

Abstract
Obese asthma is a phenotype of asthma whose occurrence is gradually increasing in both adults and children. The majority of studies have demonstrated that obesity is a major risk factor for asthma and the effect of obesity on the lungs is considerable. NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome has been previously demonstrated to serve a role in obese asthma mediated by mitochondrial reactive oxygen species (mtROS). The aim of the present in vitro study was to investigate the effect of leptin on airway epithelial cells and the protective effect of the mitochondrial-targeted antioxidant mitoquinone (mitoQ). Human normal bronchial epithelial cell lines BEAS-2 cells were used and divided into 6 groups: Control group (negative control), DMSO group (solvent control), lipopolysaccharide (LPS) group (positive control), LPS + mitoQ group, Leptin group and Leptin + mitoQ group. CCK8 assay was used to establish the optimal concentration and incubation time of the drugs. mitoTracker probe and mitoSOX reagent were used to detect the integrity of mitochondrial membranes and the content of mtROS. mRNA expression levels were detected by reverse transcription-quantitative PCR analysis. It was revealed that the mitochondrial membrane was disrupted in the Leptin group, which recovered after treatment with mitoQ. As a result, the production of mitochondrial reactive oxygen species (mtROS) in the Leptin group was significantly increased (P<0.01), but following treatment with mitoQ, this overproduction of mtROS was significantly decreased to normal levels (P<0.01). Furthermore, the expression levels of NOD-, LRR- and pyrin domain-containing protein 3 NLRP3 and caspase-1 mRNA in the leptin-pretreated BEAS-2 cells were significantly increased compared with those in the control group (P<0.01), while they were decreased following mitoQ treatment (P<0.01). Taken together, these data suggested that leptin may promote airway inflammation partially through upregulating the mtROS-NLRP3 inflammasome signaling pathway in airway epithelial cells and mitoQ may be a potential treatment for obese asthma.
Keywords: obese asthma, leptin, mitoquinone, mitochondrial reactive oxygen species, NOD-, LRR- and pyrin domain-containing protein 3 inflammasome
Introduction
Obesity and asthma are among the most significant public health problems worldwide. An expanding body of epidemiological evidence and longitudinal data suggest that there is a link between obesity and asthma in both children and adults (1-5). Furthermore, obesity is a major risk factor for asthma (6-9). However, managing patients with asthma who are also impacted by obesity is challenging, as they are frequently less likely to respond to conventional asthma therapies (10). Therefore, investigating the potential mechanisms underlying obesity-associated asthma is of great significance for the treatment of these patients.
In previous years, there has been an immense interest in the potential role of adipokines, factors secreted by adipocytes, in the development or worsening of asthma among obese individuals (11). Adipokines, including leptin, adiponectin and resistin, regulate energy homeostasis via hunger and satiety control (12). An imbalance of adipokines may promote pro-inflammatory responses and lead to inflammation. An increased leptin/adiponectin ratio may be an important mediator of type 2 diabetes and abdominal obesity-associated cardiovascular diseases (13). In obese patients, an increased serum concentration of leptin and resistin, and decreased adiponectin levels have been detected (14). In a six-year follow-up study performed in 246 obese and 532 non-obese children aged 6-11 years, Zhang et al (15) illustrated that the levels of leptin and leptin-to-adiponectin ratio in obese children were significantly higher than those in non-obese children. Given the role of the increased leptin/adiponectin ratio in systemic inflammation, it is plausible to hypothesize that there is an effect of increased leptin in the airway inflammation of respiratory diseases, such as asthma.
Using a newly developed analytic method, a longitudinal study from France demonstrated that leptin is an important factor mediating the association between high body mass index (BMI) and persistent asthma over time (16). Through collecting bronchoalveolar lavage (BAL) by bronchoscopy from lean and obese asthmatics and controls, Holguin et al (17) demonstrated that increases of the BMI were positively associated with the concentration of leptin in BAL fluid (BALF). A pilot study (18) evaluating the levels of leptin in exhaled breath condensate (EBC) from overweight asthmatic pediatric patients and normal children indicated that the leptin levels in EBC were significantly higher in the obese and asthmatic children as compared with those in healthy subjects, demonstrating that leptin may represent a non-invasive marker of airway inflammation in children. However, whether there is an effect of leptin on airway inflammation in obese patients with asthma and the way in which leptin affects airway inflammation remain to be elucidated.
The NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, a multi-protein complex consisting of NLRP3 as the sensor, apoptosis-associated speck-like protein containing caspase-recruitment domain (ASC) as the adaptor and caspase 1 as the effector, is known to be associated with a variety of responses to a wide range of microbial pathogens, inflammatory diseases, cancer and metabolic and autoimmune disorders (19). A growing body of evidence has suggested a role of the NLRP3 inflammasome in the inflammation of respiratory diseases, such as asthma and chronic obstructive pulmonary disease (COPD) (20,21). Kim et al (22) indicated that the NLRP3 inflammasome was required in the lung inflammation of obese asthmatic mice. Using an ozone-induced mouse model of acute allergic airway inflammation, Xu et al (23) demonstrated that the activation of the NLRP3 inflammasome caused by mitochondrial reactive oxygen species (mtROS) may have an important role in the pathogenesis of ozone-induced airway inflammation.
On the other hand, two recent studies have demonstrated that leptin may be a novel activator and modulator of the NLRP3 inflammasome in RAW 264.7 cells (24) and breast cancer cells (25). Bronchial epithelial cells (BECs) also express leptin receptors (26). Thus, it is reasonable to postulate that leptin may activate mtROS-NLRP3 inflammasomes in BECs, resulting in airway inflammation. Targeting the mtROS-NLRP3 inflammasome pathway may potentially be a therapeutic direction for the management of obese asthma.
Mitoquinone (mitoQ), a ubiquinone moiety linked to a lipophilic triphenylphosphonium cation by a 10-carbon alkyl chain, is a novel mitochondrial-targeted antioxidant (27). The oral administration of mitoQ is safe and numerous in vivo studies have demonstrated that mitoQ is able to protect against oxidative damage in a number of diseases, including cardiac ischemia-reperfusion injury (28), hypertension (29), sepsis (30) and metabolic syndrome (31). Dashdorj et al (32) indicated that mitoQ has anti-inflammatory effects, as the activation of the NLRP3 inflammasome and the expression of its downstream cytokines were significantly decreased in a mouse model of experimental colitis after treatment of mitoQ. Another study demonstrated that mitoQ may ameliorate diabetic nephropathy via the inhibition of mtROS/thioredoxin-interacting protein (TXNIP)/NLRP3/IL-1β axis activation (33). However, whether there is a role of mitoQ in the airway inflammation of obese asthma has remained to be elucidated.
In the present study, leptin and mitoQ were used to treat the human bronchial epithelial cell line BEAS-2 to investigate the activation of the mtROS-NLRP3 inflammasome by leptin and the potential role of mitoQ in leptin-pretreated BEAS-2 cells.
Materials and methods
Chemicals and reagents
BEAS-2B cells (no. ATCC® CRL-9609™) were obtained from the American Type Culture Collection and were tested negative for mycoplasma. DMEM-high glucose, FBS and Trypsin-0.25% EDTA were from Gibco (Thermo Fisher Scientific, Inc.). Recombinant human leptin was purchased from Novoprotein Scientific Inc. Lipopolysaccharide (LPS) was purchased from Sigma-Aldrich (Merck KGaA). mitoQ was purchased from Focus Biomolecules. DMSO and penicillin-streptomycin (pen-strep) were obtained from Solarbio. The Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies. MitoSOX™ Red mitochondrial superoxide indicator for live-cell imaging (cat. no. M36008), MitoTracker® Red CM-H2XRos (cat. no. M7513) and TRIzol® reagent were purchased from Invitrogen (Thermo Fisher Scientific, Inc.). DAPI staining solution was obtained from Beyotime Institute of Biotechnology, Inc. PrimeScript™ RT Master Mix (cat. no. RR036A) was obtained from Takara Bio, Inc. LightCycler® 480 SYBR-Green I Master Mix (cat. no. 04707516001) was purchased from Roche Diagnostics. ELISA kit (cat. no. ml058059; Shanghai Enzyme-linked Biotechnology Co., Ltd.) and all other chemicals of the highest purity available were purchased from local companies.
Cell culture
BEAS-2B cells were routinely cultured in high-glucose DMEM supplemented with 10% FBS and 1% pen-strep in an incubator at 37˚C in a humidified atmosphere containing 5% CO2. Cells were harvested using Trypsin-0.25% EDTA solution and all treatments were performed on cells at their 4th passages to ensure the stability of cells.
CCK-8 viability assay
In order to determine the optimal concentration and effect time of leptin, LPS and mitoQ, a CCK-8 assay was used. BEAS-2B cells were seeded in flat-bottom 96-well plates at 2x104 cells/well in triplicate with 100 µl of medium 24 h prior to the treatments. For the treatments, culture media was replaced with 100 µl of fresh complete medium that was supplemented with different concentrations of leptin (0, 1, 20, 40, 60, 80, 100 and 200 ng/ml) or LPS (0, 1, 2.5, 5, 10, 25, 50 and 100 µg/µl) or mitoQ (0, 100, 200, 500, 1,000, 1,500, 2,000 and 5,000 nM) in an incubator at 37˚C in a humidified atmosphere containing 5% CO2. At the end of the drug incubation periods (1, 6, 24, 48 or 72 h), the culture media were discarded and the plates were washed with PBS three times. Subsequently, 10 µl CCK-8 solution was added, followed by incubation in the incubator for 1 h at 37˚C. The optical density at 450 nm (OD450) was then determined on a microplate reader (Bio-Rad Laboratories, Inc.). The cell viability reflecting the cytotoxicity of drugs was calculated according to the instructions of the kit, using the following formula: Cell viability=(A-C)/(B-C), where A is the OD450 of the experimental group (culture medium with cells, drugs and CCK8 solution), B the OD450 of the control group (culture medium with cells and CCK8 solution) and C the OD450 of the empty group (culture medium with CCK8 solution but without cells).
Cell groups
After determining the optimal concentration and effect time of leptin, LPS and mitoQ, BEAS-2B cells were seeded in flat-bottom 6-well plates at 2x105 cells/well. According to the treatments with the drugs, the cells were divided into 6 groups: Control group (negative control), DMSO group (solvent control), LPS group (positive control), LPS + mitoQ group, Leptin group and Leptin + mitoQ group. For LPS + mitoQ group, 5 µg/µl LPS was added 6 h prior to detection and after that 200 nM mitoQ was added 1 h before detection. For Leptin + mitoQ group, 100 ng/ml leptin was added 24 h before collection and after that 200 nM mitoQ was added 1 h before collection. All the treatments were performed in an incubator at 37˚C in a humidified atmosphere containing 5% CO2.
mitoTracker measurement
After the drug treatments, the culture media were removed and prewarmed in a staining solution containing 200 nM MitoTracker® probe for 30 min at 37˚C in the dark. After staining was complete, the staining solution was replaced with fresh prewarmed PBS and cells were then observed using an inverted fluorescence microscope (magnification, x100; Ti-U; Nikon Corporation). The mean fluorescence intensity (MFI) was analyzed and calculated using ImageJ software 1.48v (National Institutes of Health).
mtROS measurement
After the treatments, the culture media were discarded and the plates were washed with PBS three times and loaded with MitoSOX™ reagent. First, 1 ml of 5 µM MitoSOX™ reagent was added as a working solution to cover the cells. Subsequently, the cells were incubated for 10 min at 37˚C in the dark. Next, cells were washed gently three times with warm PBS and fixed with 4% formaldehyde for 15 min at room temperature. Cells were washed again and covered with warm PBS. The cells were observed using an inverse fluorescence microscope (magnification, x100; Nikon Corporation). The MFI was analyzed and calculated using ImageJ software.
RNA extraction, complementary (c)DNA synthesis and reverse transcription-quantitative (RT-q)PCR analysis
Total RNA of cells was extracted using TRIzol® reagent. The quality and concentration of total RNA were determined using a Nanodrop® 2000 spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was synthesized from 500 ng RNA using the PrimeScript™ RT Master Mix, according to the manufacturer's protocol. qPCR was performed in triplicate using LightCycler® 480 SYBR-Green I Master Mix on the LightCycler® 480 PCR system (Roche). The thermocycling protocol consisted of an initial denaturing step at 95˚C for 5 min, followed by 40 cycles of 10 sec at 95˚C and an extension step at 60˚C for 60 sec. The primer sequences are listed in Table I. Relative mRNA expression levels were measured using the 2-∆∆Cq method (34) and normalized to the level of GAPDH.
Table I.
Sequence information of primers.
| Gene | Primer sequences |
|---|---|
| GAPDH | F: 5'-GGAGAAGGCTGGGGCTCAT-3' |
| R: 5'-TGGGTGGCAGTGATGGCA-3' | |
| NLRP3 | F: 5'-CATAGGACCGCTCTGCACTG-3' |
| R: 5'-CAGGTCTCGTGGTGATGAGC-3' | |
| CASP1 | F: 5'-ACAGGCATGACAATGCTGCT-3' |
| R: 5'-CAGGAACGTGCTGTCAGAGG-3' | |
| IL-1β | F: 5'-TGCCACCTTTTGACAGTGATG-3' |
| R: 5'-TGATGTGCTGCTGCGAGATT-3' |
NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; CASP, caspase; F, forward; R, reverse.
ELISA
The levels of cytokine IL-1β in the cell culture supernatants were determined by ELISA according to the manufacturer's protocol. The OD450 value was measured on a microplate reader (Bio-Rad Laboratories, Inc.) and the concentrations of IL-1β in the samples were calculated according to the OD450.
Statistical analysis
Values are expressed as the mean ± standard error of the mean of at least 3 samples. Experiments were performed in triplicate. All values were analyzed by one-way ANOVA among groups using SPSS (version 19.0; IBM Corp.). Tukey's test was used for further comparison between two groups. P<0.05 was considered to indicate a statistically significant difference. The graphs were drawn using GraphPad Prism 5 (GraphPad Software, Inc.).
Results
Determination of the optimal concentrations and incubation times for LPS, leptin and mitoQ
In order to determine the optimal concentrations and incubation times, the cells were treated with different concentrations of drugs and incubated for different time periods. The highest concentration incubated with the lowest incubation time that did not result in a significantly effect on cell viability was considered to be the optimal concentration. No significant differences were observed among the different concentrations of leptin after 1 and 6 h of treatment. After 24 h of treatment with leptin, the viability of the cells treated at the concentration of 200 ng/ml became significantly lower than that of untreated cells (P<0.05). However, there was no significant difference between cells treated with leptin at a concentration of 100 ng/ml and untreated cells after 24 h of treatment. Thus, 100 ng/ml was used as the final concentration of leptin and the incubation time selected was 24 h (Fig. 1A).
Figure 1.

Effects of leptin, LPS and mitoQ on the viability of BEAS-2 cells determined by a CCK-8 assay. (A) Effect of leptin. After 24 h of treatment with leptin at the concentration of 100 ng/ml, the cell viability became significantly lower than that of the untreated cells (P<0.05). (B) Effect of LPS. After 6 h of treatment with LPS at the concentration of 10 µg/µl, the cell viability was significantly decreased compared with that of untreated cells (P<0.05). (C) Effect of mitoQ. The cell viability was significantly decreased after 1 h of treatment with mitoQ at the concentration of 500 nM compared with that of untreated cells (P<0.05). Values are expressed as the mean ± standard error of the mean of 3 samples analyzed in triplicate. #P<0.05 vs. untreated group incubated for 48 h; *P<0.05 vs. untreated group incubated for 24 h; ∆P<0.05 vs. untreated group incubated for 6 h; &P<0.05 vs. untreated group incubated for 1 h. CCK-8, Cell Counting Kit-8; LPS, lipopolysaccharide; mitoQ, mitoquinone.
Similarly, after 6 h of incubation with LPS, the viability of cells treated at the concentration of 10 µg/µl was significantly decreased compared with that of untreated cells (P<0.01). However, the viability of cells treated at the concentration of 5 µg/µl was not significantly different compared with that of untreated cells. Furthermore, the viability of cells treated with mitoQ at the concentration of 500 nM was significantly decreased after 1 h of treatment as compared with that of untreated cells (P<0.05), while that of cells treated with 200 nM mitoQ was not significantly affected. Thus, in the subsequent experiments, cells were treated with 5 µg/µl LPS for 6 h (Fig. 1B) and 200 nM mitoQ for 1 h (Fig. 1C). Therefore, for LPS + mitoQ group, 5 µg/µl LPS was added 6 h prior to detection and 200 nM mitoQ was added 1 h before detection. For Leptin + mitoQ group, 100 ng/ml leptin was added 24 h before collection and 200 nM mitoQ was added 1 h before collection.
Effects of mitoQ on the mitochondrial membrane integrity in the LPS and leptin-pretreated BEAS-2 cells
The mitoTracker probe, a kind of mitochondrial membrane tracer, was used to detect the membrane integrity of mitochondria. As presented in Fig. 2A and B, the mitochondrial membrane of the control group and DMSO group was intact and the MFIs were 10.945±0.254 and 10.587±0.233, respectively. However, after treatment with LPS and leptin, the mitochondrial membrane was disrupted and the MFIs of the groups (4.474±0.264 and 4.416±0.267, respectively) were significantly decreased compared with those of the control and DMSO groups (P<0.01), indicating that the mitochondria were impaired. However, the MFI of the LPS + mitoQ group (10.47±0.271) or the Leptin + mitoQ group (11.14±0.294) was significantly increased compared with that in the LPS group or Leptin group (P<0.01), suggesting that the impaired mitochondrial membrane may be recovered following treatment with mitoQ.
Figure 2.

Effects of mitoQ on the mitochondrial membrane integrity in the LPS- and leptin-pretreated BEAS-2 cells. (A) Representative immunofluorescence images of samples stained with the mitoTracker probe (magnification, x100 in the upper panel and x200 in the magnified windows below). (B) Mean fluorescence intensity of the cells. Pretreatment with LPS and leptin resulted in a perforation/rupture of the mitochondrial membrane compared with the control group (P<0.01). Treatment with mitoQ significantly improved mitochondrial injury (P<0.01). Values are expressed as the mean ± standard error of the mean of 3 samples analyzed in triplicate. *P<0.01 vs. control group; ∆P<0.01 vs. DMSO group; #P<0.01 vs. LPS group; &P<0.01 vs. leptin group. LPS, lipopolysaccharide; mitoQ, mitoquinone.
Effects of mitoQ on the production of mtROS in the LPS and leptin-pretreated BEAS-2 cells
Next, to observe the influence of mitoQ on the production of mtROS, the mitoSOX probe was used to detect the production of mtROS after LPS and leptin treatment. In the LPS group as the positive control, the production of mtROS was significantly augmented (MFI, 11.282±0.286) when compared with that in the control group and DMSO group (6.585±0.271 and 6.965±0.178, respectively; P<0.01). Similarly, after treatment with leptin, the production of mtROS was also significantly increased compared with that in the control and DMSO groups (P<0.01), revealing the potential effect of leptin on the overproduction of mtROS in BEAS-2 cells. After treatment with mitoQ, the overproduction of mtROS caused by LPS or leptin was significantly decreased to normal levels (P<0.01), confirming the scavenger role of mitoQ on mtROS (Fig. 3A and B).
Figure 3.

Effects of mitoQ on the production of mtROS in the LPS- and leptin-pretreated BEAS-2 cells. (A) Representative immunofluorescence images of samples stained with the mitoSOX probe and DAPI (magnification, x100). (B) Mean fluorescence intensity of the cells. LPS and leptin pretreatment resulted in an overproduction of mtROS compared with the control group (P<0.01). Treatment with mitoQ significantly attenuated this overproduction of mtROS (P<0.01). Values are expressed as the mean ± standard error of the mean of 3 samples analyzed in triplicate. *P<0.01 vs. control group; ∆P<0.01 vs. DMSO group; #P<0.01 vs. LPS group; &P<0.01 vs. leptin group. mtROS, mitochondrial reactive oxygen species; LPS, lipopolysaccharide; mitoQ, mitoquinone.
Effects of mitoQ on the expression levels of NLRP3 and caspase-1 mRNA in the LPS and leptin-pretreated BEAS-2 cells
The NLRP3 inflammasome may be activated by the overproduction of mtROS. In accordance with the aforementioned results, the expression levels of NLRP3 and caspase-1 mRNA in the LPS- or leptin-pretreated BEAS-2 cells were ~2.5-fold higher than those in the control and DMSO groups (P<0.01). Following subsequent treatment with mitoQ, their expression levels of NLRP3 and caspase-1 were significantly decreased (P<0.01), suggesting that mitoQ may reverse BEAS-2 cells from leptin-induced injury, partly through inhibiting the mtROS-NLRP3 inflammasome signaling pathway (Fig. 4A and B).
Figure 4.

Expression levels of NLRP3 and caspase-1 mRNA in BEAS-2 cells. The mRNA expression levels of (A) NLRP3 and (B) caspase-1 in the LPS- and leptin-pretreated BEAS-2 cells were all significantly increased compared with those in the control group (P<0.01). After mitoQ treatment, the expression levels were significantly decreased (P<0.01). Values are expressed as the mean ± standard error of the mean of 3 samples analyzed in triplicate. *P<0.01 vs. control group; ∆P<0.01 vs. DMSO group; #P<0.01 vs. LPS group; &P<0.01 vs. leptin group. NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; mitoQ, mitoquinone; LPS, lipopolysaccharide.
Effects of mitoQ on the expression of IL-1β in the LPS and leptin-pretreated BEAS-2 cells
As presented in Fig. 5A, the mRNA expression levels of IL-1β in the LPS and leptin-pretreated BEAS-2 cells were ~3 times higher than those in the control and DMSO groups (P<0.01). The changes in the protein levels of IL-1β in the supernatant of the LPS and leptin groups were similar to those in mRNA expression (Fig. 5B). By contrast, the mRNA expression and protein levels in the supernatant of the LPS + mitoQ and Leptin + mitoQ groups were significantly decreased (P<0.01).
Figure 5.
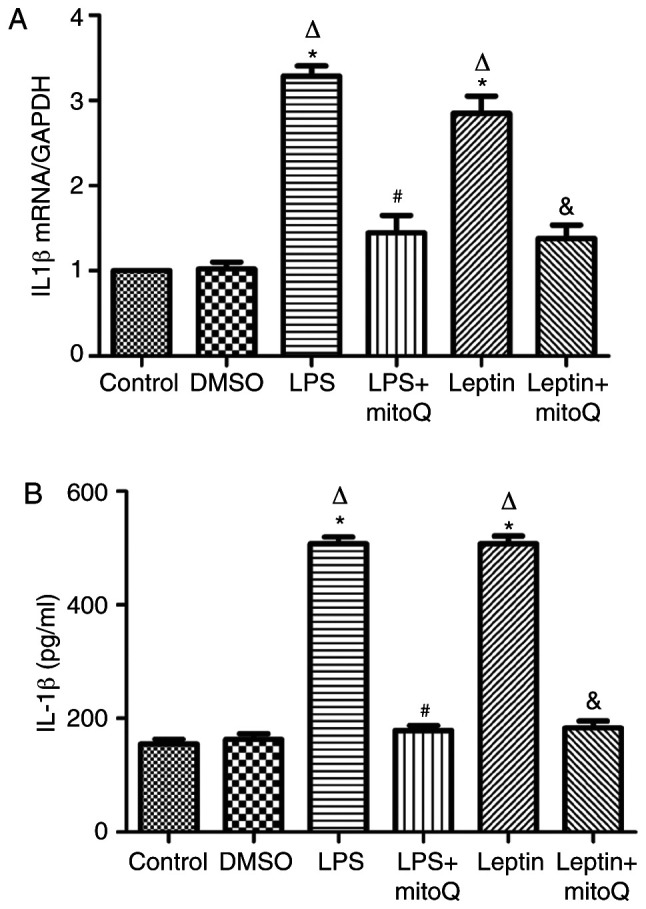
Expression of IL-1β mRNA and protein in BEAS-2 cells. (A) the IL-1β mRNA expression in the LPS group and leptin group was significantly higher than that in the control group (P<0.01). Treatment with mitoQ significantly decreased the IL-1β mRNA expression induced by LPS and leptin (P<0.01). (B) The protein levels of IL-1β in the supernatant of LPS- and leptin-pretreated BEAS-2 cells were significantly increased compared with those in the control group (P<0.01). After mitoQ treatment, their expression levels were significantly decreased (P<0.01). Values are expressed as the mean ± standard error of the mean of 3 samples analyzed in triplicate. *P<0.01 vs. control group; ∆P<0.01 vs. DMSO group; #P<0.01 vs. LPS group; &P<0.01 vs. leptin group. mitoQ, mitoquinone; LPS, lipopolysaccharide.
Discussion
Airway inflammation is the key characteristic of asthma. As a distinct asthma phenotype, the airway inflammation of obese asthma is different from that of conventional asthma, making it difficult to control the condition (11). Leptin, a 16-kDa protein derived from the obesity gene, is secreted predominantly by adipocytes (35). The serum leptin concentrations are 4-6 times greater in severely obese patients compared with those in lean patients (36). Studies have demonstrated that leptin concentrations, either in sputum or in BALF, are highly associated with serum leptin concentrations (17,37). Bruno et al (38) revealed that in the airway epithelial cells, the expression levels of leptin and its receptor LEPR were higher in normal subjects than in smokers or in patients with mild-to-severe COPD. In a previous study, it was also revealed that the expression level of leptin in the BALF was significantly increased in obese mice with asthma (39). Thus, it is conceivable that obesity-associated increases in airway leptin may have an effect on the airway inflammation of asthma.
The NLRP3 inflammasome is a multi-protein complex involved in the innate immune system. Formation of the NLRP3 inflammasome activates caspase 1, which in turn cleaves pro-IL-1β and pro-IL-18, leading to the release of IL-1β and IL-18. The NLRP3 inflammasome may be activated by a diverse series of endogenous and exogenous agonists and the generation of mtROS is thought to be an essential and proximal upstream event of NLRP3 activation (40). A growing body of evidence has suggested a role of the NLRP3 inflammasome-linked cytokine IL-1β in severe, steroid-resistant asthma (41). Using an ovalbumin- and LPS-induced mouse model of neutrophil dominant allergic airway disease, Kim et al (42) revealed that mtROS have a crucial role in the pathogenesis of allergic airway inflammation through activating the NLRP3 inflammasome in airway epithelial cells. Consistent with previous results, the present study revealed that in BEAS-2 cells treated with LPS, the mitochondrial membrane was disrupted, followed by an upregulation of mtROS. Furthermore, similar to LPS, treating cells with leptin also led to significantly increased mtROS levels, suggesting that leptin may serve as an activator of the NLRP3 inflammasome.
Next, the transcriptional expression levels of NLRP3 and caspase-1 were detected by RT-qPCR in the present study. The results demonstrated that the expression levels of NLRP3 and caspase-1 mRNA were significantly upregulated in both leptin- and LPS-pretreated groups. Furthermore, to validate the potential role of leptin in activating the NLRP3 inflammasome, leptin receptor, LEPR was knocked down in the cells by transfection with LEPR RNA interference lentivirus. The mRNA expression of NLRP3 and caspase-1 were not increased following leptin treatment compared with those in the control group (data not shown). Similarly, upregulation of IL-1β in the supernatant of leptin-pretreated cells was also detected, further indicating that leptin may promote airway inflammation through activating the NLRP3 inflammasome pathway. However, a limitation of the present study was that the protein levels of NLRP3 inflammasome were not detected. In a further study, the protein levels of NLRP3 inflammasome should be measured.
MitoQ is an orally active mitochondria-targeted antioxidant that has the ability to scavenge mtROS. Two clinical trials have been performed to investigate the benefits of mitoQ. One is a human phase II trial in Parkinson's disease, the PROTECT trial (www.clinicaltrials.gov; no. NCT00329056) (43). Although no difference has yet been observed compared with the placebo, this trial suggested that mitoQ may be safely administered to patients for 1 year. The other is the CLEAR trial on patients with chronic hepatitis C virus (clinicaltrials.gov; no. NCT00433108) (44). This study identified a decrease in markers of liver damage, indicating a clinical benefit provided by mitoQ in humans. Another study assessed the lungs of ozone-exposed mice and human airway smooth muscle cells (ASMC) isolated from bronchial biopsy specimens from patients with COPD and indicated that mitoQ reduced airway inflammation and inhibited TGF-β-induced ASMC proliferation (45).
Consistent with the results of previous studies, the present study suggested that mitoQ decreased the production of mtROS induced by leptin in BEAS-2 cells, which further inhibited the expression of NLRP3 and caspase-1 mRNA. The increase of IL-1β was also restored after treatment with mitoQ. The in vitro results of the present study provide a molecular basis for mitoQ having a potential role in managing obese asthma through targeting the mtROS-NLRP3 inflammasome pathway. However, the study did not use any group treated with mitoQ alone to observe the effect of mitoQ on the cells, which may be another limitation in the present study.
In conclusion, the results of the present study suggested that leptin may induce or worsen airway inflammation partly through activating the mtROS-NLRP3 inflammasome pathway, which may be a potential mechanism underlying obesity-associated asthma. The mitochondrial-targeted antioxidant mitoQ may have potential therapeutic effects in obese asthma. However, the present study was only performed on cells. Future in vivo studies in obese mice with asthma are required in order to further investigate the role of mitoQ in airway inflammation.
Acknowledgements
The authors thank Professor Changchong Li (Discipline of Pediatric Respiratory Medicine, the Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, China) for contributing to the design of the study and revising the manuscript.
Funding Statement
Funding: The present study was supported by Zhejiang Provincial Natural Science Foundation of China (grant no. LQ19H010002).
Availability of data and materials
All of the data used and/or analyzed in the current study are available from the corresponding author on reasonable request.
Authors' contributions
LC and GY conceived and designed the current study. LC and HL performed the experiments. LZ analyzed the data and prepared the figures. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Figueroa-Muñoz JI, Chinn S, Rona RJ. Association between obesity and asthma in 4-11 year old children in the UK. Thorax. 2001;56:133–137. doi: 10.1136/thorax.56.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beuther DA, Sutherland ER. Overweight, obesity, and incident asthma: A meta-analysis of prospective epidemiologic studies. Am J Respir Crit Care Med. 2007;175:661–666. doi: 10.1164/rccm.200611-1717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quinto KB, Zuraw BL, Poon KY, Chen W, Schatz M, Christiansen SC. The association of obesity and asthma severity and control in children. J Allergy Clin Immunol. 2011;128:964–969. doi: 10.1016/j.jaci.2011.06.031. [DOI] [PubMed] [Google Scholar]
- 4.Papoutsakis C, Priftis KN, Drakouli M, Prifti S, Konstantaki E, Chondronikola M, Antonogeorgos G, Matziou V. Childhood overweight/obesity and asthma: Is there a link? A systematic review of recent epidemiologic evidence. J Acad Nutr Diet. 2013;113:77–105. doi: 10.1016/j.jand.2012.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Jiang D, Wang L, Bai C, Chen O. Association between abdominal obesity and asthma: A meta-analysis. Allergy Asthma Clin Immunol. 2019;15(16) doi: 10.1186/s13223-019-0333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barros R, Moreira P, Padrão P, Teixeira VH, Carvalho P, Delgado L, Moreira A. Obesity increases the prevalence and the incidence of asthma and worsens asthma severity. Clin Nutr. 2017;36:1068–1074. doi: 10.1016/j.clnu.2016.06.023. [DOI] [PubMed] [Google Scholar]
- 7.Rastogi D. Quantifying the contribution of obesity to incident childhood asthma: It's about time. Pediatrics. 2018;142(e20182979) doi: 10.1542/peds.2018-2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lang JE, Bunnell HT, Hossain MJ, Wysocki T, Lima JJ, Finkel TH, Bacharier L, Dempsey A, Sarzynski L, Test M, Forrest CB. Being overweight or obese and the development of asthma. Pediatrics. 2018;142(e20182119) doi: 10.1542/peds.2018-2119. [DOI] [PubMed] [Google Scholar]
- 9.Rzehak P, Wijga AH, Keil T, Eller E, Bindslev-Jensen C, Smit HA, Weyler J, Dom S, Sunyer J, Mendez M, et al. Body mass index trajectory classes and incident asthma in childhood: Results from 8 European Birth Cohorts-a Global Allergy and Asthma European Network initiative. J Allergy Clin Immunol. 2013;131:1528–1536. doi: 10.1016/j.jaci.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 10.Mohan A, Grace J, Wang BR, Lugogo N. The effects of obesity in asthma. Curr Allergy Asthma Rep. 2019;19(49) doi: 10.1007/s11882-019-0877-z. [DOI] [PubMed] [Google Scholar]
- 11.Muc M, Mota-Pinto A, Padez C. Association between obesity and asthma-epidemiology, pathophysiology and clinical profile. Nutr Res Rev. 2016;29:194–201. doi: 10.1017/S0954422416000111. [DOI] [PubMed] [Google Scholar]
- 12.Trayhurn P, Bing C, Wood IS. Adipose tissue and adipokines-energy regulation from the human perspective. J Nutr. 2006;136 (Suppl 7):S1935–S1939. doi: 10.1093/jn/136.7.1935S. [DOI] [PubMed] [Google Scholar]
- 13.López-Jaramillo P, Gómez-Arbeláez D, López-López J, López-López C, Martínez-Ortega J, Gómez-Rodríguez A, Triana-Cubillos S. The role of leptin/adiponectin ratio in metabolic syndrome and diabetes. Horm Mol Biol Clin Investig. 2014;18:37–45. doi: 10.1515/hmbci-2013-0053. [DOI] [PubMed] [Google Scholar]
- 14.Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediators Inflamm. 2010;2010(802078) doi: 10.1155/2010/802078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang M, Cheng H, Zhao X, Hou D, Yan Y, Cianflone K, Li M, Mi J. Leptin and leptin-to-adiponectin ratio predict adiposity gain in nonobese children over a six-year period. Child Obes. 2017;13:213–221. doi: 10.1089/chi.2016.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Z, Leynaert B, Dumas O, Diaz Gil O, Garcia-Aymerich J, Fito Colomer M, Le Moual N, Pison C, Romieu I, Siroux V, et al. Role of leptin in the association between body adiposity and persistent asthma: A longitudinal study. Obesity (Silver Spring) 2019;27:894–898. doi: 10.1002/oby.22466. [DOI] [PubMed] [Google Scholar]
- 17.Holguin F, Rojas M, Brown LA, Fitzpatrick AM. Airway and plasma leptin and adiponectin in lean and obese asthmatics and controls. J Asthma. 2011;48:217–223. doi: 10.3109/02770903.2011.555033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodini A, Tenero L, Sandri M, Maffeis C, Piazza M, Zanoni L, Peroni D, Boner A, Piacentini G. Serum and exhaled breath condensate leptin levels in asthmatic and obesity children: A pilot study. J Breath Res. 2017;11(46005) doi: 10.1088/1752-7163/aa61c5. [DOI] [PubMed] [Google Scholar]
- 19.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birrell MA, Eltom S. The role of the NLRP3 inflammasome in the pathogenesis of airway disease. Pharmacol Ther. 2011;130:364–370. doi: 10.1016/j.pharmthera.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Theofani E, Semitekolou M, Morianos I, Samitas K, Xanthou G. Targeting NLRP3 inflammasome activation in severe asthma. J Clin Med. 2019;8(1615) doi: 10.3390/jcm8101615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2013;20:54–61. doi: 10.1038/nm.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, Wang X, Gong J, Zhang JJ, Adcock IM, et al. Mitochondrial ROS and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radic Res. 2019;53:780–790. doi: 10.1080/10715762.2019.1630735. [DOI] [PubMed] [Google Scholar]
- 24.Fu S, Liu L, Han L, Yu Y. Leptin promotes IL18 secretion by activating the NLRP3 inflammasome in RAW 264.7 cells. Mol Med Rep. 2017;16:9770–9776. doi: 10.3892/mmr.2017.7797. [DOI] [PubMed] [Google Scholar]
- 25.Raut PK, Kim SH, Choi DY, Jeong GS, Park PH. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem Pharmacol. 2019;161:73–88. doi: 10.1016/j.bcp.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 26.Löllmann B, Grüninger S, Stricker-Krongrad A, Chiesi M. Detection and quantification of the leptin receptor splice variants Ob-Ra, b, and, e in different mouse tissues. Biochem Biophys Res Commun. 1997;238:648–652. doi: 10.1006/bbrc.1997.7205. [DOI] [PubMed] [Google Scholar]
- 27.Murphy MP. Understanding and preventing mitochondrial oxidative damage. Biochem Soc Trans. 2016;44:1219–1226. doi: 10.1042/BST20160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 29.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cochemé HM, Murphy MP, Dominiczak AF. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 30.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic Biol Med. 2012;52:841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 32.Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, Yoon KS, Kim HJ, Park JH, Murphy MP, Kim SS. Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med. 2013;11(178) doi: 10.1186/1741-7015-11-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, Yang M, Yang S, Zhu X, Yuan S, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol. 2018;16:32–46. doi: 10.1016/j.redox.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 36.Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, et al. Leptin levels in human and rodent: Measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–1161. doi: 10.1038/nm1195-1155. [DOI] [PubMed] [Google Scholar]
- 37.Sood A, Seagrave JC, Herbert G, Harkins M, Qualls C, Schuyler M. Asthma is associated with lower adiponectin concentrations in sputum than controls. Am Thorac Soc Int Confer. 2012;185(A6502) [Google Scholar]
- 38.Bruno A, Chanez P, Chiappara G, Siena L, Giammanco S, Gjomarkaj M, Bonsignore G, Bousquet J, Vignola AM. Does leptin play a cytokine-like role within the airways of COPD patients? Eur Respir J. 2005;26:398–405. doi: 10.1183/09031936.05.00092404. [DOI] [PubMed] [Google Scholar]
- 39.Chong L, Liu L, Zhu L, Li H, Shao Y, Zhang H, Yu G. Expression levels of predominant adipokines and activations of STAT3, STAT6 in an experimental mice model of obese asthma. Iran J Allergy Asthma Immunol. 2019;18:62–71. [PubMed] [Google Scholar]
- 40.Xiao Y, Xu W, Su W. NLRP3 inflammasome: A likely target for the treatment of allergic diseases. Clin Exp Allergy. 2018;48:1080–1091. doi: 10.1111/cea.13190. [DOI] [PubMed] [Google Scholar]
- 41.Kim RY, Pinkerton JW, Essilfie AT, Robertson AAB, Baines KJ, Brown AC, Mayall JR, Ali MK, Starkey MR, Hansbro NG, et al. Role for NLRP3 inflammasome-mediated, IL-1β-dependent responses in severe, steroid-resistant asthma. Am J Respir Crit Care Med. 2017;196:283–297. doi: 10.1164/rccm.201609-1830OC. [DOI] [PubMed] [Google Scholar]
- 42.Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, Lee YC. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014;5(e1498) doi: 10.1038/cddis.2014.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Move Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. Protect Study Group. [DOI] [PubMed] [Google Scholar]
- 44.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 45.Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136:769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the data used and/or analyzed in the current study are available from the corresponding author on reasonable request.