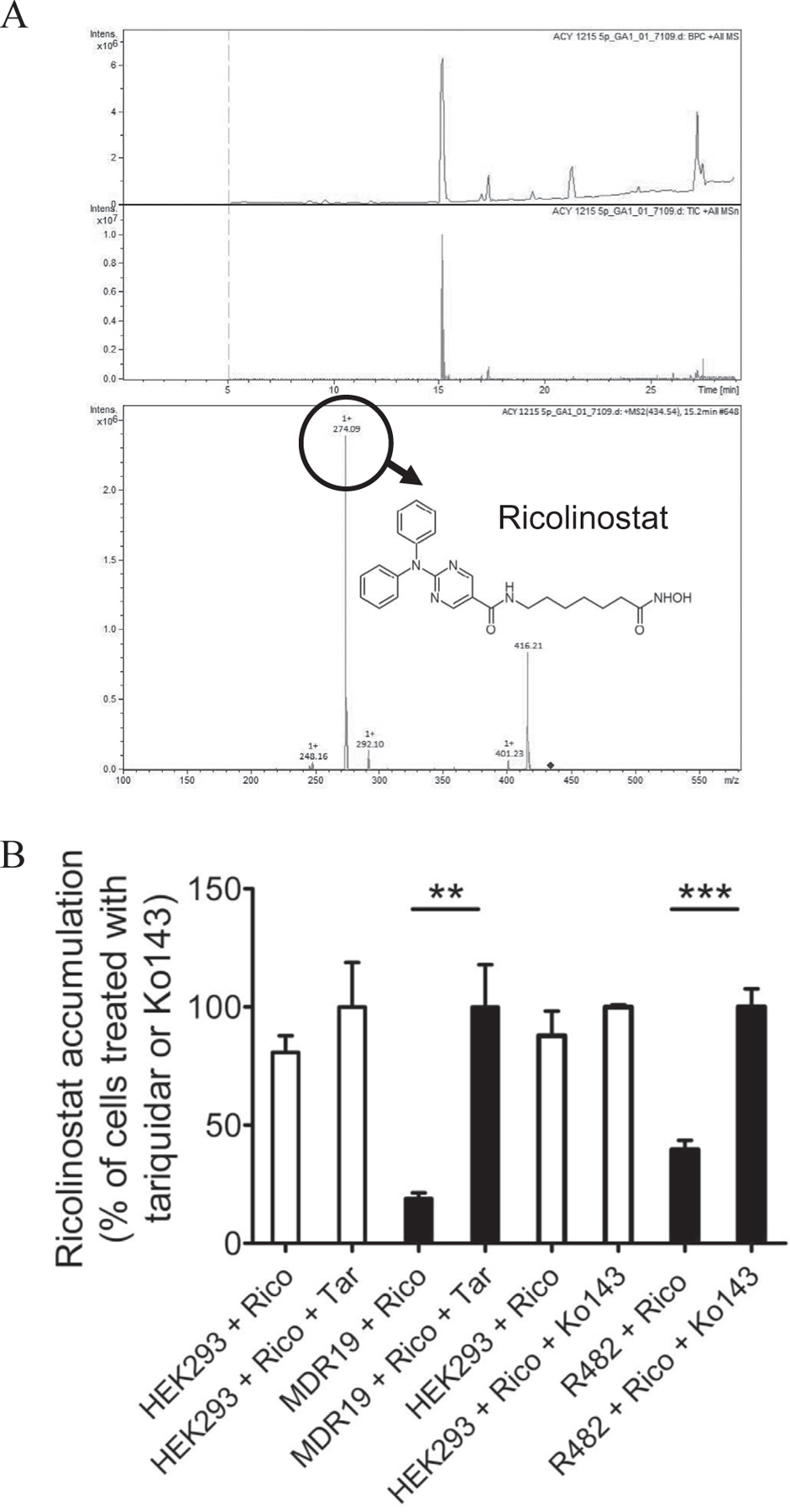
Intracellular accumulation of ricolinostat in cells overexpressing ABCB1 or ABCG2 was restored by inhibitors of ABC drug transporters. (A) The chemical structure and product ion mass spectra of ricolinostat. (B) Quantification of intracellular ricolinostat concentration in parental pcDNA-HEK293 cells (left panel, open bars) and in HEK293 cells transfected with human ABCB1, MDR19-HEK293 (left panel, filled bars), as well as in parental pcDNA-HEK293 cells (right panel, open bars) and in HEK293 cells transfected with human ABCG2, R482-HEK293 (right panel, filled bars). Cells were treated with 10 μM of ricolinostat in the presence or absence of 10 μM of tariquidar or Ko143 as indicated for 1 h before the intracellular concentration of ricolinostat was processed and quantified as described in Materials and methods. Values are presented as mean values ± S.D. calculated from at least three independent experiments. **P < 0.01; ***P < 0.001.
3.3. Ricolinostat stimulates ATPase activity of ABCB1 and ABCG2
To identify the potential binding sites and gain insight into the interaction of ricolinostat with ABCB1 and ABCG2, we examined the effect of ricolinostat on the ATPase activity of ABCB1 and ABCG2 and performed docking analysis of ricolinostat with modeled structure of ABCB1 and ABCG2 protein structure. We found that ricolinostat stimulated the vanadate-sensitive ATPase activity of ABCB1 and ABCG2 in a concentration-dependent manner. Ricolinostat produced a maximum stimulation of ABCB1 ATP hydrolysis to approximately 140% of the basal value (basal, 24.52 ± 8.13 nmole Pi/min/mg protein) and a concentration of approximately 7.5 μM required for 50% of maximum stimulation (Fig. 5A), whereas ricolinostat stimulated ABCG2 ATP hydrolysis to a maximum level of 140% of the basal value (basal, 71.31 ± 3.35 nmole Pi/min/mg protein) and a concentration of approximately 14.7 μM required for 50% of maximum stimulation (Fig. 5B). Knowing that the stimulation of ATP hydrolysis is generally coupled to substrate transport mediated by ABC transporters [43,44], our results indicate that ricolinostat behaves as a substrate for ABCB1 and ABCG2.
Fig. 5.
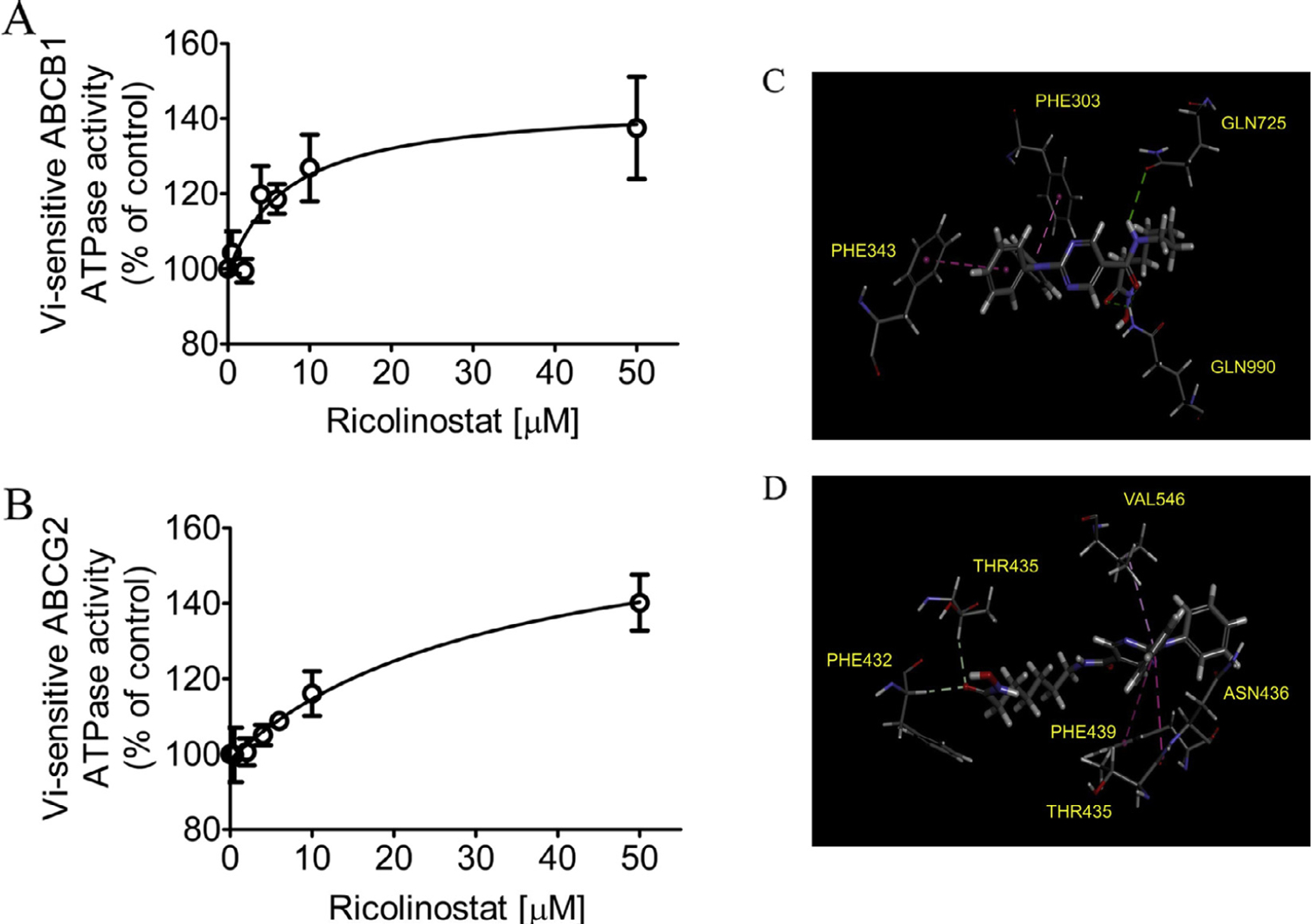
Ricolinostat stimulates the ATPase activity of ABCB1 and ABCG2. The effect of ricolinostat (0–50 µM) on (A) vanadate (Vi)-sensitive ABCB1 ATP hydrolysis and (B) vanadate (Vi)-sensitive ABCG2 ATP hydrolysis was determined by endpoint Pi assay, as described previously [70]. Points, mean from at least three independent experiments; bars, SD. Binding modes of ricolinostat with (C) homology modeled ABCB1 and (D) ABCG2 protein structure (PDB:5NJG) were predicted by Acclerys Discovery Studio 4.0 software as described in Materials and methods. Ricolinostat is shown as a molecular model with the atoms colored as carbongray, hydrogen-light gray, nitrogen-blue and oxygen-red. The same color scheme is used for interacting amino acid residues. Dotted lines indicate proposed interactions. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The homology model of human ABCB1 transporter protein was generated from online SWISS-MODEL server using mouse (Mus musculus) ABCB1 protein crystal structure (4Q9L) [37–39] as a template. Docking of ricolinostat with modeled ABCB1 protein structure revealed that this compound may bind to the substrate binding sites by interacting with 4 amino acid residues from different transmembrane helices. The phenyl rings on ricolinostat was predicted to interact with Phe303 and Phe343 via hydrophobic interaction and two possible hydrogen bonds were found between the amide moiety on ricolinostat and Gln725, Gln990 amino acid residues (Fig. 5C). The binding of ricolinostat with ABCG2 transporter protein was predicted to lie within the substrate binding cavity between two ABCG2 monomers. The carbonyl moiety on the tail of ricolinostat structure possibly formed hydrogen bonds with amino acids Thr435 and Phe432 on transmembrane domain 2 (TM2) while one aromatic ring formed hydrophobic interaction with Val546 on TM5a of the same monomer. Three amino acids residues from the other ABCG2 monomer Thr435 and Phe439 also appeared to interact with the aromatic ring on ricolinostat (Fig. 5D).
4. Discussion
HDACs, known to deacetylate lysine residues in histone and non-histone substrates [45], have emerged as an important epigenetic target in the development of anticancer agents in recent years [46,47]. Although several pan-HDAC inhibitors such as SAHA and LBH-589, and class I HDAC inhibitors such as FK-228 and PXD-101 have been tested in clinical trials, the lack of selectivity eventually led to unpredicted adverse effects in patients [48–50]. Therefore, scientists have now shifted their focus toward developing isotype-selective HDAC inhibitors [51–56]. Localized primarily in the cytoplasm, HDAC6 is responsible for the deacetylation of non-histone substrates such as α-tubulin and heat shock protein 90 (Hsp90) [41,57,58]. Studies have suggested that HDAC6-selective inhibitors should have considerably less side effects than pan-HDAC inhibitors and class I HDAC inhibitors [56,59]. Unlike non-selective pan-HDAC inhibitors vorinostat (SAHA) and panobinostat that target both class I and class IIb HDACs, ricolinostat has significantly higher activity against class IIb HDAC relative to class I HDACs [4,60]. Ricolinostat is the first selective inhibitor of HDAC6 that has been evaluated in clinical trials, both as a single agent and in combination therapy, to treat patients with multiple myeloma, relapsed chronic lymphocytic leukemia, relapsed or refractory lymphoid malignancies, unresectable or metastatic breast cancer and gynecological cancer. Therefore, it is crucial to identify the potential mechanisms of acquired resistance to ricolinostat and propose rational drug combination strategies.
Although a recent study demonstrated that upregulation of HDAC9, MAPK10, HELIOS, and FYN, or downregulation of SH3BP5 or LCK could lead to ricolinostat resistance in lymphoma cell lines [61], the potential impact of ABCB1 and ABCG2 on the efficacy of ricolinostat has not been investigated. This is surprising since the overexpression of ABCB1 or ABCG2 in cancer cells is known to confer resistance to and reduce the activity of many molecularly targeted anticancer agents [30,62–66] including HDAC inhibitors [67,68,30,34], and that exposing cancer cells to HDAC inhibitors has been shown to induce a multidrug resistance phenotype and affect treatment effectiveness [69]. Therefore, we decided to evaluate the pharmacological impact of ABCB1 and ABCG2 on the in vitro efficacy of ricolinostat in human cancer cell lines.
When treating cancer cells with ricolinostat, there is an apparent increase in acetylated tubulin levels in all cell lines tested, indicating inhibition of HDAC6 activity by ricolinostat. However, we noticed that ricolinostat was less effective in inhibiting the deacetylase activity of HDAC6 and inducing apoptosis in MDR cancer cell lines as compared to that in drug-sensitive parental cancer cell lines. By comparing the concentration of ricolinostat required to achieve 50% inhibition of the HDAC6 activity in cancer cell lines, we found that ricolinostat was 5-fold and 50-fold less effective in ABCB1-overexpressing KB-V-1 cells and ABCG2-overexpressing S1-M1–80 cells than parental KB-3–1 and S1 cells, respectively (Fig. 1). Next, we examined the cytotoxicity of ricolinostat in multiple drug-sensitive cancer cell lines and MDR cancer cells overexpressing ABCB1 or ABCG2, as well as HEK293 cells and HEK293 cells transfected with either human ABCB1 or human ABCG2. The cytotoxicity assay revealed that ABCB1- and ABCG2-overexpressing drug resistant cell lines are significantly more resistant to ricolinostat treatment than drug-sensitive parental cell lines (Table 1). More importantly, we discovered that we were able to fully restore the activity (Figs. 1 and 2) and toxicity (Table 2) of ricolinostat in ABCB1- or ABCG2-overexpressing MDR cells by co-administration of ricolinostat with an inhibitor of ABCB1 or ABCG2. To corroborate these results, we directly measured the accumulation of ricolinostat in HEK293, MDR19-HEK293 and R482-HEK293 cells, in the presence or absence of tariquidar or Ko143, respectively. We confirmed that the intracellular concentration of ricolinostat is significantly reduced in cells overexpressing ABCB1 or ABCG2, which can be restored by inhibiting the drug efflux function of ABCB1 and ABCG2 (Fig. 4). Surprisingly, based on the cytotoxicity of ricolinostat in MDR19-HEK293 and R482-HEK293 cells (Fig. 3C), we were expecting considerably lower level of ricolinostat accumulation in MDR19-HEK293 and R482-HEK293 cells than our experimental data (Fig. 4B). We suspected that non-specific binding of ricolinostat to membranes may contribute to a portion of ricolinostat accumulation detected in these cells, thus partially masking the true impact of ABCB1 and ABCG2 in MDR19-HEK293 and R482-HEK293 cells. Moreover, consistent with the cytotoxicity and drug accumulation data, we found that ricolinostat stimulated the ATPase activity of ABCB1 and ABCG2 (Fig. 5), indicating that ricolinostat is a drug substrate for both ABCB1 and ABCG2.
In summary, our results demonstrated that cells overexpressing ABCB1 or ABCG2 were significantly less sensitive to ricolinostat treatment. The inhibition of HDAC6 activity by ricolinostat and ricolinostat-induced apoptosis were substantially reduced by the transport function of ABCB1 and ABCG2 (Fig. 6). In addition, we found that the reduced sensitivity of cancer cells to ricolinostat treatment corresponds to reduced accumulation of ricolinostat in ABCB1 and ABCG2-overexpressing cells. This finding is supported by data showing that ricolinostat behaves as a substrate for both ABCB1 and ABCG2 by stimulating the ATPase activity of both ABCB1 and ABCG2. More significantly, we demonstrated that the efficacy and intracellular accumulation of ricolinostat in ABCB1- and ABCG2-overexpressing cancer cells can be fully restored by inhibiting the function of ABCB1 and ABCG2. Taken together, these findings support the conclusion that the overexpression ABCB1 or ABCG2 in cancer cells may potentially play a significant role in the development of resistance to ricolinostat in cancer cells, and should be further investigated.
Fig. 6.

A schematic diagram illustrating the impact of ABCB1 and ABCG2 on the efficacy ricolinostat in cancer cells. Efflux of ricolinostat mediated by ABCB1 or ABCG2 reduces the inhibitory effect ricolinostat on the activity of histone deacetylase 6 (HDAC6), cancer proliferation and migration.
Acknowledgments
This work was supported by funds from the Ministry of Science and Technology of Taiwan (MOST-105–2320-B-182–018 and MOST-106–2320-B-182–017), Chang Gung Medical Research Program (BMRPC17, CMRPD1D0153, CMRPD1G0112) and Taichung Veterans General Hospital (TCVGH-T1067802 and TCVGH-T1077802 to YSW). Drs. Suresh V. Ambudkar, Megumi Murakami and Shahrooz Vahedi were supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Abbreviations:
- MDR
multidrug resistance
- ABC
ATP-binding cassette
- HDAC
histone deacetylase
- FCS
fetal calf serum
- CCK-8
Cell Counting Kit-8
- MTT
3-(4,5-dimethylthiazol-yl)-2,5-diphenyllapatinibrazolium bromide
- Vi
sodium orthovanadate
- IMDM
Iscove’s Modified Dulbecco’s Medium
- RF
resistance factor
Footnotes
5. Conflict of interest
The authors declare no conflict of interest.
References
- [1].Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, et al. , Molecular sequelae of histone deacetylase inhibition in human malignant B cells, Blood 101 (2003) 4055–4062. [DOI] [PubMed] [Google Scholar]
- [2].Yoo CB, Jones PA, Epigenetic therapy of cancer: past, present and future, Nat. Rev 5 (2006) 37–50. [DOI] [PubMed] [Google Scholar]
- [3].Haggarty SJ, Koeller KM, Wong JC, Butcher RA, Schreiber SL, Multidimensional chemical genetic analysis of diversity-oriented synthesis-derived deacetylase inhibitors using cell-based assays, Chem. Biol 10 (2003) 383–396. [DOI] [PubMed] [Google Scholar]
- [4].Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL, Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation, Proc. Natl. Acad. Sci. U.S.A 100 (2003) 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, Richardson PG, et al. , Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: a multicentre phase 1b trial, Lancet Oncol 17 (2016) 1569–1578. [DOI] [PubMed] [Google Scholar]
- [6].Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, et al. , Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma, Clin. Cancer Res (2017). [DOI] [PMC free article] [PubMed]
- [7].Gillet JP, Gottesman MM, Mechanisms of multidrug resistance in cancer, Methods Mol. Biol 596 (2010) 47–76. [DOI] [PubMed] [Google Scholar]
- [8].Wu CP, Hsieh CH, Wu YS, The emergence of drug transporter-mediated multidrug resistance to cancer chemotherapy, Mol. Pharm 8 (2011) 1996–2011. [DOI] [PubMed] [Google Scholar]
- [9].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP-dependent transporters, Nat. Rev. Cancer 2 (2002) 48–58. [DOI] [PubMed] [Google Scholar]
- [10].Brozik A, Hegedus C, Erdei Z, Hegedus T, Ozvegy-Laczka C, Szakacs G, et al. , Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin. Drug Metab. Toxicol 7 (2011) 623–642. [DOI] [PubMed] [Google Scholar]
- [11].Camidge DR, Pao W, Sequist LV, Acquired resistance to TKIs in solid tumours: learning from lung cancer, Nat. Rev. Clin. Oncol 11 (2014) 473–481. [DOI] [PubMed] [Google Scholar]
- [12].Noguchi K, Katayama K, Sugimoto Y, Human ABC transporter ABCG2/BCRP expression in chemoresistance: basic and clinical perspectives for molecular cancer therapeutics, Pharmacogenomics Personalized Med 7 (2014) 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pilarski LM, Belch AR, Intrinsic expression of the multidrug transporter, P-glycoprotein 170, in multiple myeloma: implications for treatment, Leuk. Lymphoma 17 (1995) 367–374. [DOI] [PubMed] [Google Scholar]
- [14].Pilarski LM, Szczepek AJ, Belch AR, Deficient drug transporter function of bone marrow-localized and leukemic plasma cells in multiple myeloma, Blood 90 (1997) 3751–3759. [PubMed] [Google Scholar]
- [15].Schwarzenbach H, Expression of MDR1/P-glycoprotein, the multidrug resistance protein MRP, and the lung-resistance protein LRP in multiple myeloma, Med. Oncol 19 (2002) 87–104. [DOI] [PubMed] [Google Scholar]
- [16].Nakagawa Y, Abe S, Kurata M, Hasegawa M, Yamamoto K, Inoue M, et al. , IAP family protein expression correlates with poor outcome of multiple myeloma patients in association with chemotherapy-induced overexpression of multidrug resistance genes, Am. J. Hematol 81 (2006) 824–831. [DOI] [PubMed] [Google Scholar]
- [17].Tsubaki M, Satou T, Itoh T, Imano M, Komai M, Nishinobo M, et al. , Overexpression of MDR1 and survivin, and decreased Bim expression mediate multidrug-resistance in multiple myeloma cells, Leuk. Res 36 (2012) 1315–1322. [DOI] [PubMed] [Google Scholar]
- [18].Turner JG, Gump JL, Zhang C, Cook JM, Marchion D, Hazlehurst L, et al. , ABCG2 expression, function, and promoter methylation in human multiple myeloma, Blood 108 (2006) 3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hofmeister CC, Yang X, Pichiorri F, Chen P, Rozewski DM, Johnson AJ, et al. , Phase I trial of lenalidomide and CCI-779 in patients with relapsed multiple myeloma: evidence for lenalidomide-CCI-779 interaction via P-glycoprotein, J. Clin. Oncol 29 (2011) 3427–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ross DD, Karp JE, Chen TT, Doyle LA, Expression of breast cancer resistance protein in blast cells from patients with acute leukemia, Blood 96 (2000) 365–368. [PubMed] [Google Scholar]
- [21].Steinbach D, Sell W, Voigt A, Hermann J, Zintl F, Sauerbrey A, BCRP gene expression is associated with a poor response to remission induction therapy in childhood acute myeloid leukemia, Leukemia 16 (2002) 1443–1447. [DOI] [PubMed] [Google Scholar]
- [22].Uggla B, Stahl E, Wagsater D, Paul C, Karlsson MG, Sirsjo A, et al. , BCRP mRNA expression v. clinical outcome in 40 adult AML patients, Leuk. Res 29 (2005) 141–146. [DOI] [PubMed] [Google Scholar]
- [23].Matthews C, Catherwood MA, Larkin AM, Clynes M, Morris TC, Alexander HD, MDR-1, but not MDR-3 gene expression, is associated with unmutated IgVH genes and poor prognosis chromosomal aberrations in chronic lymphocytic leukemia, Leuk. Lymphoma 47 (2006) 2308–2313. [DOI] [PubMed] [Google Scholar]
- [24].Kovalev AA, Tsvetaeva DA, Grudinskaja TV, Role of ABC-cassette transporters (MDR1, MRP1, BCRP) in the development of primary and acquired multiple drug resistance in patients with early and metastatic breast cancer, Exp. Oncol 35 (2013) 287–290. [PubMed] [Google Scholar]
- [25].Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM, Targeting multidrug resistance in cancer, Nat. Rev 5 (2006) 219–234. [DOI] [PubMed] [Google Scholar]
- [26].Shen DW, Fojo A, Chin JE, Roninson IB, Richert N, Pastan I, et al. , Human multidrug-resistant cell lines: increased mdr1 expression can precede gene amplification, Science 232 (1986) 643–645. [DOI] [PubMed] [Google Scholar]
- [27].Honjo Y, Hrycyna CA, Yan QW, Medina-Perez WY, Robey RW, van de Laar A, et al. , Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells, Cancer Res 61 (2001) 6635–6639. [PubMed] [Google Scholar]
- [28].Currier SJ, Kane SE, Willingham MC, Cardarelli CO, Pastan I, Gottesman MM, Identification of residues in the first cytoplasmic loop of P-glycoprotein involved in the function of chimeric human MDR1-MDR2 transporters, J. Biol. Chem 267 (1992) 25153–25159. [PubMed] [Google Scholar]
- [29].Wu CP, Shukla S, Calcagno AM, Hall MD, Gottesman MM, Ambudkar SV, Evidence for dual mode of action of a thiosemicarbazone, NSC73306: a potent substrate of the multidrug resistance linked ABCG2 transporter, Mol. Cancer Ther 6 (2007) 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wu CP, Hsiao SH, Su CY, Luo SY, Li YQ, Huang YH, et al. , Human ATP-binding cassette transporters ABCB1 and ABCG2 confer resistance to CUDC-101, a multi-acting inhibitor of histone deacetylase, epidermal growth factor receptor and human epidermal growth factor receptor 2, Biochem. Pharmacol 92 (2014) 567–576. [DOI] [PubMed] [Google Scholar]
- [31].Ishiyama M, Tominaga H, Shiga M, Sasamoto K, Ohkura Y, Ueno K, A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet, Biol. Pharm. Bull 19 (1996) 1518–1520. [DOI] [PubMed] [Google Scholar]
- [32].Anderson HA, Maylock CA, Williams JA, Paweletz CP, Shu H, Shacter E, Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells, Nat. Immunol 4 (2003) 87–91. [DOI] [PubMed] [Google Scholar]
- [33].Ambudkar SV, Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells, Methods Enzymol 292 (1998) 504–514. [DOI] [PubMed] [Google Scholar]
- [34].Wu CP, Hsieh YJ, Hsiao SH, Su CY, Li YQ, Huang YH, et al. , Human ATP-binding cassette transporter ABCG2 confers resistance to CUDC-907, a dual inhibitor of histone deacetylase and phosphatidylinositol 3-kinase, Mol. Pharm 13 (2016) 784–794. [DOI] [PubMed] [Google Scholar]
- [35].Cihalova D, Ceckova M, Kucera R, Klimes J, Staud F, Dinaciclib, a cyclin-dependent kinase inhibitor, is a substrate of human ABCB1 and ABCG2 and an inhibitor of human ABCC1 in vitro, Biochem. Pharmacol 98 (2015) 465–472. [DOI] [PubMed] [Google Scholar]
- [36].Shen C, Chen R, Qian Z, Huang C, Meng X, Ma T, et al. , A HPLC-MS/MS method for the quantitation of free, conjugated, and total HDND-7, a novel hesperetin derivative, in rat plasma and tissues: application to the pharmacokinetic and tissue distribution study, J. Pharm. Biomed. Anal 118 (2015) 149–160. [DOI] [PubMed] [Google Scholar]
- [37].Arnold K, Bordoli L, Kopp J, Schwede T, The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling, Bioinformatics 22 (2006) 195–201. [DOI] [PubMed] [Google Scholar]
- [38].Benkert P, Biasini M, Schwede T, Toward the estimation of the absolute quality of individual protein structure models, Bioinformatics 27 (2011) 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, et al. , SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information, Nucl. Acids Res 42 (2014) W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Taylor NMI, Manolaridis I, Jackson SM, Kowal J, Stahlberg H, Locher KP, Structure of the human multidrug transporter ABCG2, Nature 546 (2017) 504–509. [DOI] [PubMed] [Google Scholar]
- [41].Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. , HDAC6 is a microtubule-associated deacetylase, Nature 417 (2002) 455–458. [DOI] [PubMed] [Google Scholar]
- [42].Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. , Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma, Blood 119 (2012) 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM, Biochemical, cellular, and pharmacological aspects of the multidrug transporter, Annu. Rev. Pharmacol. Toxicol 39 (1999) 361–398. [DOI] [PubMed] [Google Scholar]
- [44].Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM, P-glycoprotein: from genomics to mechanism, Oncogene 22 (2003) 7468–7485. [DOI] [PubMed] [Google Scholar]
- [45].Minucci S, Pelicci PG, Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer, Nat. Rev. Cancer 6 (2006) 38–51. [DOI] [PubMed] [Google Scholar]
- [46].Weichert W, HDAC expression and clinical prognosis in human malignancies, Cancer Lett 280 (2009) 168–176. [DOI] [PubMed] [Google Scholar]
- [47].Falkenberg KJ, Johnstone RW, Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders, Nat. Rev 13 (2014) 673–691. [DOI] [PubMed] [Google Scholar]
- [48].Ramalingam SS, Belani CP, Ruel C, Frankel P, Gitlitz B, Koczywas M, et al. , Phase II study of belinostat (PXD101), a histone deacetylase inhibitor, for second line therapy of advanced malignant pleural mesothelioma, J. Thoracic Oncol.: Official Publ. Int. Assoc. Study Lung Cancer 4 (2009) 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Reid T, Valone F, Lipera W, Irwin D, Paroly W, Natale R, et al. , Phase II trial of the histone deacetylase inhibitor pivaloyloxymethyl butyrate (Pivanex, AN-9) in advanced non-small cell lung cancer, Lung Cancer 45 (2004) 381–386. [DOI] [PubMed] [Google Scholar]
- [50].Fraczek J, Vanhaecke T, Rogiers V, Toxicological and metabolic considerations for histone deacetylase inhibitors, Expert Opin. Drug Metab. Toxicol 9 (2013) 441–457. [DOI] [PubMed] [Google Scholar]
- [51].Kozikowski AP, Tapadar S, Luchini DN, Kim KH, Billadeau DD, Use of the nitrile oxide cycloaddition (NOC) reaction for molecular probe generation: a new class of enzyme selective histone deacetylase inhibitors (HDACIs) showing picomolar activity at HDAC6, J. Med. Chem 51 (2008) 4370–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kalin JH, Bergman JA, Development and therapeutic implications of selective histone deacetylase 6 inhibitors, J. Med. Chem 56 (2013) 6297–6313. [DOI] [PubMed] [Google Scholar]
- [53].Olson DE, Wagner FF, Kaya T, Gale JP, Aidoud N, Davoine EL, et al. , Discovery of the first histone deacetylase 6/8 dual inhibitors, J. Med. Chem 56 (2013) 4816–4820. [DOI] [PubMed] [Google Scholar]
- [54].Thaler F, Mercurio C, Towards selective inhibition of histone deacetylase isoforms: what has been achieved, where we are and what will be next, ChemMedChem 9 (2014) 523–526. [DOI] [PubMed] [Google Scholar]
- [55].Tang G, Wong JC, Zhang W, Wang Z, Zhang N, Peng Z, et al. , Identification of a novel aminotetralin class of HDAC6 and HDAC8 selective inhibitors, J. Med. Chem 57 (2014) 8026–8034. [DOI] [PubMed] [Google Scholar]
- [56].Lin X, Chen W, Qiu Z, Guo L, Zhu W, Li W, et al. , Design and synthesis of orally bioavailable aminopyrrolidinone histone deacetylase 6 inhibitors, J. Med. Chem 58 (2015) 2809–2820. [DOI] [PubMed] [Google Scholar]
- [57].Boyault C, Sadoul K, Pabion M, Khochbin S, HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination, Oncogene 26 (2007) 5468–5476. [DOI] [PubMed] [Google Scholar]
- [58].Kekatpure VD, Dannenberg AJ, Subbaramaiah K, HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling, J. Biol. Chem 284 (2009) 7436–7445. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [59].Govindarajan N, Rao P, Burkhardt S, Sananbenesi F, Schluter OM, Bradke F, et al. , Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease, EMBO Mol. Med 5 (2013) 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zagni C, Floresta G, Monciino G, Rescifina A, The Search for potent, small-molecule HDACIs in cancer treatment: a decade after vorinostat, Med. Res. Rev (2017). [DOI] [PubMed]
- [61].Amengual JE, Prabhu SA, Lombardo M, Zullo K, Johannet PM, Gonzalez Y, et al. , Mechanisms of acquired drug resistance to the HDAC6 selective inhibitor ricolinostat reveals rational drug-drug combination with Ibrutinib, Clin. Cancer Res (2016). [DOI] [PMC free article] [PubMed]
- [62].Thomas J, Wang L, Clark RE, Pirmohamed M, Active transport of imatinib into and out of cells: implications for drug resistance, Blood 104 (2004) 3739–3745. [DOI] [PubMed] [Google Scholar]
- [63].Nakanishi T, Shiozawa K, Hassel BA, Ross DD, Complex interaction of BCRP/ ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression, Blood 108 (2006) 678–684. [DOI] [PubMed] [Google Scholar]
- [64].Hegedus C, Ozvegy-Laczka C, Szakacs G, Sarkadi B, Interaction of ABC multidrug transporters with anticancer protein kinase inhibitors: substrates and/or inhibitors? Curr. Cancer Drug Targets 9 (2009) 252–272. [DOI] [PubMed] [Google Scholar]
- [65].Wu CP, Sim HM, Huang YH, Liu YC, Hsiao SH, Cheng HW, et al. , Overexpression of ATP-binding cassette transporter ABCG2 as a potential mechanism of acquired resistance to vemurafenib in BRAF(V600E) mutant cancer cells, Biochem. Pharmacol 85 (2013) 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wu CP, Hsieh CH, Hsiao SH, Luo SY, Su CY, Li YQ, et al. , Human ATP-binding cassette transporter ABCB1 confers resistance to Volasertib (BI 6727), a selective inhibitor of polo-like Kinase 1, Mol. Pharm 12 (2015) 3885–3895. [DOI] [PubMed] [Google Scholar]
- [67].Xiao JJ, Foraker AB, Swaan PW, Liu S, Huang Y, Dai Z, et al. , Efflux of depsipeptide FK228 (FR901228, NSC-630176) is mediated by P-glycoprotein and multidrug resistance-associated protein 1, J. Pharmacol. Exp. Ther 313 (2005) 268–276. [DOI] [PubMed] [Google Scholar]
- [68].Xiao JJ, Huang Y, Dai Z, Sadee W, Chen J, Liu S, et al. , Chemoresistance to depsipeptide FK228 [(E)-(1S,4S,10S,21R)-7-[(Z)-ethylidene]-4,21-diisopropyl-2-oxa-12,13-dithia-5,8,2 0,23-tetraazabicyclo[8,7,6]-tricos-16-ene-3,6,9,22-penta-none] is mediated by reversible MDR1 induction in human cancer cell lines, J Pharmacol. Exp. Ther 314 (2005) 467–475. [DOI] [PubMed] [Google Scholar]
- [69].Hauswald S, Duque-Afonso J, Wagner MM, Schertl FM, Lubbert M, Peschel C, et al. , Histone deacetylase inhibitors induce a very broad, pleiotropic anticancer drug resistance phenotype in acute myeloid leukemia cells by modulation of multiple ABC transporter genes, Clin. Cancer Res 15 (2009) 3705–3715. [DOI] [PubMed] [Google Scholar]
- [70].Wu CP, Hsiao SH, Sim HM, Luo SY, Tuo WC, Cheng HW, et al. , Human ABCB1 (P-glycoprotein) and ABCG2 mediate resistance to BI 2536, a potent and selective inhibitor of Polo-like kinase 1, Biochem. Pharmacol 86 (2013) 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]