

Abstract
Biobanks linked to electronic health records provide rich resources for health-related research. With improvements in administrative and informatics infrastructure, the availability and utility of data from biobanks have dramatically increased. In this paper, we first aim to characterize the current landscape of available biobanks and to describe specific biobanks, including their place of origin, size, and data types.
The development and accessibility of large-scale biorepositories provide the opportunity to accelerate agnostic searches, expedite discoveries, and conduct hypothesis-generating studies of disease-treatment, disease-exposure, and disease-gene associations. Rather than designing and implementing a single study focused on a few targeted hypotheses, researchers can potentially use biobanks’ existing resources to answer an expanded selection of exploratory questions as quickly as they can analyze them. However, there are many obvious and subtle challenges with design and analysis of biobank-based studies. Our second aim is to discuss statistical issues related to biobank research such as study design, sampling strategy, phenotype identification, and missing data. We focus our discussion on biobanks that are linked to electronic health records. Some of the analytic issues are illustrated using data from the Michigan Genomics Initiative and UK Biobank, two biobanks with two different recruitment mechanisms. We summarize the current body of literature for addressing some of these challenges and discuss some standing open problems. This work complements and extends recent reviews about biobank-based research and serves as a resource catalog with analytical and practical guidance for statisticians, epidemiologists, and other medical researchers pursuing research using biobanks.
Keywords: biobanks, electronic health records, Michigan Genomics Initiative, UK Biobank, selection bias
Section 1: Introduction
Biobanks linked to detailed disease phenotype information such as electronic health records (EHR) provide rich data resources for health-related research. Biobanks, loosely defined, are biorepositories that accept, process, store and distribute biospecimen and/or associated data for use in research and clinical care.1 The rise in the number and size of biobanks across the world in recent years can be explained by improvements in biospecimen analysis and the need for large and holistic datasets to address complex diseases and conditions.1,2 Many types of biobanks exist, including commercial, single medical center, health system-based, and population-based biobanks. Some biobanks are disease- or organ-specific, while others encompass an extensive breadth of diseases.
Biospecimens are increasingly being linked with their donor’s EHR. An individual’s EHR contains basic demographic characteristics as well as data on symptoms, medical history, behavior and lifestyle factors, physical examinations, diagnoses, tests, procedures, treatments, medications, referrals, admissions, and discharges.3 In addition to the structured data, there exists clinical notes, images, and other unstructured components of an EHR. An EHR is maintained by a health care provider to primarily plan and document care and assess patient outcomes.3 EHR are distinct from medical and pharmacy claims data, which are maintained by insurance companies. Pharmacy and claims data include billing codes assigned during visits, diagnoses, tests and procedures administered (but usually not test results) from any provider an insured individual interacts with along with prescription data, including dates of when prescriptions are filled or refilled. There are ongoing efforts to link claims data with EHR data to have both a “broad” as well as “deep” view of an individual’s encounters with the health system. The possibility to link EHR with biospecimen, insurance and prescription claims, national disease registries, and death indices, creates the potential for generating an incredibly rich, longitudinal database for health researchers.
Access to such integrated data frames enables researchers to bypass expensive data collection and provide a quick, cost-effective option to explore associations related to diagnosis, patient-reported outcomes, prognosis, treatment response, and survival. While some of the questions answered using biobanks have been driven by a priori biological hypotheses, such biorepositories also allow for agnostic (“hypothesis-free”) interrogations, new discoveries, and hypothesis-generating studies. Phenome-wide association studies (PheWAS), first introduced in Denny et al. (2010), which explore the associations between a single genetic variant of interest and many EHR-derived phenotypes, are one example that highlights the power of phenotype-linked biobank data. PheWAS can be used to replicate known associations and has the potential to discover novel and previously unknown associations for further research.4
The growth and evolution of research around biobanks have led to thoughtful and accessible literature on the topic. Recent reviews briefly discuss statistical and computational considerations for studies involving genetic data,5 limitations of traditional study designs, identifying real-world phenotypes,6,7 and EHR enabled database linkages in making pharmacogenetic discoveries.8 These reviews are limited in their discussion of statistical methods related to biobank and EHR-based research and, in particular, their exploration of critical concepts such as study design, sampling, missing data, and other analytic issues.
In this paper, we complement and extend recent reviews about biobank-based research with the ultimate goal of providing an extensive catalog of resources with analytical, conceptual and practical guidance to statisticians, epidemiologists, and other medical researchers pursuing biobank-based research. We will focus on EHR-linked biobanks, but many of the topics covered are relevant to other biobanks with detailed self-reported disease history information instead of medical records.9 In Section 2, we characterize different types of biobanks and provide descriptions of specific biobanks, including their geographic location, size, data access and availability, data linkages, and more. In Section 3, we discuss general statistical issues related to EHR-linked biobank research, including study design, sampling strategy, phenotype identification, and missing data. We illustrate some of these issues using data from two biobanks: the Michigan Genomics Initiative (MGI)10,11 and the UK Biobank (UKB).12,13 In Section 4, we mention potential opportunities and promising future directions for expanded and principled biobank-based research through a discussion of novel and emerging uses of EHR data, creation of improved analytic infrastructure, and the integration of EHR with external data sources.
Section 2: A Characterization of Major Biobanks
In this section, we describe the types of biobanks that are frequently discussed in the literature and provide detailed descriptions for several existing biobanks. An in-depth discussion of the literature search algorithm used to conduct this review is in Supplementary Section S1. To get a sense of the existing landscape, Supplementary Section S2 enumerates the common health outcomes receiving attention in the biobank literature. A table summarizing the differences in target populations, potential biases, EHR quality, and inferential goals between population-based and medical center/health care system-based biobanks can be found in Supplementary Section S3. The rationale for providing this detailed supplementary material is to create a comprehensive set of resources describing features of various biobanks for a researcher interested in pursuing new lines of inquiry using such data.
Existing Biobanks
Table 1 describes some notable major biobanks with detailed disease phenotype data in terms of their size, location, type, and data access. This table extends the biobank descriptions in Wolford et al. (2018) to include additional information about data linkages and cohort characteristics, and it includes information for a broader set of biobanks.5 Many of the biobanks listed in Table 1 provide access to data for outside researchers, while some offer linkages to additional data sources, such as death registries and detailed prescription information. The biobanks in Table 1 often fall into two general categories: population-based biobanks and medical/health care system-based biobanks. While we attempt to categorize biobanks that share important characteristics, there is substantial heterogeneity within each category. As with any data source, researchers should understand who the participants are, whom the data represents, how the data were collected, and how these factors impact the breadth, depth, quality, and quantity of data.
Table 1:
Description of Selected Major Biobanks
| Biobank | Start year | Location | Age | Size | Type* | Institution | Access | Linked with prescriptions? | Linked to death registry? | Biospecimen Collected | Survey | Website |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All of Us | 2018 | USA | 18+ | 1 million (goal) | Health system | National Institutes of Health | Not yet available | Yes** | - | Blood, saliva, urine | Yes | https://www.joinallofus.org/en |
| BioBank Japan | 2003 | Japan | - | 200,000+ | Population | Ministry of Education, Culture, Sports, Science and Technology | Inquire with biobank | - | Yes | Blood (buccal swabs or nail/hair trimmings) | Yes | http://www.ims.riken.jp/english/projects/pj02.php |
| BioME | - | Mount Sinai Health System | - | 42,000+ | Health system | Mount Sinai Health System | Inquire with biobank | - | - | Blood | Yes | https://icahn.mssm.edu/research/ipm/programs/biome-biobank |
| BioVU | 2007 | Tennessee | 18+ | 250,000+ | Health system | Vanderbilt University | Inquire with biobank | No | No | Blood | No | https://victr.vanderbilt.edu/pub/biovu/?sid=194 |
| China Kadoorie Biobank | 2004 | China | 30–79 | 510,000+ | Population | University of Oxford + Chinese Academy of Medical Sciences | Application for researchers | - | Yes | Blood | Yes | http://www.ckbiobank.org/site/ |
| deCODE Genetics | 1996 | Iceland | - | ~500,000 | Commercial | deCODE (Amgen) | Inquire with biobank | - | - | - | - | https://www.decode.com/ |
| DiscovEHR | 2014 | Geisinger Health System; Regeneron Genetics Center | 18+ | 50000 | Health system | Regeneron Genetics Center + Geisinger Health System | Inquire with biobank | No | No | Blood | No | http://www.discovehrshare.com/ |
| eMERGE Network | 2007 | NHGRI | All | 126,000+ | Network of biobanks | National Human Genome Research Institute | Application for researchers | No | No | Genetic results obtained from external sources | No | https://emerge.mc.vanderbilt.edu/ |
| Generation Scotland | 2006 | Scotland | 18–65 | 30,000+ | Population | University of Edinburgh | Application for researchers | Yes | Yes | Blood, urine (saliva for some patients) | Yes | https://www.ed.ac.uk/generation-scotland |
| Guangzhou Biobank Cohort Study | 2003 | Guangzhou | 50+ | ~30,000 | Population | Universities of Birmingham and Hong Kong + The Guangzhou Occupational Diseases Prevention and Treatment Center | Inquire with biobank | No | Yes | Blood | Yes | https://www.birmingham.ac.uk/research/activity/mds/projects/HaPS/PHEB/Guangzhou/index.aspx |
| HUNT - Nord-Trøndelag Health Study | 2002 | Nord-Trøndelag County, Norway | 20+ | 125,000 | Population | Norwegian University of Science and Technology | Application for researchers | Yes | Yes | Blood (urine for some patients) | Yes | https://www.ntnu.edu/hunt/hunt-biobank |
| Kaiser Permanente Research Bank | 2008 | Kaiser Permanente | 18+ | 308,425 | Health system | Kaiser Permanente | Application for researchers | Yes | - | Blood, saliva | Yes | https://researchbank.kaiserpermanente.org/ |
| Michigan Genomics Initiative | 2012 | Michigan | 18+ | 60,000+ | Health system | University of Michigan | Inquire with biobank | No | Yes** | Blood | Yes* | https://www.michigangenomics.org |
| Million Veterans Program | 2011 | USA | - | 600,000+ | Health system | US Dept. of Veterans Affairs | Inquire with biobank | - | - | Blood | Yes | https://www.research.va.gov/mvp/ |
| MyCode Community Health Initiaitve (Geisinger) | 2007 | Geisinger Health System | 7+ | 190,000+ | Health System | Geisinger Health | Inquire with biobank | No | No | Blood or saliva | No | https://www.geisinger.org/mycode#cgg |
| Partners HealthCare Biobank | 2010 | Brigham and Women’s Hospital; Massachusetts General | 18+ | 80,000+ | Health System | Partners Healthcare | Inquire with biobank | No | No | Blood | Yes | https://biobank.partners.org/ |
| UK Biobank | 2006 | United Kingdom | 40–69 | 500,000 | Population | UK Biobank charity | Application for researchers | - | - | Blood, urine, saliva | Yes | http://www.ukbiobank.ac.uk/about-biobank-uk |
| CARTaGENE† | 2009 | Quebec | 40–69 | 43,000 | Population | CHU Sainte-Justine Research Center | Application for researchers | No | Yes | Blood, urine | Yes | https://www.cartagene.qc.ca/en/about |
| Genes for Good† | 2015 | USA | 18+ | 77,700+ | Self-initiated | University of Michigan | Inquire with biobank | No | No | Saliva | Yes | https://genesforgood.sph.umich.edu |
| Lifelines† | 2006 | Northern Netherlands | All | 167,000+ | Population | Lifelines Biobank | Application for researchers | No | No | Blood, urine | Yes | https://www.lifelines.nl/researcher |
| Trans-Omics for Precision Medicine (TopMed)† | 2014 | USA (various sites) | - | ~145,000 | Consortium of studies | Univeristy of Washington | NIH Database of Genotypes and Phenotypes (dbGaP) | No | No | Genetic results obtained from external sources | No | https://www.nhlbiwgs.org/ |
- indicates information is unknown;
we chose categories we thought best fit each biobank;
indicates we found a source saying the resource is being developed or will be available in the future;
indicates it is not connected to EHR
Note: The information in this table is ascertained to the best of our knowledge. Where it indicates ‘yes’, this means we were able to find a source that indicates this is a feature of the biobank. Where it indicates ‘no’, this means that it was either absent or there was sufficient reason to believe the resource is unavailable at the biobank. It is best to contact the biobank to confirm the availability of resources that are unknown or indicated as not available.
Population-based biobanks
Population-based biobanks are large-scale biorepositories that aim to recruit subjects reasonably representative of the source population. Population-based biobanks recruit directly from the general population (e.g., China Kadoorie Biobank), and subjects are eligible for enrollment irrespective of their disease status or healthcare utilization. Estonia,14,15 Denmark,16 Sweden,17 Saudi Arabia,18 China,19 the Republic of Korea,20,21 Qatar,22,23 and Taiwan24,25 are some of the countries that have invested in establishing population-based (or reasonably representative) biobanks. Their sampling strategy may include active recruitment for particular subpopulations; for example, BioBank Japan26 recruits patients with particular current or past disease status, and the NIH All of Us27 program targets enriched recruitment of underrepresented minorities.
Perhaps the most well-known population-based biobank is the UKB (used in illustrative examples in this paper).12 With approximately 500,000 subjects, it is one of the largest biobanks in the world. All residents aged 40–69 who lived within 25 miles of one of their 22 assessment centers (~9.2 million people) were invited to participate.13 UKB takes advantage of the UK National Health Service to obtain follow-up data (e.g., mortality, cancer registrations, hospital admissions, primary care data) and actively collect and verify conditions that are typically under-reported (e.g., cognitive function, depression).13 These data are linked with genetic, biomarker, and, for some, imaging data, all of which are accessible for research use.
Health care system or medical center-based biobanks
Another class of biobank is based on a particular medical center or health care system. In general, health system-based biobanks, such as Vanderbilt’s BioVU biobank or Geisinger Health’s MyCode Community Health and DiscovEHR initiatives, contain EHR and genotype data while others, like Partners HealthCare Biobank, also collect supplemental survey data. Some, like the large Kaiser Permanente Research Bank (KPRB), have additional linkages with detailed prescription information and feature-specific sub-cohorts (e.g., pregnancy and cancer cohorts in the case of KPRB). A notable health-system based biobank is the Million Veteran Program. With already more than 600,000 enrolled, it is one of the world’s largest genomic biobanks and also allows for the investigation of military-related diseases and conditions. Other such biobanks recruit patients from a distributed network of health centers throughout the country.
MGI (used in illustrative examples) is an academic medical center-based biobank that started at the University of Michigan in 2012. It recruits surgical patients over the age of 18 using opt-in consent (allowing re-contact for future research purposes), collects and stores blood samples, genotypes DNA samples, collects brief survey data related to pain, and is linked to EHR. This biobank can connect patient data to other data sources, including the cancer registry, prescription data, insurance claims, and the national death index. A very appealing feature of MGI is the consent of patients for future re-contact. The biobank is also undergoing an effort to implement an extensive epidemiologic questionnaire designed to be comparable to other biobank survey data, namely the UKB.
For medical center and health system-based biobanks, it is crucial to understand how the participants are recruited and what type of services the health center/system provides. Participants recruited as surgical patients in a specialized medical center will often have very different breadth and depth of data available compared to those recruited from a general clinic at an integrated health system that serves as the patient’s primary provider and offers a wide array of preventive services.
Other types of biobanks
Initially planning to become the first nationwide biobank, deCODE Genetics is now a privately-owned commercial biobank. Launched in 2007 and funded by the National Human Genome Research Institute (NHGRI), the Electronic Medical Records and Genomics (eMERGE) Network combines a network of DNA biorepositories linked with EHR as a resource for genetic analyses. Disease-specific biobanks are also common, and these biobanks may focus on rarer conditions. Two examples are PcBaSe Sweden,28 a prostate cancer cohort, and the Mayo Clinic Biobank for bipolar disorder.29 While disease-specific biobanks may be better powered than other biobank types to study certain diseases, they are typically smaller in size and do not allow us to examine the associations and disease pathways across the medical phenome.
‘Biobank’ is a broad term that includes biobanks that are not linked to EHR. Many biobanks obtain disease and phenotype status through other means (usually self-reported through surveys).9 Many of the analytic challenges discussed in this paper also apply to these non-EHR-linked biobanks that contain disease status and other behavioral and genotype data. We restrict our attention to solely EHR-linked biobanks.
In this section, we introduced the concept of biobanks, described some key characteristics of different types of biobanks while providing detail on some major biobanks, and provided summary information regarding data access and availability (Table 1). These are critical considerations for downstream statistical analysis
Section 3: Statistical Issues Related to Biobank Research
In this section, we discuss statistical issues and strategies for EHR-linked biobank data analysis following a general workflow for a well-designed research study. In Figure 1, we provide a flowchart describing the steps researchers generally take while conducting a study. The development of the research question, clarification of study goals, selection of study sample, and definition of the target population are critical stages of this process. With vast amounts of data becoming increasingly available, there is a tendency for researchers to try a large number of analyses and broadly define their research question based on an analysis that shows something interesting. This strategy is at odds with good statistical practice. We make a distinction between this strategy and large-scale agnostic hypothesis-generating studies such as PheWAS, where the research goal itself is to generate hypotheses or potential associations for future study.
Figure 1:
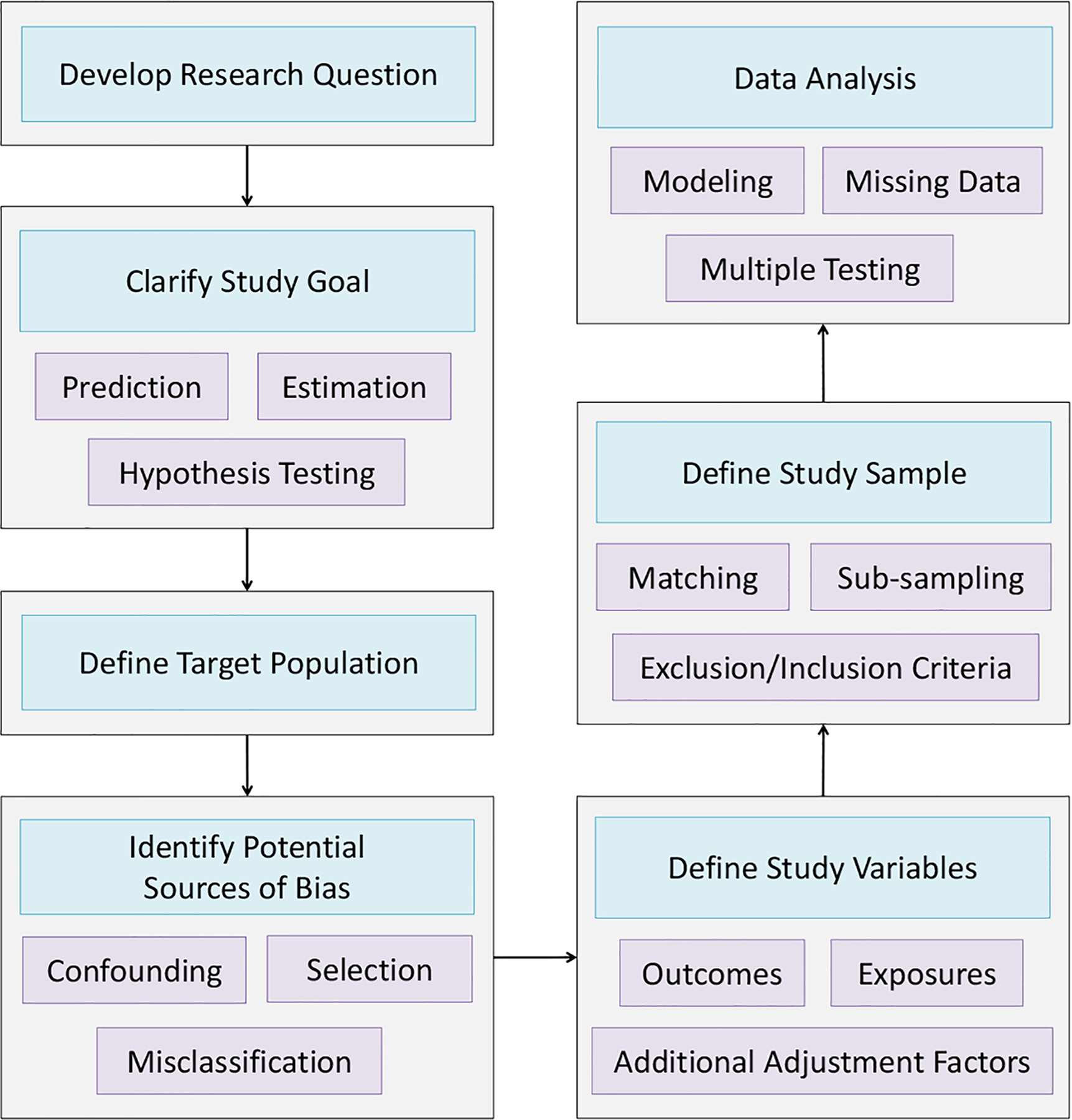
Flowchart of Study Planning, Design and Analysis
Given our research question and data availability, the next step is generally to identify potential sources of bias. In this section, we describe several particular concerns of confounding bias, selection bias, and misclassification of EHR-derived phenotype variables. We then describe challenges and strategies for study design and discuss methods for data analysis, including modeling, correction for multiple testing, and handling of missing data.
Section 3.1: Potential Sources of Bias
Selection Bias due to Non-Probability Sampling
One challenge for research using EHR-linked biobank data is that the mechanism by which a patient from the population enters the biobank and when a visit appears in the EHR is often unknown and inherently patient-driven.30,31 This phenomenon, called non-probability sampling, has been studied extensively in the statistical literature, and certain mechanisms governing self-selected patient recruitment can introduce bias.32 The extent to which the selection mechanism impacts study results depends on the estimand of interest and remains an open question.
The selection mechanism by which patient data are collected may vary widely across biobanks. Population-based biobanks are often large and obtain participants from a network of health or administrative centers across each country with the goal of being reasonably representative of the entire population. However, individual characteristics such as living near an assessment center (e.g., UKB) or living in a specific region of interest (e.g., China Kadoorie) may still impact inclusion. In contrast, medical center and health system-based biobanks attempt to recruit all patients meeting specific criteria within the center/health system, often through selected clinics. Generally, participation in these biobanks requires patients to use healthcare, which is indicative both of ability to access healthcare (e.g., ability to overcome barriers to access including transportation and insurance) and health (i.e., people with diseases and chronic conditions are more frequent users of healthcare). Compared to population-based biobanks, academic medical center-based biobanks tend to see more patients with rare or complicated diseases due to the availability of specialty care and, thus, are often useful for investigating rare conditions. For example, MGI10,11 is enriched for analyses of some cancer types, most notably melanoma of the skin, since Michigan Medicine is known for its skin cancer treatment and care. In all cases, the data generating mechanisms have the potential to induce selection and participation biases into the analysis. These biases can have implications on the generalizability of the results and impact measures of association.33 For guidelines and suggestions for diagnosing and handling non-probability sampled data, we refer the reader to AAPOR task force report on non-probability sampling.34
As a simple demonstration of the impact of different selection mechanisms, we consider prevalence rates for different disease phenotypes in two biobanks: MGI and UKB. As mentioned previously, MGI is a biobank of over 60,000 patients treated at an academic medical center. Patients in MGI were most commonly recruited through the Anesthesiology department as patients were preparing to have a surgical procedure. The UKB is a population-based collection of over 500,000 patients. Table 2 provides comparisons of the patients in MGI and UKB in terms of demographics. Disease statuses were defined for MGI and UKB using aggregated versions of ICD codes, called PheWAS codes or phecodes.35 This method of phenotype classification resulted in 1,681 phecodes that are present in both MGI and UKB. A description of the phenotype generation process can be found in Supplementary Section S5.
Table 2.
Comparison of MGI and UKB Patient Populations
| MGI (Academic Medical Center) | UKB (Population-Based) | |
|---|---|---|
| Sample Size, n | 30,702 | 408,961 |
| Females, n (%) | 16,297 (53.1) | 221,052 (54.1) |
| Mean Age, years (sd) | 54.2 (15.9) | 57.7 (8.1) |
| Median Number of Visits Per Participant | 27 | n/a* |
| Median Days Between First and Last Visit | 1,469 | n/a* |
| Mean Body Mass Index (sd) | 29.7 (7.0) | 27.4 (4.8) |
| Ever Smoked, n (%) | 17,044 (55.5) | 246,320 (60.2) |
Data unavailable for UKB
The different selection mechanisms in the various biobanks have implications for the observed disease prevalences across disease categories. Figure 2 shows the ratios of prevalences of various phenotype codes in MGI and UKB within different disease categories. We see that the majority of the prevalences are higher in MGI. In particular, prevalences for neoplasms, symptoms, endocrine/metabolic disorders, infectious diseases, and congenital anomalies are uniformly higher for MGI compared to UKB. Table 3 presents prevalences of some particular diseases in MGI and UKB along with published prevalences for their corresponding nationwide populations. MGI often captures subjects with many conditions at a higher rate than is observed in the general US population. The UKB has higher case counts than MGI for several conditions due to its size. The UKB is also often more representative of the rates observed in the population (at least for conditions common among ages 40–69, the age range of participants in UKB), with exceptions discussed in Supplementary Section S6.
Figure 2:
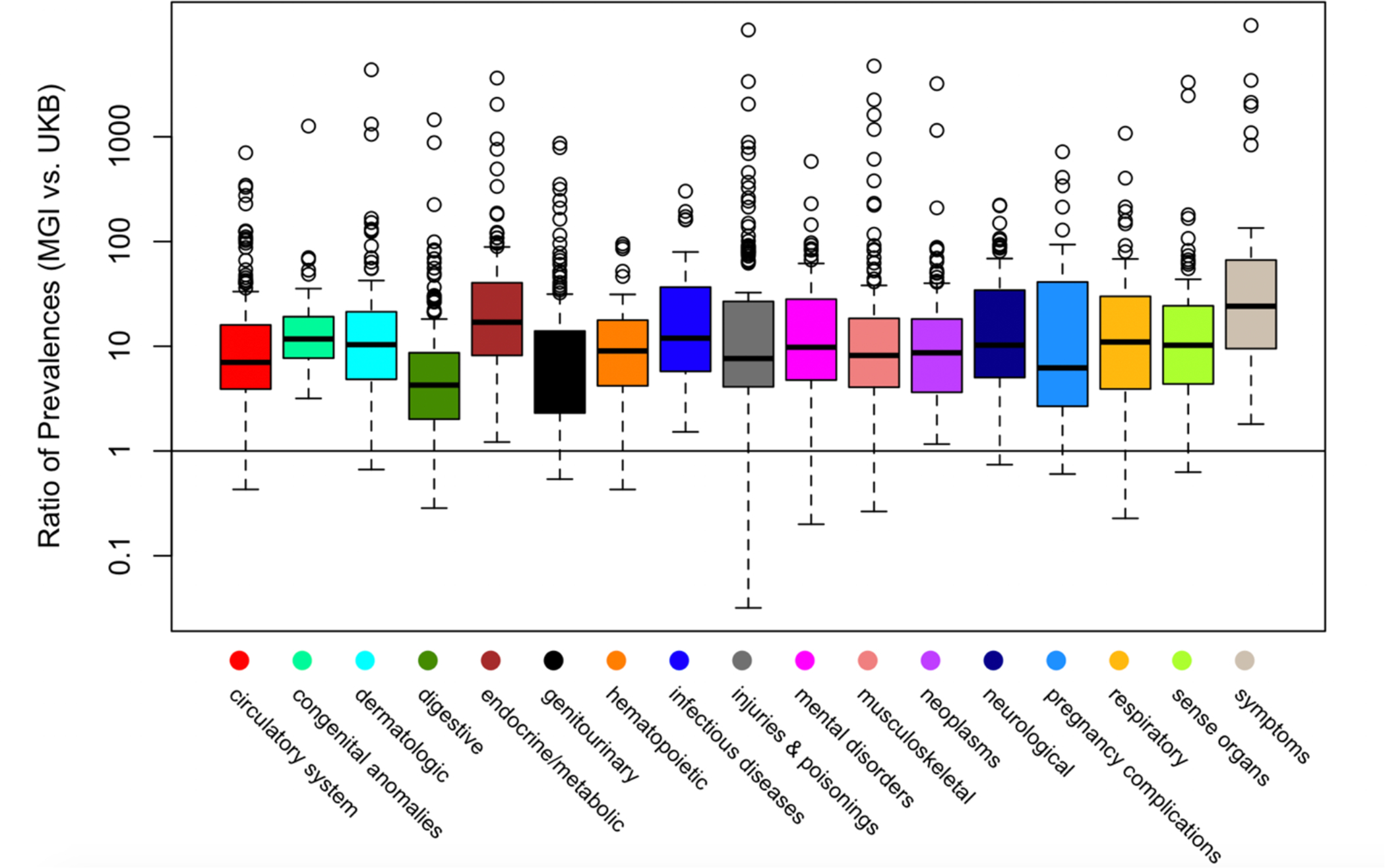
Boxplots of Ratio of PheWAS Code Prevalences in MGI vs. UK Biobank Across Phenome
Table 3:
Prevalences of Selected Conditions in the Michigan Genomics Initiative and UK Biobank along with Estimates from their Respective National Populations∇
| MGI (Academic Medical Center) | United States | UKB (Population-Based) | United Kingdom | |
|---|---|---|---|---|
| N = 30,702 | N = 408,961 | |||
| Psychiatric/Neurologic | ||||
| Depression | 21.7 (6,651) | 16.9** | 2.9 (11,918) | 3.3† |
| Alzheimer’s | 0.2 (60) | 1.6*** | 0.1 (433) | 1.3‡ |
| Anxiety* | 22.1 (6,782) | 31.2**** | 1.6 (6,945) | 5.9† |
| Schizophrenia | 0.3 (78) | .7–1.5 | 0.1 (573) | 0.2–0.59§ |
| Bipolar Disorder | 2.9 (886) | 4.4**** | 0.2 (1,064) | 2.0† |
| Cardiovascular Disease | ||||
| Atrial fibrillation | 9.5 (2,919) | 2–9 | 3.6 (14,839) | 1.2–1.3 |
| Coronary heart disease | 14.3 (4,396) | 6 | 5.0 (20,539) | 3–4 |
| Myocardial infarction | 5.5 (1,702) | 4.7** | 3.0 (12,099) | .87–2.46 |
| Obesity | 33.7 (10,351) | 39.8 | 2.6 (10,820) | 26.2 |
| Diabetes | 21.4 (6,571) | 12.6 | 5.0 (20,260) | 6.2 |
| Cancer | ||||
| Colorectal | 2.6 (806) | 4.2**** | 1.1 (4,627) | 5.3–7.1 **** |
| Breast (female) | 12.4 (2,025) | 12.4**** | 5.7 (12,680) | 12.5 **** |
| Lung | 2.3 (707) | 6.2**** | 0.5 (2,243) | 5.9–7.7 **** |
| Pancreatic | 1.0 (313) | 1.6**** | 0.2 (749) | 1.4 **** |
| Melanoma of skin | 6.2 (1,896) | 2.3**** | 0.7 (2,724) | 1.9 **** |
| Prostate (male) | 12.4 (1,794) | 11.2**** | 3.6 (6,762) | 12.5 **** |
| Bladder | 3.7 (1,147) | 2.3**** | 0.6 (2,433) | 0.9–2.6 **** |
| Non-Hodgkins lymphoma | 3.1 (937) | 2.1**** | 0.4 (1,827) | 1.7–2.1**** |
Phenotypes were defined using ICD-based PheWAS codes35 for MGI and UKB. A description of the phenotype definitions can be found in Supplementary Section S5.
Any anxiety disorder;
adults 40 and older;
adults 65 and older;
lifetime risk of developing disease/condition;
past week prevalence, refers to the presence of symptoms in the past week;
point prevalence, refers to the prevalence measured at a particular point in time (proportion of persons with a particular disease at a point in time);
estimate is from England
Notes: ranges for schizophrenia represent the minimum and maximum point estimates from several estimates included in the source material; ranges for myocardial infarction and cancer estimates provided indicate the range of sex-specific point estimates; lack of representativeness in UKB for obesity phenotype discussed in Supplementary Section S6
Sources for US and UK estimates can be found in Supplementary Table S4
Confounding Bias
Measured and unmeasured confounding are common sources of bias in observational data. Careful use of existing analytical tools can help reduce or eliminate biases resulting from confounding. Here, we define a confounder as a variable that impacts both our outcome and our predictor(s). Failure to adjust for the confounder may result in biased inference regarding the association between the predictor and the outcome. Confounding is of particular concern for EHR data as some well-established measures routinely collected in population-based studies may not be available. In the EHR setting, confounders of interest (e.g., comorbidities) may also often be crudely measured, incomplete, or not measured at all. On the other hand, many potential confounders may be extracted from an EHR database, and variable selection to identify important confounders or adjusting for a high dimensional confounder set in the analysis model are issues specific to EHR studies.36,37
There are many analytical strategies in the statistical literature for dealing with confounding. Popular methods for general observation studies include adjusting for or stratifying analyses by confounders,38 selection propensity weighting, and adjustment and matching on known confounders. Because of the large sample sizes, matching or stratification with respect to levels of confounders still may entail adequate power for a specific hypothesis, leading to new design issues to consider in such studies. Techniques in causal inference such as instrumental variable analysis can also be used to address issues of confounding in EHR.39,40 Recently, researchers have used particular genetic variants as instrumental variables in analyses relating variables such as hormone levels to phenotypes of interest.41 Mendelian randomization analysis is then used to explore potential causal relationships.42 Marginal structural models can be used to address confounding by time-dependent variables and has recently been applied to EHR in Sperrin et al. (2018).43,44 Techniques for reducing and eliminating confounding often assume that the potential confounders are measured. When key confounders are not measured, sensitivity analyses and related statistical methods can be used to explore the impact of and to correct for potential unmeasured confounding.45–48
Defining the Phenome
A central challenge for research involving EHRs is in defining phenotypes. The data available falls into two broad categories: structured and unstructured. Some examples of structured data are billing and procedure codes, numeric lab and test results, and prescription information. Some examples of unstructured data are narrative notes made by physicians/nurses, radiological/pathological notes, and images.
ICD9 and ICD10 diagnosis codes are the most common source for defining phenomes. They are universally defined, which make them appealing (although there may be differential usage across institutions).49 Incorporating other structured data, such as continuous lab values, is more challenging and may require pre-processing. The development and use of automated algorithms for making these data useful for phenotyping are essential.50 Additional expert input (e.g., through a consortium) can be used to create phenotype definitions, however, establishing a well-accepted definition requires time, careful thought, and discussion. The eMERGE Phenotype Knowledgebase51 (PheKB) details existing phenotyping algorithms for individual phenotypes that incorporate additional patient information. Due to the complexity of these phenotyping algorithms, the simpler ICD-based phenotyping method is common for PheWAS studies, but the incorporation of these external phenotyping resources may help improve phenotype definitions in the future.
Unstructured data have also been used to define phenotypes, particularly for diseases with unreliable ICD9 classifications such as some psychiatric diseases, using natural language processing methods.52–60 Such methods can also be used to obtain patient measures such as smoking status.52 Natural language processing methods mine free text such as narrative doctor’s notes for words or phrases to develop a model combining structured and unstructured data to classify each patient as having or not having the phenotype of interest.52,53 Some challenges include dealing with misspellings, tenses, alternative phrasing, negation, and defining a trained dictionary of words and phrases that may correspond to a particular phenotype. Algorithms are usually trained using expert annotations, but new methods have attempted to automate this step as well.58,59 Additional machine learning methods have also been used to define phenotypes (e.g., imaging analytics from medical imaging datasets) using a broad spectrum of patient information.61–63
Recent works propose phenotyping strategies to overcome hurdles using multiple data sources to more accurately ascertain disease status.64–72 However, future work is needed to provide statistical methods for incorporating data of different types for phenome generation. For a detailed review of phenotyping procedures, see Bush et al. (2016).7 Figure S8 provides some examples of the types of structured and unstructured EHR information that can be used to construct phenotypes.
Misclassification and Information Bias
While we have discussed methods for the assignment of phenotype status, there exist many nuanced challenges to consider when before analyzing these data. Disease status determination is usually performed across subjects who have different lengths of follow-up time, who have different numbers of visits, and who are being seen in different types of medical clinics. The EHR cannot capture future diagnoses, and information on past medical history and treatment by external providers may be incomplete. Generally, the observation process can be complicated and may be related to patient- and provider-specific information such as gender and underlying disease status (Figures S3–5).73,74 Misclassification of the disease status may depend on this observation process, where subjects followed for a longer period of time or more often may be more likely to have their disease recorded in the medical record. Some statistical tools have been developed to try to deal with outcome misclassification and related issues, but computational restrictions may make these methods difficult to apply to large-scale biobank data.57,75 Additionally, symptoms occurring between visits may not always be reported, and the use of diagnostic guidelines and assessment of the phenotype may vary from doctor to doctor.76,77 These underlying patient- and provider-specific properties are often ignored when classifying subjects as cases and controls for a particular disease.
ICD-based phenotype misclassification is common for psychiatric disorders, where a diagnosis can be particularly challenging.55,76 For diseases with burdensome treatments such as cancer, we may expect that all subjects receiving a cancer diagnosis truly do have cancer, and there may be only a few cancer cases without a corresponding ICD code. In contrast, ICD codes for psychiatric disorders such as anxiety may be sometimes attributed to some subjects that do not meet the ICD definitions for the disorder. There may also be a tendency for patients to receive ICD classifications that result in reimbursement from the insurance provider. Additionally, disease ICD codes are sometimes assigned when a disease is suspected prior to further diagnostic testing, so it may be unclear whether a given ICD code refers to the final diagnosis.7,78
Figure 3 provides a visualization of the relationship between phecode-based diagnosis and the length of follow-up in MGI within age strata for anxiety and heart attack. We observe a greater rate of anxiety diagnoses among subjects followed for a longer period of time. Many factors may contribute to this, but one explanation is that more anxiety diagnoses are missed in subjects followed for a shorter period of time. In contrast, the proportion of subjects with a heart attack phecode was not appreciably related to the length of follow-up, and these acute events are captured when they happen.
Figure 3:
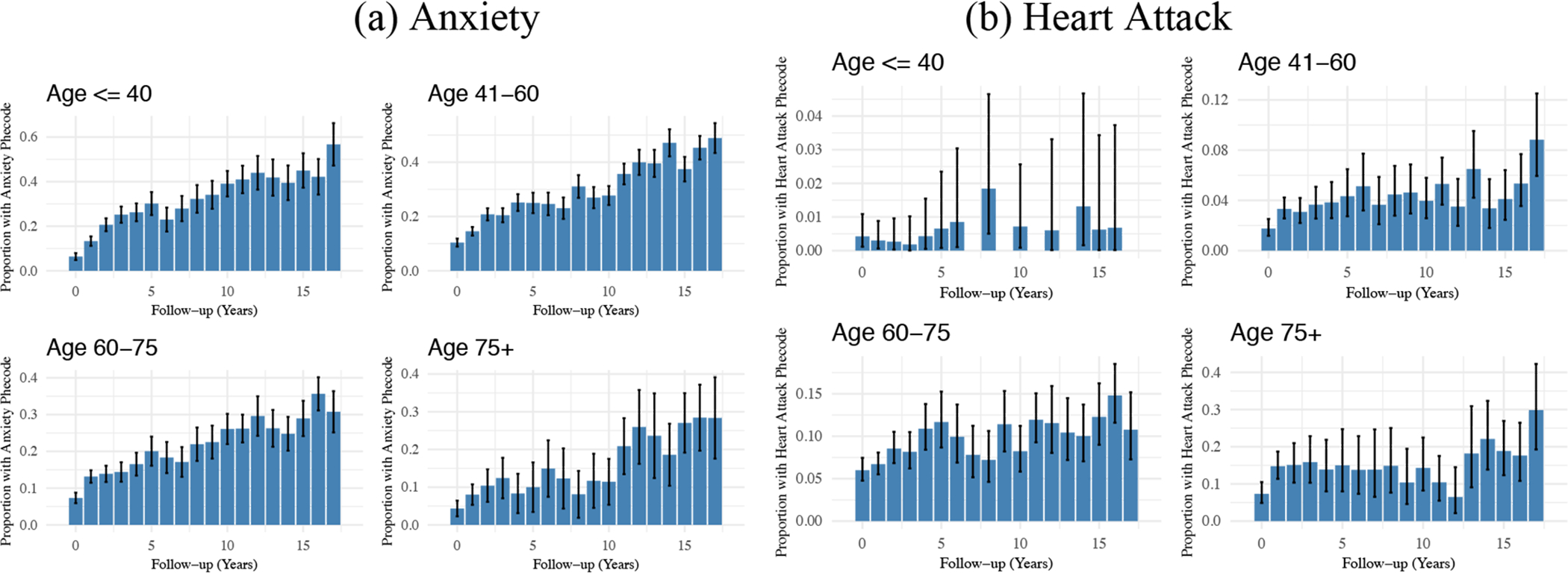
Relationship between (a) Anxiety or (b) Heart Attack Diagnosis and Length of Follow-up within Age Strata in MGI*
* Plotted intervals indicate 95% confidence intervals for each proportion.
Phenotype misclassification can result in bias (“information bias”) and negatively impact the statistical power to detect associations. Differential misclassification of disease status can also result in inflated type I error.79 The extent of misclassification can be described using quantities such as sensitivity, specificity, and negative and positive predictive values (provided a gold standard exists for comparison). Researchers have explored methods for incorporating external information about sensitivity/specificity to account for outcome misclassification.80–82 However, these quantities can vary from population to population and from phenotype to phenotype, and it is difficult to know the extent of phenotype misclassification in a particular population without performing further phenotype validation.82,83 Among other examples,57,82,84–86 Beesley et al. (2018) proposed a sensitivity analysis approach for exploring the potential impact of phenotype misclassification and disease-dependent patient selection on logistic regression effect estimates simultaneously.33
We demonstrate the potential bias induced by phenotype misclassification and disease-dependent patient selection using data from MGI in Figure 4. We consider a logistic regression model for whether the patient was diagnosed with cancer and the association of having cancer with gender. On the entire sample, we estimate the gender odds ratio as 0.89 (95% CI: 0.85, 0.93). We suppose the observed cancer diagnosis status is the truth and artificially induce misclassification and disease-dependent selection of the MGI patients. We then calculate the corresponding association between gender and the misclassified outcome in the selected patients. We impose misclassification under 90% specificity and ~70% sensitivity, and subsampling was imposed under an average 50% sampling rate for the entire cohort. If we compare the three analyses without any outcome misclassification, we see that sub-sampling dependent only on disease status does not induce bias in the association estimate (OR 0.89, 95% CI: 0.82, 0.94), but it does result in a less efficient estimate due to the smaller sample size. However, we do see bias when sub-sampling depends on both disease status and gender (OR 1.01, 95% CI: 0.95, 1.08). This provides a demonstration of biases expected under different sampling mechanisms. Additionally, when we compare the odds ratio estimates for a particular sub-sampling setting, we see that outcome misclassification is associated with bias in all settings, and this bias is not always towards the null.
Figure 4:
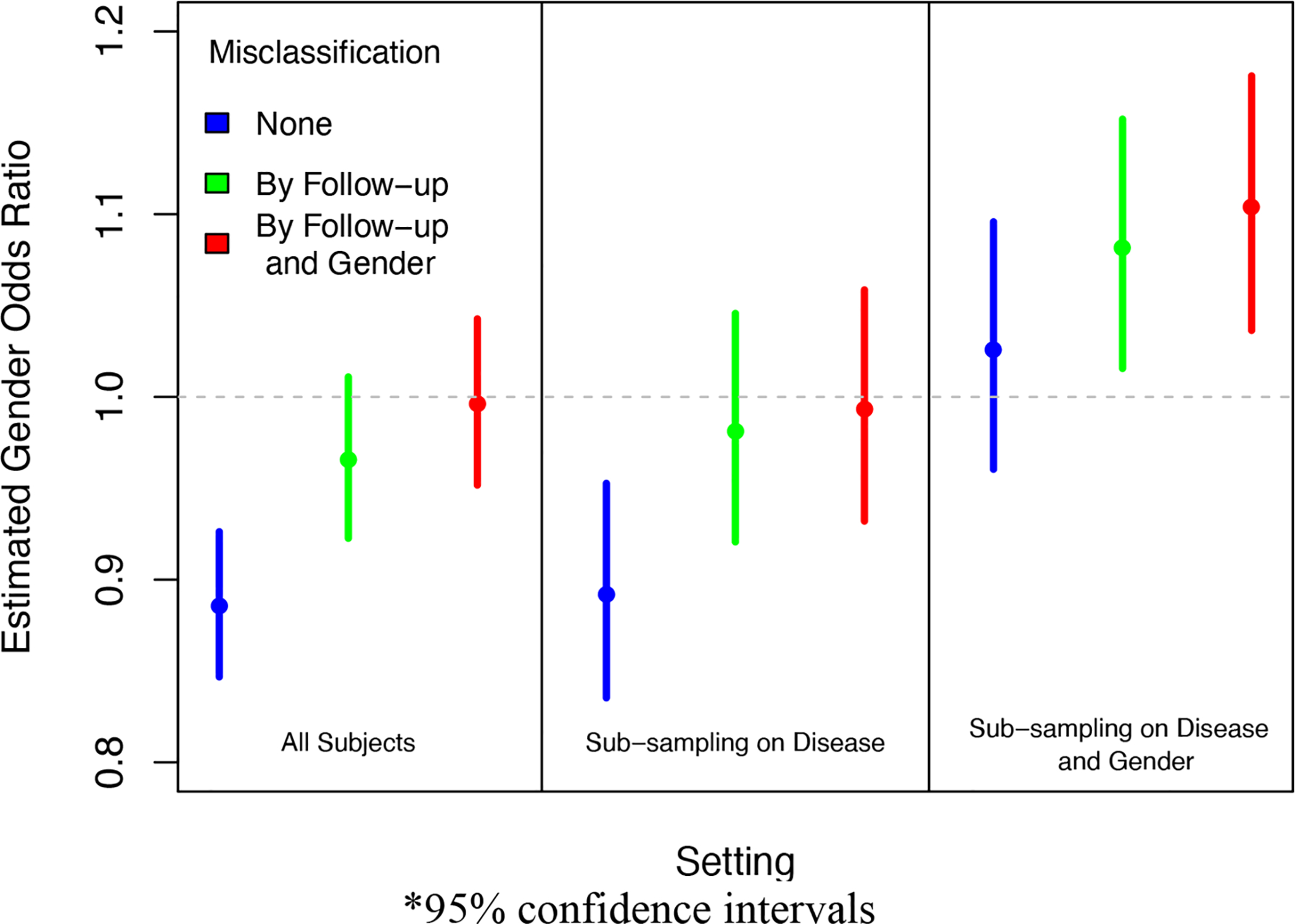
Impact of Selection Mechanism and Phenotype Misclassification on Estimated Association between Gender and Cancer Diagnosis in MGI*
*95% confidence intervals
Section 3.2: Study Design
Defining the Study Sample
A vital issue to consider when performing a biobank-based investigation is study design. Design choices can have implications for the analysis and interpretation of the study results. In this section, we describe several approaches for study design used in biobank research and describe some design-based strategies for dealing with common sources of bias.
Within pre-existing biobanks, researchers seek to sample patients for inclusion in a particular study. A common study design involves phenotype-specific case-control sampling, where all observed cases for a particular phenotype are selected and some subset of (possibly matched) controls for that phenotype are sampled from the biobank (e.g., Fritsche et al. 2018, Abana et al. 2017).10,87 Cases are often defined as subjects receiving a particular diagnosis code a prespecified number of times, e.g., twice. An advantage of case-control sampling is that it does not require additional longitudinal information and instead relies on dichotomized phenotypes, but it is heavily dependent on the “case” and “control” definitions. One crucial aspect of case-control sampled data is the validity of secondary analyses of related outcomes, and many methods exist for addressing this issue.88–91 Additionally, the choice of controls should be considered carefully. Controls might be defined as all patients without the primary phenotype, or we may exclude patients with related diseases from being included as controls. Another common practice is to restrict the analysis to patients with a certain amount of follow-up, which can bias sampling toward sicker patients.92 In the presence of many competing control definitions, one strategy is to evaluate internal validity by performing inference using many different control group definitions to “bracket” the association of interest.93,94 Another common study design is cohort sampling, where all biobank patients with available data meeting the inclusion criteria are included in the analysis (e.g., Au Yeung et al. 2014, Hall et al. 2018).38,95
Self-controlled designs in which each patient serves as his/her own control are emerging as an appealing design paradigm for some scientific problems (e.g., Kuhnert et al. 2011, Zhou et al. 2018).96,97 Two variations of self-controlled designs are the self-controlled case series design and the case-crossover design. Recently, Schuemie et al. (2016) developed an adapted self-controlled case series design that uses the notion of accumulated exposure to study long-term drug effects.98 A detailed comparison of the self-controlled case series and case cross-over designs can be found in MacClure et al. (2012),99 and additional exploration of self-controlled case series can be found in Petersen et al. (2016) and Simpson et al. (2013).100,101 An advantage of this design is that it controls for confounding due to time-invariant variables. Unlike cohort and case-control designs, however, this method requires longitudinal data to be available for all patients, which may be missing, incomplete, or insufficient in some EHR-linked databases.
Due to finite resources, some biobanks may collect data, e.g., genotype data, on a subset of their cohort. The strategy of collecting data on a subset of patients enriched for certain characteristics and related issues are explored in detail in Sun et al. (2017)102 and Schildcrout et al. (2015) and (2018).103,104 Two-phase designs also result in missing data by design, where more expensive assays or time-consuming surveys may be administered to a subset of the patients determined based on results from the first phase. Exposure-dependent (e.g., when we have rare exposures of interest) and other stratified trait-dependent sampling designs can also be used. For example, extreme phenotype sampling designs collect additional data only for patients with extreme values of a continuous variable.105,106
Another critical concept to consider when defining the study sample is the independence between patients. Longitudinal outcomes are expected to be correlated within patients, and outcomes may be correlated between patients due to relatedness, nesting within doctor or clinic, belonging to a common social network, or other reasons. The software KING (Kinship-based Inference for GWAS) uses genotype data to determine pairwise kinship between patients.107 We might then define the study sample restricted to unrelated patients and apply methods that rely on independence between patients (e.g., Firth-corrected logistic regression in Fritsche et al. 2018).10 Statistical modeling approaches such as mixed modeling (e.g., SAIGE) can also be used to account for residual correlations between individuals.108
Many variations and alternative strategies for designing the study sample exist in the statistical literature and can also be applied in the EHR setting. For a review of many general study design strategies, see Modern Epidemiology: study design and data analysis.109,110
Considerations Related to Study Design
Madigan et al. (2013) compares effect estimates resulting from several study designs in a particular setting and demonstrates that the choice of study design can have substantial impacts on effect estimates.111 These study design choices also impact the statistical power and generalizability of the results. Therefore, the study design should be considered carefully. In addition to impacting power, the method by which the patients are included in the study sample may result in biased inference (with respect to the target population), called sampling bias. Haneuse et al. (2016) provide a general framework for exploring and dealing with design-based sampling bias for EHR analyses.112 Haneuse et al. (2016) focus on characterizing the mechanism by which patients were included in the dataset by breaking it into smaller observation mechanisms, which may be impacted by different factors. Possible sources of sampling bias arising from each mechanism can be explored in detail in a sensitivity analysis framework.
There is a belief in the literature that GWAS/PheWAS study results may be less susceptible to bias resulting from the patient sampling mechanism, since the opt-in consent is not likely to depend on the value of a single genetic marker. However, bias due to genotype relationships with the sampling mechanism can still arise in certain settings.33,113,114 Additional work may help clarify settings in which bias is and is not expected in GWAS and PheWAS studies. In general, issues of sampling bias are not unique to EHR data, and many authors have explored the impact of sampling on inference. Some works exploring selection/observation biases in the EHR setting include Zheng et al. (2017), Phelan et al. (2017), Goldstein et al. (2016), and Rusanov et al. (2014).30,31,92,115 However, additional characterizations of the mechanisms by which we can have sampling bias in biobank and EHR research may help guide study design in the future.
In terms of methods designed for large-scale EHR-based studies, Schuemie et al. (2014) and Schuemie et al. (2018) propose a p-value calibration method that may be able to account for both random and systematic (e.g. confounding, sampling biases) sources of error using distributions of effect estimates believed to be null effects.116,117 Modern causal inference methods using the potential outcome/counterfactual framework are also being integrated in biobank analysis.118–120
Section 3.3: Data Analysis and Modeling
In performing statistical analysis, researchers may have a variety of goals, such as developing a prediction model, estimation (e.g., finding candidate biomarkers, hypothesis-generating studies), causal inference, or hypothesis testing (e.g., is drug A better than drug B). The analysis strategy and concerns will depend on the research goal and the data considered. In this section, we describe several common modeling challenges encountered in EHR-based data analysis, and we address specific issues, including multiple testing, handling of missing data, and comparison across different EHRs.
Modeling
EHR data present many challenges concerning modeling and inference. For example, correlation structures between variables can be complicated, the number of adjustment factors can be large, and events of interest can be rare. In this section, we describe some popular and emergent modeling strategies.
A common goal of EHR-based analyses is to study the associations between specific phenotypes and variants at a particular gene region or across the genome, and this analysis is often performed using linear or logistic regression or using mixed linear model association (MLMA) analysis.38,41,121–123 Firth-corrected logistic regression may prove useful for modeling rare binary outcomes or settings in which there is strong covariate separation, and its application to PheWAS is demonstrated in Fritsche et al. (2018).124 Recently, Dey et al. (2017) proposed a fast alternative to Firth-penalized regression to stabilize estimation for PheWAS studies using saddle-point approximation (SPA) that is useful for handling extremely unbalanced case-control data.125 These methods can be applied in many other modeling settings as well. A saddle-point approximation approach for estimating mixed models (called SAIGE) was proposed for handling highly unbalanced case-control data with additional sample relatedness, which is typical for biobank data.108 Another common target for these studies is to identify the proportion of variation in a particular phenotype that can be attributed to genetic variation, called heritability. Some popular statistical methods include polygenic profile scoring, univariate linkage disequilibrium regression, and genomic relatedness-matrix restricted maximum likelihood (GREML).38,126–130
A popular strategy for studying the aggregate association between genetic information and disease development is through polygenic risk scores (PRS). PRS involve summing the contributions of a potentially large number of genetic loci and can be used to stratify patients with respect to disease risk.131 Many strategies exist for determining the genetic loci to include in the PRS and their relative contributions. Many PRS construction strategies and software packages exist, and we will not detail these various methods here.124,132,141–143,133–140 For a recent exploration of PRS construction, we refer the reader to Choi et al. (2018).144 Recently, statistical methods have been developed to leverage published GWAS and other omics summary statistics to improve the performance of prediction algorithms and perform analyses adjusting for many genetic loci simultaneously.145–149
Researchers may also be interested in studying relationships between phenotypes or joint relationships between phenotypes and other patient-level factors such as treatments or genotypes. Existing statistical methods for dealing with correlated outcomes such as mixed modeling and generalized estimating equations (when the model coefficients are of primary interest) can often be applied. Shaddox et al. (2016) and Xue et al. (2017) propose strategies for modeling correlated rare outcomes.150,151 Recently, Bastarache et al. (2018) developed a phenotype risk score-based method to study rare genetic variants associated with Mendelian diseases.152 More generally, phenotype-based risk scores could be used to describe the combined association between secondary phenotypes and the primary phenotype and may prove useful for risk stratification in combination with PRS. However, construction of phenotype-based risk scores would involve modeling the relationship between many phenotypes, either pairwise or jointly, and this modeling would be complicated by phenotype misclassification. Additional statistical development is needed to handle many correlated, misclassified binary phenotypes.
In probabilistic phenotyping models, risk prediction models, and other modeling using EHR data, we are often interested in incorporating a broad spectrum of patient information. Variable selection and penalization methods along with sparse estimation strategies allow many predictors to be incorporated into statistical models, and there is an excellent opportunity for the use of such methods in the setting of EHR. Automated feature selection algorithms are often used within machine learning algorithms to determine which predictors to include, and this can also be combined with expert preprocessing of the candidate predictors.153,154 Regularization techniques, including LASSO, ridge regression, and elastic net, have been applied in the EHR setting.155,156
Machine learning algorithms have also gained popularity in EHR data analysis, particularly in the development of risk prediction models. Traditional machine learning methods such as support vector machines and random forests with boosting are often used.157,158 Deep learning, neural networks, and ensemble methods have emerged as attractive approaches to prediction using EHR data.158–161 For a review of deep learning methods for EHR data, see Schickel et al. (2018).158 Care must be taken when applying these machine learning techniques in the setting of rare outcomes, and additional model calibration may be needed. A disadvantage of machine learning algorithms is the difficulty in estimating prediction uncertainty. Some work has been done exploring uncertainty estimation in particular settings, but additional work is needed.162 Machine learning algorithms can have excellent performance for prediction in some settings. When the goal of the analysis is to develop a prediction model for making predictions for new patients in the same EHR, challenges such as sampling bias and confounding, may be of less concern. However, the resulting model may be susceptible to overfitting and may not always have good properties in terms of transportability to other EHRs and generalizability to other populations.
While we may conceive of many elegant modeling strategies for dealing with statistical issues for EHR data, these methods may not always scale well with respect to large samples, large numbers of variables, or a large number of repeated analyses (e.g., in a PheWAS or GWAS). Computational feasibility will be an important factor to consider for applying statistical tools at scale. While computational efficiency strategies are outside the scope of this paper, we refer the reader to Thompson and Charnigo (2015) and Prive et al. (2018) for more information on phenome-wide computing for GWAS.163–165
Missing Data
Missing data is a common issue for biobank analyses, and data may be missing for a variety of reasons. A common source of missingness in GWAS/PheWAS studies is missingness in the genotypes. This can be handled by first excluding patients with missingness rates above a particular threshold (say, 2%) and then imputing missing values for patients with lower missingness rates.38,128 Genotype imputation has improved over time due to larger and more diverse reference panels. While many of these biobank analyses reported their treatment of missing genotype data, missing information in the phenotype information or demographics is rarely discussed. Additionally, many studies define their analytical sample based on some subset of biobank participants, and it is sometimes unclear how these participants were chosen. A more transparent description of how the study sample was derived and the treatment of missing data may shed some light on the generalizability of study results.
Statistical methods for dealing with missing data in the EHR often rely on multiple imputation, a statistical approach in which the missing data is “filled in” using information from patients with observed values.166–169 Such approaches can prove extremely valuable to EHR-based research, but implicit assumptions about the missingness mechanisms should be carefully considered. A common assumption behind many statistical methods for dealing with missing data is that data are missing at random, meaning that missingness depends only on fully observed information.170 However, missingness in EHR data may often be related to a patient’s underlying health state and other unmeasured individual or facility characteristics.171 For example, healthier patients may be more likely to drop out of the EHR. Additionally, lab tests are only ordered for patients with suspected disease. This setting, called missing not at random, is more challenging to address in the statistical analysis. For a discussion of dealing with missing not at random data, see Little and Rubin (2002).170 In general, we cannot tell from the data what mechanisms generate the missingness, but additional data and subject matter experts can provide insight into the drivers of missingness. For example, Haneuse (2016) describes a survey-based strategy to explore the reasons for missingness in EHR data, which may help shed light on the validity of missingness assumptions.172 McCullough and Neuhaus (2018) proposes a strategy for exploring outcome dependence in the mechanism by which patients visit the clinic.171
A common type of “missing” data is the true phenotype state of each patient. We can view the sampling mechanism that gave rise to our study population and the mechanism behind phenotype misclassifications (which we might call the observation mechanism) in a missing data framework, as discussed in Supplementary Section S7 and Beesley et al. (2018).33 Further work should be done to explore the impact of different sampling and phenotyping mechanisms on statistical inference.
Multiple Testing of Hypotheses
GWAS/PheWAS studies and many other types of EHR-based research often involve the simultaneous testing of many hypotheses. Failure to account for multiple testing can result in inflated type I error. Some methods for controlling the type I error include Bonferroni adjustment, false discovery rate-controlling thresholds (e.g., Li et al. 2018),41,173 and Benjamini-Hochberg thresholds (e.g., Liao et al. 2017).84 However, many of these methods (in particular, the simple Bonferroni adjustment) are overly conservative when the many statistical tests are not independent. This is often the case in large-scale GWAS/PheWAS studies, where associations are explored for many related characteristics. In this setting, the goal may be to control for the effective number of independent tests rather than the number of correlated tests being performed. Such an approach may improve statistical power to detect significant associations while still controlling the type I error rate.
Several methods have been proposed to estimate the effective number of tests (e.g. Li 2012) or control for correlated tests. Good (2005) describes resampling-based testing via permutation or bootstrap to correct the p-values for multiple testing.174 Gao et al. (2008) propose the simple M method to estimate the effective number of tests, which uses a combination of principal components analysis and Bonferroni correction.175 For a PheWAS study presented in Ge et al. (2017), the effective number of tests is estimated using principal components analysis of a matrix of pairwise correlations between pairs of phenotypes.129
Similarly, heuristic approaches have been suggested to identify a maximal independent set of uncorrelated phenotypes among pairwise correlations between pairs of phenotypes.10,176 A popular method for identifying phenotypes is to aggregate ICD codes into a set of phenotype codes called “phecodes.” For example, using 1,578 phecodes in MGI, we identified a maximal set of 981 phenotypes with no pairwise Pearson correlation above 0.1. However, no general guidelines exist for multiple testing correction in the PheWAS setting. Alternative methods adjust for multiple testing using multivariate normal assumptions for the correlated test statistics (e.g., Han et al. 2009, Lin 2005, Seaman et al. 2005).177–179 In the context of correlated SNPs, some methods correct for multiple testing via analysis of the underlying linkage disequilibrium structure of the genetic data (e.g., Duggal et al. 2008).180 Johnson et al. (2010), Zhang et al. (2012) and Li et al. (2012) provide some simulations comparing the performance of different methods.181–183
An emerging challenge is the correction of multiple testing across the medical phenome × genome two-dimensional landscape. With recent work regarding phenotype risk scores, there is increasing interest in studying phenotype-phenotype associations across the phenome.184 As such, there is a need to develop a corresponding statistical methodology to correctly account for potentially strong cross-phenotype correlations, which are particularly common with hierarchically structured phenotypes.
Ultimately, the best strategy for correcting for multiple testing may depend on whether the goal is hypothesis generation/discovery or validation/hypothesis testing. In the former, we may be more willing to accept false-positive results for individual tests in exchange for higher power, while in the latter case, we may want to control the rate of false positives better.
Heterogeneity between Biobanks
Researchers often attempt to validate statistical findings from their data analysis using an independent dataset from a different population. For example, we may wish to validate results obtained using data from one biobank (e.g., MGI) by performing the same analysis for another biobank (often, UKB). Here, we make a distinction between validation and replication, where replication involves comparing results in samples drawn with few systematic differences from the same population and validation involves comparing results in samples drawn from different populations or using different sampling approaches.185 Systematic differences between the population characteristics or sampling mechanisms, however, could impact the generalizability of results between populations and impact our ability to validate findings.
In the meta-analysis literature, heterogeneity between studies is broadly grouped into three categories: clinical heterogeneity (differences in patients, interventions, and effects), methodological heterogeneity (differences in study design and sampling), and statistical heterogeneity (when the observed effects are more variable across studies than we would expect from random chance). Statistical heterogeneity may be a result of clinical and/or methodological heterogeneity.
Some analyses may be more impacted by differences between biobanks. As a demonstrative example, we compare the results of different data analyses using data from MGI and UKB. These biobanks exhibit substantial methodological heterogeneity concerning their sampling mechanisms, where MGI is based on an academic medical center and UKB is population-based. Suppose we are interested in comparing the odds ratio for having a particular phenotype based on the status of another phenotype, called phenotype co-occurrences. While prevalences will be impacted by the different sampling designs between MGI and UKB (see Figure 2), it is not clear how phenotype-phenotype associations will compare.
Figure S6 presents the estimated log-odds ratios of having a phecode diagnosis of melanoma regressed on other diagnoses in the phenome. See Supplementary Section S5 for details on the phenotype generation procedure. The estimated odds ratios from the UKB data tend to be larger in magnitude compared to the odds ratios in MGI (for 70% of diagnoses). One possible explanation for this phenomenon is that in order for patients to get a phecode in UKB, they must visit a health care provider, during which time they may get multiple codes. When we compare UKB patients who did and did not receive a particular phecode (perhaps they did not visit a health care provider or did not visit as often), we may obtain inflated odds ratios. The patients in MGI are enriched with phecodes across the board, but patients with and without a particular phenotype may have many opportunities to collect other diagnoses through their interactions with the health care provider. In this melanoma example, the odds ratios for other neoplasms did not exhibit the same differences in MGI and UKB as seen for other classes of diseases. This may be due to enhanced screening of these diseases after diagnosis of melanoma in both MGI and UKB.
We predict the heterogeneity of the sampling mechanisms may not appreciably impact some associations; for example, GWAS results. In Figure 5, we compare GWAS results in MGI and UKB for several cancers. In this figure, points represent SNPs identified as being related to the corresponding phenotype in the NHGRI-EBI GWAS catalog.186 See Supplementary Section S8 for details. While MGI and UKB have very different sampling mechanisms, the GWAS results generally appear similar.
Figure 5:
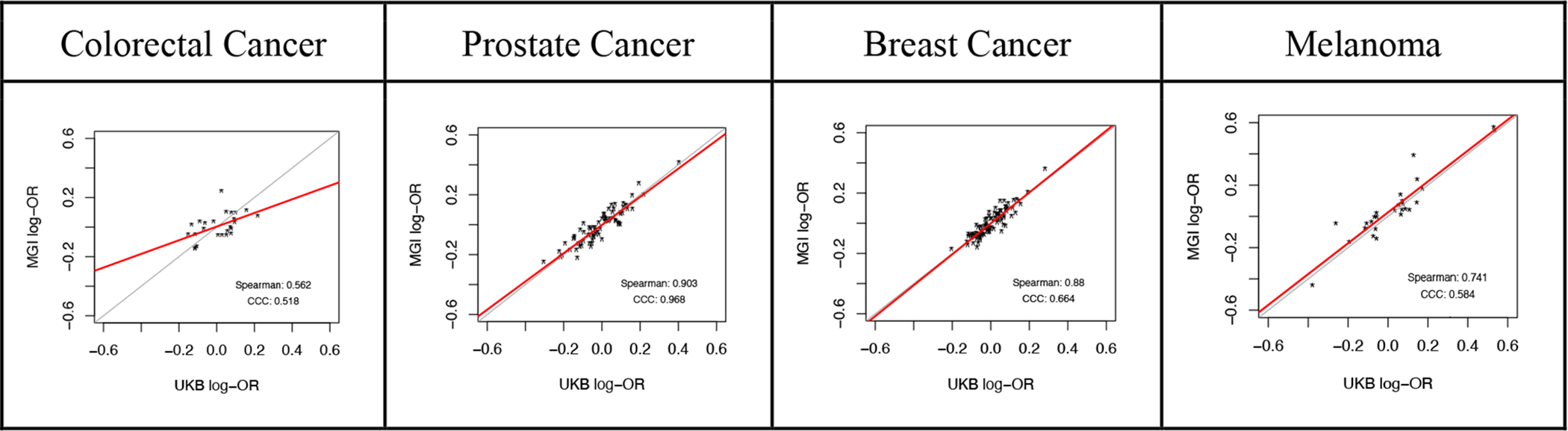
Comparison of GWAS Results in MGI and UK Biobank for Selected Cancer Phenotypes*
* Each point represents a SNP identified as being related to the corresponding phenotype in the NHGRI-EBI GWAS catalog. The point location corresponds to the log-odds ratio association between the SNP and the phenotype of interest in MGI and UK Biobank. The two lines correspond to equality of the estimates and a fitted line to the points (excluding any outlying points with absolute log-OR greater than 0.6). “Spearman” indicates the Spearman correlation and “CCC” indicates Lin’s concordance correlation coefficient, which is a measure of agreement (with 1 being perfect agreement).
In addition to methodological heterogeneity, clinical heterogeneity could impact validation of results across biobanks. Some examples of clinical heterogeneity include differences in patient demographics, or the kinds of treatments prescribed, screening practices, and whether health care is public or private. An example of clinical heterogeneity for MGI and UKB is age, where MGI consists of patients aged 18 and up, while UKB consists of patients aged 40–69. If the association of interest depends on age, we would have different marginal associations in MGI and UKB. Another notable difference between biobanks/EHRs is how physicians encode diagnoses within the ICD framework. For a given patient, physicians in one EHR may tend to enter diagnosis A, while physicians in another EHR may enter related diagnosis B. This presents a problem for researchers seeking to validate diagnosis code-based phenotype associations across biobanks. Additionally, we may be interested in using biomarker or lab value measurements across biobank datasets, and these may be measured with different degrees of error.187 When comparing this association overall between two different populations, a failure to adjust for the clinical heterogeneity across the two populations could result in biased inference.
In the presence of this heterogeneity between study populations, we may explore statistical methods to improve our ability to compare between different populations. There is a body of statistical literature for quantifying and handling between-study heterogeneity via meta-analysis.188–191 Weighting-based and resampling-based methods for dealing with heterogeneity have also been explored.192–194 The large number of subjects and the large number of available adjustment factors in EHR data provide an opportunity to effectively address more refined questions such as the relationship between treatment and molecular subgroups of disease (inherently a question of interactions) directly, potentially allowing clinical heterogeneity to be handled directly through a redefinition of the quantity of interest.195 Recently, Shi et al. (2018) developed a spherical regression-based method for handling heterogeneity in ICD code designation across different EHR systems.196 Methodology in the data integration literature may also prove useful for addressing these challenges.197 Future work may explore resampling-based methods to make studies more comparable in the presence of heterogeneity with respect to the sampling mechanism.
Section 4: Emerging Uses of Electronic Health Record Data and Combination with External Data
There is a tremendous opportunity to incorporate additional data to enrich EHR and enhance the scope of research. For example, we may link cancer and death registry information to the EHR to study survival and disease-related outcomes after clinical diagnosis. Local and national surgical registries offer opportunities for studying more granular health-related outcomes. When registry data is not available, claims data may also provide some insight for survival and disease-related research.198 Recent work has developed methods for defining the exposome based on clinical narrative information or additional patient-level measurements.199,200 Geo-coded data can provide a wealth of exposure information including social determinants of health, neighborhood characteristics, socioeconomic status, and pollution information.201–206 Freely available resources like the eICU Collaborative Research Database207 are becoming more common and increasingly accessible, allowing for additional exploration of data and aggregation for larger analyses.
Longitudinal data within the EHR and beyond also offer many opportunities for research. Mobile fitness tracking devices provide an opportunity to incorporate longitudinal health metrics or even use text messages or game performance to define phenotypes.208,209 Noren et al. (2010), Noren et al. (2013), and Boland et al. (2015) use longitudinal health data to discover and adjust for temporal patterns.210–212 Longitudinal EHR data has proven to be extremely useful in the fields of pharmacovigilance, pharmacoepidemiology, and pharmacogenomics.211,213–217 Additional work leverages large-scale medical data to study potential new indications for existing drugs, called drug repurposing or repositioning.218 Longitudinal EHR data can also be used to develop dynamic predictions for patient prognosis, adverse events, etc. over time.219–222
When combining data from multiple disparate sources, several problems arise. Most notably are issues regarding patient privacy. Additionally, we must consider issues such as data processing and rules for linking records for a single patient. Many statistical methods have been developed for linking records corresponding to individual patients across data sources, and many of these methods explicitly address issues of privacy.223–227 Statistical methods have also been developed for combining data across distributed data sources where data from individual patients are not accessible.228,229 Yang et al. (2013) developed methods for performing meta-analysis based on existing GWAS, and similar methods should be developed for PheWAS studies in the future.230
Large biobank datasets also provide an opportunity to study different treatment pathways and their corresponding outcomes.231 Additional components such as treatment nonresponse and treatment adherence can also be explored.54,232 While such studies are certainly not new, the wealth of information provided through EHRs provides opportunities to study treatment-related outcomes at scale. Additionally, these data sources provide a clearer look at treatment-related outcomes in practice, which may not always align with outcomes under more ideal settings of a clinical trial. These data can be used to analyze and/or predict various outcomes to treatments, medications, and/or dosages (sometimes stratified by patient characteristics).
EHR have also been used for disease forecasting, where researchers use electronic health records to determine population rates of disease and forecast future rates.233,234 Disease forecasting is a challenging problem, and EHR-informed forecasts can prove extremely useful for medical staffing, vaccine production, and policymaking.235
Section 5: Conclusion
Biobanks linked to EHR provide rich data resources for health-related research, and scientific interest in biobank-based research has grown dramatically in recent years. As more researchers become interested in using biobank data to explore a spectrum of scientific questions, resources guiding the data access, design, and analysis of biobank-based studies will be crucial. This work serves to complement and extend recent publications about biobank-based research (e.g., Wolford et al. 2018, Glicksberg et al. 2018, Bush et al. 2016, Ohno-Machado et al. 2018) and aims to provide some statistical and practical guidance to statisticians, epidemiologists, and other medical researchers pursuing biobank-based research.5–8
In this paper, we provide a detailed characterization of many of the major EHR-linked biobanks to facilitate researchers’ ability to obtain and investigate research-quality biobank data with some understanding of the associated population, sampling mechanism, and data linkages. This characterization provides a useful starting point for understanding the type of biobank data available and for requesting and accessing data. We also survey biobank-based papers that have been published. Future research can utilize increasingly large EHR-linked biobank cohorts to study a broad range of diseases. Biobank data also present an exciting opportunity to explore treatment and therapy schedules, drug repurposing, or gene-by-treatment interactions in the future. Such explorations can also be used to inform dynamic, patient-centric predictions for monitoring and treating future patients.
When using biobank data for health-related research, it is essential that researchers understand the statistical and practical issues that accompany such analyses and have resources to address them. There is a great need for statistical developments to address the many varied issues that go hand in hand with EHR-based research. Our discussion is structured to address statistical issues and strategies that researchers encounter when following a typical research study structure (see Figure 1).
Given our research question and data availability, the next step is generally to identify potential sources of bias. In this paper, we describe several particular concerns of confounding bias, selection bias, and misclassification of EHR-derived phenotype variables. Researchers should carefully consider issues of phenotype misclassification both in terms of ICD code-based phenotyping and in terms of the limitations of the EHR as a whole. A better understanding of the mechanisms governing misclassification (in terms of under- and over-reporting of disease) may help shed light on the limitations of the EHR data and how to deal with potential information biases that result. Biases, in terms of patient selection into the biobank/EHR and in terms of study design using EHR data, need to be carefully considered. Many statistical methods exist for addressing issues of non-probability sampling in particular, and additional work looking into the mechanisms driving patient selection for EHR may help researchers better generalize results to their target populations.
Historically, a large body of statistical work has focused on studying how we can most efficiently use available data to estimate our quantity of interest. As the size of the data grows, however, efficiency becomes less and less of a concern and characterization of bias becomes critical.236 This is particularly important in the study of EHR, where many possible sources of bias can come into play and the data generation mechanisms are often difficult to characterize. The recent push away from p-values and dichotomization of study results in the statistical community reflects these changing perceptions. Increased emphasis must be placed on reproducibility and scientific rigor, particularly when large repositories of data are being made widely accessible.
Given a large pool of EHR and biobank data, the next step is to design our study using the data available. One considerable challenge involves defining the phenome, and future work can explore ways to incorporate a broader spectrum of EHR information into phenotype classification. Defining exposure and outcome variables can be particularly challenging for EHR-based data. For example, suppose we are interested in studying relationships between genetics and smoking behavior. Smoking behavior may not be directly recorded in the EHR, and careful thought is needed to determine how we can use EHR information to extract these data and the possible implications for the veracity of resulting statistical inference. We also need to clarify which patients we will include in our analyses. In many cases, this may consist of all available patients, but careful sub-sampling of the large pool of available to define our study dataset can also be used to help mitigate possible sources of bias, can reduce computational burdens of large data, and can identify subjects for additional data collection.
Once we have designed our study, the next general step is data analysis. Many issues need to be considered, including how we want to model the data, correction for multiple testing, and handling of missing data. The treatment of missing data in EHR-based studies is an area in particular need of additional statistical development. For example, analyses wishing to include lab values as predictors need to reconcile somehow the inherent relationship between missingness (whether a given test was ordered) and the test results. Data can be missing for a variety of reasons, and the mechanism generating the missingness can have serious implications on inference. Statistical methods tailored to handling issues of missing data in EHR could prove extremely useful. In general, reporting of how missingness was handled needs to be more explicit in studies using EHR. Additional statistical methods are also needed to handle multiple testing adjustment for studies involving many correlated phenotypes or studies exploring the phenome × genome landscape. In general, there is a strong need for the development of statistical methods to address the many and varied challenges we face when analyzing EHR-linked biobank data.
The combination of genetic and phenotypic information (for example, through polygenic and phenotype risk scores) presents a big opportunity for improving risk prediction, and future work can attempt to interrogate these different types of patient-level information to untangle the genetic and environmental factors related to disease generation and risk. With an increase in the volume and variety of data becoming available, emphasis should be placed on methods for incorporating data from external sources and emerging data streams (for example, geo-coded data, longitudinal biomonitoring data, mobile data, registry data, genomics/metabolomics data, imaging data, ecologic data, etc.). Such analyses can widen the scope of scientific questions we can address, and they necessitate a new wave of related statistical methods.
Supplementary Material
Acknowledgements
Part of this research has been conducted using the UK Biobank Resource under application number 24460. The research is supported by NCI P30 CA046592 and NSF DMS 1712933.
Abbreviations:
- EHR
electronic health record
- eMERGE
Electronic Medical Records and Genomics Network
- GREML
genomic relatedness-matrix restricted maximum likelihood
- GWAS
genome-wide association study
- ICD
International Classification of Diseases
- KPRB
Kaiser Permanente Research Bank
- MGI
Michigan Genomics Initiative
- NIH
National Institutes of Health
- MLMA
mixed linear model association analysis
- NHGRI
National Human Genome Research Institute
- PheWAS
phenome-wide association study
- SNP
single nucleotide polymorphism
- UKB
UK Biobank
References
- 1.De Souza YG & Greenspan JS Biobanking past, present and future. AIDS 27, 303–312 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greely HT The Uneasy Ethical and Legal Underpinnings of Large-Scale Genomic Biobanks. Annu. Rev. Genomics Hum. Genet 8, 343–364 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Hayrinen K, Saranto K & Nyk P Definition, structure, content, use and impacts of electronic health records: A review of the research literature. Int. J. Med. Inform 7, 291–304 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Denny JC et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 26, 1205–1210 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolford BN, Willer CJ & Surakka I Electronic health records: The next wave of complex disease genetics. Hum. Mol. Genet 27, R14–R21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glicksberg BS, Johnson KW & Dudley JT The next generation of precision medicine: Observational studies, electronic health records, biobanks and continuous monitoring. Hum. Mol. Genet 27, R56–R62 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Bush WS, Oetjens MT & Crawford DC Unravelling the human genome–phenome relationship using phenome-wide association studies. Nat. Rev. Genet 17, 129–145 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Ohno-Machado L, Kim J, Gabriel RA, Kuo GM & Hogarth MA Genomics and electronic health record systems. Hum. Mol. Genet 27, R48–R55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brieger K et al. Genes for Good: Engaging the Public in Genetics Research via Social Media. Am. J. Hum. Genet 105, 65–77 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fritsche LG et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. Am. J. Hum. Genet 205021 (2018). doi: 10.1016/j.ajhg.2018.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michigan Genomics Initiative Website. Available at: https://www.michigangenomics.org.
- 12.UK Biobank Website. Available at: http://www.ukbiobank.ac.uk.
- 13.Allen N et al. UK Biobank: Current status and what it means for epidemiology. Heal. Policy Technol 1, 123–126 (2012). [Google Scholar]
- 14.Estonian Genome Center. Available at: https://www.geenivaramu.ee/en/access-biobank.
- 15.Leitsalu L et al. Cohort profile: Estonian biobank of the Estonian genome center, university of Tartu. Int. J. Epidemiol 44, 1137–1147 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Danish National Biobank. Available at: http://www.biobankdenmark.dk.
- 17.Biobank Sweden. Available at: http://biobanksverige.se/research/.
- 18.Saudi Biobank. Available at: http://kaimrc.med.sa.
- 19.China National GeneBank. Available at: https://www.cngb.org/home.html.
- 20.National Biobank of Korea. Available at: http://www.nih.go.kr/NIH/cms/content/eng/14/65714_view.html.
- 21.Cho SY et al. Opening of the National Biobank of Korea as the Infrastructure of Future Biomedical Science in Korea. Osong Public Heal. Res. Perspect 3, 177–184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qatar Biobank. Available at: https://www.qatarbiobank.org.qa.
- 23.Al Kuwari H et al. The Qatar Biobank: background and methods. BMC Public Health 15, 1208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin E et al. Association and interaction effects of Alzheimer’s disease-associated genes and lifestyle on cognitive aging in older adults in a Taiwanese population. Oncotarget 8, 24077–24087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taiwan Biobank. Available at: https://www.twbiobank.org.tw/new_web_en/index.php.
- 26.Nagai A et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol 27, S2–S8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Institutes of Health. The All of Us Research Program: Operational Protocol. (2018).
- 28.PcBaSe Sweden Website. Available at: http://www.surgsci.umu.se/english/sections/urology-and-andrology/research/pcbase/?languageId=1.
- 29.Mayo Clinic Biobank for Bipolar Disorder Website. Available at: https://www.mayo.edu/research/centers-programs/bipolar-disorder-biobank/overview.
- 30.Phelan M, Bhavsar N & Goldstein BA Illustrating Informed Presence Bias in Electronic Health Records Data: How Patient Interactions with a Health System Can Impact Inference. eGEMs (Generating Evid. Methods to Improv. patient outcomes) 5, 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein BA, Bhavsar NA, Phelan M & Pencina MJ Controlling for informed presence bias due to the number of health encounters in an electronic health record. Am. J. Epidemiol 184, 847–855 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keiding N & Louis TA Perils and potentials of self-selected entry to epidemiological studies and surveys. J. R. Stat. Soc. Ser. A Stat. Soc 179, 319–376 (2016). [Google Scholar]
- 33.Beesley LJ, Fritsche LG & Mukherjee B A Modeling Framework for Exploring Sampling and Observation Process Biases in Genome and Phenome-wide Association Studies using Electronic Health Records. bioRXiv 1–19 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Baker R et al. Report of the AAPOR Task Force on Non-Probability Sampling. (2013).
- 35.Carroll RJ, Bastarache L & Denny JC R PheWAS: Data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–2376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tian Y, Schuemie MJ & Suchard MA Evaluating large-scale propensity score performance through real-world and synthetic data experiments. Int. J. Epidemiol 2005–2014 (2018). doi: 10.1093/ije/dyy120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schneeweiss S et al. High-dimensional Propensity Score Adjustment in Studies of Treatment Effects Using Health Care Claims Data. Epidemiology 20, 512–522 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall LS et al. Genome-wide meta-analyses of stratified depression in Generation Scotland and UK Biobank. Transl. Psychiatry 8, 9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MIT Critical Data. Secondary Analysis of Electronic Health Records. (Springer, 2016). [PubMed] [Google Scholar]
- 40.Salmasi L & Capobianco E Use of instrumental variables in electronic health record-driven models. Stat. Methods Med. Res 27, 607–621 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Li X et al. MR-PheWAS: exploring the causal effect of SUA level on multiple disease outcomes by using genetic instruments in UK Biobank. Ann. Rheum. Dis 77, 1039–1047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burgess S, Timpson NJ, Ebrahim S & Smith GD Mendelian randomization: Where are we now and where are we going? Int. J. Epidemiol 44, 379–388 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Robins JM & Miguel A Marginal Structural Models and Causal Inference in. Epidemiology 11, 550–560 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Sperrin M, Martin GP, Peek N, Buchan I & Pate A Using marginal structural models to adjust for treatment drop-in when developing clinical prediction models. Stat. Med 37, 4142–4154 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carnegie NB, Harada M & Hill JL Assessing Sensitivity to Unmeasured Confounding Using a Simulated Potential Confounder. J. Res. Educ. Eff 9, 395–420 (2016). [Google Scholar]
- 46.Uddin MJ et al. Methods to control for unmeasured confounding in pharmacoepidemiology: an overview. Int. J. Clin. Pharm 38, 714–723 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Faries DE, Li H, Stamey JD & Imbens GW Addressing unmeasured confounding in comparative observational research. Pharmacoepidemiol. Drug Saf 27, 373–382 (2018). [DOI] [PubMed] [Google Scholar]
- 48.VanderWeele TJ & Ding P Sensitivity Analysis in Observational Research: Introducing the E-Value. Ann. Intern. Med 167, 268 (2017). [DOI] [PubMed] [Google Scholar]
- 49.ICD Code Informational Website. Available at: https://www.cdc.gov/nchs/icd/index.htm.
- 50.Pendergrass SA & Ritchie MD Phenome-wide association studies: leveraging comprehensive phenotypic and genotypic data for discovery. Curr. Genet. Med. Rep 42, 407–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.eMERGE PheKB Website. Available at: https://phekb.org.
- 52.Liao KP et al. Methods to develop an electronic medical record phenotype algorithm to compare the risk of coronary artery disease across 3 chronic disease cohorts. PLoS One (2015). doi: 10.1371/journal.pone.0136651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao KP et al. Development of phenotype algorithms using electronic medical records and incorporating natural language processing. BMJ (2015). doi: 10.1136/bmj.h1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ananthakrishnan AN et al. Identification of Nonresponse to Treatment Using Narrative Data in an Electronic Health Record Inflammatory Bowel Disease Cohort. Inflamm. Bowel Dis (2016). doi: 10.1097/MIB.0000000000000580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Castro V et al. Identification of subjects with polycystic ovary syndrome using electronic health records. Reprod. Biol. Endocrinol (2015). doi: 10.1186/s12958-015-0115-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy TH et al. Genome-wide Association Study of Dimensional Psychopathology Using Electronic Health Records. Biol. Psychiatry (2018). doi: 10.1016/j.biopsych.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sinnott JA et al. Improving the power of genetic association tests with imperfect phenotype derived from electronic medical records. Hum. Genet 133, 1369–1382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu S et al. Surrogate-assisted feature extraction for high-throughput phenotyping. J. Am. Med. Inform. Assoc 24, e143–e149 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yu S et al. Enabling phenotypic big data with PheNorm. J. Am. Med. Informatics Assoc 25, 54–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castro VM et al. Large-scale identification of patients with cerebral aneurysms using natural language processing. Neurology 88, 164–168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kermany DS et al. Identifying Medical Diagnoses and Treatable Diseases by Image-Based Deep Learning. Cell 172, 1122–1131.e9 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Teixeira PL et al. Evaluating electronic health record data sources and algorithmic approaches to identify hypertensive individuals. J. Am. Med. Informatics Assoc 24, 162–171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gan M, Li W, Zeng W, Wang X & Jiang R Mimvec: a deep learning approach for analyzing the human phenome. BMC Syst. Biol 11, 76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hubbard RA et al. A Bayesian latent class approach for EHR-based phenotyping. Stat. Me 38, 74–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu C, Wang F, Hu J & Xiong H Temporal Phenotyping from Longitudinal Electronic Health Records: A Graph Based Framework Categories and Subject Descriptors. in Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 705–714 (2015). [Google Scholar]
- 66.Zhao J et al. Learning from Longitudinal Data in Electronic Health Record and Genetic Data to Improve Cardiovascular Event Prediction. Sci. Rep 9, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sikorska K et al. GWAS with longitudinal phenotypes: performance of approximate procedures. Eur. J. Hum. Genet 23, 1384–1391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiu Y, Justice AE & Melton PE Longitudinal analytical approaches to genetic data. BMC Genet. 17, 25–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pivovarov R Automated methods for the summarization of electronic health records. J. Am. Med. Informatics Assoc 938–947 (2015). doi: 10.1093/jamia/ocv032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Albers DJ et al. Estimating summary statistics for electronic health record laboratory data for use in high-throughout phenotyping algorithms. J. Biomed. Inform 78, 87–101 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang H et al. From phenotype to genotype: an association study of longitudinal phenotypic markers to Alzheimer’s disease relevant SNPs. Bioinformatics 28, 619–625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xu Z, Shen X, Pan W & Neuroimaging D Longitudinal Analysis Is More Powerful than Cross- Sectional Analysis in Detecting Genetic Association with Neuroimaging Phenotypes. PLoS One 9, 1–13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Agniel D, Kohane IS & Weber GM Biases in electronic health record data due to processes within the healthcare system: retrospective observational study. BMJ Open 361, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lange JM, Hubbard RA, Inoue LYT & Minin VN A joint model for multistate disease processes and random informative observation times, with applications to electronic medical records data. Biometrics (2015). doi: 10.1111/biom.12252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bergeron PJ, Asgharian M & Wolfson DB Covariate bias induced by length-biased sampling of failure times. J. Am. Stat. Assoc 103, 737–742 (2008). [Google Scholar]
- 76.Castro VM et al. Validation of electronic health record phenotyping of bipolar disorder cases and controls. Am. J. Psychiatry 172, 363–372 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baiardini I, Braido F, Bonini M, Compalati E & Canonica GW Why do doctors and patients not follow guidelines? Curr. Opin. Allergy Clin. Immunol 9, 228–233 (2009). [DOI] [PubMed] [Google Scholar]
- 78.Ritchie MD et al. Robust Replication of Genotype-Phenotype Associations across Multiple Diseases in an Electronic Medical Record. Am. J. Hum. Genet 86, 560–572 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Y, Wang J, Chubak J & Hubbard RA Inflation of type I error rates due to differential misclassification in EHR ‐ derived outcomes: Empirical illustration using breast cancer recurrence. Pharmacoepidemiol. Drug Saf 28, 264–268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luan X, Pan W, Gerberich SG & Carlin BP Does it always help to adjust for misclassiÿcation of a binary outcome in logistic regression? Stat. Med 24, 2221–2234 (2005). [DOI] [PubMed] [Google Scholar]
- 81.Carroll RJ, Ruppert D, Stefanski LA & Crainiceanu CM Measurement Error in Nonlinear Models: A Modern Perspective. (Chapman and Hall, 2006). [Google Scholar]
- 82.Huang J et al. PIE: A prior knowledge guided integrated likelihood estimation method for bias reduction in association studies using electronic health records data. J. Am. Med. Informatics Assoc (2018). doi: 10.1093/jamia/ocx137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hubbard RA et al. Classification accuracy of claims-based methods for identifying providers failing to meet performance targets. Stat. Med (2015). doi: 10.1002/sim.6318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liao KP et al. Phenome-Wide Association Study of Autoantibodies to Citrullinated and Noncitrullinated Epitopes in Rheumatoid Arthritis. Arthritis Rheumatol. (2017). doi: 10.1002/art.39974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang L et al. Phenotype validation in electronic health records based genetic association studies. Genet Epidemiol. 41, 790–800 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duffy SW et al. A simple model for potential use with a misclassified binary outcome in epidemiology. J. Epidemiol. Community Health 58, 712–717 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Abana CO et al. IL-6 variant is associated with metastasis in breast cancer patients. PLoS One 12, e0181725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ghosh A, Wright FA & Zou F Unified Analysis of Secondary Traits in Case-Control Association Studies. J. Am. Stat. Assoc 108, 566–576 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang Y, Scott AJ & Wild CJ Secondary analysis of case-control data. Stat. Med 25, 1323–1339 (2006). [DOI] [PubMed] [Google Scholar]
- 90.Tchetgen EJT A general regression framework for a secondary outcome in case – control studies. Biostatistics 15, 117–128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang J & Shete S Estimation of Odds Ratios of Genetic Variants for the Secondary Phenotypes Associated with Primary Disease. Genet. Epidemiol 35, 190–200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rusanov A, Weiskopf NG, Wang S & Weng C Hidden in plain sight: Bias towards sick patients when sampling patients with sufficient electronic health record data for research. BMC Med. Inform. Decis. Mak 14, 1–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Reynolds KIMD & West SG A multiplist strategy for strengthening nonequivalent control group designs. Evluation Rev. 11, 691–714 (1987). [Google Scholar]
- 94.West SG et al. Alternatives to the Randomized Controlled Trial. Res. Innov. Recomm 98, 1359–1366 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Au Yeung SL et al. Aldehyde dehydrogenase 2—a potential genetic risk factor for lung function among southern Chinese: evidence from the Guangzhou Biobank Cohort Study. Ann. Epidemiol 24, 606–611 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Kuhnert R et al. A modified self-controlled case series method to examine association between multidose vaccinations and death. Stat. Med 30, 666–677 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Zhou X, Douglas IJ, Shen R & Bate A Signal Detection for Recently Approved Products: Adapting and Evaluating Self-Controlled Case Series Method Using a US Claims and UK Electronic Medical Records Database. Drug Saf. 41, 523–536 (2018). [DOI] [PubMed] [Google Scholar]
- 98.Schumie MJ, Trifiro G, Coloma PM, Ryan PB & Madigan D Detecting Adverse drug reactions following long-term exposure in longitudinal observational data: the exposure-adjusted self-controlled case series. Stat. Methods Med. Res 25, 2577–2592 (2016). [DOI] [PubMed] [Google Scholar]
- 99.Maclure M et al. When should case-only designs be used for safety monitoring of medical products? Pharmacoepidemiol. Drug Saf (2012). doi: 10.1002/pds.2330 [DOI] [PubMed] [Google Scholar]
- 100.Simpson SE et al. Multiple self-controlled case series for large-scale longitudinal observational databases. Biometrics (2013). doi: 10.1111/biom.12078 [DOI] [PubMed] [Google Scholar]
- 101.Petersen I, Douglas I & Whitaker H Self controlled case series methods: an alternative to standard epidemiological study designs. BMJ 354, i4515 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Sun Z, Mukherjee B, Estes JP, Vokonas PS & Park SK Exposure enriched outcome dependent designs for longitudinal studies of gene–environment interaction. Stat. Med 36, 2947–2960 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schildcrout JS, Rathouz PJ, Zelnick LR, Garbett SP & Heagerty PJ Biased sampling designs to improve research efficiency: Factors influencing pulmonary function over time in children with asthma. Ann. Appl. Stat 9, 731–753 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schildcrout JS, Schisterman EF, Mercaldo ND, Rathouz PJ & Heagerty PJ Extending the case-control design to longitudinal data: stratified sampling based on repeated binary outcomes. Epidemiology 29, 67–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li D, Lewinger JP, Gauderman WJ, Murcray CE & Conti D Using Extreme Phenotype Sampling to Identify the Rare Causal Variants of Quantitative Traits in Association Studies. Genet. Epidemiol 35, 790–799 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bjørnland T, Bye A & Ryeng E Improving power of genetic association studies by extreme phenotype sampling: a review and some new results. arXiv 1–26 (2017). [Google Scholar]
- 107.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou W et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Woodward M Epidemiology: Study Design and Data Analysis. (Chapman and Hall, 2013). [Google Scholar]
- 110.Rothman KJ, Lash TL & Greenland S Modern Epidemiology. (Wilters Kluwer, 2012). [Google Scholar]
- 111.Madigan D, Ryan PB & Schuemie MJ Does design matter? Systematic evaluation of the impact of analytical choices on effect esitmates in observational studies. Ther. Adv. Drug Saf 4, 53–62 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Haneuse S, Chan HTH & Daniels M A General Framework for Considering Selection Bias in EHR-Based Studies: What Data are Observed and Why? doi: 10.13063/2327-9214.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Smith GD & Ebrahim S ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol 32, 1–22 (2003). [DOI] [PubMed] [Google Scholar]
- 114.Avery CL, Monda KL & North KE Genetic association studies and the effect of misclassification and selection bias in putative confounders. BMC Proc. 3, S48 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zheng K, Gao J, Ngiam KY, Ooi BC & Yip WLJ Resolving the Bias in Electronic Medical Records. Proc. 23rd ACM SIGKDD Int. Conf. Knowl. Discov. Data Min. - KDD ‘17 2171–2180 (2017). doi: 10.1145/3097983.3098149. [DOI] [Google Scholar]
- 116.Schuemie MJ, Ryan PB, Dumouchel W, Suchard MA & Madigan D Interpreting observational studies: Why empirical calibration is needed to correct p-values. Stat. Med 33, 209–218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schuemie MJ, Hripcsak G, Ryan PB, Madigan D & Suchard MA Empirical confidence interval calibration for population-level effect estimation studies in observational healthcare data. Proc. Natl. Acad. Sci 115, 2571–2577 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Johnson KW, Glicksberg BS, Hodos RA, Shameer K & Dudley JT Causal inference on electronic health records to assess blood pressure treatment targets: an application of the parametric g formula. in Biocomputing 2018 23, 180–191 (WORLD SCIENTIFIC, 2018). [PMC free article] [PubMed] [Google Scholar]
- 119.Kleinberg S & Hripcsak G A review of causal inference for biomedical informatics. J. Biomed. Inform 44, 1102–1112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stuart EA, DuGof E, Abrams M, Salkever D & Steinwachs D Estimating Causal Effects in Observational Studies Using Electronic Health Data: Challenges and (some) Solutions. eGEMs (Generating Evid. Methods to Improv. patient outcomes) 1, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beaumont RN et al. Genome-wide association study of offspring birth weight in 86 577 women identifies five novel loci and highlights maternal genetic effects that are independent of fetal genetics. Hum. Mol. Genet 27, 742–756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Klarin D et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat. Genet 49, 1392–1397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yang J, Zaitlen NA, Goddard ME, Visscher PM & Price A Advantages and pitfalls in the application of mized model association methods. Nat. Genet 46, 100–106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fritsche LG et al. Exploring Various Polygenic Risk Scores for Basal Cell Carcinoma, Cutaneous Squamous Cell Carcinoma and Melanoma in the Phenomes of the Michigan Genomics Initiative and the UK Biobank. bioRxiv (2018). doi: 10.1101/384909 [DOI] [Google Scholar]
- 125.Dey R, Schmidt EM, Abecasis GR & Lee S A Fast and Accurate Algorithm to Test for Binary Phenotypes and Its Application to PheWAS. Am. J. Hum. Genet 101, 37–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bulik-Sullivan B et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Purcell SM et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460, 748–752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hagenaars SP et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N=112 151) and 24 GWAS consortia. Mol. Psychiatry 21, 1624–1632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ge T, Chen C-Y, Neale BM, Sabuncu MR & Smoller JW Phenome-wide heritability analysis of the UK Biobank. PLOS Genet. 13, e1006711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yang J et al. A commentary on ‘common SNPs explain a large proportion of the heritability for human height’ by Yang et al. (2010). Twin Res. Hum. Genet 13, 517–524 (2010). [DOI] [PubMed] [Google Scholar]
- 131.Torkamani A, Wineinger NE & Topol EJ The personal and clinical utility of polygenic risk scores. Nat Rev Genet 19, 581–590 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Ge T, Chen C, Ni Y, Feng YA & Smoller JW Polygenic Prediction via Bayesian Regression and Continuous Shrinkage Priors. bioRXiv 1–30 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Euesden J, Lewis CM & Reilly PFO Genome analysis PRSice: Polygenic Risk Score software. Bioinformatics 31, 1466–1468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vlaming R. De & Groenen PJF The Current and Future Use of Ridge Regression for Prediction in Quantitative Genetics. Biomed Res Int 1–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.So H & Sham PC Improving polygenic risk prediction from summary statistics by an empirical Bayes approach. Sci. Rep 7, 1–11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Paré G, Mao S & Deng WQ A machine-learning heuristic to improve gene score prediction of polygenic traits. Sci. Rep 7, 12665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mak TSH, Sheung J, Kwan H & Dedalus D Local True Discovery Rate Weighted Polygenic Scores Using GWAS Summary Data. Behav. Genet 46, 573–582 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Lloyd-Jones LR et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. bioRXiv 1–39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wray NR, Lee SH, Mehta D, Vinkhuyzen AAE & Middeldorp CM Research Review: Polygenic methods and their application to psychiatric traits. J. Child Psychol. Psychiatry 55, 1068–1087 (2014). [DOI] [PubMed] [Google Scholar]
- 140.Dudbridge F Power and predictive accuracy of polygenic risk scores. PLoS Genet 9, e1003348 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Neale B Neale Lab Website for GWAS Summary Statistics. (2019). Available at: http://www.nealelab.is.
- 142.Vihjalmsson BJ et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet 97, 576–592 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mak TSH, Sham PC, Porsch RM, Choi SW & Zhou X Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol 41, 469–480 (2017). [DOI] [PubMed] [Google Scholar]
- 144.Choi SW, Shin T, Mak H & Reilly PFO A guide to performing Polygenic Risk Score analyses. bioRXiv (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wu Y et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat. Commun 9, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lloyd-Jones LR et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. bioRxiv 1–41 (2019). doi: 10.1101/522961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhu X & Stephens M Bayesian large-scale multiple regression with summary statistics from genome-wide association studies. Ann. Appl. Stat 11, 1561–1592 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Turley P et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet 50, 229–237 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Maier RM et al. Improving genetic prediction by leveraging genetic correlations among human diseases and traits. Nat. Commun 9, 1–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shaddox TR, Ryan PB, Schuemie MJ, Madigan D & Suchard MA Hierarchical Models for Multiple, Rare Outcomes using Massive Observational Databases. Stat Anal Data Min2 9, 260–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xue X, Kim MY, Wang T, Kuniholm MH & Strickler HD A statistical methods for studying correlated rare events and their risk factors. Stat Methods Med Res 26, 1416–1428 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bastarache L et al. Phenotype risk scores identify pations with unrecognized Mendelian disease patterns. Science (80-. ). 359, 1233–1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gronsbell J, Minnier J, Yu S, Liao K & Cai T Gronsbell2018.pdf. Biometrics (2018). [DOI] [PubMed] [Google Scholar]
- 154.Scheurwegs E, Cule B, Luyckx K, Luyten L & Daelemans W Selecting relevant features from the electronic health record for clinical code prediction. J. Biomed. Inform 74, 92–103 (2017). [DOI] [PubMed] [Google Scholar]
- 155.Steele AJ, Denaxas SC, Shah AD & Hemingway H Machine learning models in electronic health records can outperform conventional survival models for predicting patient mortality in coronary artery disease. PLoS One 13, 1–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu Y et al. Quantifying predictive capability of electronic health records for the most harmful breast cancer. in Proc SPIE Int Soc Opt Eng 1–15 (2018). doi: 10.1117/12.2293954.Quantifying [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wu J et al. Prediction Modeling Using EHR Data Challenges, Strategies, and a Comparison of Machine Learning Approaches. Med. Care 48, S106–S113 (2010). [DOI] [PubMed] [Google Scholar]
- 158.Shickel B, Tighe PJ, Bihorac A & Rashidi P Deep EHR: A Survey of Recent Advances in Deep Learning Techniques for Electronic Health Record. arXiv 1–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Rajkomar A et al. Scalable and accurate deep learning with electronic health records. Digit. Med 18, 1–10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Adkins DE Machine learning and electronic health records: A paradigm shift. Am. J. Psychiatry 174, 93–94 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Garg R, Dong S, Shah S & Jonnalagadda SR A Bootstrap Machine Learning Approach to Identify Rare Disease Patients from Electronic Health Records. arXiv 1–8 (2016). [Google Scholar]
- 162.Harang R & Rudd EM Towards Principled Uncertainty Estimation for Deep Neural Networks. arXiv (2018). [Google Scholar]
- 163.Thompson K & Charnigo R Parallel Computing in Genome-Wide Association Studies Journal of Biometrics & Biostatistics. J. Biometrics Biostat 6, 1–3 (2015). [Google Scholar]
- 164.Prive F, Aschard H, Ziyatdinov A, Blum MGB & Timc-imag L Efficient analysis of large-scale genome-wide data with two R packages: bigstatsr and bigsnpr. Bioinformatics 34, 2781–2787 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Berger B, Peng J & Singh M Computational solutions for omics data. Nat. Rev. Genet 14, 333–346 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wells BJ, Chagin KM, Nowacki AS & Kattan MW Strategies for handling missing data in electronic health record derived data. EGEMS (Washington, DC) 1, 1035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hormozdiari F et al. Imputing Phenotypes for Genome-wide Association Studies. Am. J. Hum. Genet 99, 89–103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Beaulieu-Jones BK & Moore JH Missing Data Imputation in the Electronic Health Record Using Deeply Learned Autoencoders. Biocomput. 2017 207–218 (2017). doi: 10.1142/9789813207813_0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Beaulieu-Jones BK et al. Characterizing and Managing Missing Structured Data in Electronic Health Records: Data Analysis. JMIR Med. Informatics 6, e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Little RJA & Rubin DB Statistical Analysis with Missing Data. (John Wiley and Sons, Inc, 2002). doi: 10.1002/9781119013563 [DOI] [Google Scholar]
- 171.Mcculloch CE & Neuhaus JM Diagnostic methods for uncovering outcome dependent visit processes. Biostatistics 1–16 (2018). doi: 10.1093/biostatistics/kxy068 [DOI] [PubMed] [Google Scholar]
- 172.Haneuse S et al. Learning about Missing Data Mechanisms in Electronic Health Records-based Research: A Survey-based Approach. Epidemiology 27, 82–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brzyski D et al. Controlling the Rate of GWAS False Discoveries. Genetics 205, 61–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Good P Permutation, Parametric and Bootstrap Tests of Hypotheses. (Springer, 2005). [Google Scholar]
- 175.Gao X, Starmer J & Martin ER A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet. Epidemiol 32, 361–369 (2008). [DOI] [PubMed] [Google Scholar]
- 176.Abraham KJ & Diaz C Identifying large sets of unrelated individuals and unrelated markers. Source Code Biol. Med 9, 1–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Han B, Kang HM & Eskin E Rapid and accurate multiple testing correction and power estimation for millions of correlated markers. PLoS Genet. 5, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lin DY An efficient Monte Carlo approach to assessing statistical significance in genomic studies. Bioinformatics 21, 781–787 (2005). [DOI] [PubMed] [Google Scholar]
- 179.Seaman SR & Müller-Myhsok B Rapid simulation of P values for product methods and multiple-testing adjustment in association studies. Am. J. Hum. Genet 76, 399–408 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Duggal P, Gillanders EM, Holmes TN & Bailey-Wilson JE Establishing an adjusted p-value threshold to control the family-wide type 1 error in genome wide association studies. BMC Genomics 9, 1–8 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Johnson RC, Nelson GW, Troyer JL, Lautenberger JA & Winkler CA Accounting for multiple comparisons in a genome- wide association study (GWAS). 0–20 (2010). doi: 10.1186/1471-2164-11-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang X, Huang S, Sun W & Wang W Rapid and robust resampling-based multiple-testing correction with application in a genome-wide expression quantitative trait loci study. Genetics 190, 1511–1520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Li MX, Yeung JMY, Cherny SS & Sham PC Evaluating the effective numbers of independent tests and significant p-value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet 131, 747–756 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Bastarache L et al. Phenotype risk scores identify patients with unrecognized Mendelian disease patterns. Science (80-. ). 359, 1233–1239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Inke BI & Andreas RK What Do We Mean by ‘Replication’ and ‘Validation’ in Genome-Wide Association Studies? Hum. Hered 67, 66–68 (2009). [DOI] [PubMed] [Google Scholar]
- 186.NHGRI-EBI GWAS catalog. Available at: https://www.ebi.ac.uk/gwas/.
- 187.Long Q, Flanders WD, Fedirko V & Bostick RM Robust Statistical Methods for Analysis of Biomarkers Measured with Batch/Experiment Specific Errors. Stat. Med 29, 361–370 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Thompson SG Systematic Review: Why sources of heterogeneity in meta-analysis should be investigated. Bmj 309, 1351–1355 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fletcher J What is heterogeneity and is it important? British Medical Journal (2007). doi: 10.1136/bmj.39057.406644.68 [DOI] [Google Scholar]
- 190.Higgins JPT, Thompson SG, Deeks JJ & Altman DG Measuring inconsistency in meta-analyses Need for consistency. BMJ 327, 557–560 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kriston L Dealing with clinical heterogeneity in meta-analysis. Assumptions, methods, interpretation. Int. J. Methods Psychiatr. Res 22, 1–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Li Y & Ghosh D Assumption weighting for incorporating heterogeneity into meta-analysis of genomic data. Bioinformatics 28, 807–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Grimmer J, Messing S & Westwood SJ Estimating Heterogeneous Treatment Effects and the Effects of Heterogeneous Treatments with Ensemble Methods. Polit. Anal 25, 413–434 (2017). [Google Scholar]
- 194.Gagnier JJ, Moher D, Boon H, Bombardier C & Beyene J An empirical study using permutation-based resampling in meta-regression. Syst. Rev 1, 1–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Altman RB & Ashley EA Using “Big Data” to Dissect Clinical Heterogeneity. Circulation 131, 232–233 (2015). [DOI] [PubMed] [Google Scholar]
- 196.Shi X, Li X & Cai T Spherical Regression under Mismatch Corruption with Application to Automated Knowledge Translation. arXiv 1–45 (2018). [Google Scholar]
- 197.Tang L Statistical Methods of Data Integration, Model Fusion, and Heterogeneity Detection in Big Biomedical Data Analysis. (University of Michigan-Ann Arbor, 2018). [Google Scholar]
- 198.Chubak J, Onega T, Zhu W, Buist DSM & Hubbard RA An Electronic Health Record-based Algorithm to Ascertain the Date of Second Breast Cancer Events. Med. Care (2017). doi: 10.1097/MLR.0000000000000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Manrai AK et al. Informatics and Data Analytics to Support Exposome-Based Discovery for Public Health. Annu. Rev. Public Heal 38, 279–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fan JW, Li J & Lussier YA Semantic Modeling for Exposomics with Exploratory Evaluation in Clinical Context. J. Healthc. Eng (2017). doi: 10.1155/2017/3818302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Baek J et al. Methods to study variation in associations between food store availability and body mass in the multi-ethnic study of atherosclerosis. Epidemiology 28, 403–411 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bazemore AW et al. ‘Community vital signs’: Incorporating geocoded social determinants into electronic records to promote patient and population health. J. Am. Med. Informatics Assoc 23, 407–412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Christine PJ et al. Exposure to Neighborhood Foreclosures and Changes in Cardiometabolic Health: Results from MESA. Am. J. Epidemiol 185, 106–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Frederickson Comer K, Grannis S, Dixon BE, Bodenhamer DJ & Wiehe SE Incorporating Geospatial Capacity within Clinical Data Systems to Address Social Determinants of Health. Public Health Rep. 3, 54–61 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sánchez BN, Sanchez-Vaznaugh EV, Uscilka A, Baek J & Zhang L Differential associations between the food environment near schools and childhood overweight across race/ethnicity, gender, and grade. Am. J. Epidemiol 175, 1284–1293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Xie S, Greenblatt R, Levy MZ & Himes BE Enhancing Electronic Health Record Data with Geospatial Information. in AMIA Jt Summits Transl Sci Proc 123–132 (2017). [PMC free article] [PubMed] [Google Scholar]
- 207.Pollard TJ et al. The eICU Collaborative Research Database, a freely available multi-center database for critical care research. Sci. Data 5, 180178 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Al-Azwani IK & Aziz HA Integration of Wearable Technologies into Patients’ Electronic Medical Records. Qual. Prim. Care 24, 151–155 (2016). [Google Scholar]
- 209.Polzer N & Gewald H A Structured Analysis of Smartphone Applications to Early Diagnose Alzheimeŕs Disease or Dementia. Procedia Comput. Sci 113, 448–453 (2017). [Google Scholar]
- 210.Norén GN, Hopstadius J, Bate A, Star K & Edwards IR Temporal pattern discovery in longitudinal electronic patient records. Data Min. Knowl. Discov (2010). doi: 10.1007/s10618-009-0152-3 [DOI] [Google Scholar]
- 211.Norén GN et al. Empirical performance of the calibrated self-controlled cohort analysis within temporal pattern discovery: Lessons for developing a risk identification and analysis system. Drug Saf. (2013). doi: 10.1007/s40264-013-0095-x [DOI] [PubMed] [Google Scholar]
- 212.Boland MR, Shahn Z, Madigan D, Hripcsak G & Tatonetti NP Birth month affects lifetime disease risk: A phenome-wide method. J. Am. Med. Informatics Assoc (2015). doi: 10.1093/jamia/ocv046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Liu M et al. Comparative analysis of pharmacovigilance methods in the detection of adverse drug reactions using electronic medical records. J Am Med Inf. Assoc 20, 420–426 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ramirez AH et al. Predicting warfarin dosage in European-Americans and African-Americans using DNA samples linked to an electronic health record. Pharmacogenomics (2012). doi: 10.2217/pgs.11.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Peterson JF et al. Electronic Health Record Design and Implementation for Pharmacogenomics: a Local Perspective HHS Public Access. Genet Med 15109, 833–841 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Madigan D & Shin J Drospirenone-containing oral contraceptives and venous thromboembolism: an analysis of the FAERS database. Open Access J. Contracept 9, 29–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Shuldiner AR et al. The pharmacogenomics research network translational pharmacogenetics program: Overcoming challenges of real-world implementation. Clin. Pharmacol. Ther 94, 207–210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kuang Z et al. Computational Drug Repositioning Using Continuous Self-Controlled Case Series. 491–500 (2017). doi: 10.1145/2939672.2939715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Paige E et al. Landmark Models for Optimizing the Use of Repeated Measurements of Risk Factors in Electronic Health Records to Predict Future Disease Risk. Am. J. Epidemiol 187, 1530–1538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Goldstein BA, Navar AM, Pencina MJ & Ioannidis JPA Opportunities and challenges in developing risk prediction models with electronic health records data: a systematic review. J. Am. Med. Informatics Assoc 24, 198–208 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Caballero K & Akella R Dynamic Estimation of the Probability of Patient Readmission to the ICU using Electronic Medical Records. AMIA Annu. Symp. Proc 2015, 1831–40 (2015). [PMC free article] [PubMed] [Google Scholar]
- 222.Aczon M et al. Dynamic Mortality Risk Predictions in Pediatric Critical Care Using Recurrent Neural Networks. 1–18 (2017). [Google Scholar]
- 223.Steorts RC, Hall R & Fienberg SE A Bayesian Approach to Graphical Record Linkage and Deduplication. J. Am. Stat. Assoc 111, 1660–1672 (2016). [Google Scholar]
- 224.Sayers A, Ben-Shlomo Y, Blom AW & Steele F Probabilistic record linkage. Int. J. Epidemiol 45, 954–964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Vatsalan D, Christen P & Verykios VS A taxonomy of privacy-preserving record linkage techniques. Inf. Syst 38, 946–969 (2013). [Google Scholar]
- 226.Mamun A. Al, Aseltine R & Rajasekaran S Efficient record linkage algorithms using complete linkage clustering. PLoS One 11, 1–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Schmidlin K, Clough-Gorr KM & Spoerri A Privacy Preserving Probabilistic Record Linkage (P3RL): A novel method for linking existing health-related data and maintaining participant confidentiality. BMC Med. Res. Methodol 15, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Long Q Statistical Methods for Handling Missing Data in Distributed Health Data Networks. in Joint Statistical Meetings (2018). [Google Scholar]
- 229.Tang L, Zhou L & Song PX-K Method of Divide-and-Combine in Regularised Generalised Linear Models for Big Data. (2016).
- 230.Yang J et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet 44, 1–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Hripcsak G et al. Characterizing treatment pathways at scale using the OHDSI network. Proc. Natl. Acad. Sci (2016). doi: 10.1073/pnas.1510502113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Simon GE, Peterson D & Hubbard R Is treatment adherence consistent across time, across different treatments and across diagnoses? Gen. Hosp. Psychiatry (2013). doi: 10.1016/j.genhosppsych.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Santillana M et al. Cloud-based Electronic Health Records for Real-time, Region-specific Influenza Surveillance. Sci. Rep 25732, 1–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Yang S et al. Using electronic health records and Internet search information for accurate influenza forecasting. BMC Infect. Dis 17, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Moran KR et al. Epidemic Forecasting is Messier Than Weather Forecasting: The Role of Human Behavior and Internet Data Streams in Epidemic Forecast. J. Infect. Dis 214, 404–408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Meng XL Statistical paradises and paradoxes in big data (I): Law of large populations, big data paradox, and the 2016 us presidential election. Ann. Appl. Stat 12, 685–726 (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.