

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a genetically heterogeneous, biologically aggressive malignancy with a uniformly poor prognosis. While most pancreatic cancers arise sporadically, a small subset of PDACs develop in patients with hereditary and familial predisposition. Detailed studies of the rare hereditary syndromes have led to identification of specific genetic abnormalities that contribute to malignancy. For example, germline mutations involving BRCA1, BRCA2, PRSS1, and mismatch repair genes predispose patients to PDAC. While patients with Lynch syndrome develop a rare “medullary” variant of adenocarcinoma, intraductal papillary mucinous tumors are observed in patients with McCune-Albright syndrome. It is now well established that PDACs originate via a multistep progression from microscopic and macroscopic precursors due to cumulative genetic abnormalities. Improved knowledge of tumor genetics and oncologic pathways has contributed to a better understanding of tumor biology with attendant implications on diagnosis, management, and prognosis. In this article, the genetic landscape of PDAC and its precursors will be described, the hereditary syndromes that predispose to PDAC will be reviewed, and the current role of imaging in screening and staging assessment, as well as the potential role of molecular tumor-targeted imaging for evaluation of patients with PDAC and its precursors, will be discussed.
Keywords: Abdomen/GI, Genetic Defects, Oncology, Pancreas
Supplemental material is available for this article.
© RSNA, 2020
Summary
Understanding the genetic landscape and molecular biology of sporadic and hereditary pancreatic ductal adenocarcinoma may permit early diagnosis, optimal management, and precise prognostication.
Essentials
■ Pancreatic adenocarcinoma (PDAC), the most lethal pancreatic malignancy, arises from one of the three main precursor lesions, namely pancreatic intraepithelial neoplasms, intraductal papillary mucinous neoplasms, or mucinous cystic tumors.
■ Ninety percent of PDACs occur in sporadic forms and up to 10% occur in familial or hereditary syndromes.
■ Four key driver gene abnormalities (KRAS, p16, TP53, and SMAD4) are characteristically found in most PDACs in addition to several other chromosomal alterations and epigenetic modifications; specific genetic changes are associated with characteristic subtypes of PDACs and their precursors.
■ A deeper understanding of the precursor lesions of familial and sporadic PDAC with their imaging appearances, key clinical parameters, and genetic abnormalities may be useful to develop strategies for screening and early detection of PDAC.
■ Better knowledge of tumor genetics and oncologic pathways may lead to molecular diagnostics and targeted therapeutics; for example, patients with BRCA mutations respond exquisitely to poly (adenosine diphosphate–ribose) polymerase inhibitors.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common pancreatic malignancy (90%). Although PDAC accounts for 2% of all cancer cases, it constitutes the fourth leading cause of cancer-related deaths in the United States with a 5-year survival of 5%–10% (1). PDAC is also projected to be the second leading cause of cancer-related death by 2030 (2). Ninety percent of PDACs are sporadic in origin; around 10% of cases occur in hereditary and familial predisposition syndromes. To date, certain high-risk factors such as smoking, alcohol use, and chronic pancreatitis have been strongly implicated (3). The risk for PDAC increases with age; more than 80% of cases occur between ages 60 and 80 years (4). The mean size of PDAC is approximately 3.1 cm, and approximately 80% of patients manifest distant metastases or locally advanced disease at presentation, which make them ineligible for surgical cure. Rapid advances in precision medicine and immunotherapy have contributed little to early detection with only modest improvements in patient outcomes (3). Recent advances in high-throughput sequencing technologies have allowed for a better understanding of the PDAC genomic landscape, defined precursor lesions, and delineated genetic alterations that lead to tumorigenesis (5). Moreover, improved knowledge of genetic causes of PDAC may improve prognostication and targeted treatment in select cases (6). In this article, we will review current knowledge about genetic abnormalities of precursor lesions that contribute to PDAC development. Imaging features of PDAC and precursor lesions, the potential role of molecular imaging, and select hereditary syndromes associated with a high risk of PDAC development will also be discussed. Finally, we will review the current guidelines for screening of PDAC and surveillance of precursor lesions.
Role of Imaging and Brief Overview of Staging in PDAC
Multiphasic CT and multiparametric MRI play crucial roles in diagnosing, staging, assessing treatment response, and detecting recurrence of PDAC. Multiphasic CT is considered the diagnostic test of choice on account of consistently superior and reproducible image quality as well as wide availability. CT allows fast acquisition of thin-section, isovolumetric data that permits high-resolution images and multiplanar reconstructions. Multiphasic MRI complemented by MR cholangiopancreatography and diffusion-weighted imaging allows for robust staging evaluation of PDAC as well (7). A detailed account of imaging techniques, protocols, and manifestations of PDAC is beyond the scope of this article. PDAC manifests as an infiltrative, hypoenhancing mass that is best observed on the pancreatic parenchymal phase of CT and MR images. The mass is associated with abrupt duct cutoff with upstream ductal dilatation of the pancreatic and biliary ductal system (depending on tumor location) as well as variable parenchymal atrophy of the distal pancreas (Fig 1). Involvement of adjacent vasculature determines surgical resectability as well as potential for tumor recurrence (7,8). A distinctive feature of PDAC is its dense desmoplastic stroma that is partly reflected in the hypoenhancement (on parenchymal phase) and delayed enhancement of tumors. PDAC also exhibits remarkable proclivity for perineural invasion (53%–100% of tumors) and invasion of small veins (65% of surgically resected tumors), which are factors thought to be responsible for aggressive local-regional spread, frequent hepatic metastases, and high rates of recurrence (7,9,10).
Figure 1:

Metastatic pancreatic ductal adenocarcinoma in a 65-year-old man. Coronal contrast-enhanced reformatted CT image from CT examination performed for evaluation of refractory vomiting and epigastric discomfort revealed the pancreatic head mass (arrowhead) with distal pancreatic atrophy and upstream dilatation of the pancreatic duct, causing duodenal obstruction (not shown) with marked gastric dilatation (black arrows). There are also peripancreatic lymphadenopathy (✰) and rim-enhancing hepatic nodules (white arrows). This was proven to be metastatic pancreatic ductal adenocarcinoma with nodal and hepatic metastases on further workup.
Multidetector CT is highly accurate for assessing tumor extent, vascular invasion, resectability, and evaluating distant metastases. Vascular enhancement of a PDAC, the most important parameter determining tumor resectability, is better assessed with multidetector CT than MRI. However, CT cannot reliably depict isodense tumors (11%–27% of tumors) or small tumors of less than 1.5 cm in size (7). MRI has sensitivity of 85%–93% and specificity of 72%–79% for diagnosis of PDAC, and it is preferred for small or isodense tumors (8). MR cholangiopancreatography can depict subtle ductal narrowing due to a small mass (7). A radiation-dose-neutral technique, dual-energy CT, allows viewing CT images at multiple energy levels and generation of iodine images and virtual noncontrast (water only) CT images. Iodine images with high contrast-to-noise ratio enhance lesion conspicuity and detection, making this modality useful for depicting small and isoattenuating cancers (7,11). CT angiograms can also be generated from low energy or iodine data sets improving staging of pancreatic cancer (7). Endoscopic US-guided fine-needle aspiration has very high sensitivity and specificity of up to 86% and 98%, respectively, and is found superior to multidetector CT for detection of solid pancreatic lesions of less than 2 cm in size with accuracy of about 92% (12). Endoscopic US-guided fine-needle aspiration also allows for tissue sampling, although with added invasiveness and risks. PET/CT is useful in whole-body imaging and staging, detecting viable malignancy, predicting tumor aggressiveness and survival, and assessing treatment response. PET/CT has sensitivity and specificity of about 93% and 76% for PDAC diagnosis compared with about 86% and 71% with multidetector CT, but PET/CT also can have false-positive findings (8).
The TNM staging system is the most commonly used staging method to assess tumor status (T), lymph nodes (N), and metastasis (M). While providing important prognostic information, TNM staging does not assess tumor feasibility of surgical resection. The most desired result of staging is to segregate the resectable, borderline resectable, locally advanced, and metastatic tumors. TNM stage I and II are typically resectable. These tumors show normal tissue planes between the tumor and visceral arteries, or these tumors may abut the superior mesenteric vein or portal vein without distortion. TNM stage IV is characterized by distant metastasis (Fig 1); consequently, these are directed to palliative therapy options and will not benefit from surgery. TNM stage III tumor receives further attention, as it is a localized tumor with major vessel involvement. TNM stage III tumors need subcategorization into either locally advanced unresectable or borderline resectable tumors. If the tumor encases (>180 degrees circumference) the celiac, superior mesenteric artery, or hepatic artery or occludes the superior mesenteric vein or portal vein without a viable option for venous reconstruction, it is designated as unresectable (Fig 2). If the tumor just abuts (< 180 degrees circumference) the celiac, superior mesenteric artery, or hepatic artery and if venous reconstruction is possible despite invasion of the superior mesenteric vein or portal vein, then it is regarded as borderline resectable cancer. If preceded and downstaged by neoadjuvant treatment, a borderline resectable pancreatic cancer may benefit more from resection (13).
Figure 2:

Locally advanced unresectable pancreatic ductal adenocarcinoma in a 37-year-old man. Axial arterial phase 70-keV image from dual-energy CT of the abdomen performed for the workup of new onset diabetes, abdominal pain, and rapid weight loss revealed infiltrative pancreatic head mass (arrowhead) completely encasing the proximal superior mesenteric artery (arrow) and its jejunal branch. This was proven to be pancreatic ductal adenocarcinoma at biopsy and was unresectable due to vascular involvement.
However, despite multiple important roles of imaging in management of PDAC, the currently used conventional imaging and surgical methods have limited impact. Currently, only 15%–25% of PDAC are detected at early stages (optimal for resection) with 70%–80% of these tumors achieving negative margins at resection with pathologically complete resection (R0). When a 1-mm free margin was used to define R0 resection, the rate of R0 resection dropped to about 15%–50% in several reports (14,15). Standard imaging techniques such as intraoperative US, endoscopic US, MRI, or PET, also do not detect the several below-described mutations and/or molecular features needed for early detection of premalignant or malignant pancreatic lesions. Imaging techniques such as fluorescence, photoacoustic, and Raman optical imaging may open new opportunities to determine tumor extent and delineation for complete tumor resection and assessing metastases and nodal status. These technologies are at early stages of clinical translation.
Precursor Lesions
Detailed pathologic studies incorporating advanced, genetic, and molecular techniques have expanded our understanding of histogenesis and pathogenesis of PDAC. It is now well-known that most PDACs originate from microscopic (pancreatic intraepithelial neoplasia [PanIN]) and macroscopic precursor lesions. Conservative time estimates for transformation of PanIN to infiltrating cancer, development of metastatic subclone in the carcinoma, and dissemination of metastatic subclone to death, are thought to be on average 11.7 years, 6.8 years, and 2.7 years, respectively (16). While PanINs contribute to most (about 90%) PDACs, a significant proportion of PDACs arise from macroscopic mucinous neoplasms such as intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms (Fig 3) (17–19). These precursor lesions share many genetic alterations found in PDACs. Improved understanding of cancer precursors sheds light on the importance of early detection of such lesions, particularly in high-risk patients.
Figure 3:

Precursors, genomic landscape, and approximate timelines in the course of development of pancreatic ductal adenocarcinoma (PDAC). T1, T2, T3 are approximate successive time intervals from initiation of pancreatic intraepithelial neoplasm (PanIN) to gaining invasive ability, from attaining metastatic potential to death. HG-IPMN = high-grade intraductal papillary mucinous neoplasm, HG-MCN = high-grade mucinous cystic neoplasm, HG-PanIN = high-grade PanIN, LG-IPMN = low-grade intraductal papillary mucinous neoplasm, LG-MCN = low-grade mucinous cystic neoplasm, LG-PanIN = low-grade PanIN.
PanIN and PDAC
Most PDACs arise from PanINs. PanIN refers to microscopic, intraductal neoplasms (by definition <5 mm) lined by gastric-foveolar epithelia of varying degrees of architectural and cytologic atypia. These cannot be detected by current imaging modalities (20). According to nuclear and cellular atypia, PanINs are classified into three categories: PanIN-1, PanIN-2, and PanIN-3. The first two are now considered low-grade tumors and may be found in the normal pancreas after the age of 40 years, while PanIN-3 is a high-grade lesion that almost always occurs with concomitant cancer (95% of PanIN-3) (Fig 4) (21).
Figure 4a:

Pancreatic ductal adenocarcinoma in a 61-year-old man. (a, b) Axial contrast-enhanced CT images of the pancreas show a hypovascular infiltrative mass (arrows) in the uncinate process with associated upstream ductal dilatation (arrowheads). Histopathologic findings following Whipple surgery revealed adenocarcinoma against background chronic pancreatitis and high-grade pancreatic intraepithelial neoplasm (microscopic precursor).
Figure 4b:

Pancreatic ductal adenocarcinoma in a 61-year-old man. (a, b) Axial contrast-enhanced CT images of the pancreas show a hypovascular infiltrative mass (arrows) in the uncinate process with associated upstream ductal dilatation (arrowheads). Histopathologic findings following Whipple surgery revealed adenocarcinoma against background chronic pancreatitis and high-grade pancreatic intraepithelial neoplasm (microscopic precursor).
Intraductal Papillary Mucinous Neoplasm
IPMNs are macroscopic (>1 cm by definition) cystic tumors characterized by intraductal growth of papillary lesions with typically copious and thick mucin production (22). IPMNs commonly arise in the pancreatic head and are more common in elderly men (mean age, 65 years). Most patients with IPMNs are asymptomatic; some patients may have nonspecific symptoms of abdominal pain, jaundice, as well as symptoms due to exocrine and/or endocrine pancreatic insufficiency (23). IPMNs may be multifocal and may predominantly involve branch ducts, main ducts, or both. The pathologic nomenclature of IPMNs is complex and still evolving. Histologically, IPMNs are classified according to their epithelial lining into gastric, intestinal, pancreatobiliary, and oncocytic subtypes. However, there may be considerable histologic overlap and “mixed” or unclassifiable type tumors that are observed in a significant proportion of patients (24). The oncocytic type was recently considered a separate entity as well (21). IPMNs may also be classified into low-grade or high-grade tumors based on the degree of epithelial dysplasia. The incidence and type of invasive malignancy developing in IPMNs differ from one histologic type to another (25). Gastric-type IPMNs typically involve branch ducts and are least likely to harbor associated invasive malignancy. Colloid carcinoma almost always arises in association with intestinal-type IPMNs (26) and carries a better prognosis than tubular carcinoma that is associated with pancreatobiliary-type IPMNs (27). Branch-duct IPMNs appear as unilocular or multilocular cystic lesions that communicate with a branch duct. Mixed-type IPMN also involves the main pancreatic duct with associated ductal dilatation in addition to branch duct cysts. Pancreatic ductal communication is the key feature differentiating these from other cystic neoplasms (Fig 5). Main-duct IPMN is characterized by cystic dilatation of the main duct, variable ductal wall thickening, and thick, abundant mucin that distends and obstructs the duct (Fig 6).
Figure 5a:

Side-branch intraductral pancreatic mucinous neoplasm in a 78-year-old man. (a) Axial fat-saturated T2-weighted fast-spin-echo image and (b) coronal maximum intensity projection image of a three-dimensional MR cholangiopancreatography show dominant multilocular cystic lesion in the pancreatic head (arrow) and multiple unilocular cystic lesions in distal pancreas (arrowheads) communicating with the nondilated pancreatic duct.
Figure 6:

Main duct intraductal papillary mucinous neoplasm with high-grade dysplasia in a 72-year-old woman. Coronal maximum intensity projection image of a three-dimensional MR cholangiopancreatography shows diffuse dilatation of the main pancreatic duct and cystic dilatation of multiple side branches (arrows), along with diffuse dilatation of the biliary system.
Figure 5b:

Side-branch intraductral pancreatic mucinous neoplasm in a 78-year-old man. (a) Axial fat-saturated T2-weighted fast-spin-echo image and (b) coronal maximum intensity projection image of a three-dimensional MR cholangiopancreatography show dominant multilocular cystic lesion in the pancreatic head (arrow) and multiple unilocular cystic lesions in distal pancreas (arrowheads) communicating with the nondilated pancreatic duct.
MRI and MR cholangiopancreatography are superior to CT in the assessment of ductal communication of the cystic lesions as well as depiction of internal characteristics such as septations and mural nodules (Fig 7), although both modalities are equivalent in early detection and characterization. Moreover, MRI is preferable to CT for imaging surveillance of cystic lesions, particularly in young patients because of cumulative deleterious effects of ionizing radiation with CT (28). Thickened irregular septae, enhancing mural nodule, or solid components within the IPMN are suspicious for malignancy (Fig 8). Specific, worrisome imaging features that strongly suggest malignancy include main duct diameter of greater than 1 cm, enhancing mural nodule greater than 5 mm (at endoscopic US), cystic growth rate of greater than 5 mm per year, and cyst greater than 4 cm in diameter (29). IPMNs are multicentric in 20%–40% of cases, which emphasizes the importance of follow-up of patients after surgical resection (24).
Figure 7a:

Malignant main-duct-type intraductal papillary mucinous neoplasm (IPMN) in a 58-year-old man. (a) Coronal contrast-enhanced CT and (b) T2-weighted MR images of the pancreas show a heterogeneous solid-cystic pancreatic head mass (arrows) and upstream dilatation (✰) of the main pancreatic duct, suspicious for malignant IPMN. Communication (long arrow in b) of the mass with the main pancreatic duct is clearly seen better at MR cholangiopancreatography compared with CT. Main-duct-type IPMN was found with dysplasia and microscopic focus of mucinous adenocarcinoma at pathologic findings following Whipple surgery.
Figure 8a:

Colloid adenocarcinoma in the pancreatic neck and body arising from intraductal papillary mucinous neoplasm (IPMN) in a 66-year-old man. (a) Axial and (b) coronal contrast-enhanced CT images show markedly dilated pancreatic duct with irregular nodular soft tissue along its wall consistent with malignancy in the main duct IPMN (arrows). Diagnosis confirmed following radical surgery. Colloid carcinomas frequently show GNAS mutations. (c) Histopathologic (hematoxylin-eosin stain) slide shows moderate to poorly differentiated carcinoma cells (long arrows) and signet cells forming small acini floating in mucin pool (short arrows), consistent with colloid carcinoma.
Figure 7b:

Malignant main-duct-type intraductal papillary mucinous neoplasm (IPMN) in a 58-year-old man. (a) Coronal contrast-enhanced CT and (b) T2-weighted MR images of the pancreas show a heterogeneous solid-cystic pancreatic head mass (arrows) and upstream dilatation (✰) of the main pancreatic duct, suspicious for malignant IPMN. Communication (long arrow in b) of the mass with the main pancreatic duct is clearly seen better at MR cholangiopancreatography compared with CT. Main-duct-type IPMN was found with dysplasia and microscopic focus of mucinous adenocarcinoma at pathologic findings following Whipple surgery.
Figure 8b:

Colloid adenocarcinoma in the pancreatic neck and body arising from intraductal papillary mucinous neoplasm (IPMN) in a 66-year-old man. (a) Axial and (b) coronal contrast-enhanced CT images show markedly dilated pancreatic duct with irregular nodular soft tissue along its wall consistent with malignancy in the main duct IPMN (arrows). Diagnosis confirmed following radical surgery. Colloid carcinomas frequently show GNAS mutations. (c) Histopathologic (hematoxylin-eosin stain) slide shows moderate to poorly differentiated carcinoma cells (long arrows) and signet cells forming small acini floating in mucin pool (short arrows), consistent with colloid carcinoma.
Figure 8c:

Colloid adenocarcinoma in the pancreatic neck and body arising from intraductal papillary mucinous neoplasm (IPMN) in a 66-year-old man. (a) Axial and (b) coronal contrast-enhanced CT images show markedly dilated pancreatic duct with irregular nodular soft tissue along its wall consistent with malignancy in the main duct IPMN (arrows). Diagnosis confirmed following radical surgery. Colloid carcinomas frequently show GNAS mutations. (c) Histopathologic (hematoxylin-eosin stain) slide shows moderate to poorly differentiated carcinoma cells (long arrows) and signet cells forming small acini floating in mucin pool (short arrows), consistent with colloid carcinoma.
Mucinous Cystic Neoplasm
Mucinous cystic neoplasms are the least common macroscopic precursor lesion that can lead to PDAC (19). Mucinous cystic neoplasms differ from IPMNs in that they do not communicate with the ductal system and they are almost always solitary. In addition, 98% of mucinous cystic neoplasms occur in perimenopausal women with a distinct proclivity to involve the distal pancreas (> 90% occur in the tail region) (22) and histologically characterized by a pathognomonic spindle cell ovarian-type stroma (22).
Mucinous cystic neoplasms commonly appear as large, complex cystic masses with variable CT density due to hemorrhage, necrosis, and calcifications (Fig 9). MRI and MR cholangiopancreatography exquisitely depict the internal septations, variable fluid signal intensity, and solid components of multilocular cystic masses. As with IPMNs, the malignant mucinous cystic neoplasms show heterogeneity, thickened septations, and enhancing solid components (Fig 10). Surgical resection is recommended in mucinous cystic neoplasms of greater than 4 cm, symptomatic tumors, and mucinous cystic neoplasms with features suspicious for malignancy such as enhancing mural nodules. European evidence-based guidelines suggest that asymptomatic or smaller mucinous cystic neoplasms without suspicious features need long-term surveillance (every 6 months for 1st year and then annual) as long as they do not have surgical contraindications (29). Also, mucinous cystic neoplasms must be monitored more closely in pregnant women as faster growth rates have been documented during pregnancy (29). The risk of malignant transformation of mucinous cystic neoplasms is lower than that of IPMN; approximately 16% of mucinous cystic neoplasms are associated with invasive malignancy. When malignancy develops, it is typically of the tubular carcinoma variant of PDAC, but lymph node metastasis is usually not seen (30). Mucinous cystic neoplasms share the same genetic alterations found in PDAC, and on average there are 16 nonsynonymous mutations per tumor (31).
Figure 9a:

(a) Mucinous cystic neoplasm with low-grade dysplasia in a 62-year-old woman. Axial contrast-enhanced CT image of the pancreas shows a unilocular cystic lesion (arrow) involving the pancreatic body without mural nodule. (b) Mucinous cystic neoplasm with high-grade dysplasia in a 25-year-old woman. Axial contrast-enhanced CT image of the pancreas shows a unilocular cystic lesion involving the pancreatic body with wall calcifications (arrow).
Figure 10a:

Mucinous cystadenocarcinoma in a 34-year-old woman. (a) Axial contrast-enhanced CT image and (b) US image of the pancreas show a large, complex cystic mass in the pancreatic tail (arrow) with enhancing solid nodules (arrowhead) consistent with mucinous cystadenocarcinoma.
Figure 9b:

(a) Mucinous cystic neoplasm with low-grade dysplasia in a 62-year-old woman. Axial contrast-enhanced CT image of the pancreas shows a unilocular cystic lesion (arrow) involving the pancreatic body without mural nodule. (b) Mucinous cystic neoplasm with high-grade dysplasia in a 25-year-old woman. Axial contrast-enhanced CT image of the pancreas shows a unilocular cystic lesion involving the pancreatic body with wall calcifications (arrow).
Figure 10b:

Mucinous cystadenocarcinoma in a 34-year-old woman. (a) Axial contrast-enhanced CT image and (b) US image of the pancreas show a large, complex cystic mass in the pancreatic tail (arrow) with enhancing solid nodules (arrowhead) consistent with mucinous cystadenocarcinoma.
Genetic and Epigenetic Landscape of PDAC and Its Precursor Lesions
PDAC is a genetically heterogeneous neoplasm. It is now being recognized that the progression of invasive PDAC from normal pancreatic cells involves successive accumulation of genetic and epigenetic alterations in core signaling pathways within tumor cells over a period of up to 15–20 years (17,32). Understanding and exploiting the pathogenesis of these genetic and/or epigenetic alterations and associated precursor lesions will facilitate new screening tools for early detection of PDAC before invasion and metastasis and development of targeted, molecular therapeutics. Whole-exome sequencing of tumor cells has revealed an average of 48 nonsynonymous somatic mutations per tumor, altering a core set of 12 signaling pathways; many of these mutations are also found in the precursor lesions (33). Barring some exceptions, four driver mutations are consistently found in the majority of PDACs; these include an activating mutation of the KRAS oncogene and subsequent inactivating mutations of three tumor suppressor genes (CDKN2A/P16, TP53, and SMAD4) (Fig 1) (34). Each of these mutations causes deregulation of a specific signaling pathway, resulting in initiation or evolution of cancer clones. KRAS mutation and telomere shortening are the earliest genetic alterations during PDAC development, seen in greater than 90% of low-grade PanINs (Fig 1) (34). In addition to the frequent genetic alterations that IPMNs share with PDAC, IPMNs also carry distinct mutations (32). Approximately 75% of IPMNs harbor an inactivating mutation in the tumor suppressor gene, RNF43, which encodes a ubiquitin ligase (31). Also, an activating mutation at a hotspot codon in the oncogene GNAS has been found in 60% of IPMNs (35). GNAS encodes Gas protein, and its mutation causes constitutive activation of adenylyl cyclase with downstream effects driving proliferation. It is thus not so surprising then, that patients with McCune-Albright syndrome, an autosomal dominant syndrome with GNAS activating mutations, have an increased risk for IPMN (36). The colloid carcinoma variant, which is related to intestinal IPMNs, also harbors GNAS mutations more frequently than conventional PDAC (35). Medullary carcinoma, a rare variant associated with Lynch syndrome, shows KRAS mutation much less frequently, and rather shows the characteristic microsatellite instability related to Lynch syndrome (37). Adenosquamous carcinoma is reported to harbor distinct mutations including amplification of the MYC oncogene and mutations of UPF1 gene, which encodes for a core component in the nonsense-mediated RNA decay pathway (38). The four most common key mutations are described in detail below.
KRAS
KRAS mutation plays a pivotal role in PDAC development; it is the most frequently mutated oncogene and the earliest genetic alteration to occur. Mutations in KRAS occur in 95% of PDACs and 80% of IPMNs (34). KRAS proto-oncogene encodes a small GTPase molecule that acts as a transducer for growth factor receptors present on the cell surface (34,39). KRAS mutations dysregulate intrinsic GTPase activity resulting in constitutively active KRAS with persistent stimulation of downstream pathways such as PI3K/AKT/mTOR and RAF/MAP kinase pathways. Activation of these pathways drives uncontrolled cellular proliferation, angiogenesis, suppression of apoptosis, and evasion of the immune response (34,39). Attempts to target mutated KRAS protein in the clinic have failed or resulted in excess toxicity (34,40).
CDKN2A/p16
CDKN2A/p16 tumor suppressor gene is altered in about 95% of PDACs; it is the most commonly inactivated tumor suppressor gene during PDAC carcinogenesis (39). Inactivation of the p16 protein, a regular of the retinoblastoma signaling pathway (39), results in unchecked cell cycle progression through the G1/S (DNA synthesis) checkpoint, with enhanced cell proliferation (41). P16 inactivation may be observed in lesions as early as at PanIN-1B stage (42). Germline mutation of the CDKN2A gene is seen in familial atypical multiple mole melanoma syndromes, an inherited susceptibility to melanoma and PDAC development (43,44).
TP53
TP53 tumor suppressor gene inactivation is a later event in PDAC, appearing at PanIN-3 stage. Mutations in TP53 occur in 75% of PDACs (17,42). TP53 encodes for p53, the master guardian of the genome, which plays a central role in DNA repair, cell cycle arrest, and induction of apoptosis in response to DNA damage or cellular stress (18). Inactivation of p53 (loss of function mutation) allows DNA damage to go unchecked with failed apoptosis and unregulated G1/S cell cycle transition. Studies have shown that many resultant mutant p53 proteins not only lose the tumor suppressive activities of endogenous wild-type p53, but also gain new independent pro-oncogenic activities (gain-of-function mutation) promoting cell proliferation, survival, angiogenesis, and metastases. Recently, another type of TP53 mutation called truncating or separation of function mutation has been described, which affects only certain biochemical properties of wild-type p53 and is described elsewhere. TP53 mutations occur in many human malignancies and also in Li-Fraumeni cancer predisposition syndrome (Fig 11). Patients with Li-Fraumeni syndrome with gain-of-function TP53 germline mutation have increased chances of developing cancers in childhood and adolescence, while those with loss-of-function TP53 mutations have more propensity for cancers later in life (45,46).
Figure 11a:
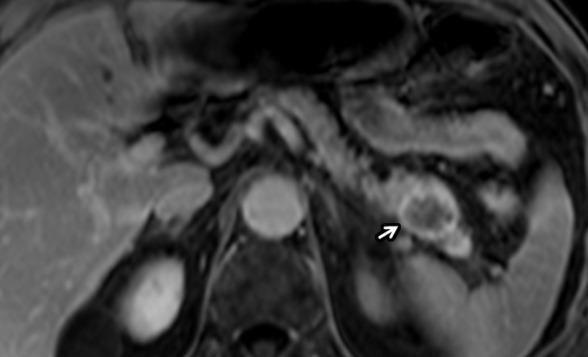
Pancreatic adenocarcinoma in a 67-year-old man with Li-Fraumeni syndrome (TP53 mutations) with prior history of lung and prostate cancers. (a) Axial postcontrast T1-weighted and (b) diffusion-weighted images show incidentally detected hypovascular mass (arrow) in the pancreatic tail with restricted diffusion during screening MRI. This was proven to be PDAC following distal pancreatectomy.
Figure 11b:

Pancreatic adenocarcinoma in a 67-year-old man with Li-Fraumeni syndrome (TP53 mutations) with prior history of lung and prostate cancers. (a) Axial postcontrast T1-weighted and (b) diffusion-weighted images show incidentally detected hypovascular mass (arrow) in the pancreatic tail with restricted diffusion during screening MRI. This was proven to be PDAC following distal pancreatectomy.
SMAD4
SMAD4 inactivation occurs in approximately 50% of PDACs as a late event during progression. Mutations in SMAD4 classically are not present in low-grade PanINs or IPMNs (17,33). SMAD4 inactivation results in loss of its encoded protein, Smad4, which is a downstream effector of transforming growth factor-beta (TGF-β) signaling pathway (18). Loss of Smad4 promotes cancer progression by alleviating the early growth inhibitory effect of the TGF-β pathway and is associated with higher rates of metastasis and poorer prognosis (47).
Epigenetic Control and Tumor Microenvironment
Epigenetic events have been increasingly recognized in development of PDAC (48), and epigenetic changes potentially can even serve as biomarkers or even targets for therapy. Noncoding RNA (known for epigenetic control of gene expression), such as micro-RNA-34 (miR-34) (49), is deleted or significantly downregulated in PDAC (50). Overexpression of miR-21 correlates with poor prognosis in PDAC (49). miR-21 levels are also found to be elevated in precursor lesions (51). Interestingly, many targets of miR-21 are negative regulators of KRAS (49,51), which further enhances signaling by this oncogene. Emerging evidence also suggests that pancreatic cancer cells induce epigenetic alterations in stromal fibroblasts to promote their own growth (42), promoting desmoplastic stromal reaction in the tumor microenvironment hampering the passive transport of chemotherapeutics (52).
Potential Role of Molecular Tumor-targeted Imaging
Several strategies using imaging agents to target the molecular features or specific receptors are being explored for precise and early detection of PDAC or its precursors. Detailed discussion of these is beyond the scope of this article. Briefly, specific biomarkers resulting from above altered signaling pathways may serve as targets for tumor-specific imaging. For example, highly expressed claudin-4 in high-grade PanIn lesions (53), and similarly upregulated claudin-4 and prostate stem cell antigen in most PDACs (54) represent attractive candidates for targeted imaging aimed at early detection. Contrast-enhanced US with microbubbles developed to recognize vascular endothelial growth factor shows promise to detect tumor angiogenesis at US (55). MRI accuracy may be enhanced by strategies delivering gadolinium in a tumor-targeted nanocomplex against biomarkers such as plectin-1 and epidermal growth factor receptor (8). Investigators have also developed dual-modality imaging agents such as targeting mucin-1 with MRI and near-infrared fluorescence imaging (8,56). Similarly, dual-mode imaging using PET and near-infrared targeting the CA 19–9 tumor marker appears to be promising in mapping sentinel lymph nodes and detection of metastases on early animal studies. Different PET imaging tracers targeting cell surface receptor integrin αvβ6 or transmembrane glycoprotein tissue factor, which are overexpressed in PDAC, are other attractive imaging targets (8).
Hereditary Syndromes Associated with PDAC
Up to 10% of PDAC cases are hereditary and/or familial in origin (57). These disorders may be syndromic or nonsyndromic familial aggregations with increased risk of PDAC development. Known hereditary syndromes with predisposition for PDAC include Lynch syndrome, Peutz-Jeghers syndrome, familial adenomatous polyposis syndrome, ataxia telangiectasia, Li-Fraumeni syndrome, hereditary breast and ovarian cancer syndrome, familial atypical multiple mole melanoma, hereditary pancreatitis, and cystic fibrosis (43). For simplicity, these entities can be segregated into those associated with chronic pancreatitis, gastrointestinal tract cancers, other solid cancers, or those with unspecific familial predisposition (Table 1). There are no recognized genetic alterations in the majority of the patients with familial pancreatic cancer disorder; the cause of familial clustering of PDAC in these cases is still unclear (58). While imaging features of hereditary PDAC do not differ much from the sporadic form, they tend to have precursor lesions more frequently than the sporadic counterparts. PanINs and IPMNs have been shown to occur with higher frequency in many of the above-mentioned syndromes. A study showed that 18% of familial pancreatic cancer syndromes were associated with high-grade IPMNs, whereas 10% of sporadic cancers were associated with such lesions (59).
Table 1:
Pancreatic Ductal Adenocarcinoma Predisposition Syndromes

Familial Pancreatic Cancer Syndrome
Familial pancreatic cancer syndrome, which represents 90% of all hereditary PDAC cases, is defined as pedigrees with two or more first-degree relatives affected by PDAC and who do not meet the criteria for a pancreatic cancer predisposition syndrome (such as those mentioned later) (58,60). The risk for developing PDAC (Fig 12) is proportional to the number of affected first-degree relatives; it is 6.4% when two first-degree relatives are affected but goes up to 32% when three are affected (60). As mentioned previously, the genetic mutation(s) responsible for familial pancreatic cancer syndrome is still unclear. However, segregation analysis models have supported a rare autosomal-dominant gene to be responsible, which seems to put 0.4%–0.7% of the population at risk for developing PDAC (61). Although smoking is a well-documented risk factor for sporadic PDAC, it remains unclear whether or how environmental factors influence PDAC risk in familial pancreatic cancer syndrome families (58).
Figure 12:

Metastatic pancreatic cancer in a 57-year-old man with familial pancreatic cancer syndrome. Patient had a very strong family history of pancreatic cancer, with all his maternal great aunts, maternal grandmother, and mother affected with pancreatic cancer. Axial contrast-enhanced CT image of the abdomen revealed subtle hypoenhancing pancreatic head mass (black arrow) compared with adjacent normal enhancing pancreatic parenchyma (arrowhead). Multiple enlarged retroperitoneal nodes (white arrows) are seen, some with low attenuation center, suspicious for nodal metastases. This was proven to be pancreatic ductal adenocarcinoma with retroperitoneal nodal metastases. Patient also had multiple hepatic and pulmonary metastases (not shown).
Hereditary Pancreatitis
Patients with hereditary pancreatitis have a 50- to 70-fold relative risk for developing PDAC compared with the general population (62). Smokers with hereditary pancreatitis are particularly more vulnerable to develop PDAC. About 30%–40% of all hereditary pancreatitis carriers will develop PDAC by the age of 70 years (Fig E1 [supplement]). However, PDAC tends to develop earlier in smokers with hereditary pancreatitis (up to 20 years earlier than the average age of occurrence) (63). Hereditary pancreatitis is caused by a germline gain-of-function mutation in PRSS1 (autosomal-dominant) and/or loss of function mutation of SPINK1 (autosomal-recessive) genes (64). The gene PRSS1 encodes for trypsinogen, whereas SPINK1 encodes for a trypsin inhibitor. Genetic alterations of PRSS1 and SPINK1 result in either premature activation or reduced inhibition of trypsin within the pancreas, respectively, resulting in tissue injury and chronic pancreatitis. Most patients develop symptoms of pancreatitis before the age of 20 years (43,64).
Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome has the highest risk for developing PDAC of any tumor predisposition syndrome, although its rarity makes the incidence of Peutz-Jeghers syndrome–associated PDAC low (65). Patients with Peutz-Jeghers syndrome have roughly a 130-fold increase risk for PDAC development compared with the general population; their lifetime risk of PDAC is 11%–32%. Peutz-Jeghers syndrome is caused by an autosomal dominant germline inactivation of the STK11/LKB1 gene (43,66). The syndrome presents with benign gastrointestinal hamartomatous polyps and mucocutaneous pigmentation in all patients, in addition to the risk for developing different malignancies, including gastrointestinal, lung, breast, ovarian, and uterine cancers. Increased risk of IPMNs has also been reported (67).
Lynch Syndrome
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome, is an autosomal dominant disorder that carries about 8.5-fold increase risk for PDAC development (Fig E2 [supplement]) (68,69). Lynch syndrome is caused by a germline mutation in any of the four genes involved in DNA mismatch repair process, namely, MLH1, MSH2, MSH6, and PMS2 (70). One other mechanism includes upstream epithelial cell adhesion molecule (EPCAM) gene deletions, which results in silencing of the neighboring MSH2 gene by promoter hypermethylation. Methylation of the MSH2 promoter reduces the expression of MSH2 gene and its protein which is important for repairing DNA errors (71). Errors in DNA repair could be particularly seen in areas of repetitive DNA sequences resulting in microsatellite instability, a characteristic feature of Lynch syndrome–associated malignancies. This feature can be a possible target for new immunotherapy regimens such as programmed cell death protein 1 inhibitors (72). The most common malignancies in patients with Lynch syndrome are colorectal cancer (with 50%–80% penetrance) and endometrial cancer (with 25%–60% penetrance) (73,74). Variability in penetrance seems to depend on the type of mismatch repair gene mutated, which also might be responsible for the variability in PDAC incidence in these cases (43). PDACs in patients with Lynch syndrome are usually of the conventional type, but poorly differentiated medullary variants may rarely occur (37).
Hereditary Breast and Ovarian Cancer Syndrome
The precise risk associated with hereditary breast and ovarian cancer syndrome for PDAC development varies widely in the literature and differs according to the specific mutated gene (43,66). Hereditary breast and ovarian cancer syndrome is an autosomal-dominant disorder caused by a germline inactivation of BRCA1, BRCA2, or PALB2 tumor suppressor genes, which are involved in double-stranded DNA break repair. BRCA1 and BRCA2 are strongly associated with the development of breast cancer (90% and 41% by age 80 years, respectively) and ovarian cancer (24% and 8.4% by age 80 years, respectively) (75). The BRCA2 mutation is associated with three- to 10-fold increased risk for PDAC development (Fig E3 [supplement]) and probably accounts for the highest percentage of hereditary pancreatitis with known genetic cause (76). In one retrospective study of 204 patients with BRCA mutations, 17% and 4% of BRCA2 patients developed IPMN and PDAC, respectively, whereas 8% of BRCA1 patients developed IPMN (77). An association between BRCA1 mutation and PDAC is still debatable in the literature (78). Association between PALB2 mutation and PDAC development has been recognized (79), and PALB2 heterozygotes are six times more likely to have a relative with PDAC (80). Nonpancreatic BRCA-associated tumors have shown response to platinum agents and poly (adenosine diphosphate–ribose) polymerase inhibitors, which are also being tested in the treatment for PDAC in this patient cohort (81).
Familial Atypical Multiple Mole Melanoma Syndrome
The relative risk for PDAC development in familial atypical multiple mole melanoma syndrome varies by the type of genetic alteration. Germline variants that cause familial atypical multiple mole melanoma syndrome include CDKN2A, CDK4, and MITF (44). The variant CDKN2A, which encodes the p16 protein, discussed above, causes 40% of familial atypical multiple mole melanoma syndrome cases and is the only germline variant associated with PDAC development; the risk varies widely between 13- and 65-fold increase (43,66). Even different CDKN2A mutations (eg, P16-Leiden mutation) carry variable penetrance for PDAC development. Familial atypical multiple mole melanoma syndrome is characterized by multiple nevi and an increased risk for developing melanoma (60%–90% risk by age 80 years in CDKN2A/P16 carriers) (44). PDAC incidence is second only to melanomas.
Familial Adenomatous Polyposis Syndrome
Primary PDAC development in patients with familial adenomatous polyposis syndrome is rare, and the incidence is not clearly defined yet (66). However, familial adenomatous polyposis syndrome is roughly associated with 4.5-fold increase risk of periampullary malignancies. These are mostly duodenal and ampullary cancers, rather than primary PDAC, but the latter may also occur (43). Familial adenomatous polyposis syndrome is caused by an autosomal-dominant inactivating mutation of the tumor suppressor gene, APC, which is responsible for β-catenin degradation through the Wnt/β-catenin pathway (66). High-level cytoplasmic expression and nuclear localization of accumulated β-catenin induces oncogenic cellular traits, promotes cancer cell proliferation and survival, and also facilitates tumor progression by suppressing T-cell immune response (82). Familial adenomatous polyposis syndrome is characterized by the development of innumerable colonic polyps that proceed to cancer by the age of 40 years (73). Although patients undergo prophylactic colectomy at a young age to reduce the risk of colon cancer, there are currently no clear guidelines for PDAC screening in these cases.
PDAC Screening
Late diagnosis of PDAC from lack of symptoms early in the disease course is one of the major contributing factors to the lethal biologic behavior of the disease (83). Although consensus exists for some aspects of PDAC screening, the exact benefit of screening remains unclear (84,85). Screening of the general population is currently not feasible due to the low disease incidence and high costs (86). The main goal of screening, therefore, is early detection of asymptomatic high-grade precursor lesions and noninvasive PDAC through targeted screening of high-risk populations, such as patients with a family history of PDAC or a cancer susceptibility syndrome, to enable detection of resectable lesions (83,84). The lethal behavior of PDAC and early metastases still limit outcomes (87).
Imaging remains the best available screening option. Biomarkers such as CA 19–9 have limitations as screening tools (84,86,88). CA 19–9 suffers low sensitivity (70%–80%) and specificity (80%–90%) for PDAC in addition to an abysmal positive predictive value of 59% (88). Other diagnostic tools involving gene expression profiles, cell-free DNA, and microRNA are still not available for standard clinical use (89). The most consistently used imaging modalities for screening and follow-up are endoscopic US, MRI, and MR cholangiopancreatography which have demonstrated high accuracy for depicting small cystic lesions, with an excellent concordance of lesion location, number, and size between both modalities (Fig 9) (84,86,90). Endoscopic US is considered a more invasive procedure than MRI, but it has an advantage in that suspicious tissue can be sampled during the procedure (86). Although endoscopic US typically identifies more lesions, it also carries a higher risk than MRI for incorrect diagnosis due to operator dependency (84). This may lead to overtreatment, a major concern of the screening process. Also, endoscopic US may have limitations for evaluating solid pancreatic lesions. CT is much less utilized for PDAC screening due to the radiation exposure (90). It is important to remember that PanIN are microscopic lesions and cannot be visualized at imaging; some studies suggest an association between lobulocentric pancreatic atrophy and the presence of PanIN, but this relation is still weak (91). Some authors have suggested that endoscopic US features of chronic pancreatitis, which are highly prevalent in high-risk patients (for pancreatic cancer), correlate with lobulocentric atrophy due to PanIN seen on pathologic findings (92).
The International Cancer of the Pancreas Screening consortium recommended certain clinical criteria to select candidates for screening or further workup for pancreatic predisposition cancer syndromes and to initiate screening (Table 2) (84). Selections are mostly performed based on family history. However, patients with Peutz-Jeghers syndrome hereditary pancreatitis, and CDKN2A mutation carriers should be screened regardless of family history (84). There is no consensus about the age to start screening or screening intervals. Most start screening at the age of 40–50 years, or 10 years younger than the earliest PDAC in the family (93). Suggested screening intervals are 12 months in the absence of pancreatic abnormalities at the baseline examination, an interval of 6–12 months for patients with a nonsuspicious pancreatic cyst, or 3 months for indeterminate solid lesions and indeterminate main duct strictures. Because most of the detected lesions at screening are low-risk branch-duct IPMN, the panel suggested that IPMNs in patients with hereditary pancreatitis should be managed similar to the guidelines for sporadic and/or incidental IPMNs (22); branch-duct IPMNs should be resected if they become symptomatic, contain mural nodules, and/or if their size is greater than 3 cm; whereas all main-duct IPMNs should be resected because of their risk of malignant progression (22).
Table 2:
Hereditary-Familial Pancreatic Ductal Adenocarcinoma: Risk Assessment and Screening

Conclusion
Pancreatic cancer is a biologically aggressive, highly lethal malignancy that frequently presents with metastases despite a well-to-moderately differentiated histomorphology. Recent advances in cytogenetics and molecular biology offer unique insights into the precursor lesions, tumor genetics, pathogenesis, and differences in the tumor biology of PDAC subtypes. Several hereditary syndromes that predispose patients to development of PDAC are characterized by specific pathogenetic abnormalities; screening and surveillance paradigms are being developed in these patients. In addition to refinements in pancreatic surgery, imaging techniques and therapeutic agents aimed at specific molecular targets are being developed for early detection and treatment of patients with pancreatic cancer.
SUPPLEMENTAL FIGURES
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Authors declared no funding for this work.
Disclosures of Conflicts of Interest: A.C.M. disclosed no relevant relationships. A.K.H. disclosed no relevant relationships. N.S.R. disclosed no relevant relationships. V.S.K. disclosed no relevant relationships. S.Y. disclosed no relevant relationships. A.K.D. disclosed no relevant relationships. S.R.P. disclosed no relevant relationships.
Abbreviations:
- IPMN
- intraductal papillary mucinous neoplasm
- PanIN
- pancreatic intraepithelial neoplasia
- PDAC
- pancreatic adenocarcinoma
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67(1):7–30. [DOI] [PubMed] [Google Scholar]
- 2.Are C, Chowdhury S, Ahmad H, et al. Predictive global trends in the incidence and mortality of pancreatic cancer based on geographic location, socio-economic status, and demographic shift. J Surg Oncol 2016;114(6):736–742. [DOI] [PubMed] [Google Scholar]
- 3.McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol 2018;24(43):4846–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosetti C, Bertuccio P, Negri E, La Vecchia C, Zeegers MP, Boffetta P. Pancreatic cancer: overview of descriptive epidemiology. Mol Carcinog 2012;51(1):3–13. [DOI] [PubMed] [Google Scholar]
- 5.Amundadottir LT. Pancreatic Cancer Genetics. Int J Biol Sci 2016;12(3):314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang X, Hao HX, Growney JD, et al. Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2013;110(31):12649–12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pietryga JA, Morgan DE. Imaging preoperatively for pancreatic adenocarcinoma. J Gastrointest Oncol 2015;6(4):343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tummers WS, Willmann JK, Bonsing BA, Vahrmeijer AL, Gambhir SS, Swijnenburg RJ. Advances in Diagnostic and Intraoperative Molecular Imaging of Pancreatic Cancer. Pancreas 2018;47(6):675–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolfgang CL, Herman JM, Laheru DA, et al. Recent progress in pancreatic cancer. CA Cancer J Clin 2013;63(5):318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hong SM, Goggins M, Wolfgang CL, et al. Vascular invasion in infiltrating ductal adenocarcinoma of the pancreas can mimic pancreatic intraepithelial neoplasia: a histopathologic study of 209 cases. Am J Surg Pathol 2012;36(2):235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNamara MM, Little MD, Alexander LF, Carroll LV, Beasley TM, Morgan DE. Multireader evaluation of lesion conspicuity in small pancreatic adenocarcinomas: complimentary value of iodine material density and low keV simulated monoenergetic images using multiphasic rapid kVp-switching dual energy CT. Abdom Imaging 2015;40(5):1230–1240. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Shpaner A, Krishna SG, et al. Use of EUS-FNA in diagnosing pancreatic neoplasm without a definitive mass on CT. Gastrointest Endosc 2013;78(1):73–80. [DOI] [PubMed] [Google Scholar]
- 13.McIntyre CA, Winter JM. Diagnostic evaluation and staging of pancreatic ductal adenocarcinoma. Semin Oncol 2015;42(1):19–27. [DOI] [PubMed] [Google Scholar]
- 14.Chandrasegaram MD, Goldstein D, Simes J, et al. Meta-analysis of radical resection rates and margin assessment in pancreatic cancer. Br J Surg 2015;102(12):1459–1472. [DOI] [PubMed] [Google Scholar]
- 15.Ansari D, Tingstedt B, Andersson B, et al. Pancreatic cancer: yesterday, today and tomorrow. Future Oncol 2016;12(16):1929–1946. [DOI] [PubMed] [Google Scholar]
- 16.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut 2012;61(7):1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernard V, Fleming J, Maitra A. Molecular and Genetic Basis of Pancreatic Carcinogenesis: Which Concepts May be Clinically Relevant? Surg Oncol Clin N Am 2016;25(2):227–238. [DOI] [PubMed] [Google Scholar]
- 18.Hosoda W, Wood LD. Molecular Genetics of Pancreatic Neoplasms. Surg Pathol Clin 2016;9(4):685–703. [DOI] [PubMed] [Google Scholar]
- 19.Mazur PK, Siveke JT. Genetically engineered mouse models of pancreatic cancer: unravelling tumour biology and progressing translational oncology. Gut 2012;61(10):1488–1500. [DOI] [PubMed] [Google Scholar]
- 20.Basturk O, Hong SM, Wood LD, et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am J Surg Pathol 2015;39(12):1730–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JY, Hong SM. Precursor Lesions of Pancreatic Cancer. Oncol Res Treat 2018;41(10):603–610. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka M, Fernández-del Castillo C, Adsay V, et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology 2012;12(3):183–197. [DOI] [PubMed] [Google Scholar]
- 23.Tran Cao HS, Kellogg B, Lowy AM, Bouvet M. Cystic neoplasms of the pancreas. Surg Oncol Clin N Am 2010;19(2):267–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castellano-Megías VM, Andrés CI, López-Alonso G, Colina-Ruizdelgado F. Pathological features and diagnosis of intraductal papillary mucinous neoplasm of the pancreas. World J Gastrointest Oncol 2014;6(9):311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yopp AC, Allen PJ. Prognosis of invasive intraductal papillary mucinous neoplasms of the pancreas. World J Gastrointest Surg 2010;2(10):359–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seidel G, Zahurak M, Iacobuzio-Donahue C, et al. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: a study of 39 cases. Am J Surg Pathol 2002;26(1):56–63. [DOI] [PubMed] [Google Scholar]
- 27.Takasu N, Kimura W, Moriya T, et al. Intraductal papillary-mucinous neoplasms of the gastric and intestinal types may have less malignant potential than the pancreatobiliary type. Pancreas 2010;39(5):604–610. [DOI] [PubMed] [Google Scholar]
- 28.Bhosale P, Balachandran A, Tamm E. Imaging of benign and malignant cystic pancreatic lesions and a strategy for follow up. World J Radiol 2010;2(9):345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.European Study Group on Cystic Tumours of the Pancreas . European evidence-based guidelines on pancreatic cystic neoplasms. Gut 2018;67(5):789–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crippa S, Salvia R, Warshaw AL, et al. Mucinous cystic neoplasm of the pancreas is not an aggressive entity: lessons from 163 resected patients. Ann Surg 2008;247(4):571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A 2011;108(52):21188–21193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wood LD, Hruban RH. Pathology and molecular genetics of pancreatic neoplasms. Cancer J 2012;18(6):492–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321(5897):1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014;111(5):817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 2011;3(92):92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gaujoux S, Salenave S, Ronot M, et al. Hepatobiliary and Pancreatic neoplasms in patients with McCune-Albright syndrome. J Clin Endocrinol Metab 2014;99(1):E97–E101. [DOI] [PubMed] [Google Scholar]
- 37.Wilentz RE, Goggins M, Redston M, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am J Pathol 2000;156(5):1641–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6(1):6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grant TJ, Hua K, Singh A. Molecular Pathogenesis of Pancreatic Cancer. Prog Mol Biol Transl Sci 2016;144:241–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kessler D, Gmachl M, Mantoulidis A, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci U S A 2019;116(32):15823–15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol 2001;2(10):731–737. [DOI] [PubMed] [Google Scholar]
- 42.Khan MA, Azim S, Zubair H, et al. Molecular Drivers of Pancreatic Cancer Pathogenesis: Looking Inward to Move Forward. Int J Mol Sci 2017;18(4):E779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Connor AA, Gallinger S. Hereditary Pancreatic Cancer Syndromes. Surg Oncol Clin N Am 2015;24(4):733–764. [DOI] [PubMed] [Google Scholar]
- 44.Aoude LG, Wadt KA, Pritchard AL, Hayward NK. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res 2015;28(2):148–160. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Zhang C, Feng Z. Tumor suppressor p53 and its gain-of-function mutants in cancer. Acta Biochim Biophys Sin (Shanghai) 2014;46(3):170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller M, Shirole N, Tian R, Pal D, Sordella R. The Evolution of TP53 Mutations: From Loss-of-Function to Separation-of-Function Mutants. J Cancer Biol Res 2016;4(4):1091. [PMC free article] [PubMed] [Google Scholar]
- 47.Yamada S, Fujii T, Shimoyama Y, et al. SMAD4 expression predicts local spread and treatment failure in resected pancreatic cancer. Pancreas 2015;44(4):660–664. [DOI] [PubMed] [Google Scholar]
- 48.Silverman BR, Shi J. Alterations of Epigenetic Regulators in Pancreatic Cancer and Their Clinical Implications. Int J Mol Sci 2016;17(12):E2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frampton AE, Krell J, Jamieson NB, et al. microRNAs with prognostic significance in pancreatic ductal adenocarcinoma: A meta-analysis. Eur J Cancer 2015;51(11):1389–1404. [DOI] [PubMed] [Google Scholar]
- 50.Ji Q, Hao X, Zhang M, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009;4(8):e6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Abue M, Yokoyama M, Shibuya R, et al. Circulating miR-483-3p and miR-21 is highly expressed in plasma of pancreatic cancer. Int J Oncol 2015;46(2):539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR. Stromal reengineering to treat pancreas cancer. Carcinogenesis 2014;35(7):1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nichols LS, Ashfaq R, Iacobuzio-Donahue CA. Claudin 4 protein expression in primary and metastatic pancreatic cancer: support for use as a therapeutic target. Am J Clin Pathol 2004;121(2):226–230. [DOI] [PubMed] [Google Scholar]
- 54.Iacobuzio-Donahue CA, Ashfaq R, Maitra A, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res 2003;63(24):8614–8622. [PubMed] [Google Scholar]
- 55.Pysz MA, Machtaler SB, Seeley ES, et al. Vascular endothelial growth factor receptor type 2-targeted contrast-enhanced US of pancreatic cancer neovasculature in a genetically engineered mouse model: potential for earlier detection. Radiology 2015;274(3):790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Medarova Z, Pham W, Kim Y, Dai G, Moore A. In vivo imaging of tumor response to therapy using a dual-modality imaging strategy. Int J Cancer 2006;118(11):2796–2802. [DOI] [PubMed] [Google Scholar]
- 57.Permuth-Wey J, Egan KM. Family history is a significant risk factor for pancreatic cancer: results from a systematic review and meta-analysis. Fam Cancer 2009;8(2):109–117. [DOI] [PubMed] [Google Scholar]
- 58.Petersen GM. Familial pancreatic cancer. Semin Oncol 2016;43(5):548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi C, Klein AP, Goggins M, et al. Increased Prevalence of Precursor Lesions in Familial Pancreatic Cancer Patients. Clin Cancer Res 2009;15(24):7737–7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res 2004;64(7):2634–2638. [DOI] [PubMed] [Google Scholar]
- 61.Schneider R, Slater EP, Sina M, et al. German national case collection for familial pancreatic cancer (FaPaCa): ten years experience. Fam Cancer 2011;10(2):323–330. [DOI] [PubMed] [Google Scholar]
- 62.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol 2010;24(3):349–358. [DOI] [PubMed] [Google Scholar]
- 63.Lowenfels AB, Maisonneuve P, Whitcomb DC, Lerch MM, DiMagno EP. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. JAMA 2001;286(2):169–170. [DOI] [PubMed] [Google Scholar]
- 64.Stram M, Liu S, Singhi AD. Chronic Pancreatitis. Surg Pathol Clin 2016;9(4):643–659. [DOI] [PubMed] [Google Scholar]
- 65.van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol 2010;105(6):1258–1264; author reply 1265. [DOI] [PubMed] [Google Scholar]
- 66.Pittman ME, Brosens LA, Wood LD. Genetic Syndromes with Pancreatic Manifestations. Surg Pathol Clin 2016;9(4):705–715. [DOI] [PubMed] [Google Scholar]
- 67.Sato N, Rosty C, Jansen M, et al. STK11/LKB1 Peutz-Jeghers gene inactivation in intraductal papillary-mucinous neoplasms of the pancreas. Am J Pathol 2001;159(6):2017–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302(16):1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geary J, Sasieni P, Houlston R, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam Cancer 2008;7(2):163–172. [DOI] [PubMed] [Google Scholar]
- 70.Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 2015;15(3):181–194. [DOI] [PubMed] [Google Scholar]
- 71.Kuiper RP, Vissers LE, Venkatachalam R, et al. Recurrence and variability of germline EPCAM deletions in Lynch syndrome. Hum Mutat 2011;32(4):407–414. [DOI] [PubMed] [Google Scholar]
- 72.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 2015;372(26):2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Al-Sukhni W, Aronson M, Gallinger S. Hereditary colorectal cancer syndromes: familial adenomatous polyposis and lynch syndrome. Surg Clin North Am 2008;88(4):819–844, vii. [DOI] [PubMed] [Google Scholar]
- 74.Talseth-Palmer BA, Wijnen JT, Grice DM, Scott RJ. Genetic modifiers of cancer risk in Lynch syndrome: a review. Fam Cancer 2013;12(2):207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Risch HA, McLaughlin JR, Cole DE, et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: a kin-cohort study in Ontario, Canada. J Natl Cancer Inst 2006;98(23):1694–1706. [DOI] [PubMed] [Google Scholar]
- 76.Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK and Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269-275. Cancer 2015;121(14):2474–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roch AM, Schneider J, Carr RA, et al. Are BRCA1 and BRCA2 gene mutation patients underscreened for pancreatic adenocarcinoma? J Surg Oncol 2019;119(6):777–783. [DOI] [PubMed] [Google Scholar]
- 78.Moran A, O’Hara C, Khan S, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer 2012;11(2):235–242. [DOI] [PubMed] [Google Scholar]
- 79.Tischkowitz MD, Sabbaghian N, Hamel N, et al. Analysis of the gene coding for the BRCA2-interacting protein PALB2 in familial and sporadic pancreatic cancer. Gastroenterology 2009;137(3):1183–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res 2011;71(6):2222–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lowery MA, Kelsen DP, Stadler ZK, et al. An emerging entity: pancreatic adenocarcinoma associated with a known BRCA mutation: clinical descriptors, treatment implications, and future directions. Oncologist 2011;16(10):1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shang S, Hua F, Hu ZW. The regulation of β-catenin activity and function in cancer: therapeutic opportunities. Oncotarget 2017;8(20):33972–33989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Poruk KE, Firpo MA, Adler DG, Mulvihill SJ. Screening for pancreatic cancer: why, how, and who? Ann Surg 2013;257(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Canto MI, Harinck F, Hruban RH, et al. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62(3):339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vasen H, Ibrahim I, Ponce CG, et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J Clin Oncol 2016;34(17):2010–2019. [DOI] [PubMed] [Google Scholar]
- 86.Lindquist CM, Miller FH, Hammond NA, Nikolaidis P. Pancreatic cancer screening. Abdom Radiol (NY) 2018;43(2):264–272. [DOI] [PubMed] [Google Scholar]
- 87.Yu J, Blackford AL, Dal Molin M, Wolfgang CL, Goggins M. Time to progression of pancreatic ductal adenocarcinoma from low-to-high tumour stages. Gut 2015;64(11):1783–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fong ZV, Winter JM. Biomarkers in pancreatic cancer: diagnostic, prognostic, and predictive. Cancer J 2012;18(6):530–538. [DOI] [PubMed] [Google Scholar]
- 89.Chen L, Zhang Y, Cheng Y, Zhang D, Zhu S, Ma X. Prognostic value of circulating cell-free DNA in patients with pancreatic cancer: A systemic review and meta-analysis. Gene 2018;679:328–334. [DOI] [PubMed] [Google Scholar]
- 90.Canto MI, Hruban RH, Fishman EK, et al. Frequent detection of pancreatic lesions in asymptomatic high-risk individuals. Gastroenterology 2012;142(4):796–804; quiz e14–e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Del Chiaro M, Segersvärd R, Lohr M, Verbeke C. Early detection and prevention of pancreatic cancer: is it really possible today? World J Gastroenterol 2014;20(34):12118–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Béchade D. Strategies for screening for pancreatic adenocarcinoma in high-risk patients: the place of endoscopic ultrasound [in French]. Presse Med 2011;40(3):230–238. [DOI] [PubMed] [Google Scholar]
- 93.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110(2):223–262; quiz 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.