

Abstract
The terms autoimmune dementia and autoimmune encephalopathy may be used interchangeably; autoimmune dementia is used here to emphasize its consideration in young-onset dementia, dementia with a subacute onset, and rapidly progressive dementia. Given their potential for reversibility, it is important to distinguish the rare autoimmune dementias from the much more common neurodegenerative dementias. The presence of certain clinical features [e.g. facio-brachial dystonic seizures that accompany anti-leucine-rich-glioma-inactivated-1 (LGI1) encephalitis that can mimic myoclonus] can be a major clue to the diagnosis. When possible, objective assessment of cognition with bedside testing or neuropsychological testing is useful to determine the degree of abnormality and serve as a baseline from which immunotherapy response can be judged. Magnetic resonance imaging (MRI) head and cerebrospinal fluid (CSF) analysis are useful to assess for inflammation that can support an autoimmune etiology. Assessing for neural autoantibody diagnostic biomarkers in serum and CSF in those with suggestive features can help confirm the diagnosis and guide cancer search in paraneoplastic autoimmune dementia. However, broad screening for neural antibodies in elderly patients with an insidious dementia is not recommended. Moreover, there are pitfalls to antibody testing that should be recognized and the high frequency of some antibodies in the general population limit their diagnostic utility [e.g., anti-thyroid peroxidase (TPO) antibodies]. Once the diagnosis is confirmed, both acute and maintenance immunotherapy can be utilized and treatment choice varies depending on the accompanying neural antibody present and the presence or absence of cancer. The target of the neural antibody biomarker may help predict treatment response and prognosis, with antibodies to cell-surface or synaptic antigens more responsive to immunotherapy and yielding a better overall prognosis than those with antibodies to intracellular targets. Neurologists should be aware that autoimmune dementias and encephalopathies are increasingly recognized in novel settings, including post herpes virus encephalitis and following immune-checkpoint inhibitor use.
Keywords: autoimmune cognitive impairment, autoimmune encephalitis, central nervous system autoimmunity, immune check point inhibitors, limbic encephalitis/encephalopathy
Rationale
Autoimmunity is a cause of cognitive decline that is potentially reversible, and, in the case of paraneoplastic syndromes, can lead to discovery of an underlying cancer. It is important for neurologists recognize these syndromes and know when testing is appropriate as early treatment typically results in better outcomes.
Introduction
When evaluating a patient with a cognitive disorder, one of the most important aspects is to distinguish reversible from irreversible causes. The presentation of immune-mediated brain dysfunction can range from acute encephalitis and status epilepticus to an insidious cognitive disorder. Therefore, given the potential for good response to immunotherapy, it is essential that neurologists evaluating a patient with new onset dementia consider autoimmune causes. Major advances in antibody biomarker discovery have allowed us to better identify these immune-mediated cognitive disorders. These disorders were initially recognized to occur as an idiopathic autoimmune phenomenon or as a cancer-related immune response (paraneoplastic disorder). It is now recognized that autoimmune dementias can occur in other novel settings, such as post-herpes virus encephalitis,1 post-transplant,2 or after cancer treatment with immune checkpoint inhibitors (ICI).3 The expanding range of clinical scenarios in which autoimmune dementia can arise emphasizes the need for all neurologists to recognize and understand these disorders. In this article, we will highlight the current understanding of autoimmune dementia, its clinical presentation, diagnostic biomarkers, and treatments.
The overlap and differences between autoimmune dementia and encephalopathy
Dementia has been defined by the World Health Organization (WHO) as a syndrome, usually chronic and progressive, with deteriorating cognitive function beyond what is expected with normal aging.4 The syndrome can affect memory, thinking, orientation, comprehension, calculation, learning, capacity, language and judgment, emotional control, social behavior, and motivation, and is without impairment in consciousness.4 A recent updated Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-V) criteria uses the terminology major neurocognitive disorder to describe this syndrome.5 The term encephalopathy, strictly speaking, is defined as a disease process that affects brain function. The term encephalitis is used when an inflammatory cause, which can be autoimmune or infectious, is at play.6 While grave alterations in consciousness have historically been associated with encephalopathy/encephalitis, and do occur with many of these patients [e.g. new onset refractory status epilepticus (NORSE) or florid limbic encephalitis], it is also recognized that many patients with autoimmune encephalitis/encephalopathy will not have alteration in consciousness and have a subacute/rapidly progressive dementia syndrome in which they present for evaluation to an outpatient clinic rather than acutely to a hospital. Moreover, some characteristic dementia syndromes, such as Lewy Body Disease, are recognized to have prominent fluctuations in arousal, which makes distinction between neurodegenerative and autoimmune etiologies of dementia more difficult.7 This article will focus on patients who present with more of a subacute dementia syndrome, realizing that patients with the same disease [e.g., anti-leucine-rich-glioma-inactivated-1 (LGI1) encephalitis/dementia] may present as a rapidly progressive dementia without altered consciousness or acutely to the hospital with severe encephalopathy and impaired consciousness. The difficulties in distinguishing these syndromes is highlighted by up to a third of patients with autoimmune etiologies of dementia initially given a diagnosis of an irreversible neurodegenerative dementia or prion disorder.8
Nomenclature
Autoimmune dementia is a term that describes cognitive decline of an autoimmune basis.8 Neurologists are most familiar with dementia occurring with an insidious onset of cognitive decline in older patients – a situation in which the vast majority of causes are degenerative (e.g., Alzheimer’s disease). However, the likelihood of encountering a cognitive decline of immune-mediated origin is higher in specific clinical settings. For instance, autoimmune etiologies of dementia represent a higher proportion of cases in young onset (<45 years) and early onset (<65 years) dementia (see below).9,10 Also, rapidly progressive dementia that has a subacute onset, with worsening over weeks to months, is a category in which autoimmune etiologies are over-represented.11–13 However, it should be noted that autoimmune etiologies can also occur in the elderly, and up to one-third of these can be misdiagnosed with an irreversible neurodegenerative disorder.8 When the autoimmune dementia occurs in the setting of an immune-mediated response to an underlying cancer, the term paraneoplastic dementia/encephalitis may be used. A number of autoantibody biomarkers of immune-mediated cognitive disorders have been discovered, and, thus, these disorders may be named by the antibody they associate with [anti-N-methyl-d-aspartate-receptor (NMDAR) encephalitis]. The age, sex, and associated cancers vary by antibody and are summarized in Table 1. An autoimmune dementia may also occur without an accompanying autoantibody, and negative autoantibody testing does not exclude autoimmune dementia.
Table 1.
Demographic and clinical characteristics of the main CNS autoantibodies targeting cell-surface antigens associated with cognitive impairment/encephalopathy according to the largest series reported.
| Cell-surface target | Female sex (%) | Typical age of onset (years) | Cognitive impairment (%) | Common clinical accompaniments | Cancer risk (cancer types) |
|---|---|---|---|---|---|
| AMPAR102,103 | 65–90 | 60–70 | 100 | LE; hyponatremia | 64% (small-cell lung) |
| CASPR2104 | 10–25 | 60–70 | 40–80 | Encephalopathy; Morvan’s/Isaac’s syndrome; ataxia | 10–20% (thymoma) |
| DPPX105 | 10–40 | 50–60 | 80–100 | GI symptoms (diarrhea, episodic severe weight loss); sleep disturbances | 10% (hematologic malignancies) |
| GABAAR51 | 50 | 40–50 | 67 | Seizures/status epilepticus; movement disorders | 40% (thymoma) |
| GABABR106,107,108 | 40–65 | 60–70 | 80–100 | LE; status epilepticus | 50% (small cell lung) |
| mGluR5109 | 45 | 20–30 | 90 | LE; viral-like prodromes; seizures | 64% (Hodgkin’s lymphoma) |
| GlyRα1110 | 45 | 40–50 | 30 | SPS; PERM | 10% (thymoma, seminoma) |
| IgLON5111 | 50 | 60–70 | 30–40 | Sleep disturbances; bulbar symptoms; ataxia | Rare |
| LGI128,79 | 35–40 | 60–70 | 90–100 | LE; FBDS; hyponatremia | 1–10% (thymoma) |
| Neurexin 3α112 | 80 | 40–50 | 60 | Encephalitis; viral-like prodrome; oro-facial dyskinesia; central hypoventilation; positive ANA | Unknown |
| NMDAR96 | 80–90 | 20–30 | 90–100 | LE; psychosis; viral-like prodrome; dyskinesias; central hypoventilation | 40–60% (teratoma, usually ovarian) |
AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ANA, anti-nuclear antibody; CASPR2, contactin associated protein 2; CNS, central nervous system; DPPX, dipeptidyl aminopeptidase-like protein 6; FBDS, facio-brachial dystonic seizures; GABAAR, γ-aminobutyric acid type-A receptor; GABABR, γ-aminobutyric acid type-B receptor; GI, gastrointestinal; GlyRα1, glycine receptor subunit alpha-1; IgLON5, immunoglobulin-like cell adhesion molecule IgLON family member 5; LE, limbic encephalitis; LGI1, leucine-rich glioma inactivated 1; mGluR5, metabotropic glutamate receptor 5; NMDAR, N-methyl-D-aspartate receptor; ON, optic neuritis; PERM, progressive encephalopathy with rigidity and myoclonus; SPS, stiff-person syndrome. (Modified with permission from Table 1 in ref.75).
Pathophysiology
Antibodies can be stratified into two categories based on their antigenic target location within the cell14:
Antibodies that bind to cell-surface/synaptic antigens (Table 1). In patients with these antibodies (e.g. anti-NMDAR encephalitis), it is suspected or has been confirmed that the antibody is directly pathogenic.15,16,17 The binding of the antibody may result in dysfunction of the receptor, internalization, antibody mediated cytotoxicity, or activation of complement. The frequency of cancer is less (~ 50%) and these disorders tend to respond well to immunotherapy, particularly antibody depleting therapy [e.g., plasma exchange (PLEX),18 rituximab].19
Antibodies that bind intracellular antigens (Table 2). These encompass what were traditionally referred to as the classical paraneoplastic disorders, where antigens within the cytoplasm or nucleus are targets of the antibody. The exact pathophysiologic mechanisms for autoantibodies targeting intracellular antigens is unknown. In vitro studies suggest that the antibody is not directly pathogenic but rather a biomarker of a cytotoxic CD8-T-cell mediated process [e.g., anti-neuronal nuclear antibodies type-1 (ANNA-1)/anti-HU associated limbic encephalitis],20,21 although these findings have not yet been reproduced in animal models. Postmortem findings may also support a cytotoxic damage in these patients.22 These disorders tend to be associated more frequently with an accompanying cancer (>80%) and respond less to immunotherapy.
Table 2.
Demographic and clinical characteristics of the main CNS autoantibodies targeting intracellular antigens associated with cognitive impairment/encephalopathy according to the largest series reported.
| Intracellular target | Female sex (%) | Typical age of onset | Cognitive impairment (%) | Common clinical accompaniments | Cancer risk (cancer types) |
|---|---|---|---|---|---|
| AK5113 | 30 | 60–70 | 100 | LE | 0% |
| Amphiphysin114 | 60 | 60–70 | 30 | Encephalopathy; peripheral neuropathy; myelitis; SPS | 80% (breast, small cell lung) |
| ANNA-1 (Hu)115,116 | 55–65 | 60–70 | 10–20 | LE; sensory neuronopathy; autoimmune GI dysmotility | 85–90% (small cell lung) |
| ANNA-2 (Ri)39 | 65 | 60–70 | 10–20 | Brainstem symptoms; opsoclonus-myoclonus; laryngospasm; jaw opening dystonia; ataxia | 75% (small cell lung, breast) |
| ANNA-3117 | 50–60 | 50–60 | 10–20 | Limbic encephalitis; cerebellar ataxia; peripheral neuropathy; myelopathy | 80–90% (small cell lung) |
| CRMP5 (CV2)118,119 | 30–60 | 60–70 | 40 | Chorea; optic neuropathy/retinopathy; peripheral neuropathy; myelitis | 90% (small cell lung, thymoma) |
| GAD-65120,121 | 75–85 | 50–60 | 3–5 | LE; SPS; ataxia; seizures | 8% (small cell lung) |
| GFAP42 | 68 | 50–60 | 15–60 | Meningo-encephalo-myelitis or limited forms; optic disc edema; tremor; viral-like prodrome | 35% (teratoma) |
| ITPR1122 | 70 | 60–70 | 21 | Cerebellar ataxia; peripheral neuropathy; encephalitis with seizures; myelopathy | 30–40% (breast cancer) |
| Kelch11123 | 0 | 40–50 | 5–10 | Ataxia; brainstem dysfunction with hearing loss; encephalopathy | 100% (testicular seminoma) |
| Ma2 (Ta)45 | 32 | 60–70 | 68 | LE; diencephalic syndrome (narcolepsy/cataplexy); brainstem symptoms | 90% (testicular tumors) |
| NfL124 | 50 | 60–70 | 33 | Ataxia; encephalopathy; myelitis | 76% [neuroendocrine (small cell lung, Merkel cell)] |
| PCA-2/MAP1B125 | 70 | 60–70 | 30 | Ataxia; brainstem symptoms; peripheral neuropathy | 90% (small cell lung) |
AK5, adenylate kinase 5; ANNA-1/2, anti-neuronal nuclear antibodies type-1/2; CNS, central nervous system; CRMP5, collapsin response-mediator protein-5; GAD-65, glutamic acid decarboxylase-65; GFAP, glial fibrillary acidic protein; GI, gastrointestinal; LE, limbic encephalitis; NfL, neurofilament light chain; PCA-2/MAP1B, Purkinje cells antigens-2/microtubule-associated protein 1B; SPS, stiff-person syndrome. (Modified with permission from Table 2 in in ref.75).
Epidemiology
Autoimmune dementia and encephalopathy is less common than other causes of cognitive decline, such as neurodegenerative diseases. A study evaluating the causes of dementia in young patients (ages 17–45 years) found that neurodegenerative dementia was the most common cause (31.1%), followed by autoimmune or inflammatory etiologies (21.3%).10 A more recent study found immune-mediated diseases accounted for 46% of rapidly progressive dementia in Brazil,12 while it was lower at 18% in India, where infectious diseases predominated.11 In a separate study from Spain, autoimmune/paraneoplastic causes accounted for only 4%.13 The frequency of autoimmune dementia as a proportion of all dementia is lower at an older age as neurodegenerative etiologies become more common, but certain antibodies do have a preponderance for older individuals (e.g., anti-LGI1 autoantibodies). In patients with rapidly progressive dementia that was not secondary to prion disease, autoimmunity was a cause of 9–13% of cases in tertiary referral centers.23,24 Therefore, although it is critical to recognize autoimmunity as a cause of dementia, it is important to know it is not as common as other etiologies, and the diagnosis should be made carefully. However, some epidemiologic studies are based on older data and autoimmune dementias may be underdiagnosed in these cohorts. Gender differences in autoimmune dementia are recognized and vary depending on the accompanying autoantibody. For instance, anti-NMDAR encephalitis predominate in females, while anti-LGI1 encephalitis is more common in males.
Clinical features
Certain clinical features that accompany cognitive decline may be a clue to an autoimmune dementia, and these are summarized in Tables 1 and 2. In addition, clinical features that give a clue to autoimmune dementia are summarized in Table 3. The early prodromal phase in autoimmune dementia may consist of non-specific or non-neurologic symptoms. Episodes of severe weight loss (median of 10–20 kg), with or without diarrhea, may precede encephalopathy/dementia, and have been reported with dipeptidyl aminopeptidase-like protein 6 (DPPX) and occassionally with glial fibrillary acidic protein (GFAP) autoantibodies, the latter often preceded by a viral prodrome or vaccination.25,26 The onset of cognitive decline in autoimmune cases is typically acute to subacute, and worsens rapidly with the memory domain impacted disproportionately.8 A prior study comparing patients with autoimmune dementia to those with other causes of dementia, a subacute onset, fluctuating course, tremor, and headache were symptoms that were more likely to suggest an autoimmune cause.8 Short-term memory loss developing subacutely without severe loss of consciousness can occur with limbic encephalitis.27 Following the onset of the cognitive/behavioral symptoms, the development of seizures early in the course is a clue to an autoimmune etiology and is rare with neurodegenerative dementia, usually occurring at the end-stage.8,27 Facio-brachial dystonic seizures are a unique and characteristic manifestation of anti-LGI1 encephalitis and can be mistaken for myoclonus or non-epileptic phenomenon, and, hence, up to half of patients with these episodes are initially diagnosed as having Creutzfeldt Jakob Disease (CJD) or psychiatric disease.28 The hallmark feature consists of short-lived episodes of dystonic posturing of the face, arm, or both, with the leg also sometimes impacted.28-30 Patients often retain awareness and may complain of dropping items from their hand. The episodes occur frequently (multiple per hour is typical) and can alternate from side to side. Paroxysmal dizzy spells are also recognized in association with anti-LGI1 encephalitis.31 Psychosis is another common feature of autoimmune encephalitis, particularly with anti-NMDAR antibodies.16 Other presentations have been reported including a case mimicking fronto-temporal dementia32; the subacute onset, rapid progression, and absence of a family history would help distinguish an autoimmune etiology from a neurodegenerative cause. Sleep disorders are increasingly recognized; parasomnias and sleep-disordered breathing is a major feature of immunoglobulin-like cell adhesion molecule IgLON family member (IgLON5) antibodies, which has a chronic presentation and may be accompanied by a bulbar disorder and gait dysfunction sometimes mimicking progressive supranuclear palsy.33,34 A rapidly progressive dementia with insomnia, hyperhidrosis, and peripheral nerve hyper-excitability (painful cramps and fasciculations) should raise suspicion for Morvan’s syndrome, which is associated with contactin-associated protein 2 (CASPR2) antibodies.35 While isolated Parkinsonism is uncommon, a clinical syndrome of progressive encephalomyelitis, rigidity, and myoclonus (PERM) is recognized (often with accompanying hyperekplexia) and occurs in association with GAD65 or glycine receptor antibodies.36,37 Other movement disorders that can be encountered include autoimmune/paraneoplastic chorea [e.g. collapsin response mediator protein 5 (CRMP5)/anti-CV2],38 jaw dystonia [anti-neuronal nuclear antibody type-2 (ANNA-2)/anti-Ri],39 and oro-facial dyskinesias (anti-NMDAR encephalitis).16 Episodic ataxia has been reported with CASPR2 antibodies,40 while many other antibodies can associate with a progressive ataxia combined with an autoimmune dementia (Table 1). Opsoclonus-myoclonus, jaw dystonia, and laryngospasm may associate with ANNA-2/anti-Ri antibodies.41 Visual dysfunction can occur in autoimmune dementia with optic disc edema (GFAP-IgG),42 or retinopathy (CRMP5-IgG/anti-CV2).43
Table 3.
Features on history indicative of a higher risk of autoimmune or paraneoplastic dementia.
| Demographics |
| Young onset |
| Clinical features |
| Subacute onset and rapidly progressive |
| Fluctuating course |
| Headaches |
| Early psychosis |
| Seizures (particularly faciobrachial dystonic seizures which may mimic myoclonus) |
| Medical history |
| Known cancer, particularly tumors associated with paraneoplastic syndromes |
| Medications |
| Immune checkpoint inhibitor use |
| Family history |
| Strong family history of cancer or autoimmunity |
| Social history |
| Cigarette smoking |
Diagnostic testing
Cognitive assessment
Bedside cognitive testing, involving tests such as the Short Test of Mental Status (Kokmen) or Mini Mental Status Exam are useful when possible to determine the severity of dementia and to monitor the progression over time.8 Formal neuropsychological testing can be useful to better identify abnormalities and provide a baseline from which immunotherapy response can be judged (see below). On both assessments, short-term memory loss appears to be the most severely impacted cognitive domain.8 However, some patients may be too unwell to participate acutely, depending on the severity of the cognitive disorder.
Imaging
Magnetic resonance imaging (MRI) is helpful to diagnose autoimmune etiologies of dementia and encephalopathy and exclude other causes of cognitive impairment. Unilateral or bilateral fluid attenuated inversion recovery (FLAIR) or T2-hyperintensity of mesial temporal lobes with or without gadolinium enhancement is characteristic of limbic encephalitis (Figure 1a,c) but can also be seen with epilepsy and herpes simplex virus (HSV) encephalitis.44 Basal ganglia T1 and T2 hyperintensities can be seen in some patients with facio-brachial dystonic seizures and anti-LGI1 encephalitis28; basal ganglia T2-hyperintensities may be seen in association with multiple other antibodies including anti-NMDAR, anti-Ma2, or anti-CRMP5.45-47 Anti-Ma2 antibodies may also have diencephalic involvement, particularly around the surface of the third ventricle.45 Signal abnormalities within the basal ganglia and thalamus, particularly when accompanied by restricted diffusion, are recognized with non-autoimmune dementia/encephalopathy such as CJD or Wernicke encephalopathy.48-50 γ-Aminobutyric acid type-A receptor (GABAAR) antibodies may have multiple cortical and subcortical T2-hyperintensities.51 Radial perivascular enhancement is typical of GFAP-IgG (Figure 1b).42 Susac syndrome is an immune mediated vascular disorder that may reveal “snow-ball” and “spoke” type lesions in the center of the corpus callosum, or punctate restricted diffusion that may form a “string of pearls” sign along the internal capsule.52 Restricted diffusion and micro-hemorrhages illustrated by darkening on gradient echo or susceptibility weighted imaging often accompanied by leptomeningeal enhancement may be encountered in central nervous system (CNS) vasculitis or amyloid beta related angiitis, and should be considered in the appropriate setting.53 CNS vasculitis is recognized to occur in a paraneoplastic context in association with lymphoma.54 Most neurodegenerative disorders have accompanying atrophy; moderate-to-severe atrophy without signal abnormality should raise suspicion for a non-autoimmune dementia.
Figure 1.
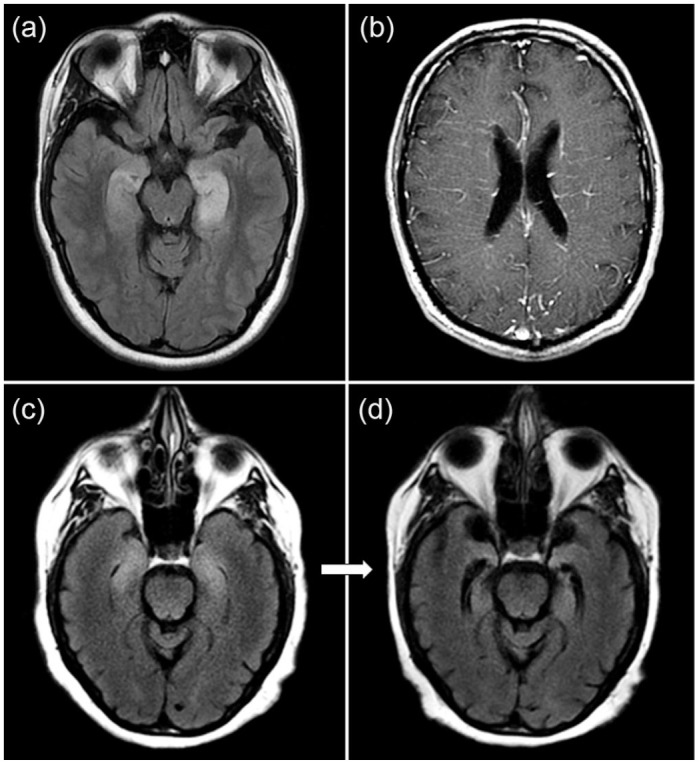
MRI examples of autoimmune dementia. (a) Bilateral T2-hyperintensities in the mesial temporal lobe are shown in a patient with autoimmune limbic encephalitis associated with GAD65 autoantibodies. (b) Radial perivascular enhancement is shown in a patient with GFAP autoantibodies. (c) Bilateral mesial temporal T2-hyperintensities consistent with limbic encephalitis and occurring after immune checkpoint inhibitor use with an accompanying unclassified neural autoantibody detected with follow-up MRI 1 month later revealing bilateral temporal lobe atrophy (d).
MRI, magnetic resonance imaging.
Electroencephalogram
Electroencephalogram (EEG) may assist in the diagnosis of patients with suspected autoimmune dementia. In anti-NMDAR encephalitis, 30% of patients have a pattern of extreme delta brush with rhythmic delta (1–3 Hz) activity with bursts of beta (20–30 Hz) superimposed on the delta waves.55 Occasionally, this pattern may also be seen in patients with other conditions including: non-autoimmune mesial temporal lobe epilepsy, hypoxic ischemic encephalopathy, and brain tumors. Other findings on EEG in autoimmune dementia are non-specific, including diffuse or focal slowing and epileptiform activity,56 the latter favoring an autoimmune over most neurodegenerative etiologies.57 Of note, facio-brachial-dystonic seizures rarely have any ictal EEG correlate.28 EEG can also provide clues to alternative etiologies, with periodic sharp waves suggestive of CJD,58 and periodic slow wave complexes suggestive of subacute sclerosing pan-encephalitis post-measles infection.59
Laboratory testing
Laboratory evaluation, including serum and cerebrospinal fluid (CSF) testing are important to evaluate patients with suspected autoimmune dementia. An optimal panel of routine serum tests include a complete blood count; sedimentation rate; C-reactive protein; markers of liver, renal, and thyroid function; electrolytes; vitamins (especially group B-vitamins like B1 and B12)60; and, as appropriate, markers or toxic/metabolic encephalopathies (e.g., benzodiazepines, opioids); and cancer CSF assessment (cytology, flow cytometry). Neurologic involvement of a rheumatologic disorder can occasionally, initially or during its course, manifest with a subacute dementia syndrome.61,62 Thus, serum testing for non-neural antibodies such as antinuclear antibody, double-stranded DNA antibodies, antibodies to the extractable nuclear antigen, anti-neutrophilic cytoplasmic antibodies (ANCA), rheumatoid factor, phospholipid antibodies, beta 2 glycoprotein-1 antibodies, lupus anticoagulant, and IgG4 can be considered when clinical features are suggestive to assess for neurologic involvement of rheumatologic disorders.63
CSF analysis may show evidence of inflammation, including pleocytosis, elevated oligoclonal bands, and elevated IgG index, in up to 50% of patients.8 It is very rare for CSF pleocytosis and elevated oligoclonal bands to be encountered in neurodegenerative disorders.64,65 It is important to note that an inflammatory CSF does not confirm the diagnosis of autoimmune dementia nor does a normal CSF exclude it; anti-LGI1 encephalitis and other autoantibody-associated encephalitides may have a normal CSF profile, and, in those situations, detection of the antibody is particularly useful.66 Other CSF testing for alternative causes of dementia can be helpful, including levels and ratio of phospho-tau and amyloid-β-42 to evaluate for Alzheimer’s dementia,67 and real-time quaking-induced conversion (RT-QuIC) to evaluate for prion disorders.68
Extensive screening for viral, bacterial, and fungal/parasitic infections is recommended in both serum and CSF, especially in immunocompromised patients or patients with recent history of travel to a region where infectious encephalitis is endemic.
Other diagnostic investigations
Evaluation for infectious, neoplastic, and toxic or metabolic etiologies should be undertaken prior to initiating immunotherapy. Brain fludeoxy-glucose-positron-emission-tomography (FDG-PET) in patients with autoimmune encephalitis can show focal or diffuse areas of hypo- or hyper-metabolic activity.69 In anti-NMDAR encephalitis, medial occipital hypometabolism is recognized.70 In patients who do not respond to immunotherapy, or if the diagnosis remains unclear, brain biopsy may be considered, and the presence of inflammatory infiltrates may support the diagnosis of autoimmune encephalitis, but is not diagnostic. Typically brain biopsy is most helpful in ruling out other causes of cognitive impairment such as a primary brain tumor or lymphoma.
Neural autoantibody testing
Utility
Testing of neural antibodies in patients with appropriate clinical history can confirm the diagnosis of autoimmune dementia. It is important to test for antibodies prior to initiating treatment as testing after starting therapy may lead to false positive (e.g. IVIg),71 or false negative results [e.g., PLEX (unpublished observations from E.P.F)]. The sample with optimal sensitivity for autoantibody detection varies; CSF is preferable for anti-NMDAR and anti-GFAP antibodies, while serum is preferable for anti-LGI1.6 In many cases where an autoimmune etiology is suspected, testing both serum and CSF is preferred.72 In most cases it is best to test a panel of antibodies as most clinical syndromes may be caused by a wide variety of autoantibodies (Table 1), and, in rare patients with multiple positive results, the profile of antibodies may help refine the cancer search. The range of antibodies that associate with autoimmune dementia are summarized and referenced in Tables 1 and 2.
Antibody testing methodology
Older generation techniques using immunoprecipitation have an increased risk of false positivity, where up to 5% of normal or disease controls can be positive.73, 74 Newer generation techniques using cell-based assays have reduced the risk of false positivity to approximately 0.2%.74 It is also recognized that tissue immunofluorescence/immunohistochemistry in combination with western blot (mostly used for antibodies targeting intracellular antigens), or in combination with cell-based assays (generally used only for antibodies directed against cell-surface antigens), is optimal, and caution is advised when using western blot alone where there is a high risk of false positivity.75 An initial screening with tissue immunofluorescence/immunohistochemistry in specialized laboratories is generally recommended as it may reveal unexpected, or as yet not classified, antibodies.
Pitfalls
Given the relative rarity of autoimmune dementias in the population, it is important to first exclude other more common etiologies (e.g., neurodegenerative or metabolic disorders).76 Testing for antibodies in very low probability situations (e.g., classical dementia due to Alzheimer’s disease) increases the risk of false positives, particularly with antibodies detected by older generation techniques such as immunoprecipitation.74 Refinement of the antibody targets has also led to improved categorization of some antibody-associated cognitive disorders. For example, voltage-gated-potassium-channel-complex antibodies were initially identified as the antibody target in some patients with autoimmune dementia/encephalitis, but it was later shown that only antibodies bound to certain proteins that associate with the channel (LGI1, CASPR2) were clinically relevant, and antibodies that bind to the channel but not LGI1 or CASPR2 were of no clinical significance.77,78 With some autoantibodies, the titer may be useful; for example, glutamic acid decarboxylase 65 (GAD-65) antibodies may be found at low titer in up to 8% of the general population,80 while high titers can be associated with PERM,81 limbic encephalitis, and can occasionally mimic CJD.82 Further, clinically significant antibodies are typically of the IgG isotype. Anti-thyroid peroxidase (TPO) antibodies are not specific and are seen in anywhere from 10% to 25% of the normal population.83,84 These antibodies have been associated with a disorder termed Hashimoto’s encephalopathy, and this syndrome has been reported to occasionally manifest with a rapidly progressive dementia mimicking CJD.85 However, caution is advised as the high frequency of these antibodies in the general population precludes their diagnostic utility, and many patients can be assigned with this diagnosis when an alternative etiology for their cognitive impairment is present.86
Cancer investigations
Cancer investigation should be directed to particular antibody positivity status, as outlined in Table 1.87 Age- and sex-appropriate cancer screening should be up to date, including colonoscopies and mammograms. Cancer screening typically starts with a computed tomography (CT) chest, abdomen, and pelvis. Whole body FDG-PET has increased sensitivity for cancer in paraneoplastic disorders, and should be considered if the initial cancer screen is unrevealing, particularly when antibodies with high positive predictive value for cancer are detected.88 Ovarian or testicular ultrasounds and skin evaluation for melanoma should also be considered as applicable. In patients with antibodies that have a strong association with cancer such as ANNA-1/anti-Hu antibodies if the initial cancer evaluation is negative, cancer screening should be repeated at 6 months and 1 year as occult cancers may occasionally emerge in follow-up imaging.
Emerging clinical settings
Besides the traditional idiopathic and paraneoplastic forms, novel clinical scenarios are increasingly recognized to be at higher risk for development of autoimmune dementia and encephalopathies:
In patients with HSV encephalitis, about one-third of patients develop a subsequent autoimmune encephalitis in the following 4–6 weeks, likely triggered by neural epitopes released during the viral infection. Neural autoantibody testing is highly recommended after HSV encephalitis in patients who relapse or worsen after an initial improvement, as the probability of an autoimmune etiology is high. Anti-NMDAR antibodies are the most common accompaniment.1
Post-transplant patients may paradoxically develop autoimmunity despite being on maintenance anti-rejection immunosuppressive medications and manifestations can include autoimmune dementia/encephalopathy. This is thought to be due to an imbalance between T and B cell immunosuppression that may result in systemic or neurological autoimmunity.2
ICI are increasingly recognized as an effective treatment for many types of cancer. However, the augmented immune system activation induced by ICI may result in autoimmunity directed against any organ, including the central and peripheral nervous systems.3 In patients with CNS involvement, encephalopathy represents the most common manifestation, variably including brainstem/limbic encephalitis, slowly progressive cognitive impairment mimicking a neurodegenerative disorder,89 and posterior reversible encephalopathy syndrome (PRES).90 Clinical and MRI features may be indistinguishable from their idiopathic counterparts (Figure 1c,d).3 Encephalopathy secondary to tumefactive demyelination,91 granulomatosis, or cerebral vasculitis has also been described.92 Specific neural autoantibodies can be detected in the serum and/or CSF in about 50% of cases, although the frequency of novel or yet unidentified antibodies detectable only in highly specialized laboratories seems higher compared with idiopathic cases.3 Neurological symptoms typically manifest within the first 3 months of ICI treatment but can be observed up to 1 year after treatment discontinuation.3 Symptoms should carefully be differentiated from other more common causes of encephalopathy seen in cancer patients (e.g., brain metastases, metabolic/medication-related). Care is also needed in patients with a pre-existing paraneoplastic encephalitis/dementia as use of immune-checkpoint inhibitors in such patients may accelerate the autoimmune neurologic disease leading to severe morbidity or death.3
Treatment
Treatment and management of autoimmune dementias is multifaceted and requires treatment of the underlying autoimmune disease, management of symptoms and sequelae, and treatment of associated cancers when present. If there is underlying cancer, treatment of the malignancy may result in improvement in the neurologic syndrome; however, adjuvant immunotherapy is often necessary. The general principle guiding therapy is to utilize antibody or B-cell depleting agents for antibodies to cell-surface antigens and T-cell depleting agents for antibodies to intracellular antigens (see section Pathophysiology). Randomized controlled clinical trials in acute and maintenance treatment of autoimmune dementia are generally lacking, and therefore treatment recommendations are typically based on results from retrospective analyses and expert opinion. While specific studies on autoimmune dementia alone do not exist, studies of autoimmune or paraneoplastic syndromes that include antibodies that can result in autoimmune dementia have been undertaken. An open label trial compared the efficacy of plasmapheresis plus conventional cancer chemotherapy versus plasmapheresis plus oral cyclophosphamide in 20 patients with new-onset paraneoplastic neurological syndromes, and showed improvement or stabilization in 40% and 60% of cases, respectively.93 Recently, a small randomized trial on 17 patients with anti-LGI1 or anti-CASPR2 encephalitis associated showed superiority of intravenous immunoglobulins over placebo for 50% reduction in seizure frequency from baseline, but did not assess cognitive outcomes.94 A randomized, double-blind, placebo controlled trial on the efficacy of ocrelizumab (anti-CD20 monoclonal antibody) in patients with autoimmune encephalitis is ongoing.95
Acute treatment
Acute immunotherapy typically includes steroids, intravenous immunoglobulin (IVIg), and PLEX. Methylprednisolone may be given intravenously at 1 g daily for 5 days, and sometimes continued with 1 g intravenously once weekly for 6–12 weeks. IVIg and PLEX may be considered as an alternative, or as an adjunct to, therapy in severe cases. IVIg is dosed at 0.4 g/kg/day for 5 days and then 0.4 g/kg once weekly for 6–12 weeks, and PLEX can be done every other day for 5–7 sessions.
In a prior Mayo Clinic study focused specifically on autoimmune dementia, most patients (64%) had improvement with first-line therapy, mostly within the first week of therapy.8 Individual antibody types respond differently to therapy. Patients with anti-LGI1 encephalitis often respond well to initial corticosteroid therapy with resolution of cognitive impairment in the first few weeks.30 Patients with anti-NMDAR encephalitis were shown to improve with first-line therapy in the first 4 weeks in half of cases, but returning to baseline function typically takes months to years. In this same study, half of the patients that did not improve received a second-line immunotherapy with subsequent improvement in final outcome. Therefore, patients with anti-NMDAR encephalitis are frequently initiated on second-line therapies such as rituximab or cyclophosphamide.96
In patients with autoimmune dementia triggered by ICI, the recommended treatment consists of discontinuation of the causative drug, plus administration of high-dose intravenous corticosteroids, IVIg, and/or PLEX, and generally results in a favorable outcome.3 However, a more aggressive approach with addition of rituximab or cyclophosphamide could be necessary in specific subsets of patients with worse prognosis (e.g., patients with neurological autoimmunity pre-existing to ICI treatment).3 The decision to withdraw ICI should be evaluated carefully for each patient based on cancer response to treatment and severity of the neurological manifestations. Improvement has been reported in some cases with ICI-related neurological manifestations of moderate severity treated with immunotherapy only (without ICI discontinuation).3 Depending on available cancer treatment options, ICI re-challenge may be considered, and seems safer in those with mild/moderate acute manifestations.97 Further discussion on ICI-related toxicity is beyond the scope of this manuscript, but specific recommendations have been published.98
Maintenance therapy
In patients that respond to acute immunotherapy a variety of maintenance steroid-sparing immunosuppressants may be considered in an attempt to try prevent relapses. Such treatments include azathioprine, methotrexate, mycophenolate, and rituximab. The decision for which maintenance immunotherapy to use depends on the antibody type, immunosuppressant medication availability, cost, and speed of onset (rituximab begins working within a few weeks, while mycophenolate and azathioprine may take 3–6 months to take effect). Patients should be regularly monitored (every 6–8 months) for disease-relapses and opportunistic infections that can occur in case of immunosuppression. Often, maintenance immunosuppressants are utilized for 2–5 years and, at that stage, weaning off of immunotherapy with observation may be considered.
Clinical course and prognosis
The prognosis for patients with autoimmune dementia varies based on antibody and associated cancers. In a large case series of autoimmune dementia, a mean of 9 point improvement on the 38 point Kokmen Short Test of Mental Status was noted.8 Improvements were most notable in learning and memory. The clinical improvements are generally followed by resolution of MRI, EEG, and CSF abnormalities. The type of antibody accompanying the dementia syndrome can help predict prognosis with antibodies to cell surface targets having a much better prognosis than those with antibodies to intracellular antigens.
Among patients with autoantibodies targeting cell-surface antigens, a study of 38 patients with anti-LGI1 encephalitis showed a 2-year fatality rate of 19%, but 80% of patients had substantial response to immunotherapy, with seizures responding quickly and cognitive function improving more gradually. Approximately one-third of patients experienced relapse.79 A study evaluating 577 patients with anti-NMDAR encephalitis found that 30 patients died in the first year, but about half of patients had improvement with first line treatment or tumor removal in the first 4 weeks, and those that did not respond well to first-line treatment did better if they received a second-line therapy.96 In a study of 38 patients with anti-NMDAR encephalitis, predictors of poor outcomes were abnormal findings on MRI, sensorimotor deficits, and treatment delayed by more than 4 weeks.99 Scoring systems have been developed to assist in predicting clinical outcomes. The antibody prevalence in epilepsy and encephalopathy (APE) score has high sensitivity (99%) and specificity (93%) for predicting neural specific antibodies, while the Response-to-immunotherapy-in-epilepsy-and-Encephalopathy (RITE) score has high sensitivity (96%) and specificity of 86% for predicting favorable response to initial immunotherapy for patients with any autoimmune encephalitis.100 The anti-NMDAR Encephalitis 1-Year Functional Status (NEOS) Score has a strong association for poor functional status at 1 year.101
Conclusion
Clinicians should have an understanding of, and recognize the potential for, autoimmune causes of cognitive decline as these syndromes are potentially reversible, and early recognition and treatment can dramatically improve patient outcomes. Novel antibody biomarkers of autoimmune cognitive decline assist with diagnosis and prognosis. Current acute and treatment strategies are based mostly on retrospective studies and expert opinion. Future prospective randomized controlled trials are needed to help guide treatment decisions.
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
Funding: The authors received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Eoin P. Flanagan  https://orcid.org/0000-0002-6661-2910
https://orcid.org/0000-0002-6661-2910
Contributor Information
Samantha A. Banks, Department of Neurology, Mayo Clinic, Rochester, MN, USA
Elia Sechi, Department of Neurology, Mayo Clinic, Rochester, MN, USA.
Eoin P. Flanagan, Departments of Neurology, Mayo Clinic, 200 First Street SW, Rochester, MN 55905, USA; Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester, MN 55905, USA.
References
- 1. Armangue T, Spatola M, Vlagea A, et al. Frequency, symptoms, risk factors, and outcomes of autoimmune encephalitis after herpes simplex encephalitis: a prospective observational study and retrospective analysis. Lancet Neurol 2018; 17: 760–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cohen DA, Lopez-Chiriboga AS, Pittock SJ, et al. Posttransplant autoimmune encephalitis. Neurol Neuroimmunol Neuroinflamm 2018; 5: e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sechi E, Markovic SN, McKeon A, et al. Neurological autoimmunity and immune checkpoint inhibitors: autoantibody profiles and outcomes. Neurology 2020; 95: e2442–e2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization. Dementia, https://www.who.int/news-room/fact-sheets/detail/dementia (2020, accessed 20 October 2020).
- 5. American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th ed. American Psychiatric Association, 2013. [Google Scholar]
- 6. Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 2016; 15: 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McKeith IG, Ferman TJ, Thomas AJ, et al. Research criteria for the diagnosis of prodromal dementia with Lewy bodies. Neurology 2020; 94: 743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85: 991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gable MS, Sheriff H, Dalmau J, et al. The frequency of autoimmune N-methyl-D-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California Encephalitis Project. Clin Infect Dis 2012; 54: 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelley BJ, Boeve BF, Josephs KA. Young-onset dementia: demographic and etiologic characteristics of 235 patients. Arch Neurol 2008; 65: 1502–1508. [DOI] [PubMed] [Google Scholar]
- 11. Anuja P, Venugopalan V, Darakhshan N, et al. Rapidly progressive dementia: an eight year (2008–2016) retrospective study. PLoS One 2018; 13: e0189832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Studart Neto A, Soares Neto HR, Simabukuro MM, et al. Rapidly progressive dementia: prevalence and causes in a neurologic unit of a tertiary hospital in Brazil. Alzheimer Dis Assoc Disord 2017; 31: 239–243. [DOI] [PubMed] [Google Scholar]
- 13. Sala I, Marquie M, Sanchez-Saudinos MB, et al. Rapidly progressive dementia: experience in a tertiary care medical center. Alzheimer Dis Assoc Disord 2012; 26: 267–271. [DOI] [PubMed] [Google Scholar]
- 14. Pittock SJ, Vincent A. Introduction to autoimmune neurology. Handb Clin Neurol 2016; 133: 3–14. [DOI] [PubMed] [Google Scholar]
- 15. Martinez-Hernandez E, Horvath J, Shiloh-Malawsky Y, et al. Analysis of complement and plasma cells in the brain of patients with anti-NMDAR encephalitis. Neurology 2011; 77: 589–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol 2008; 7: 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ramberger M, Berretta A, Tan JMM, et al. Distinctive binding properties of human monoclonal LGI1 autoantibodies determine pathogenic mechanisms. Brain 2020; 143: 1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rössling R, Prüss H. Apheresis in autoimmune encephalitis and autoimmune dementia. Clin Med 2020; 9: 2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nepal G, Shing YK, Yadav JK, et al. Efficacy and safety of rituximab in autoimmune encephalitis: a meta-analysis. Acta Neurol Scand 2020; 142: 449–459. [DOI] [PubMed] [Google Scholar]
- 20. Roberts WK, Deluca IJ, Thomas A, et al. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuD-specific type 2 CD8+ T cells. J Clin Invest 2009; 119: 2042–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka M, Tanaka K, Tokiguchi S, et al. Cytotoxic T cells against a peptide of Yo protein in patients with paraneoplastic cerebellar degeneration and anti-Yo antibody. J Neurol Sci 1999; 168: 28–31. [DOI] [PubMed] [Google Scholar]
- 22. Bien CG, Vincent A, Barnett MH, et al. Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 2012; 135(Pt 5): 1622–1638. [DOI] [PubMed] [Google Scholar]
- 23. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol 2008; 64: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chitravas N, Jung RS, Kofskey DM, et al. Treatable neurological disorders misdiagnosed as Creutzfeldt-Jakob disease. Ann Neurol 2011; 70: 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tobin WO, Lennon VA, Komorowski L, et al. DPPX potassium channel antibody: frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014; 83: 1797–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sechi E, Morris PP, McKeon A, et al. Glial fibrillary acidic protein IgG related myelitis: characterisation and comparison with aquaporin-4-IgG myelitis. J Neurol Neurosurg Psychiatry 2019; 90: 488–490. [DOI] [PubMed] [Google Scholar]
- 27. Gultekin SH, Rosenfeld MR, Voltz R, et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain 2000; 123(Pt 7): 1481–1494. [DOI] [PubMed] [Google Scholar]
- 28. Flanagan EP, Kotsenas AL, Britton JW, et al. Basal ganglia T1 hyperintensity in LGI1-autoantibody faciobrachial dystonic seizures. Neurol Neuroimmunol Neuroinflamm 2015; 2: e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69: 892–900. [DOI] [PubMed] [Google Scholar]
- 30. Thompson J, Bi M, Murchison AG, et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 2018; 141: 348–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gadoth A, Pittock SJ, Dubey D, et al. Expanded phenotypes and outcomes among 256 LGI 1/CASPR 2-IgG-positive patients. Ann Neurol 2017; 82: 79–92. [DOI] [PubMed] [Google Scholar]
- 32. McKeon A, Marnane M, O’Connell M, et al. Potassium channel antibody associated encephalopathy presenting with a frontotemporal dementia like syndrome. Arch Neurol 2007; 64: 1528–1530. [DOI] [PubMed] [Google Scholar]
- 33. Sabater L, Gaig C, Gelpi E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol 2014; 13: 575–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Honorat JA, Komorowski L, Josephs KA, et al. IgLON5 antibody: neurological accompaniments and outcomes in 20 patients. Neurol Neuroimmunol Neuroinflamm 2017; 4: e385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Irani SR, Pettingill P, Kleopa KA, et al. Morvan syndrome: clinical and serological observations in 29 cases. Ann Neurol 2012; 72: 241–255. [DOI] [PubMed] [Google Scholar]
- 36. Carvajal-Gonzalez A, Leite MI, Waters P, et al. Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 2014; 137(Pt 8): 2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKeon A, Robinson MT, McEvoy KM, et al. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol 2012; 69: 230–238. [DOI] [PubMed] [Google Scholar]
- 38. Vernino S, Tuite P, Adler CH, et al. Paraneoplastic chorea associated with CRMP-5 neuronal antibody and lung carcinoma. Ann Neurol 2002; 51: 625–630. [DOI] [PubMed] [Google Scholar]
- 39. Pittock SJ, Lucchinetti CF, Lennon VA. Anti-neuronal nuclear autoantibody type 2: paraneoplastic accompaniments. Ann Neurol 2003; 53: 580–587. [DOI] [PubMed] [Google Scholar]
- 40. Joubert B, Gobert F, Thomas L, et al. Autoimmune episodic ataxia in patients with anti-CASPR2 antibody-associated encephalitis. Neurol Neuroimmunol Neuroinflamm 2017; 4: e371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pittock SJ, Parisi JE, McKeon A, et al. Paraneoplastic jaw dystonia and laryngospasm with antineuronal nuclear autoantibody type 2 (anti-Ri). Arch Neurol 2010; 67: 1109–1115. [DOI] [PubMed] [Google Scholar]
- 42. Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol 2017; 81: 298–309. [DOI] [PubMed] [Google Scholar]
- 43. Cohen DA, Bhatti MT, Pulido JS, et al. Collapsin response-mediator protein 5-associated retinitis, vitritis, and optic disc edema. Ophthalmology 2020; 127: 221–229. [DOI] [PubMed] [Google Scholar]
- 44. Oyanguren B, Sánchez V, González F, et al. Limbic encephalitis: a clinical-radiological comparison between herpetic and autoimmune etiologies. Eur J Neurol 2013; 20: 1566–1570. [DOI] [PubMed] [Google Scholar]
- 45. Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain 2004; 127: 1831–1844. [DOI] [PubMed] [Google Scholar]
- 46. Kinirons P, Fulton A, Keoghan M, et al. Paraneoplastic limbic encephalitis (PLE) and chorea associated with CRMP-5 neuronal antibody. Neurology 2003; 61: 1623–1624. [DOI] [PubMed] [Google Scholar]
- 47. Zhang T, Duan Y, Ye J, et al. Brain MRI characteristics of patients with anti-N-methyl-D-aspartate receptor encephalitis and their associations with 2-year clinical outcome. Am J Neuroradiol 2018; 39: 824–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mittal S, Farmer P, Kalina P, et al. Correlation of diffusion-weighted magnetic resonance imaging with neuropathology in Creutzfeldt-Jakob disease. Arch Neurol 2002; 59: 128–134. [DOI] [PubMed] [Google Scholar]
- 49. Zuccoli G, Santa Cruz D, Bertolini M, et al. MR imaging findings in 56 patients with Wernicke encephalopathy: nonalcoholics may differ from alcoholics. AJNR Am J Neuroradiol 2009; 30: 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sechi E, Addis A, Fadda G, et al. Teaching NeuroImages: subacute encephalopathy in a young woman with THTR2 gene mutation. Neurology 2015; 85: e108–e109. [DOI] [PubMed] [Google Scholar]
- 51. Spatola M, Petit-Pedrol M, Simabukuro MM, et al. Investigations in GABAA receptor antibody associated encephalitis. Neurology 2017; 88(11): 1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dorr J, Krautwald S, Wildemann B, et al. Characteristics of Susac syndrome: a review of all reported cases. Nat Rev Neurol 2013; 9: 307–316. [DOI] [PubMed] [Google Scholar]
- 53. Salvarani C, Hunder GG, Morris JM, et al. Aβ-related angiitis: comparison with CAA without inflammation and primary CNS vasculitis. Neurology 2013; 81: 1596–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salvarani C, Brown RD, Jr, Christianson TJH, et al. Primary central nervous system vasculitis associated with lymphoma. Neurology 2018; 90: e847–e855. [DOI] [PubMed] [Google Scholar]
- 55. Baykan B, Gungor Tuncer O, Vanli-Yavuz EN, et al. Delta brush pattern is not unique to NMDAR encephalitis: evaluation of two independent long-term EEG cohorts. Clin EEG Neurosci 2018; 49: 278–284. [DOI] [PubMed] [Google Scholar]
- 56. Schäuble B, Castillo PR, Boeve BF, et al. EEG findings in steroid-responsive encephalopathy associated with autoimmune thyroiditis. Clin Neurophysiol 2003; 114: 32–37. [DOI] [PubMed] [Google Scholar]
- 57. Kaplan PW, Sutter R. Electroencephalography of autoimmune limbic encephalopathy. Clin Neurophys 2013; 30: 490–504. [DOI] [PubMed] [Google Scholar]
- 58. Wieser HG, Schindler K, Zumsteg D. EEG in Creutzfeldt–Jakob disease. Clin Neurophysiol 2006; 117: 935–951. [DOI] [PubMed] [Google Scholar]
- 59. Westmoreland BF, Blume WT, Gomez MR. Generalized sharp and slow wave and electrodecremental seizure pattern in subacute sclerosing panencephalitis. Mayo Clin Proc 1976; 51: 107–111. [PubMed] [Google Scholar]
- 60. Sechi G, Sechi E, Fois C, et al. Advances in clinical determinants and neurological manifestations of B vitamin deficiency in adults. Nutr Rev 2016; 74: 281–300. [DOI] [PubMed] [Google Scholar]
- 61. Erten-Lyons D, Oken B, Woltjer RL, et al. Relapsing polychondritis: an uncommon cause of dementia. J Neurol Neurosurg Psychiatry 2008; 79: 609–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abraham P, Neel I, Bishay S, et al. Central nervous system systemic lupus erythematosus (CNS-SLE) vasculitis mimicking Lewy body dementia: a case report emphasizing the role of imaging with an analysis of 33 comparable cases from the scientific literature. J Geriatr Psychiatry Neurol 2020; 34: 128–141. [DOI] [PubMed] [Google Scholar]
- 63. Lapides DA, McDonald MM. Inflammatory manifestations of systemic diseases in the central nervous system. Curr Treat Options Neurol 2020; 22: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jesse S, Brettschneider J, Süssmuth SD, et al. Summary of cerebrospinal fluid routine parameters in neurodegenerative diseases. J Neurol 2011; 258: 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Janssen J, Godbolt A, Ioannidis P, et al. The prevalence of oligoclonal bands in the CSF of patients with primary neurodegenerative dementia. J Neurol 2004; 251: 184–188. [DOI] [PubMed] [Google Scholar]
- 66. Escudero D, Guasp M, Ariño H, et al. Antibody-associated CNS syndromes without signs of inflammation in the elderly. Neurology 2017; 89: 1471–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hansson O, Zetterberg H, Buchhave P, et al. Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol 2006; 5: 228–234. [DOI] [PubMed] [Google Scholar]
- 68. McGuire LI, Peden AH, Orrú CD, et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt–Jakob disease. Ann Neurol 2012; 72: 278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moreno-Ajona D, Prieto E, Grisanti F, et al. 18F-FDG-PET imaging patterns in autoimmune encephalitis: impact of image analysis on the results. Diagnostics (Basel) 2020; 10: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Probasco JC, Solnes L, Nalluri A, et al. Decreased occipital lobe metabolism by FDG-PET/CT: an anti-NMDA receptor encephalitis biomarker. Neurol Neuroimmunol Neuroinflamm 2018; 5: e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Grüter T, Ott A, Meyer W, et al. Effects of IVIg treatment on autoantibody testing in neurological patients: marked reduction in sensitivity but reliable specificity. J Neurol 2020; 267: 715–720. [DOI] [PubMed] [Google Scholar]
- 72. McKeon A, Pittock SJ, Lennon VA. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology 2011; 76: 1108–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zalewski NL, Lennon VA, Lachance DH, et al. P/Q-and N-type calcium-channel antibodies: oncological, neurological, and serological accompaniments. Muscle Nerve 2016; 54: 220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lang K, Prüss H. Frequencies of neuronal autoantibodies in healthy controls: estimation of disease specificity. Neurol Neuroimmunol Neuroinflamm 2017; 4: e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ruiz-García R, Martínez-Hernández E, Saiz A, et al. The diagnostic value of onconeural antibodies depends on how they are tested. Front Immunol 2020; 11: 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sechi E, Flanagan EP. Diagnosis and management of autoimmune dementia. Curr Treat Options Neurol 2019; 21: 11. [DOI] [PubMed] [Google Scholar]
- 77. Lang B, Makuch M, Moloney T, et al. Intracellular and non-neuronal targets of voltage-gated potassium channel complex antibodies. J Neurol Neurosurg Psychiatry 2017; 88: 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Van Sonderen A, Schreurs MW, De Bruijn MA, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology 2016; 86: 1692–1699. [DOI] [PubMed] [Google Scholar]
- 79. Van Sonderen A, Thijs RD, Coenders EC, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology 2016; 87: 1449–1456. [DOI] [PubMed] [Google Scholar]
- 80. Walikonis JE, Lennon VA. (eds). Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus. Mayo Clin Proc 1998; 73: 1161–1166. [DOI] [PubMed] [Google Scholar]
- 81. Meinck H, Faber L, Morgenthaler N, et al. Antibodies against glutamic acid decarboxylase: prevalence in neurological diseases. J Neurol Neurosurg Psychiatry 2001; 71: 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chang C-C, Eggers SD, Johnson JK, et al. Anti-GAD antibody cerebellar ataxia mimicking Creutzfeldt-Jakob disease. Clin Neurol Neurosurg 2007; 109: 54–57. [DOI] [PubMed] [Google Scholar]
- 83. Amouzegar A, Gharibzadeh S, Kazemian E, et al. The prevalence, incidence and natural course of positive antithyroperoxidase antibodies in a population-based study: Tehran thyroid study. PLoS One 2017; 12: e0169283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pedersen IB, Knudsen N, Jorgensen T, et al. Thyroid peroxidase and thyroglobulin autoantibodies in a large survey of populations with mild and moderate iodine deficiency. Clin Endocrinol (Oxf) 2003; 58: 36–42. [DOI] [PubMed] [Google Scholar]
- 85. Gauthier AC, Baehring JM. Hashimoto’s encephalopathy mimicking Creutzfeldt-Jakob disease. J Clin Neurosci 2017; 35: 72–73. [DOI] [PubMed] [Google Scholar]
- 86. Mattozzi S, Sabater L, Escudero D, et al. Hashimoto encephalopathy in the 21st century. Neurology 2020; 94: e217–e224. [DOI] [PubMed] [Google Scholar]
- 87. Pittock SJ, Kryzer TJ, Lennon VA. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol 2004; 56: 715–719. [DOI] [PubMed] [Google Scholar]
- 88. McKeon A, Apiwattanakul M, Lachance DH, et al. Positron emission tomography-computed tomography in paraneoplastic neurologic disorders: systematic analysis and review. Arch Neurol 2010; 67: 322–329. [DOI] [PubMed] [Google Scholar]
- 89. Williams TJ, Benavides DR, Patrice K-A, et al. Association of autoimmune encephalitis with combined immune checkpoint inhibitor treatment for metastatic cancer. JAMA Neurol 2016; 73: 928–933. [DOI] [PubMed] [Google Scholar]
- 90. LaPorte J, Solh M, Ouanounou S. Posterior reversible encephalopathy syndrome following pembrolizumab therapy for relapsed Hodgkin’s lymphoma. J Oncol Pharm Pract 2017; 23: 71–74. [DOI] [PubMed] [Google Scholar]
- 91. Maurice C, Schneider R, Kiehl T-R, et al. Subacute CNS demyelination after treatment with nivolumab for melanoma. Cancer Immunol Res 2015; 3: 1299–1302. [DOI] [PubMed] [Google Scholar]
- 92. Daxini A, Cronin K, Sreih AG. Vasculitis associated with immune checkpoint inhibitors—a systematic review. Clin Rheumatol 2018; 37: 2579–2584. [DOI] [PubMed] [Google Scholar]
- 93. Vernino S, O’Neill BP, Marks RS, et al. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro Oncol 2004; 6: 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dubey D, Britton J, McKeon A, et al. Randomized placebo-controlled trial of intravenous immunoglobulin in autoimmune LGI1/CASPR2 epilepsy. Ann Neurol 2020; 87: 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. S. V. efficacy of ocrelizumab in autoimmune encephalitis. ClinicalTrialsgov Identifier: NCT03835728, 2020. [Google Scholar]
- 96. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12: 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Dubey D, David WS, Reynolds KL, et al. Severe neurological toxicity of immune checkpoint inhibitors: growing spectrum. Ann Neurol 2020; 87: 659–669. [DOI] [PubMed] [Google Scholar]
- 98. Puzanov I, Diab A, Abdallah K, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer 2017; 5: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bartels F, Krohn S, Nikolaus M, et al. Clinical and magnetic resonance imaging outcome predictors in pediatric anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 2020; 88: 148–159. [DOI] [PubMed] [Google Scholar]
- 100. Dubey D, Kothapalli N, McKeon A, et al. Predictors of neural-specific autoantibodies and immunotherapy response in patients with cognitive dysfunction. J Neuroimmunol 2018; 323: 62–72. [DOI] [PubMed] [Google Scholar]
- 101. Balu R, McCracken L, Lancaster E, et al. A score that predicts 1-year functional status in patients with anti-NMDA receptor encephalitis. Neurology 2019; 92: e244–e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hoftberger R, Van Sonderen A, Leypoldt F, et al. Encephalitis and AMPA receptor antibodies: novel findings in a case series of 22 patients. Neurology 2015; 84: 2403–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65: 424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Van Sonderen A, Arino H, Petit-Pedrol M, et al. The clinical spectrum of Caspr2 antibody-associated disease. Neurology 2016; 87: 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hara M, Arino H, Petit-Pedrol M, et al. DPPX antibody-associated encephalitis: main syndrome and antibody effects. Neurology 2017; 88: 1340–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hoftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and GABAB receptor antibodies: novel findings in a new case series of 20 patients. Neurology 2013; 81: 1500–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jeffery OJ, Lennon VA, Pittock SJ, et al. GABAB receptor autoantibody frequency in service serologic evaluation. Neurology 2013; 81: 882–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Spatola M, Sabater L, Planaguma J, Martinez- 759 Hernandez E, Armangue T, Pruss H, et al. Encephalitis 760 with mGluR5 antibodies: symptoms and antibody ef- 761 fects. Neurology. 2018;90(22):e1964–e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. McKeon A, Martinez-Hernandez E, Lancaster E, et al. Glycine receptor autoimmune spectrum with stiff-man syndrome phenotype. JAMA Neurol 2013; 70: 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gaig C, Graus F, Compta Y, et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017; 88: 1736–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gresa-Arribas N, Planaguma J, Petit-Pedrol M, et al. Human neurexin-3α antibodies associate with encephalitis and alter synapse development. Neurology 2016; 86: 2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Do LD, Chanson E, Desestret V, et al. Characteristics in limbic encephalitis with anti-adenylate kinase 5 autoantibodies. Neurology 2017; 88: 514–524. [DOI] [PubMed] [Google Scholar]
- 114. Pittock SJ, Lucchinetti CF, Parisi JE, et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol 2005; 58: 96–107. [DOI] [PubMed] [Google Scholar]
- 115. Dalmau J, Graus F, Rosenblum MK, et al. Anti-Hu—associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine 1992; 71: 59–72. [DOI] [PubMed] [Google Scholar]
- 116. Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology 1998; 50: 652–657. [DOI] [PubMed] [Google Scholar]
- 117. Chan KH, Vernino S, Lennon VA. ANNA-3 anti-neuronal nuclear antibody: marker of lung cancer-related autoimmunity. Ann Neurol 2001; 50: 301–311. [DOI] [PubMed] [Google Scholar]
- 118. Honnorat J, Cartalat-Carel S, Ricard D, et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry 2009; 80: 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yu Z, Kryzer TJ, Griesmann GE, et al. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol 2001; 49: 146–154. [PubMed] [Google Scholar]
- 120. Pittock SJ, Yoshikawa H, Ahlskog JE, et al. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin Proc 2006; 81: 1207–1214. [DOI] [PubMed] [Google Scholar]
- 121. Saiz A, Blanco Y, Sabater L, et al. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: diagnostic clues for this association. Brain 2008; 131(Pt 10): 2553–2563. [DOI] [PubMed] [Google Scholar]
- 122. Alfugham N, Gadoth A, Lennon VA, et al. ITPR1 autoimmunity: frequency, neurologic phenotype, and cancer association. Neurol Neuroimmunol Neuroinflamm 2018; 5: e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Mandel-Brehm C, Dubey D, Kryzer TJ, et al. Kelch-like protein 11 antibodies in seminoma-associated paraneoplastic encephalitis. N Engl J Med 2019; 381: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Basal E, Zalewski N, Kryzer TJ, et al. Paraneoplastic neuronal intermediate filament autoimmunity. Neurology 2018; 91: e1677–e1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Vernino S, Lennon VA. New Purkinje cell antibody (PCA-2): marker of lung cancer-related neurological autoimmunity. Ann Neurol 2000; 47: 297–305. [PubMed] [Google Scholar]