

Abstract
Peroxyacetyl nitrate (PAN) formed in the atmospheric oxidation of non-methane volatile organic compounds (NMVOCs) is the principal tropospheric reservoir for nitrogen oxide radicals (NOx = NO + NO2). PAN enables the transport and release of NOx to the remote troposphere with major implications for the global distributions of ozone and OH, the main tropospheric oxidants. Simulation of PAN is a challenge for global models because of the dependence of PAN on vertical transport as well as complex and uncertain NMVOC sources and chemistry. Here we use an improved representation of NMVOCs in a global 3-D chemical transport model (GEOS-Chem) and show that it can simulate PAN observations from aircraft campaigns worldwide. The immediate carbonyl precursors for PAN formation include acetaldehyde (44% of the global source), methylglyoxal (30 %), acetone (7 %), and a suite of other isoprene and terpene oxidation products (19 %). A diversity of NMVOC emissions is responsible for PAN formation globally including isoprene (37 %) and alkanes (14 %). Anthropogenic sources are dominant in the extratropical Northern Hemisphere outside the growing season. Open fires appear to play little role except at high northern latitudes in spring, although results are very sensitive to plume chemistry and plume rise. Lightning NOx is the dominant contributor to the observed PAN maximum in the free troposphere over the South Atlantic.
1. Introduction
Peroxyacetic nitric anhydride (CH3COO2NO2), commonly known by its misnomer peroxyacetyl nitrate (PAN), is the principal tropospheric reservoir species for nitrogen oxide radicals (NOx = NO + NO2) with important implications for the production of tropospheric ozone (O3) and the hydroxyl radical OH (the main atmospheric oxidant) (Singh and Hanst, 1981). PAN is formed by oxidation of non-methane volatile organic compounds (NMVOCs) in the presence of NOx. NMVOCs and NOx have both natural and anthropogenic sources. Fossil fuel combustion is the principal NOx source, with additional contributions from biomass burning, lightning and soils (van der A et al., 2008). The organic side of PAN formation involves many stages of NMVOC oxidation. Most NMVOCs can serve as PAN precursors, but the yields vary widely (Roberts, 2007).
PAN enables the long-range transport of NOx at cold temperatures, and PAN decomposition releases NOx in the remote troposphere where it is most efficient at producing O3 and OH (Singh and Hanst, 1981; Hudman et al., 2004; Fischer et al., 2010; Singh, 1987). NOx abundance controls the balance of O3 production and destruction. Without PAN formation the distributions of tropospheric NOx, O3 and OH would be very different, with higher values in NOx source regions and lower values in the remote troposphere (Kasibhatla et al., 1993; Moxim et al., 1996; Wang et al., 1998a). PAN chemistry can also be important for oxidant formation on a regional scale. In polluted environments, PAN formation is a sink for both NOx and hydrogen oxide radicals (HOx). Observations show that O3 concentrations increase when temperature increases, and this has been in part related to PAN thermal instability (Sillman and Samson, 1995). Observations also show that the production of PAN becomes more efficient relative to O3 in highly polluted air masses (Roberts et al., 1995). Thus a comprehensive understanding of PAN is needed to understand oxidant distributions on a spectrum of scales.
A large body of PAN observations worldwide has accumulated over the years, including in particular from aircraft platforms and mountaintop sites. There have also been recent retrievals of PAN concentrations in the upper troposphere (UT) from satellites (Glatthor et al., 2007; Tereszchuk et al., 2013). Concentrations vary from pptv levels in warm remote locations such as tropical oceans to ppbv levels in polluted source regions. Despite the relatively large database of measurements compared to other photochemical indicators, simulation of PAN in global chemical transport models (CTMs) has been a difficult challenge because of the complexity of PAN chemistry. Recent model intercomparisons show very large difference among themselves and with observations in many regions of the atmosphere (Thakur et al., 1999; Singh et al., 2007; von Kuhlmann et al., 2003; Sudo et al., 2002), but confirm the very important role for PAN in sustaining O3 production in remote air (Zhang et al., 2008; Hudman et al., 2004).
Here we exploit a worldwide collection of PAN observations to improve the PAN simulation in the GEOS-Chem (Goddard Earth Observing System) CTM, which has been used extensively in global studies of tropospheric oxidants (Bey et al., 2001; Sauvage et al., 2007; Murray et al., 2012). The earliest global models that included PAN chemistry (Kasibhatla et al., 1993; Moxim et al., 1996) relied on highly simplified NMVOC budgets. Our improvements involve new treatments of NMVOC sources and chemistry, a well-known weakness even in current CTMs (Williams et al., 2013; Ito et al., 2007). Our new simulation, which captures the major features of the existing observations, affords a new opportunity to understand the factors driving the global PAN distribution and the essential chemistry that needs to be described. A detailed analysis of how PAN shapes the global distributions of the atmospheric oxidants and nitrogen deposition will be the focus of a subsequent paper.
2. Model description
We use the GEOS-Chem global 3-D CTM including detailed ozone–NOx–VOC–aerosol chemistry (version 9.01.01, www.geos-chem.org) with significant modifications as described below.
2.1. Chemistry
GEOS-Chem uses a chemical scheme originally described by Horowitz et al. (1998) and Bey et al. (2001), with recent updates outlined in Mao et al. (2010). Following Marais et al. (2012) we have updated the rate coefficients for the reactions of HO2 with the > C2 peroxy radicals to Eq. (iv) in Saunders et al. (2003). We also include nighttime reactions of organic peroxy radicals with NO3 following Stone et al. (2013). To implement the Stone et al. (2013) nighttime chemistry, we went through each of the RO2 + NO reactions in the GEOS-Chem chemical mechanism, copied each of these reactions, and changed the RO2 reactants to react with NO3 rather than NO. The Master Chemical Mechanism (MCM) considers three different reactions rates for this class, one for CH3O2, one for RC(O)O2 and one for all other RO2. There is no temperature dependence included, and all products are assumed to be the same as the corresponding reaction of the RO2 radical with NO (Bloss et al., 2005). We replaced the isoprene chemical mechanism with one based on Paulot et al. (2009a, b), as described by Mao et al. (2013b).
PAN is produced reversibly by reaction of the peroxyacetyl (PA) radical CH3C(O)OO with NO2:
| (R1) |
where M is a third body (typically N2 or O2). The dominant sources of CH3C(O)OO are the oxidation of acetaldehyde (CH3CHO) and the photolysis of acetone (CH3C(O)CH3) and methylglyoxal (CH3COCHO):
| (R2) |
| (R3) |
| (R4) |
PAN can also be produced at night via reaction of acetaldehyde with the nitrate radical. Acetaldehyde, acetone and methylglyoxal are all directly emitted (“primary” sources) and produced in the atmosphere from oxidation of primary emitted NMVOCs (“secondary” sources). These different sources will be discussed below. There are also other minor sources of the PA radical, again to be discussed below.
Higher acyl peroxy nitrates (RC(O)OONO2) are similarly formed from the oxidation of NMVOCs, but their yields are much lower than that for PAN. We focus on PAN because observations show that it typically accounts for 75–90% of total acyl peroxy nitrates (Roberts, 2007; Roberts et al., 1998, 2002; Wolfe et al., 2007) and there are an abundance of measurements of PAN. Closure on measurements of total reactive nitrogen oxides (NOy) confirms the dominant role of PAN as an organic nitrate reservoir for NOx (Roberts et al., 1995; Bertram et al., 2013).
The main sink of PAN is thermal decomposition (reaction 1), and the effective PAN lifetime depends on whether the released PA radical reacts with NO2 to return PAN, or with another species (mainly NO or HO2) leading to permanent loss. To describe this chemistry, GEOS-Chem uses the recommendation from Sander et al. (2011), which is taken from Bridier et al. (1991). The parameters recommended by Bridier et al. (1991) are consistent with later studies of PAN decomposition by Roberts and Bertman (1992), Orlando et al. (1992), and Grosjean et al. (1994). The rate coefficient for the reaction of the PA radical with NO is also from Tyndall et al. (2001).
Primary NMVOCs in the standard GEOS-Chem mechanism that contribute to PAN formation include ethane, propane, > C3 alkanes (lumped), > C2 alkenes (lumped), isoprene, acetaldehyde, methylglyoxal, acetone, and > C3 ketones (lumped). Our extended mechanism adds several additional primary NMVOCS, including ethanol, benzene, toluene and ethylbenzene (lumped), xylenes and trimethylbenzenes (lumped), and monoterpenes (lumped). The additions were partially motivated by the work of Ito et al. (2007), who extended the GEOS-Chem mechanism within the Integrated Massively Parallel Atmospheric Chemical Transport (IMPACT) CTM to include a broader suite of NMVOCs. One result of this exercise was a dramatic increase in PAN formation through methylglyoxal and hydroxyacetone. Liu et al. (2010) found aromatics to be a major source of PAN in urban China through the production of methylglyoxal. We calculate the associated yield of methylglyoxal using recommended values for the individual aromatic species (toluene, o-xylene, m-xylene, p-xylene, 1,2,3-trimethylbenzene, 1,2,4-trimethylbenzene, and 1,3,5-trimethylbenzene) from Nishino et al. (2010) and the observed mean aromatic speciation for Chinese cities from Barletta et al. (2006).
We adopted the treatment of monoterpene oxidation from the RACM2 chemical mechanism (Goliff et al., 2013), lumping terpenes with one double bond (alpha-pinene, beta-pinene, sabinene and delta-3-carene) into one proxy. Unlike Ito et al. (2007), hydroxyacetone is not a product of terpene oxidation in the revised RACM2 mechanism used here. The gas phase oxidation of monoterpenes is highly unconstrained. The RACM2 mechanism is primarily based on Atkinson and Avery (2003). The yields of the immediate PAN precursors resulting from terpene degradation as described by RACM2 embedded in GEOS-Chem are given in Table 1. The mechanism produces methyl ethyl ketone and acetone, both of which can serve as PA radical precursors. The addition of this lumped terpene increases PAN in the model. The largest surface changes for PAN are for eastern Europe and western Russia, where there are high biogenic terpene emissions but there is little PAN data for comparison.
Table 1.
Global contributions of primary NMVOCs to PAN formationa.
| Sources, Tg C a−1 | Molar yields of immediate PAN precursors | PAN contribution (%)e | ||||||
|---|---|---|---|---|---|---|---|---|
| Primary NMVOC | Lifetime (days)b | Fuel and industryc | Open fires | Biogenic | Acetaldehyded | Acetone | Methyl-glyoxal | |
| Isoprene | 0.10 | – | - | 427 | 0.019 | – | 0.32 | 37f |
| Terpenes | 0.46g | – | 1.3 | 65g | 0.025h | 0.017h | 0.050h | 9 |
| > C3 alkanesi | 5 | 24 | 0.67 | – | 1.07 | 0.30 | – | 9 |
| Acetone | 14 | 0.45 | 1.7 | 69j | – | 1 | 0.14k | 9 |
| Acetaldehyde | 0.8 | 1.1 | 1.6 | 44j | 1 | – | – | 8 |
| Ethane | 60 | 8.5 | 1.9 | – | 0.78 | – | – | 6 |
| Propane | 14 | 17 | 0.77 | – | 0.30 | 0.75 | – | 5 |
| > C2 alkenesl | 0.38 | 3.9 | 2.7 | 12 | 0.85 | – | – | 4 |
| Ethanol | 2.8 | 1.0 | 0.04 | 12 | 0.95 | – | – | 4 |
| Methyl-glyoxal | 0.067 | – | 2.6 | – | 0.35m | – | 1 | < 1 |
| Xylenesn | 0.58 | 11 | 0.73 | – | – | – | 0.21 | < 1 |
| Tolueneo | 2.2 | 14 | 0.26 | – | – | – | 0.46 | < 1 |
| Hydroxy-acetone | 2.1 | – | 0.65 | – | – | – | 0.82p | < 1 |
| Methyl ethyl ketone | 4.3 | 0.34 | 0.99 | – | 0.002 | – | – | < 1 |
Global primary emitted NMVOC sources of PAN and their estimated yields for the three most important immediate carbonyl PAN precursors: acetaldehyde, acetone, and methylglyoxal. Details of sources can be found in Sect. 2.2.
Global annual mean tropospheric lifetime. Lifetimes were calculated from global annual average burdens and loss rates.
Includes biofuel use.
Assumes 1 ppbv NOx from Millet et al. (2010) unless otherwise noted.
The contribution to the global annual PAN burden from individual NMVOCs is calculated by simulations with corresponding emissions turned off. To avoid large nonlinear effect in the case of isoprene, emissions were reduced by 20 %, and the difference between that simulation and the standard simulation was multiplied by 5.
PAN production from isoprene involves additional precursors other than acetaldehyde and methylglyoxal, including methyl vinyl ketone, methacrolein and other short-lived oxidation intermediates.
34 Tg C as α-pinene, 16 Tg C as β-pinene, 7.3 Tg C as sabinene, and 6.1 Tg C as δ−3-carene; lifetime is calculated as a lumped species.
Calculated using difference between global simulations with and without terpene chemistry.
> C3 alkanes are emitted as a mixed butane–pentane lumped species on a carbon-weighted basis (Lurmann et al., 1986).
Includes primary terrestrial and ocean sources.
From Fu et al. (2008)
> C2 alkenes are emitted as propene on a carbon-weighted basis.
Photolysis of methylglyoxal produces acetaldehyde in GEOS-Chem. Calculation assumes each of two absorption bands is responsible for half of the photolysis.
> Lumped species including o-xylene, m-xylene, p-xylene, 1,2,3-trimethylbenzene, 1,2,4-trimethylbenzene and1,2,5-trimethylbenzene with the reactivity of m-xylene.
Also including ethylbenzene with the reactivity of toluene.
Chemical yield from photolysis and reaction with OH of hydroxyacetone is unity, but 18% of hydroxyacetone is removed by wet deposition.
In addition to thermal decomposition, we include minor sinks for PAN from dry deposition and photolysis. The dry deposition velocity for PAN is simulated using a standard resistance-in-series approach (Wesely, 1989) as implemented in GEOS-Chem by Wang et al. (1998b). We assume that the PAN reactivity with surfaces is as strong as that of O3 (Shepson et al., 1992), but we explore the sensitivity to this choice. Photolysis and deposition account globally for 1.8% and 1.2% of the global PAN sink, respectively. Photolysis of PAN is important in the upper troposphere, where the lifetime against loss via photolysis is on the order of a month (Talukdar et al., 1995). We find that assuming PAN reactivity with surfaces is more similar to O3 rather than NO2 decreases surface PAN concentrations over Northern Hemisphere continents by 15–20% in spring. Reaction of PAN with both the OH radical and Cl atoms is slow, and these are both minor loss pathways, unnecessary to include in global models (Wallington et al., 1990; Talukdar et al., 1995). Uptake on ice particles in convective clouds (Marecal et al., 2010) and on organic aerosols (Roberts, 2005) are both thought to be negligible and are not included here. PAN is only sparingly soluble, but hydrolysis of the PA radical is thought to explain observed PAN loss in fog (Villalta et al., 1996; Roberts et al., 1996). We do not consider this to be a significant loss process for PAN on a global scale (Jacob, 2000).
2.2. Emissions
Production of PAN can be limited by the supply of either NOx or NMVOCs, as discussed below. Global fossil fuel emissions of NOx in GEOS-Chem are from the EDGAR inventory (Oliver and Berdowski, 2001) and are overwritten with regional inventories for Europe (EMEP) (Vestreng and Klein, 2002), Mexico (BRAVO) (Kuhns et al., 2003), eastern Asia (Zhang et al., 2009), Canada (NPRI, http://www.ec.gc.ca/inrp-npri/), and the United States (EPA/NEI2005, http://www.epa.gov/ttnchie1/net/2005inventory.html). All anthropogenic NOx emissions are scaled to 2008 based on energy statistics (van Donkelaar et al., 2008). Soil NOx emissions in GEOS-Chem are based on Yienger and Levy (1995) as implemented by Wang et al. (1998b). Lightning NOx emissions are described by Sauvage et al. (2007).
Table 1 lists the global emissions of all NMVOCs that contribute to PAN formation. We use the RETRO (REanalysis of the TROpospheric chemical composition) emission inventory (van het Bolscher et al., 2008) as the global default for anthropogenic NMVOC emissions aside from ethane and propane. Ethane and propane emissions in RETRO were far too low compared to the GEOS-Chem inventories from Xiao et al. (2008), which are unbiased relative to observations. Emissions of both species appeared to be missing from the major natural gas production region in Russia. We used the ethane and propane emission inventories which were developed as in Xiao et al. (2008). The RETRO emission inventory is for 2000, and we scaled it to 2008 following van Donkelaar et al. (2008). RETRO includes anthropogenic emissions for benzene, xylene and toluene. Based on the observed CO-to-benzene ratio for TRACE-P, we increased benzene emissions over China by 25 %. We then scaled xylene and toluene emissions to benzene based on measurements from 43 Chinese cities from Barletta et al. (2006). Thus RETRO emissions of toluene were increased by a factor of 4 over China to create our lumped toluene, and RETRO emissions of xylene were increased by a factor of 8 over China to create our lumped xylene species. Observations show large abundances of reactive aromatics over southern and eastern China (Ran et al., 2009; Wang et al., 2002, 2013; Zhang et al., 2007a).
Terrestrial biogenic emissions of acetone, acetaldehyde, isoprene, ethanol, terpenes, and > C2 alkenes from metabolism and decay are calculated locally using the Model of Emissions of Gases and Aerosols from Nature (MEGAN v2.0) (Guenther et al., 2006). Specific other sources and sinks for acetone and acetaldehyde are described in Fischer et al. (2012) and Millet et al. (2010), respectively. The atmospheric budget of ethanol is also as described by Millet et al. (2010).
New estimates indicate that fires emit significantly more NMVOCs than previously thought (Wiedinmyer et al., 2011), and there is a large contribution from oxygenated species, many of which are unidentified (Warneke et al., 2011). Observations show rapid conversion of NOx to PAN in fire plumes, seemingly due to the oxidation of very short-lived NMVOCs (Jacob et al., 1992). We use 2008 Global Fire Emissions Database (GFED3) monthly biomass burning emissions for NOx and NMVOCs (van der Werf et al., 2010) with updated emission factors for NMVOCs and NOx from extratropical forests, savannas and agricultural fires from Akagi et al. (2011). The updated NOx emission factor for extratropical fires is approximately a factor of three lower, and the emission factors for the NMVOCs are generally higher. Following Alvarado et al. (2010) we partition 40% and 20% NOx emissions from fires directly to PAN and HNO3, respectively. The Alvarado et al. (2010) partitioning is based on observations of fresh boreal fire plumes, but we apply it here to all fire types. Our additional NMVOC budgets include the addition of biomass burning emissions as given in Table 1.
The standard version of GEOS-Chem releases all fire emissions in the boundary layer; however, previous studies have pointed out that a significant fraction of biomass burning emissions can be injected to the free troposphere because of buoyancy (Turquety et al., 2007; Val Martin et al., 2010). This is especially important for PAN because lower temperatures above the boundary layer enhance its stability. Val Martin et al. (2010) showed that a substantial fraction of plumes from North American fires are injected into the free troposphere. Smoke plumes over the boreal region reached the highest altitudes. Analysis of smoke clouds, which are a later stage of plume evolution, indicated that ~35% were above the boundary layer. Here we distribute 35% of biomass burning emissions by mass in the 10 sigma layers (4 km) above the boundary layer, and this improves our comparison with PAN observations at high latitudes. The PAN simulation is sensitive to this choice.
Kaiser et al. (2012) and Yue et al. (personal communication) find that GFED3 underestimates fire emissions by not accounting for small fires, particularly at boreal latitudes. Following their work, we increased wild fire emissions by 60% in northern Asia (30–75° N, 60–190° E), 25% in Canada and 50% in Alaska. Increasing fire emissions over Russian and North American boreal regions improves the PAN simulation over the Arctic, particularly above the surface
2.3. Model configuration
In our work GEOS-Chem is driven by NASA GEOS-5 assimilated meteorological data with 0.5° × 0.67° horizontal resolution, 47 levels in the vertical, and 3–6 h temporal resolution. We degrade the horizontal resolution to 2° × 2.5° for input into our GEOS-Chem simulation. We use a 1-year simulation for 2008, preceded by a 1-year spin-up to remove the effect of initial conditions. We also present a number of sensitivity simulations conducted at 4° × 5° horizontal resolution, which yield results very similar to the 2° × 2.5° resolution. The largest differences in the two resolutions occur over regions of biomass burning. Over these locations, the finer horizontal resolution produces 10–20% more PAN. The likely explanation is that vertical transport is faster at higher resolution because eddies are not averaged out. This was first shown by Wang et al. (2004) using a nested simulation for CO over Asia.
Throughout the paper we present results using 2008 GEOS-5 assimilated meteorology. However we have compared results using both GEOS-4 and GEOS-5 for 2006, the last year of overlap for these two meteorological data sets. The lifetime of PAN doubles for every 4 K decrease in temperature. PAN is also sensitive to biogenic emissions, lightning NOx emissions and vertical transport (Labrador et al., 2005), parameters that also depend on the underlying meteorological field. We found that differences in the monthly mean PAN produced using different assimilated meteorological grids are substantial in some locations (> 100 pptv). Higher upper-tropospheric PAN mixing ratios over the tropical Pacific in GEOS-5 appear to be driven by slower convective overturning in GEOS-5 than GEOS-4. GEOS-4 agrees better with data from PEM-Tropics B (Maloney et al., 2001). The PEM-Tropics B data set suggests very low (< 50 pptv) PAN mixing ratios in the Pacific tropical UT. Differences between the simulated and observed O3 profiles for the PEM-Tropics B regions are consistent with the view that differences in vertical motion are driving the PAN differences. There are limited opportunities to chemically constrain convective overturning in data assimilation models. However, existing analyses suggest substantial differences between overturning rates derived from in situ measurements and those in GEOS-4 (Bertram et al., 2007). Mitoviski et al. (2012) assessed the impact of convection on O3 in GEOS-Chem and found tropical upper-tropospheric O3 biases driven by the parameterized vertical transport in both GEOS-4 and GEOS-5.
3. Global PAN distribution
We used a large database of recent PAN observations from surface sites and airborne campaigns to evaluate the model, and these are presented in Table 2. For comparison to the model, we averaged the aircraft observations over the coherent regions in Fig. 1. The measurements either relied on gas chromatography with electron capture detector (GC-ECD) (Flocke et al., 2005) or thermal decomposition chemical ionization mass spectrometry (TD-CIMS) (Zheng et al., 2011).
Table 2.
Global PAN measurements used for model evaluation listed in order of map regions labeled in Fig. 2.
| Aircraft missions | ||||
|---|---|---|---|---|
| Experiment | Time frame | Location | Fig. 1 map regions | Reference |
| TRACE-P | Mar–Apr 2001 | W Pacific | 1,2,5,6 | Talbot et al. (2003) |
| PEM-West B | Feb–Mar 1994 | W Pacific | 3 | Singh et al. (1998) |
| PEM-West A | Sep–Oct 1991 | W Pacific | 4, 7, 8 | Singh et al. (1996b) |
| PEM-Tropics B | Mar–Apr 1999 | Tropical Pacific | 9–13 | Maloney et al. (2001) |
| PEM-Tropics A | Aug–Oct 1996 | S Pacific | 14–17 | Talbot et al. (2000) |
| INTEX-B | Mar–May 2006 | E Pacific | 18–20 | Singh et al. (2009) |
| PHOBEA | Mar–Apr 1999 | E Pacific | 21 | Kotchenruther et al. (2001) |
| ITCT-2K2 | Apr–May 2002 | E Pacific | 22 | Roberts et al. (2004) |
| MILAGRO | Mar–May 2006 | Mexico | 23 | Singh et al. (2009) |
| CITE-2 | Aug–Sep 1986 | W US | 24–25 | Singh et al. (1990a) |
| INTEX-A | Jul–Aug 2004 | Eastern N America | 26–28 | Singh et al. (2006) |
| SONEX | Oct–Nov 1997 | N Atlantic | 29, 42 | Talbot et al. (1999) |
| ABLE-2B | Apr–May 1987 | Amazon | 30 | Singh et al. (1990b) |
| TRACE-A | Sep–Oct 1992 | S Atlantic | 31–32, 43–45 | Singh et al. (1996a) |
| ABLE-3A | Jul–Aug 1988 | Alaska | 33 | Singh et al. (1992) |
| ABLE-3B | Jul–Aug 1990 | E Canada | 34 – 35 | Singh et al. (1994) |
| ARCTAS | Apr–Jul 2008 | N American Arctic | 36–38 | Alvarado et al. (2010) |
| ARCPAC | Mar–Apr 2008 | Alaska | 39 | Slusher et al. (2004) |
| POLARCAT | July 2008 | Greenland | 40 | Roiger et al. (2011) |
| TOPSE | Feb–Mar 2000 | N American Arctic | 41 | Atlas et al. (2003) |
| AMMA | Aug 2006 | West Africa | 46 | Stewart et al. (2008) |
| Surface measurements | ||||
| Site name | Time frame | Location | Elevation | Reference |
| Mount Bachelor | Mar–May 2008–2010 | 44° N, 122° W | 2.7 km | Fischer et al. (2010) |
| Jungfraujoch | 1997–1998, 2005–2006, 2008 | 47° N, 9° E | 3.6 km | Balzani Loov et al. (2008); Whalley et al. (2004); Zellweger et al. (2000); Pandey Deolal et al. (2013) |
| Hohenpeissenberg | 2003–2008 | 48° N, 1° E | 985 m | http://ds.data.jma.go.jp/gmd/wdcgg/ |
| Schauinsland | 1995–2010 | 48° N, 8° E | 1.2 km | http://ds.data.jma.go.jp/gmd/wdcgg/ |
| Zugspitze | 2004–2008 | 47° N, 11° E | 2.7 km | http://ds.data.jma.go.jp/gmd/wdcgg/ |
| Waliguan | Jul–Aug 2006 | 36° N, 101° E | 3.8 km | Xue et al. (2011) |
| Bush Estate | 1994–1998 | 56° N, 3° W | 200 m | McFadyen and Cape (2005) |
| Rishiri | 1999 | 45° N, 141° E | 35 m | Tanimoto et al. (2002) |
| Poker Flat | Mar–May 1993, 1995 | 65° N, 148° W | 470 m | Beine et al. (1996) |
| Alert | Jan–Apr 1992, 1998, 2000 | 82° N, 62° W | 200 m | Dassau et al. (2004); Worthy et al. (1994) |
| Zeppelin | 1994–1998 | 78° N, 16° W | 474 m | Beine et al. (1997); Beine and Krognes (2000) |
| Polarstern Cruise | May–Jun 1998 | 52° N–17° S, 7° E–19° W | Sea level | Jacobi et al. (1999) |
| Thompson Farm | 2005–2007 | 43° N, 71° W | 25 m | Robert Talbot, Ryan Chartier, unpublished data |
| Summit, Greenland | Jun–Jul 1998, Jan 1999 | 47° N, 9° E | 3.2 km | Ford et al. (2002) |
| Pico Mountain | Jul–Sep 2008, Mar–Jul 2009 | 38° N, 28° W | 2.2 km | Katja Dzepina, Jim Roberts, unpublished data |
Fig. 1.
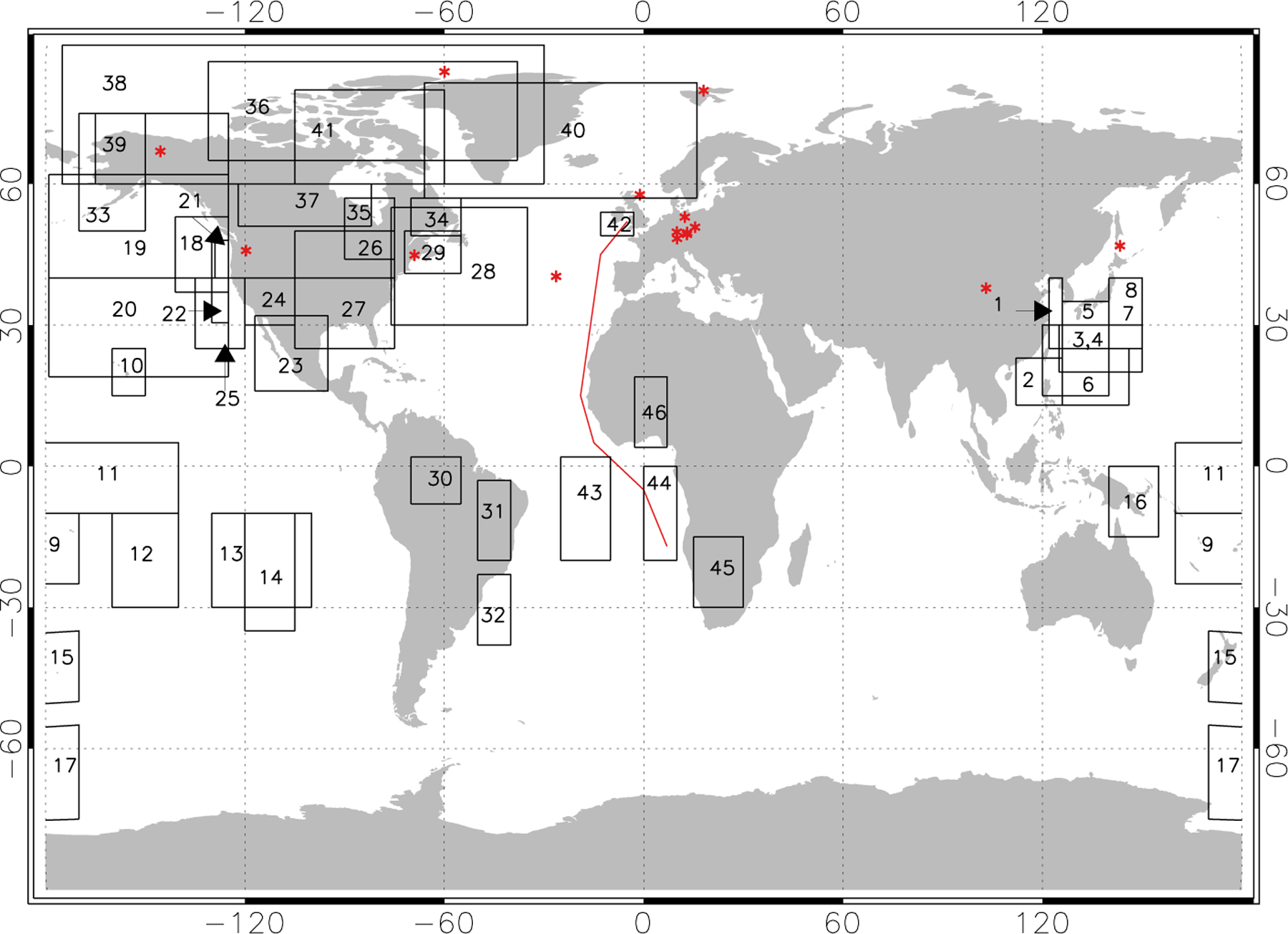
Locations of PAN observations used in our analysis (Table 2): surface sites (red asterisk); aircraft missions (black boxes) with region numbers indicated; and one cruise (red line).
Figures 2 and 3 compare the observed global distribution of PAN to that simulated by GEOS-Chem. Mean observations from the studies compiled in Table 2 are shown as filled circles. Model fields are background contours. Full vertical profiles (median and mean) for the aircraft campaigns and seasonal cycles for several European mountaintop data sets are in the supplementary materials. We compare model output from 2008 to observations collected over many years. Interannual variability in the model is smaller than other sources of error. There are relatively few in situ observations that can be used to assess interannual variability in PAN (Bottenheim et al., 1994). Observations at Mount Bachelor (Oregon, USA) indicate interannual variability of 20% during the spring maximum (Fischer et al., 2011). Recent trends in PAN in many regions of the atmosphere are also hard to assess given a paucity of consistent data (Parrish et al., 2004). As shown later PAN is highly sensitive to NOx and NMVOC emissions, both of which have changed considerably in some regions (Pollack et al., 2013).
Fig. 2.
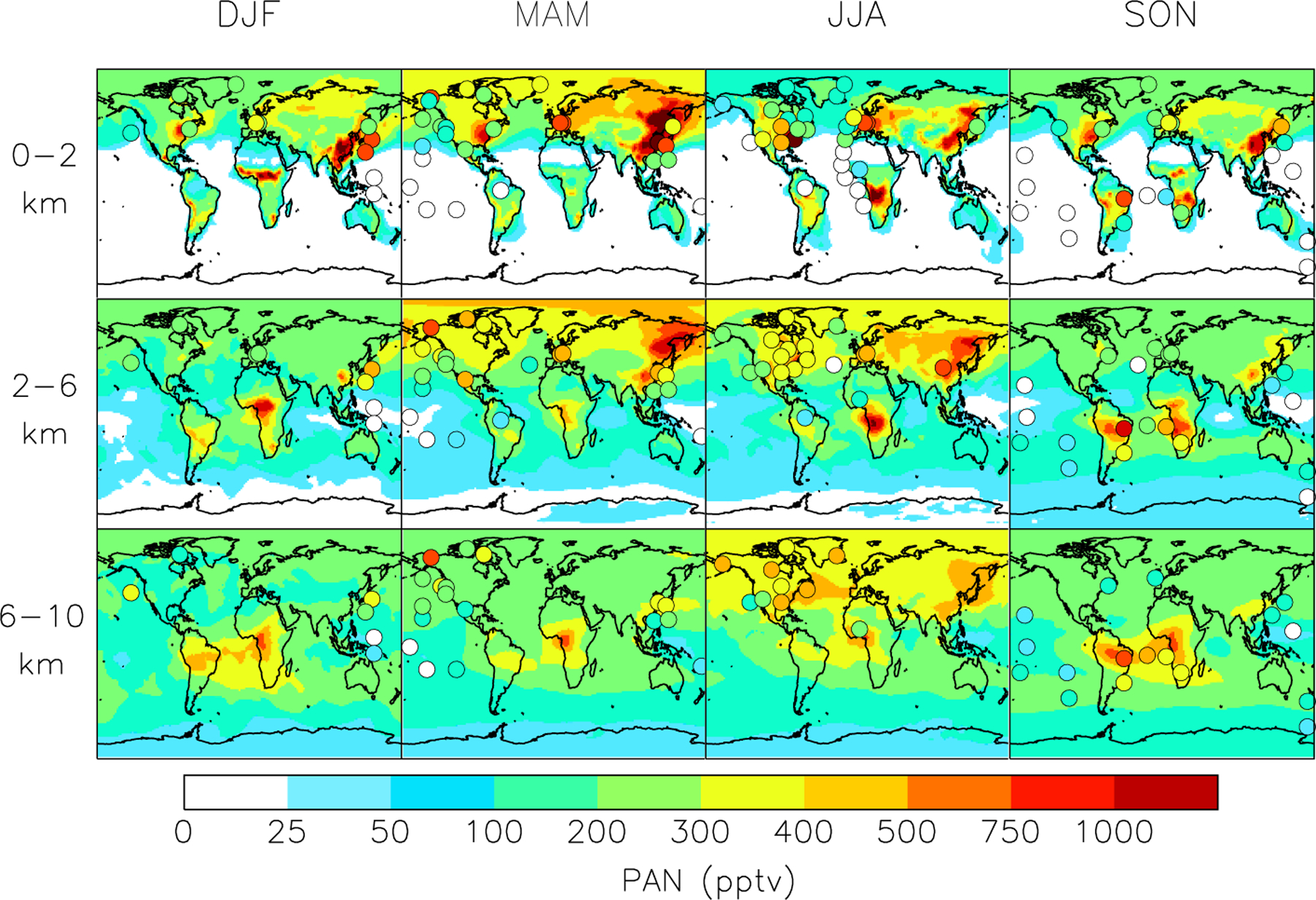
Global mean distribution of PAN for different seasons and altitude ranges. Model results for 2008 (background solid contours) are compared to observations from Table 2 for all years (filled circles). Aircraft observations are averaged vertically and horizontally over the coherent regions of Fig. 2.
Fig. 3.
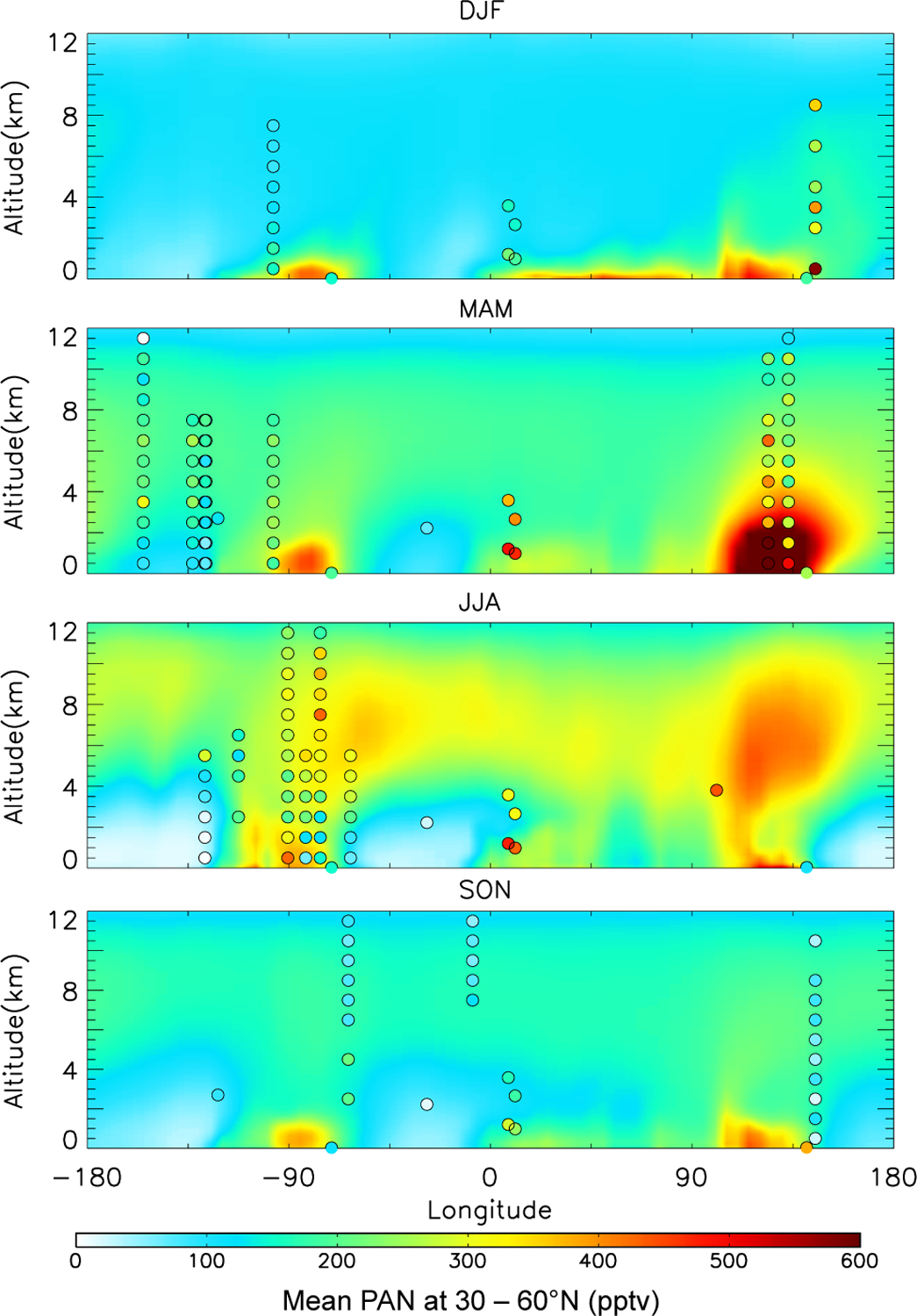
Longitudinal cross-section of seasonal mean PAN concentrations at northern midlatitudes (30–60° N) as a function of altitude. Model results for 2008 (background solid contours) are compared to observations from many years in Table 2 (filled circles). Circles are placed at the mean longitude of the coherent regions (Fig. 2) that fall between (30–60° N).
Figure 2 indicates that spring and summer Northern Hemisphere average PAN abundances below 6 km are comparable over polluted continental regions. The Northern Hemisphere springtime maximum, previously attributed to photochemical production at a time when PAN has a long thermal lifetime (Penkett and Brice, 1986; Brice et al., 1988), is primarily a feature of remote air. Long-term PAN measurements from the Hohenpeissenberg and Schauinsland European mountaintop observatories, both primarily within the atmospheric boundary layer, show either spring or summer maxima depending on the year (Supplement Fig. 2). Pandey Deolal et al. (2013) found that the PAN spring maximum at Jungfraujoch is mainly attributable to air masses advected from the polluted European boundary layer, and PAN formation in the free troposphere does not play a dominant role. Both the model and surface observations indicate that the springtime maximum is pronounced over the Arctic, and this has previously been attributed to transport of northern midlatitudes pollution (Moxim et al., 1996). We find that springtime fires in Russia and China also contribute to this feature, and this is discussed later in the context of our sensitivity simulations.
Successful simulation of PAN in Asian outflow is contingent on the inclusion of emissions of aromatic species. These account for 30% of the PAN in that region in the model. Even with the addition of aromatics, the model is biased low for this region. This could suggest missing NMVOC emissions in China, as suggested by Fu et al. (2007), or unrealistically low PA radical yields from aromatics in the chemical scheme. The model largely reproduces the average vertical profiles observed during TRACE-P (see vertical profiles in Supplement Fig. 1), but these were collected in 2001 and the model output is for 2008. Chinese NOx and NMVOCs emissions have increased by more than 55 and 29% over this period, respectively (Zhang et al., 2007b, 2009).
PAN is also sensitive to the parameterization of the uptake of the hydroperoxyl radical (HO2) by aerosols. Recent work (Mao et al., 2013a) suggests that the reactive uptake of HO2 is a much more efficient sink of HOx than previously thought or than is implemented here (Thornton et al., 2008). We tested the impact of more efficient uptake of HO2 by aerosols on PAN by setting the reactive uptake coefficient of HO2 to 1 and eliminating conversion of HO2 to H2O2 on aerosols. We found that the faster uptake of HO2 drastically reduced (50%) springtime PAN over eastern Asia. The faster uptake produces springtime PAN outflow in the model that is inconsistent with observations in that region, and would imply a large missing source of PAN.
Though the differences are smaller, PAN observations from European mountaintop sites also suggest missing PAN sources there. These PAN observations have not been used to justify emissions changes as observations from both Zugspitze (2658 m) and Jungfraujoch (3580 m) reflect terrain-induced injections of PAN-rich boundary layer air (Zanis et al., 2003, 2007; Zellweger et al., 2000; Carpenter et al., 2000; Pandey Deolal et al., 2013), and this transport scale is not captured in the model. However, Figure 3 indicates that the observations are also higher than the model output below the altitude of the measurements.
In Northern Hemisphere summer, both the model and observations show a strong contrast between high concentrations over source continents and adjacent oceans (Fig. 3), reflecting the short lifetime of PAN against thermal decomposition. PAN concentrations in the model are generally higher aloft, consistent with INTEX-A aircraft observations over the eastern US (90° W–45° W, panel 3, Fig. 3) and measurements from the Azores (Val Martin et al., 2008), reflecting the longer PAN lifetime. The INTEX-A observations indicate that PAN mixing ratios begin to decrease with altitude above 8 km over the northeastern US and the western Atlantic, but not over the southeastern US where lightning and convection support PAN production aloft (Hudman et al., 2007).
The lowest three panels of Fig. 3 show that outside of winter months there is a reservoir of 200–400 pptv PAN between 5 and 8 km over northern midlatitudes. A similar PAN reservoir aloft has also been observed over the Arctic during aircraft campaigns in spring and summer (Singh et al., 1994). PAN can be 80–90% of total NOy in the cold Arctic atmosphere (Atlas et al., 2003; Jaffe et al., 1997; Bottenheim et al., 1986). Liang et al. (2011) note that the 2008 ARCTAS PAN observations are not notably different from either the 1988 ABLE or 2000 TOPSE observations, despite dramatic changes to NOx emissions in the major anthropogenic source regions. In the upper troposphere, Northern Hemisphere PAN mixing ratios peak in summer, with contributions from anthropogenic sources, biomass burning and lightning. This summertime upper-tropospheric maximum is consistent with MIPAS retrievals for 300–150 hPa which indicate the highest Northern Hemisphere PAN concentrations in August and the lowest PAN from October to January (Moore and Remedios, 2010).
The Polarstern Cruise data from Germany to South Africa in summer reveal a sharp meridional gradient, with mixing ratios dropping below the detection limit (25 pptv) outside northern midlatitudes (Fig. 2) (Jacobi et al., 1999). The meridional gradient is much less defined in the free troposphere, reflecting biogenic and fire contributions in the southern tropics with efficient convective lofting (Fig. 2).
We see from the SON seasonal mean plots in Fig. 2 that the Southern Hemisphere features a spring PAN maximum in the upper troposphere, similar to the remote northern extratropics. Moore and Remedios (2010) observed a spring PAN maximum in the upper troposphere at 0–35° S from MIPAS retrievals and attributed it to seasonal biomass burning over central Africa. Moxim et al. (1999) also simulated the Southern Hemisphere springtime free-tropospheric PAN maximum, but suggested that it is driven by convective transport rapidly mixing PAN upward from continental surface production regions. As discussed below, our model suggests that much of the PAN in the austral free troposphere is due to continental convective injection of biogenic NMVOCs together with the lightning NOx source.
4. Contributions of different NMVOCs to PAN formation
PAN depends on NMVOCs and NOx in nonlinear ways. To diagnose this dependence and identify the most critical precursor, we conducted two sensitivity studies where NOx and NMVOC emissions were separately reduced by 20% across all sectors. The results are presented in Fig. 4. We see that PAN concentration depends in general more strongly on NMVOC than NOx emissions. Exceptions are fire-dominated regions at northern high latitudes, reflecting the very low NOx / NMVOCs emission ratio from fires. This result is also partially an artifact of partitioning 40% of GFED fire NOx emissions directly to PAN. A remarkable result is that PAN responds supra-linearly to NMVOC emissions in many locations, with the strongest effect over the North Pacific in spring and over the Arctic in summer. In both of these regions PAN is a principal source of NOx (Singh et al., 1992; Zhang et al., 2008), so that reducing PAN causes decreases in O3, in turn decreasing the [NO2] / [NO] ratio and thus reducing the effective lifetime of PAN. This chemical feedback amplifies the sensitivity of PAN to NMVOC emission changes. Another chemical feedback in source regions is that reducing NMVOC emissions increases the concentration of OH and hence the conversion of NOx to nitric acid.
Fig. 4.
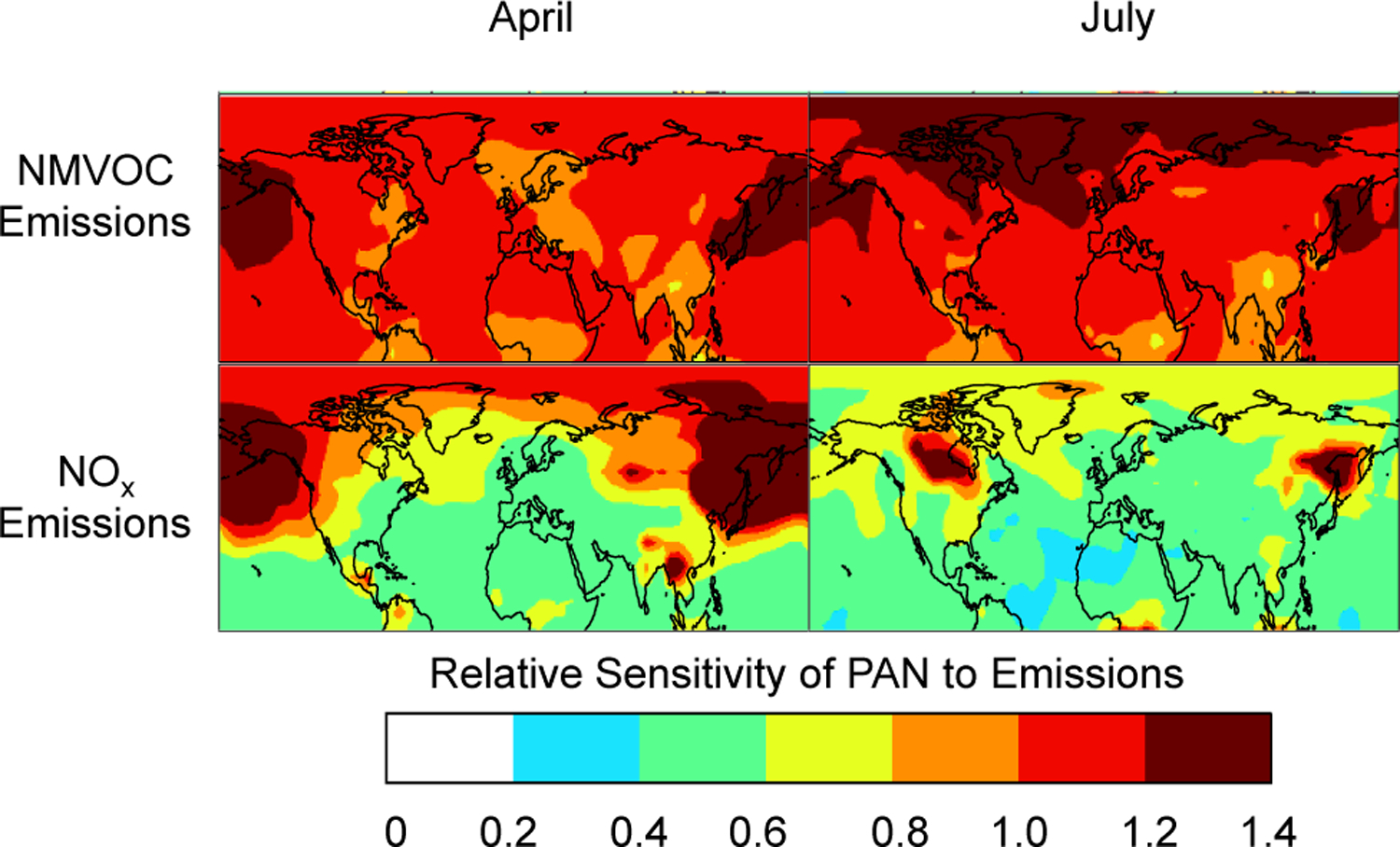
Relative sensitivity of total column PAN concentrations to emissions of NOx and NMVOCs in April and July. The sensitivity is diagnosed as ∆PAN / ∆E, where ∆PAN is the change in monthly mean PAN column concentrations resulting from a 20% decrease ∆E in global emissions of either NMVOCs (top) or NOx (bottom), including all sources and sustained year-round. Zero indicates no sensitivity, while one indicates 1 : 1 sensitivity.
In order to understand the contributions of different NMVOC precursors to PAN formation, we conducted 14 sensitivity simulations where the emissions of each precursor in the leftmost column of Table 1 were turned off individually. The change in the total burden of PAN was compared to a standard simulation with all emissions switched on. In the case of isoprene where the effect is large, we reduced emissions by 20% (and multiplied the change by 5) in order to minimize nonlinear effects. Figure 5 presents a schematic of the relative contributions of individual NMVOCs to global PAN formation through the major carbonyl species (acetaldehyde, acetone, methylglyoxal) serving as precursors of PAN (Reactions (R2)–(R4)). The absolute contributions are in Table 1. Anthropogenic, biogenic and biomass burning emissions make significant contributions to all three of the most important immediate PAN precursors (acetaldehyde, acetone, and methylglyoxal). We track PA radical formation via four different chemical pathways, from acetaldehyde, acetone, methylglyoxal and all other intermediate species. The bottom pie chart in Fig. 5 summarizes the relative importance of these four pathways for global annual total PA radical production.
Fig. 5.
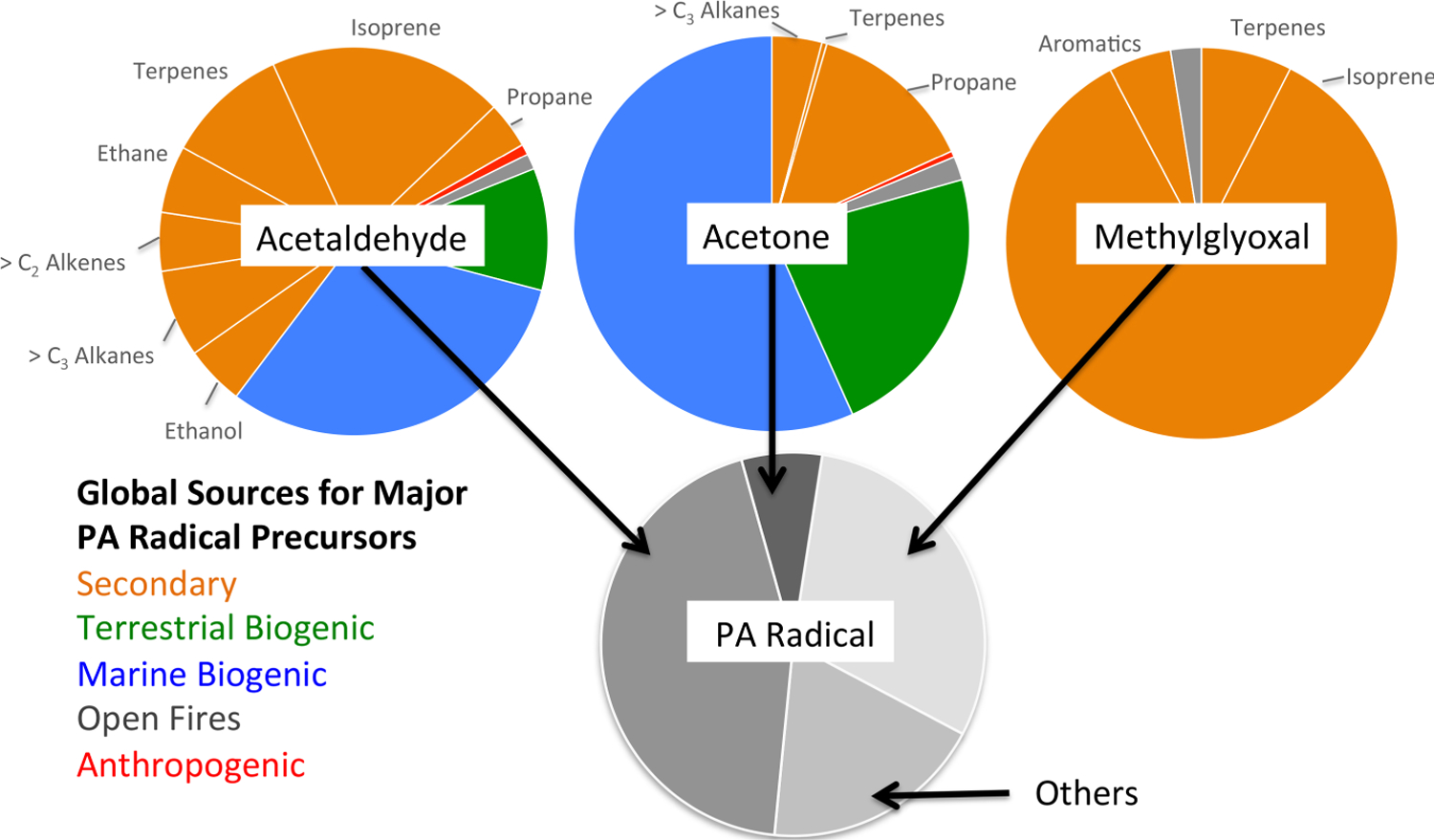
Global contributions of individual NMVOCs to PAN formation, expressed as the relative contributions to the major carbonyl species producing the peroxyacetyl radical (PA), and from there the relative contributions of the carbonyl species to global PA production. Values are from Table 1. The geographical and vertical distribution of total PA radical production is given in Fig. 6.
Figure 6 summarizes the geographical distribution of annual total PA radical production for the lower, mid- and upper troposphere. PA production is strongest in NMVOC source regions, propagating to the free troposphere in the tropics through deep convection. The patterns in Fig. 6 reflect the dominant sources and lifetime for each PA radical precursor: mean lifetimes 1–2 h for methylglyoxal, 0.8 days for acetaldehyde and 14 days for acetone. The bottom row of Fig. 6 shows total PA radical production from other pathways, mainly via isoprene and monoterpene oxidation intermediates, including methyl vinyl ketone and methacrolein. These latter species contribute to PA radical formation predominantly via photolysis. We traced PA radical formation via these species together with all other intermediates.
Fig. 6.
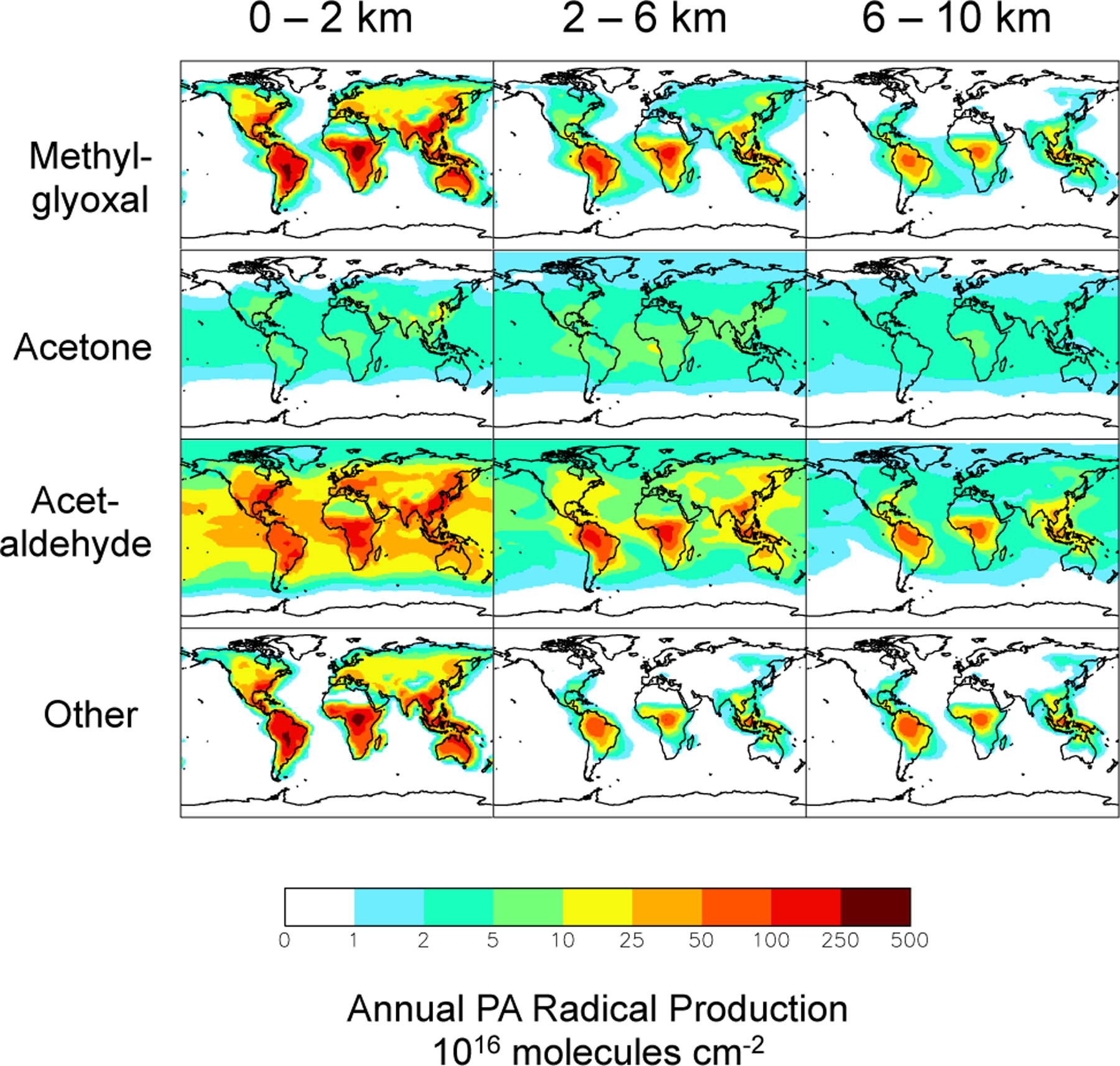
Annual total PA radical production for three altitude ranges contributed by the immediate precursors methylglyoxal, acetone, and acetaldehyde. The other precursors include a number of species produced in the oxidation of isoprene.
Figures 5 and 6 reveal that acetaldehyde is the most important PA radical precursor globally, responsible for ~40% of total PA radical production at all altitudes. Photochemical production is the dominant source of acetaldehyde, with large contributions from both biogenic and anthropogenic primary emissions (Fig. 5). There is also PA production from acetaldehyde in the marine boundary layer, partially reflecting the ocean acetaldehyde source (Millet et al., 2010). The alkanes, > C2 alkenes and ethanol all have high molar yields for acetaldehyde (Table 1). Though most originate over continents, the lifetimes of the primary precursors of acetaldehyde range from hours (isoprene) to months (ethane). Thus there is significant production of the PA radical from acetaldehyde at all altitudes over both continental regions and the downwind oceans (Millet et al., 2010).
Based on global simulations with and without acetone, Singh et al. (1995) estimated that up to 50% of observed PAN in the mid–upper troposphere could be formed from acetone. However, they assumed a photolysis rate for acetone now known to be too high. Using a similar approach and the acetone budget from Fischer et al. (2012), we find that the contribution of acetone to PAN is 25% in the upper troposphere over the Northern Hemisphere during summer and less under other conditions. Acetone is the most important PA precursor only in the most remote regions of the upper troposphere.
Isoprene and monoterpenes are also important precursors for PAN formation through methylglyoxal and other intermediates. Due to relatively short lifetimes, their role is largest in continental boundary layers (Fig. 6). von Kuhlmann et al. (2004) showed that PAN formation in models is highly sensitive to the treatment of isoprene chemistry, and there have been a number of more recent advances regarding the oxidation chemistry of isoprene (Lelieveld et al., 2008; Paulot et al., 2009b; Peeters et al., 2009; Mao et al., 2012). There are also ongoing efforts to determine appropriate yields for methylglyoxal and other important intermediates under the high-NOx conditions most relevant for PAN formation (Galloway et al., 2011). Implementation of the Paulot et al. (2009a, b) oxidation scheme in GEOS-Chem improves the simulation of summertime observations over the southeastern US (Mao et al., 2013b). It also substantially increases surface PAN mixing ratios over the Amazon and central Africa, where there are very few observational data (Angelo, 2012). In these regions surface PAN increases by 100–300 pptv with the Paulot et al. (2009a, b) scheme, but the impact is more modest above the boundary layer, generally less than 50 pptv. In the model, most of the free-tropospheric PAN in convective regions is produced above the boundary layer.
5. Contributions from different source types to PAN formation
Figure 7 presents the sensitivity of PAN concentrations to different emission types, as diagnosed by the relative decrease in a sensitivity simulation with that emission type shut off. Contributions do not add up to 100% because of nonlinearity.
Fig. 7.
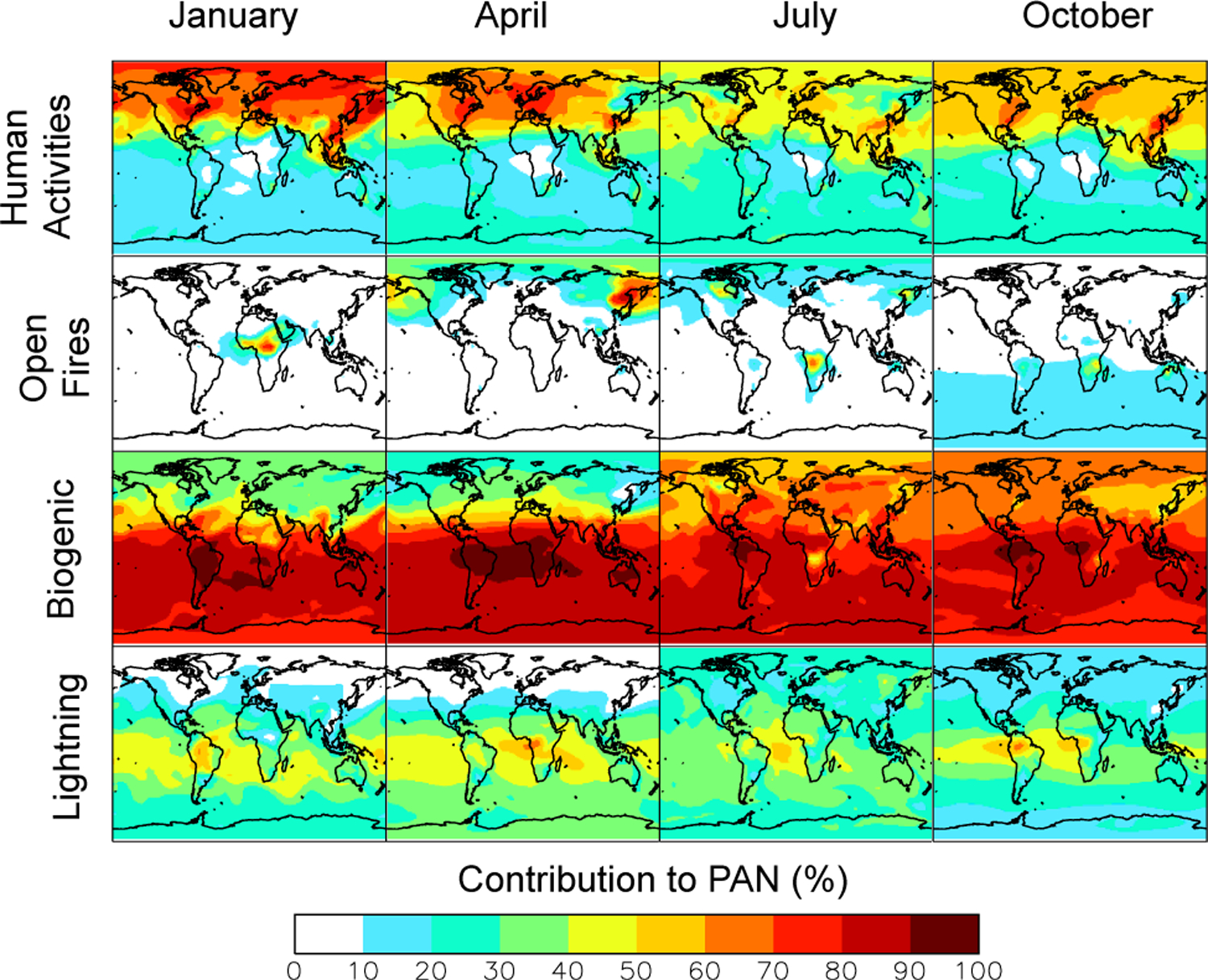
Sensitivity of PAN to different emission types. Results are shown as relative decreases of monthly mean total PAN columns in sensitivity simulations with individual emission types shut off. Biogenic signifies NMVOCs only.
During Northern Hemisphere spring, shutting off anthropogenic emissions decreases the integrated PAN burden by ~50%. Alkanes are the most important class of anthropogenic NMVOC precursors for PAN in northern midlatitudes. Their role is more important in spring when NMVOC emissions from the biosphere are smaller. In spring, the time of the surface PAN maximum, biogenic and anthropogenic NMVOCs species each support ~50% of the PAN burden.
Though most biomass burning occurs primarily in the tropics, the effect of fires on PAN appears to be largest at northern latitudes. Shutting off emissions from springtime fires located in Russia and China decreases the hemispheric burden by ~25%, but the decrease in PAN mixing ratios is 30–40% at high latitudes. These springtime fires, which exhibit strong variability in magnitude and location, contribute to the observed spring PAN maximum. Russian fires likely accentuated this feature in April 2008, an unusually strong fire season (Vivchar, 2010; Warneke et al., 2009, 2010). PAN in fire plumes from the Russian Federation was shown to support efficient O3 production over the northeast Pacific during April 2008 (Fischer et al., 2010). Enhancements in O3 of up to 20 ppbv were observed during this time from Alaska to California (Oltmans et al., 2010). Spring 2008 was an extreme burning year, but Macdonald et al. (2011) also attribute elevated monthly mean O3 concentrations at Whistler Mountain, BC, in fall 2002 and spring 2003 to fires in the Russian Federation.
As stated earlier, the treatment of PAN formation in fires plays an important role in determining the global impact of this PAN source. Past model studies have found that reproducing observed free-tropospheric CO and O3 downwind from boreal fires requires injecting a fraction of the emissions above the boundary layer (Leung et al., 2007; Turquety et al., 2007; Generoso et al., 2007; Colarco et al., 2004). Tereszchuk et al. (2013) show that PAN in the upper troposphere at high latitudes is mainly from large boreal fires in summer. Emitting a fraction of the smoke above the boundary layer is an important model update that improves the simulation of the 2–6 km PAN reservoir at high latitudes. The fraction of NOx in the springtime Russian fires that is immediately partitioned to PAN also has a large impact on springtime PAN over high latitudes because PAN has a long lifetime during this season. The combination of model updates chosen here best reproduces the evolution of the springtime PAN profile as observed during TOPSE (Wang et al., 2003). Specifically, PAN remains relatively constant (150–200 pptv) with altitude in February and March, and the 2–6 km PAN reservoir forms in April. Springtime PAN in the model is acutely sensitive to the amount of NOx that is immediately partitioned to PAN in fires. Given that O3 production in the Arctic lower troposphere is sensitive to the abundance of PAN (Walker et al., 2012; Beine et al., 1997), more work is warranted to determine the best way to incorporate the chem istry that rapidly produces PAN in fires.
We find that biogenic species drive PAN production in summer and fall. From June to October, shutting off biogenic emissions decreases the Northern Hemisphere integrated PAN burden by ~75%. In summer, the contribution to PAN from other biogenic NMVOCs (terpenes, acetone, acetaldehyde, ethanol and higher alkenes) is ~50% that of isoprene. Consistent with our analysis, Roberts et al. (2006) estimated that the isoprene contribution to PAN formation is 1.6 to 4 times larger than the anthropogenic NMVOC contribution in the northeastern US in summer.
The austral spring mid–upper-tropospheric PAN maximum (> 400 pptv) spanning the Atlantic (Fig. 2) is also apparent in MIPAS PAN retrievals (Glatthor et al., 2007; Moore and Remedios, 2010; Wiegele et al., 2012). Figure 7 shows that this feature is more sensitive to emissions of NOx from lightning than emissions from either biomass burning or anthropogenic sources. Biomass burning takes place from July to October in the part of Africa located in the Southern Hemisphere. Singh et al. (1996a) found that PAN correlated with tracers of biomass combustion in the eastern South Atlantic in the lower and middle troposphere, but not in the upper troposphere. To explain observed NOx at higher altitudes, they had to invoke a large contribution from lightning (Smyth et al., 1996). Our simulation reproduces the TRACE-A vertical PAN profiles for the South Atlantic (Supplement Fig. 1, panels 43–45) and the correlation between PAN and CO (not shown). We find that fires are responsible for approximately 30% of the PAN over the tropical Atlantic between 2 and 4 km. Above 6 km, the contribution from fires is small. In the upper troposphere, the oxidation of biogenic NMVOCs (lifted by convection (Murphy et al., 2010; Bechara et al., 2010; Warneke et al., 2001)) in the presence of lightning NOx is a large source of PAN (Tie et al., 2001; Labrador et al., 2005). Compared to the previous version of GEOS-Chem, the sensitivity of upper-tropospheric PAN to lightning is reduced by 30 %. We attribute this change to increased OH in the boundary layer through the use of the Paulot et al. (2009a, b) isoprene scheme, which reduces the amount of NMVOC injected into the free troposphere (Paulot et al., 2012). Boundary layer and upper-tropospheric chemistry in the tropics are tightly coupled (Paulot et al., 2012). Hence the simulation of upper-tropospheric PAN is sensitive to the representation of boundary layer chemistry, which remains very uncertain (Hewitt et al., 2010).
6. Conclusions
We utilized a worldwide collection of observations to improve a global simulation of PAN in the GEOS-Chem model. This new simulation, which includes an improved representation of numerous NMVOCs and a different treatment of biomass burning emissions, affords the opportunity to understand the factors driving the PAN distribution on the global scale.
We find that PAN is generally more sensitive to NMVOC emissions than NOx emissions. In many regions of the atmosphere, changes to NMVOC emissions produce a supra-linear change in PAN through feedbacks to remote NOx and O3 budgets. A different mixture of NMVOCs supports PAN formation in each region and season. Considerable improvement of the PAN simulation for the Asian outflow region is achieved by including aromatics. Our results stress the need for global CTMs, which can yield different results for PAN (Singh et al., 2007), to include and evaluate budgets for many NMVOCs that are routinely ig nored.
In order to reproduce the observed PAN reservoir at 3–6 km over high northern latitudes, we have changed the way emissions from fires are incorporated into the model. We increased the simulated PAN reservoir over high latitudes by (1) adding biomass burning emissions of shorter-lived NMVOCs (monoterpenes, aromatics), (2) emitting a fraction of the biomass burning NOx directly as PAN (Alvarado et al., 2010), (3) emitting a portion of the smoke above the boundary layer, (4) updating the emission factors for NMVOCs and NOx (Akagi et al., 2011), and (5) increasing emissions to account for undetected small fires at high latitudes (Kaiser et al., 2012). We find that PAN over the Arctic is very sensitive to fires, and particularly sensitive to the amount of NOx that is immediately partitioned to PAN in fires and to the altitude of the emissions. Given that O3 production in the Arctic lower troposphere is very sensitive to NOx abundance (Stroud et al., 2004; Walker et al., 2012), more work is warranted to determine the best way to incorporate the plume chemistry that rapidly produces PAN into CTMs.
The principal carbonyl precursors of PAN are acetaldehyde (44% of the global source), methylglyoxal (30%) and acetone (7%). Acetaldehyde is produced by a large suite of NMVOCs and also directly emitted. Methylglyoxal is mostly from isoprene. Isoprene oxidation products, other than methylglyoxal, are also significant. With updated (lower) photolysis yields, acetone is a substantially less important pathway for PAN formation than previously thought (Singh et al., 1995).
Isoprene accounts for 37% of the global PAN burden. Many other NMVOC emissions contribute to the balance, with no single species contributing more than 10% (Table 1). At Northern Hemisphere midlatitudes, alkanes contribute to a third of PAN formation during the springtime maximum.
A springtime upper troposphere PAN maximum across the tropical Atlantic is the major feature of the Southern Hemisphere PAN distribution. Lightning is the most important NOx source for PAN formation in this region of the atmosphere. A cascade of isoprene oxidation products, delivered to the upper troposphere by deep convection, provides the PA radical source. This finding is sensitive to the description of boundary layer chemistry under low-NOx conditions.
The work presented here has increased confidence in our ability to simulate the observed distribution of PAN within the GEOS-Chem CTM. In a follow-up paper we will examine the importance of PAN in affecting global tropospheric O3 and OH, and the implications for intercontinental transport of pollution, the oxidizing power of the atmosphere, and climate forcing.
Supplementary Material
Acknowledgements.
This work was supported by the NASA Atmospheric Composition Modeling and Analysis Program. Support for E. V. Fischer was provided by the NOAA Climate and Global Change Postdoctoral Fellowship Program, administered by UCAR, and by a Harvard University Center for the Environment Postdoctoral Fellowship. The contribution of PAN data from the GAW Global Station Hohenbeissenberg by Stefan Gilge, German Meteorological Service, is greatly acknowledged. The contribution of PAN data from the Jungfraujoch Mountain Site by Christoph Zellweger (EMPA) is greatly acknowledged. We thank Hiroshi Tanimoto for providing the data from Rishiri, Japan. Pico PAN data were collected under the leadership of Richard Honrath with funding from the National Science Foundation grant ATM-0720955. Funding for the analysis of the Pico PAN measurements by Katja Dzepina, Jim Roberts and Lynn Mazzoleni was provided by the National Science Foundation through grant AGS-1110059. We also appreciate the contribution of unpublished PAN data from the Thompson Farm AIRMAP Site by Ryan Chartier. Finally, we thank Martin Steinbacher and Jim Roberts for helpful comments on the manuscript.
Footnotes
Supplementary material related to this article is available online at http://www.atmos-chem-phys.net/14/2679/2014/acp-14-2679-2014-supplement.pdf.
References
- Akagi SK, Yokelson RJ, Wiedinmyer C, Alvarado MJ, Reid JS, Karl T, Crounse JD, and Wennberg PO: Emission factors for open and domestic biomass burning for use in atmospheric models, Atmos. Chem. Phys, 11, 4039–4072, 10.5194/acp-11-4039-2011, 2011. [DOI] [Google Scholar]
- Alvarado MJ, Logan JA, Mao J, Apel E, Riemer D, Blake D, Cohen RC, Min KE, Perring AE, Browne EC, Wooldridge PJ, Diskin GS, Sachse GW, Fuelberg H, Sessions WR, Harrigan DL, Huey G, Liao J, Case-Hanks A, Jimenez JL, Cubison MJ, Vay SA, Weinheimer AJ, Knapp DJ, Montzka DD, Flocke FM, Pollack IB, Wennberg PO, Kurten A, Crounse J, Clair JMS, Wisthaler A, Mikoviny T, Yantosca RM, Carouge CC, and Le Sager P: Nitrogen oxides and PAN in plumes from boreal fires during ARCTAS-B and their impact on ozone: an integrated analysis of aircraft and satellite observations, Atmos. Chem. Phys, 10, 9739–9760, 10.5194/acp-10-9739-2010, 2010. [DOI] [Google Scholar]
- Angelo C: Amazon fire analysis hits new heights, Nature News, 10.1038/nature.2012.11467, 2012. [DOI]
- Atkinson R and Arey J: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ, 37 Supplement No. 2, S197–S219, 2003. [Google Scholar]
- Atlas EL, Ridley BA, and Cantrell CA: The Tropospheric Ozone Production about the Spring Equinox (TOPSE) Experiment: Introduction, J. Geophys. Res, 108, 8353, 10.1029/2002jd003172, 2003. [DOI] [Google Scholar]
- Balzani Loov JM, Henne S, Legreid G, Staehelin J, Reimann S, Prevut ASH, Steinbacher M, and Vollmer MK: Estimation of background concentrations of trace gases at the Swiss Alpine site Jungfraujoch (3580m asl), J. Geophys. Res, 113, D22305, 10.1029/2007jd009751, 2008. [DOI] [Google Scholar]
- Barletta B, Meinardi S, Simpson IJ, Sherwood Rowland F, Chan C-Y, Wang X, Zou S, Chan LY, and Blake DR: Ambient halocarbon mixing ratios in 45 Chinese cities, Atmos. Environ, 40, 7706–7719, 10.1016/j.atmosenv.2006.08.039, 2006. [DOI] [Google Scholar]
- Bechara J, Borbon A, Jambert C, Colomb A, and Perros PE: Evidence of the impact of deep convection on reactive Volatile Organic Compounds in the upper tropical troposphere during the AMMA experiment in West Africa, Atmos. Chem. Phys, 10, 10321–10334, 10.5194/acp-10-10321-2010, 2010. [DOI] [Google Scholar]
- Beine H, Jaffe D, Herring J, Kelley J, Krognes T, and Stordal F: High-Latitude Springtime Photochemistry. Part I: NOx, PAN and Ozone Relationships, J. Atmos. Chem, 27, 127–153, 10.1023/a:1005869900567, 1997. [DOI] [Google Scholar]
- Beine HJ and Krognes T: The seasonal cycle of peroxyacetyl nitrate (PAN) in the European Arctic, Atmos. Environ, 34, 933–940, 10.1016/s1352-2310(99)00288-5, 2000. [DOI] [Google Scholar]
- Beine HJ, Jaffe DA, Blake DR, Atlas E, and Harris J: Measurements of PAN, alkyl nitrates, ozone, and hydrocarbons during spring in interior Alaska, J. Geophys. Res, 101, 12613–12619, 10.1029/96jd00342, 1996. [DOI] [Google Scholar]
- Bertram TH, Perring AE, Wooldridge PJ, Crounse JD, Kwan AJ, Wennberg PO, Scheuer E, Dibb J, Avery M, Sachse G, Vay SA, Crawford JH, McNaughton CS, Clarke A, Pickering KE, Fuelberg H, Huey G, Blake DR, Singh HB, Hall SR, Shetter RE, Fried A, Heikes BG, and Cohen RC: Direct Measurements of the Convective Recycling of the Upper Troposphere, Science, 315, 816–820, 10.1126/science.1134548, 2007. [DOI] [PubMed] [Google Scholar]
- Bertram TH, Perring AE, Wooldridge PJ, Dibb J, Avery MA, and Cohen RC: On the export of reactive nitrogen from Asia: NOx partitioning and effects on ozone, Atmos. Chem. Phys, 13, 4617–4630, 10.5194/acp-13-4617-2013, 2013. [DOI] [Google Scholar]
- Bey I, Jacob DJ, Yantosca RM, Logan JA, Field BD, Fiore AM, Li Q, Liu HY, Mickley LJ, and Schultz MG: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res, 106, 23073–23095, 10.1029/2001jd000807, 2001. [DOI] [Google Scholar]
- Bloss C, Wagner V, Jenkin ME, Volkamer R, Bloss WJ, Lee JD, Heard DE, Wirtz K, Martin-Reviejo M, Rea G, Wenger JC, and Pilling MJ: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys, 5, 641–664, 10.5194/acp-5-641-2005, 2005. [DOI] [Google Scholar]
- Bottenheim JW, Gallant AG, and Brice KA: Measurements of NOY species and O3 at 82° N latitude, Geophys. Res. Lett, 13, 113–116, 10.1029/GL013i002p00113, 1986. [DOI] [Google Scholar]
- Bottenheim JW, Sirois A, Brice KA, and Gallant AJ: Five years of continuous observations of PAN and ozone at a rural location in eastern Canada, J. Geophys. Res, 99, 5333–5352, 10.1029/93jd02716, 1994. [DOI] [Google Scholar]
- Brice KA, Bottenheim JW, Anlauf KG, and Wiebe HA: Long-term measurements of atmospheric peroxyacetylnitrate (PAN) at rural sites in Ontario and Nova Scotia; seasonal variations and long-range transport, Tellus B, 40B, 408–425. 1988. [Google Scholar]
- Bridier I, Caralp F, Loirat H, Lesclaux R, Veyret B, Becker KH, Reimer A, and Zabel F: Kinetic and theoretical studies of the reactions of acetylperoxy + nitrogen dioxide + M <–> acetyl peroxynitrate + M between 248 and 393 K and between 30 and 760 torr, J. Phys. Chem, 95, 3594–3600, 10.1021/j100162a031, 1991. [DOI] [Google Scholar]
- Carpenter LJ, Green TJ, Mills GP, Bauguitte S, Penkett SA, Zanis P, Schuepbach E, Schmidbauer N, Monks PS, and Zellweger C: Oxidized nitrogen and ozone production efficiencies in the springtime free troposphere over the Alps, J. Geophys. Res, 105, 14547–14559, 10.1029/2000jd900002, 2000. [DOI] [Google Scholar]
- Colarco PR, Schoeberl MR, Doddridge BG, Marufu LT, Torres O, and Welton EJ: Transport of smoke from Canadian forest fires to the surface near Washington, D.C.: Injection height, entrainment, and optical properties, J. Geophys. Res, 109, D06203, 10.1029/2003jd004248, 2004. [DOI] [Google Scholar]
- Dassau TM, Shepson PB, Bottenheim JW, and Ford KM: Peroxyacetyl nitrate photochemistry and interactions with the Arctic surface, J. Geophys. Res, 109, D18302, 10.1029/2004jd004562, 2004. [DOI] [Google Scholar]
- Fischer EV, Jaffe DA, Reidmiller DR, and Jaegle L: Meteorological controls on observed peroxyacetyl nitrate at Mount Bachelor during the spring of 2008, J. Geophys. Res, 115, D03302, 10.1029/2009jd012776, 2010. [DOI] [Google Scholar]
- Fischer EV, Jaffe DA, and Weatherhead EC: Free tropospheric peroxyacetyl nitrate (PAN) and ozone at Mount Bachelor: causes of variability and timescale for trend detection, Atmos. Chem. Phys 11, 5641–5654, 10.5194/acp-11-5641-2011, 2011. [DOI] [Google Scholar]
- Fischer EV, Jacob DJ, Millet DB, Yantosca RM, and Mao J: The role of the ocean in the global atmospheric budget of acetone, Geophys. Res. Lett, 39, L01807, 10.1029/2011gl050086, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flocke F, Weinheimer A, Swanson A, Roberts J, Schmitt R, and Shertz S: On the Measurement of PANs by Gas Chromatography and Electron Capture Detection, J. Atmos. Chem, 52, 19–43, 10.1007/s10874-005-6772-0, 2005. [DOI] [Google Scholar]
- Ford KM, Campbell BM, Shepson PB, Bertman SB, Honrath RE, Peterson M, and Dibb JE: Studies of Peroxyacetyl nitrate (PAN) and its interaction with the snowpack at Summit, Greenland, J. Geophys. Res, 107, 4102, 10.1029/2001jd000547, 2002. [DOI] [Google Scholar]
- Fu T-M, Jacob DJ, Palmer PI, Chance K, Wang YX, Barletta B, Blake DR, Stanton JC, and Pilling MJ: Space-based formaldehyde measurements as constraints on volatile organic compound emissions in east and south Asia and implications for ozone, J. Geophys. Res, 112, D06312, 10.1029/2006jd007853, 2007. [DOI] [Google Scholar]
- Fu T-M, Jacob DJ, Wittrock F, Burrows JP, Vrekoussis M, and Henze DK: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res, 113, D15303, 10.1029/2007JD009505, 2008. [DOI] [Google Scholar]
- Galloway MM, Huisman AJ, Yee LD, Chan AWH, Loza CL, Seinfeld JH, and Keutsch FN: Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high-NOx conditions, Atmos. Chem. Phys 11, 10779–10790, 10.5194/acp-11-10779-2011, 2011. [DOI] [Google Scholar]
- Generoso S, Bey I, Attié J-L, and Bréon F-M: A satellite- and model-based assessment of the 2003 Russian fires: Impact on the Arctic region, J. Geophys. Res, 112, D15302, 10.1029/2006jd008344, 2007. [DOI] [Google Scholar]
- Glatthor N, von Clarmann T, Fischer H, Funke B, Grabowski U, Hopfner M, Kellmann S, Kiefer M, Linden A, Milz M, Steck T, and Stiller GP: Global peroxyacetyl nitrate (PAN) retrieval in the upper troposphere from limb emission spectra of the Michelson Interferometer for Passive Atmospheric Sounding (MIPAS), Atmos. Chem. Phys, 7, 2775–2787, 10.5194/acp-7-2775-2007, 2007. [DOI] [Google Scholar]
- Goliff WS, Stockwell WR, and Lawson CV: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ, 68, 174–185, 10.1016/j.atmosenv.2012.11.038, 2013. [DOI] [Google Scholar]
- Grosjean D, Grosjean E, and Williams EL: Thermal Decomposition of PAN, PPN and Vinyl-PAN, Air Waste, 44, 391–396, 10.1080/1073161x.1994.10467260, 1994. [DOI] [Google Scholar]
- Guenther A, Karl T, Harley P, Wiedinmyer C, Palmer PI, and Geron C: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys, 6, 3181–3210, 10.5194/acp-6-3181-2006, 2006. [DOI] [Google Scholar]
- Hewitt CN, Lee JD, MacKenzie AR, Barkley MP, Carslaw N, Carver GD, Chappell NA, Coe H, Collier C, Commane R, Davies F, Davison B, DiCarlo P, Di Marco CF, Dorsey JR, Edwards PM, Evans MJ, Fowler D, Furneaux KL, Gallagher M, Guenther A, Heard DE, Helfter C, Hopkins J, Ingham T, Irwin M, Jones C, Karunaharan A, Langford B, Lewis AC, Lim SF, MacDonald SM, Mahajan AS, Malpass S, McFiggans G, Mills G, Misztal P, Moller S, Monks PS, Nemitz E, Nicolas-Perea V, Oetjen H, Oram DE, Palmer PI, Phillips GJ, Pike R, Plane JMC, Pugh T, Pyle JA, Reeves CE, Robinson NH, Stewart D, Stone D, Whalley LK, and Yin X: Overview: oxidant and particle photochemical processes above a south-east Asian tropical rainforest (the OP3 project): introduction, rationale, location characteristics and tools, Atmos. Chem. Phys, 10, 169–199, 10.5194/acp-10-169-2010, 2010. [DOI] [Google Scholar]
- Horowitz LW, Liang J, Gardner GM, and Jacob DJ: Export of reactive nitrogen from North America during summer-time: Sensitivity to hydrocarbon chemistry, J. Geophys. Res, 103, 13451–13476, 10.1029/97jd03142, 1998. [DOI] [Google Scholar]
- Hudman RC, Jacob DJ, Cooper OR, Evans MJ, Heald CL, Park RJ, Fehsenfeld F, Flocke F, Holloway J, Hübler G, Kita K, Koike M, Kondo Y, Neuman A, Nowak J, Oltmans S, Parrish D, Roberts JM, and Ryerson T: Ozone production in transpacific Asian pollution plumes and implications for ozone air quality in California, J. Geophys. Res, 109, D23S10, 10.1029/2004jd004974, 2004. [DOI] [Google Scholar]
- Hudman RC, Jacob DJ, Turquety S, Leibensperger EM, Murray LT, Wu S, Gilliland AB, Avery M, Bertram TH, Brune W, Cohen RC, Dibb JE, Flocke FM, Fried A, Holloway J, Neuman JA, Orville R, Perring A, Ren X, Sachse GW, Singh HB, Swanson A, and Wooldridge PJ: Surface and lightning sources of nitrogen oxides over the United States: Magnitudes, chemical evolution, and outflow, J. Geophys. Res, 112, D12S05, 10.1029/2006jd007912, 2007. [DOI] [Google Scholar]
- Ito A, Sillman S, and Penner JE: Effects of additional nonmethane volatile organic compounds, organic nitrates, and direct emissions of oxygenated organic species on global tropospheric chemistry, J. Geophys. Res, 112, D06309, 10.1029/2005jd006556, 2007. [DOI] [Google Scholar]
- Jacob DJ: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ, 34, 2131–2159, 2000. [Google Scholar]
- Jacob DJ, Wofsy SC, Bakwin PS, Fan SM, Harriss RC, Talbot RW, Bradshaw JD, Sandholm ST, Singh HB, Browell EV, Gregory GL, Sachse GW, Shipham MC, Blake DR, and Fitzjarrald DR: Summertime photochemistry of the troposphere at high northern latitudes, J. Geophys. Res, 97, 16421–16431, 10.1029/91jd01968, 1992. [DOI] [Google Scholar]
- Jacobi HW, Weller R, Bluszcz T, and Schrems O: Latitudinal distribution of peroxyacetyl nitrate (PAN) over the Atlantic Ocean, J. Geophys. Res, 104, 26901–26912, 10.1029/1999jd900462, 1999. [DOI] [Google Scholar]
- Jaffe DA, Berntsen TK, and Isaksen ISA: A global three-dimensional chemical transport model 2. Nitrogen oxides and nonmethane hydrocarbon results, J. Geophys. Res, 102, 21281–21296, 10.1029/96jd03400, 1997. [DOI] [Google Scholar]
- Kaiser JW, Heil A, Andreae MO, Benedetti A, Chubarova N, Jones L, Morcrette JJ, Razinger M, Schultz MG, Suttie M, and van der Werf GR: Biomass burning emissions estimated with a global fire assimilation system based on observed fire radiative power, Biogeosci, 9, 527–554, 10.5194/bg-9-527-2012, 2012. [DOI] [Google Scholar]
- Kasibhatla PS, Levy H, and Moxim WJ: Global NOx, HNO3, PAN, and NOy distributions from fossil fuel combustion emissions: A model study, J. Geophys. Res, 98, 7165–7180, 10.1029/92jd02845, 1993. [DOI] [Google Scholar]
- Kotchenruther RA, Jaffe DA, and Jaeglé L: Ozone photochemistry and the role of peroxyacetyl nitrate in the springtime northeastern Pacific troposphere: Results from the Photochemical Ozone Budget of the Eastern North Pacific Atmosphere (PHOBEA) campaign, J. Geophys. Res, 106, 28731–28742, 10.1029/2000jd000060, 2001. [DOI] [Google Scholar]
- Kuhns H, Green M, and Etyemezian V: Big Bend Regional Aerosol and Visibility Observational (BRAVO) Study Emissions Inventory, Desert Research Institute, Las Vegas, NV, 2003. [Google Scholar]
- Labrador LJ, von Kuhlmann R, and Lawrence MG: The effects of lightning-produced NOx and its vertical distribution on atmospheric chemistry: sensitivity simulations with MATCH-MPIC, Atmos. Chem. Phys, 5, 1815–1834, 10.5194/acp-5-1815-2005, 2005. [DOI] [Google Scholar]
- Lelieveld J, Butler TM, Crowley JN, Dillon TJ, Fischer H, Ganzeveld L, Harder H, Lawrence MG, Martinez M, Taraborrelli D, and Williams J: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, 2008. [DOI] [PubMed] [Google Scholar]
- Leung F-YT, Logan JA, Park R, Hyer E, Kasischke E, Streets D, and Yurganov L: Impacts of enhanced biomass burning in the boreal forests in 1998 on tropospheric chemistry and the sensitivity of model results to the injection height of emissions, J. Geophys. Res, 112, D10313, 10.1029/2006jd008132, 2007. [DOI] [Google Scholar]
- Liang Q, Rodriguez JM, Douglass AR, Crawford JH, Olson JR, Apel E, Bian H, Blake DR, Brune W, Chin M, Colarco PR, da Silva A, Diskin GS, Duncan BN, Huey LG, Knapp DJ, Montzka DD, Nielsen JE, Pawson S, Riemer DD, Weinheimer AJ, and Wisthaler A: Reactive nitrogen, ozone and ozone production in the Arctic troposphere and the impact of stratosphere-troposphere exchange, Atmos. Chem. Phys, 11, 13181–13199, 10.5194/acp-11-13181-2011, 2011. [DOI] [Google Scholar]
- Liu Z, Wang Y, Gu D, Zhao C, Huey LG, Stickel R, Liao J, Shao M, Zhu T, Zeng L, Liu S-C, Chang C-C, Amoroso A, and Costabile F: Evidence of Reactive Aromatics As a Major Source of Peroxy Acetyl Nitrate over China, Environ. Sci. Tech, 44, 7017–7022, 10.1021/es1007966, 2010. [DOI] [PubMed] [Google Scholar]
- Lurmann FW, Lloyd AC, and Atkinson R: A Chemical Mechanism for Use in Long-Range Transport/Acid Deposition Computer Modeling, J. Geophys. Res, 91, 10905–10936, 10.1029/JD091iD10p10905, 1986. [DOI] [Google Scholar]
- Macdonald AM, Anlauf KG, Leaitch WR, Chan E, and Tarasick DW: Interannual variability of ozone and carbon monoxide at the Whistler high elevation site: 2002–2006, Atmos. Chem. Phys, 11, 11431–11446, 10.5194/acp-11-11431-2011, 2011. [DOI] [Google Scholar]
- Maloney JC, Fuelberg HE, Avery MA, Crawford JH, Blake DR, Heikes BG, Sachse GW, Sandholm ST, Singh H, and Talbot RW: Chemical characteristics of air from different source regions during the second Pacific Exploratory Mission in the Tropics (PEM-Tropics B), J. Geophys. Res, 106, 32609–32625, 10.1029/2001jd900100, 2001. [DOI] [Google Scholar]
- Mao J, Jacob DJ, Evans MJ, Olson JR, Ren X, Brune WH, Clair JMS, Crounse JD, Spencer KM, Beaver MR, Wennberg PO, Cubison MJ, Jimenez JL, Fried A, Weibring P, Walega JG, Hall SR, Weinheimer AJ, Cohen RC, Chen G, Crawford JH, McNaughton C, Clarke AD, Jaegle L, Fisher JA, Yantosca RM, Le Sager P, and Carouge C: Chemistry of hydrogen oxide radicals (HOx) in the Arctic troposphere in spring, Atmos. Chem. Phys, 10, 5823–5838, 10.5194/acp-10-5823-2010, 2010. [DOI] [Google Scholar]
- Mao J, Ren X, Zhang L, Van Duin DM, Cohen RC, Park JH, Goldstein AH, Paulot F, Beaver MR, Crounse JD, Wennberg PO, DiGangi JP, Henry SB, Keutsch FN, Park C, Schade GW, Wolfe GM, Thornton JA, and Brune WH: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys, 12, 8009–8020, 10.5194/acp-12-8009-2012, 2012. [DOI] [Google Scholar]
- Mao J, Fan S, Jacob DJ, and Travis KR: Radical loss in the atmosphere from Cu-Fe redox coupling in aerosols, Atmos. Chem. Phys, 13, 509–519, 10.5194/acp-13-509-2013, 2013a. [DOI] [Google Scholar]
- Mao J, Paulot F, Jacob DJ, Cohen RC, Crounse JD, Wennberg PO, Keller CA, Hudman RC, Barkley MP, and Horowitz LW: Ozone and organic nitrates over the eastern United States: Sensitivity to isoprene chemistry, J. Geophys. Res. Atmos, 118, 11256–11268, 10.1002/jgrd.50817, 2013b. [DOI] [Google Scholar]
- Marais EA, Jacob DJ, Kurosu TP, Chance K, Murphy JG, Reeves C, Mills G, Casadio S, Millet DB, Barkley MP, Paulot F, and Mao J: Isoprene emissions in Africa inferred from OMI observations of formaldehyde columns, Atmos. Chem. Phys. Discuss, 12, 7475–7520, 10.5194/acpd-12-7475-2012, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marecal V, Pirre M, Rivire ED, Pouvesle N, Crowley JN, Freitas SR, and Longo KM: Modelling the reversible uptake of chemical species in the gas phase by ice particles formed in a convective cloud, Atmos. Chem. Phys, 10, 4977–5000, 10.5194/acp-10-4977-2010, 2010. [DOI] [Google Scholar]
- McFadyen GG and Cape JN: Peroxyacetyl nitrate in eastern Scotland, Sci. Total Environ, 337, 213–222, 2005. [DOI] [PubMed] [Google Scholar]
- Millet DB, Guenther A, Siegel DA, Nelson NB, Singh HB, de Gouw JA, Warneke C, Williams J, Eerdekens G, Sinha V, Karl T, Flocke F, Apel E, Riemer DD, Palmer PI, and Barkley M: Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys, 10, 3405–3425, 10.5194/acp-10-3405-2010, 2010. [DOI] [Google Scholar]
- Mitovski T, Folkins I, Martin RV, and Cooper M: Testing convective transport on short time scales: Comparisons with mass divergence and ozone anomaly patterns about high rain events, J. Geophys. Res, 117, D02109, 10.1029/2011jd016321, 2012. [DOI] [Google Scholar]
- Moore DP and Remedios JJ: Seasonality of Peroxyacetyl nitrate (PAN) in the upper troposphere and lower stratosphere using the MIPAS-E instrument, Atmos. Chem. Phys, 10, 6117–6128, 10.5194/acp-10-6117-2010, 2010. [DOI] [Google Scholar]
- Moxim WJ, Levy H II, and Kasibhatla PS: Simulated global tropospheric PAN: Its transport and impact on NOx, J. Geophys. Res, 101, 12621–12638, 10.1029/96JD00338, 1996. [DOI] [Google Scholar]
- Murphy JG, Oram DE, and Reeves CE: Measurements of volatile organic compounds over West Africa, Atmos. Chem. Phys, 10, 5281–5294, 10.5194/acp-10-5281-2010, 2010. [DOI] [Google Scholar]
- Murray LT, Jacob DJ, Logan JA, Hudman RC, and Koshak WJ: Optimized regional and interannual variability of lightning in a global chemical transport model constrained by LIS/OTD satellite data, J. Geophys. Res, 117, D20307, 10.1029/2012JD017934, 2012. [DOI] [Google Scholar]
- Nishino N, Arey J, and Atkinson R: Formation Yields of Glyoxal and Methylglyoxal from the Gas-Phase OH Radical-Initiated Reactions of Toluene, Xylenes, and Trimethylbenzenes as a Function of NO2 Concentration, J. Phys. Chem. A, 114, 10140–10147, 10.1021/jp105112h, 2010. [DOI] [PubMed] [Google Scholar]
- Oltmans SJ, Lefohn AS, Harris JM, Tarasick DW, Thompson AM, Wernli H, Johnson BJ, Novelli PC, Montzka SA, Ray JD, Patrick LC, Sweeney C, Jefferson A, Dann T, Davies J, Shapiro M, and Holben BN: Enhanced ozone over western North America from biomass burning in Eurasia during April 2008 as seen in surface and profile observations, Atmos. Environ, 44, 4497–4509, 10.1016/j.atmosenv.2010.07.004, 2010. [DOI] [Google Scholar]
- Orlando JJ, Tyndall GS, and Calvert JG: Thermal decomposition pathways for peroxyacetyl nitrate (PAN): Implications for atmospheric methyl nitrate levels, Atmos. Environ. Part A. General Topics, 26, 3111–3118, 10.1016/0960-1686(92)90468-Z, 1992. [DOI] [Google Scholar]
- Pandey Deolal S, Staehelin J, Brunner D, Cui J, Steinbacher M, Zellweger C, Henne S, and Vollmer MK: Transport of PAN and NOy from different source regions to the Swiss high alpine site Jungfraujoch, Atmos. Envron, 64, 103–115, 10.1016/j.atmosenv.2012.08.021, 2013. [DOI] [Google Scholar]
- Parrish DD, Dunlea EJ, Atlas EL, Schauffler S, Donnelly S, Stoud V, Goldstein AH, Millet DB, McKay M, Jaffe DA, Price HU, Hess PG, Flocke F, and Roberts JM: Changes in the photochemical environment of the temperature North Pacific troposphere in response to increased Asian emissions, J. Geophys. Res, 109, 10.1029/2004JD004978, 2004. [DOI] [Google Scholar]
- Paulot F, Crounse JD, Kjaergaard HG, Kroll JH, Seinfeld JH, and Wennberg PO: Isoprene photooxidation: new insights into the production of acids and organic nitrates, Atmos. Chem. Phys, 9, 1479–1501, 10.5194/acp-9-1479-2009, 2009a. [DOI] [Google Scholar]
- Paulot F, Crounse JD, Kjaergaard HG, Kürten A, Clair JM St., Seinfeld JH, and Wennberg PO: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–733, 10.1126/science.1172910, 2009b. [DOI] [PubMed] [Google Scholar]
- Paulot F, Henze DK, and Wennberg PO: Impact of the isoprene photochemical cascade on tropical ozone, Atmos. Chem. Phys, 12, 1307–1325, 10.5194/acp-12-1307-2012, 2012. [DOI] [Google Scholar]
- Peeters J, Nguyen TL, and Vereecken L: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys, 11, 5935–5939, 2009. [DOI] [PubMed] [Google Scholar]
- Penkett SA and Brice KA: The spring maximum in photo-oxidants in the Northern Hemisphere troposphere, Nature, 319, 655–657, 1986. [Google Scholar]
- Pollack IB, Ryerson TB, Trainer M, Neuman JA, Roberts JM, and Parrish DD: Trends in ozone, its precursors, and related secondary oxidation products in Los Angeles, California: A synthesis of measurements from 1960 to 2010, J. Geophys. Res, 118, 5893–5911, 10.1002/jgrd.50472, 2013. [DOI] [Google Scholar]
- Ran L, Zhao C, Geng F, Tie X, Tang X, Peng L, Zhou G, Yu Q, Xu J, and Guenther A: Ozone photochemical production in urban Shanghai, China: Analysis based on ground level observations, J. Geophys. Res, 114, D15301, 10.1029/2008JD010752, 2009. [DOI] [Google Scholar]
- Roberts JM: Measurement of the Henry’s law coefficient and first order loss rate of PAN in n-octanol, Geophys. Res. Lett, 32, L08803, 10.1029/2004gl022327, 2005. [DOI] [Google Scholar]
- Roberts JM: PAN and Related Compounds, in: Volatile Organic Compounds in the Atmosphere, Blackwell Publishing Ltd, 221–268, 2007. [Google Scholar]
- Roberts JM and Bertman SB: The thermal decomposition of peroxyacetic nitric anhydride (PAN) and peroxymethacrylic nitric anhydride (MPAN), Int. J. Chem. Kin, 24, 297–307, 10.1002/kin.550240307, 1992. [DOI] [Google Scholar]
- Roberts JM, Tanner RL, Newman L, Bowersox VC, Bottenheim JW, Anlauf KG, Brice KA, Parrish DD, Fehsenfeld FC, Buhr MP, Meagher JF, and Bailey EM: Relationships between PAN and ozone at sites in eastern North America, J. Geophys. Res, 100, 22821–22830, 10.1029/95jd01221, 1995. [DOI] [Google Scholar]
- Roberts JM, Parrish DD, Norton RB, Bertman SB, Holloway JS, Trainer M, Fehsenfeld FC, Carroll MA, Albercook GM, Wang T, and Forbes G: Episodic removal of NOy species from the marine boundary layer over the North Atlantic, J. Geophys. Res, 101, 28947–28960, 10.1029/96jd02632, 1996. [DOI] [Google Scholar]
- Roberts JM, Williams J, Baumann K, Buhr MP, Goldan PD, Holloway J, Hübler G, Kuster WC, McKeen SA, Ryerson TB, Trainer M, Williams EJ, Fehsenfeld FC, Bertman SB, Nouaime G, Seaver C, Grodzinsky G, Rodgers M, and Young VL: Measurements of PAN, PPN, and MPAN made during the 1994 and 1995 Nashville Intensives of the Southern Oxidant Study: Implications for regional ozone production from biogenic hydrocarbons, J. Geophys. Res, 103, 22473–22490, 10.1029/98jd01637, 1998. [DOI] [Google Scholar]
- Roberts JM, Flocke F, Stroud CA, Hereid D, Williams E, Fehsenfeld F, Brune W, Martinez M, and Harder H: Ground-based measurements of peroxycarboxylic nitric anhydrides (PANs) during the 1999 Southern Oxidants Study Nashville Intensive, J. Geophys. Res, 107, ACH 1–1–ACH 1–10, 10.1029/2001jd000947, 2002. [DOI] [Google Scholar]
- Roberts JM, Flocke F, Chen G, de Gouw J, Holloway JS, Hübler G, Neuman JA, Nicks DK Jr., Nowak JB, Parrish DD, Ryerson TB, Sueper DT, Warneke C, and Fehsenfeld FC: Measurement of peroxycarboxylic nitric anhydrides (PANs) during the ITCT 2K2 aircraft intensive experiment, J. Geophys. Res, 109, D23S21, 10.1029/2004jd004960, 2004. [DOI] [Google Scholar]
- Roberts JM, Marchewka M, Bertman SB, Goldan P, Kuster W, de Gouw J, Warneke C, Williams E, Lerner B, Murphy P, Apel E, and Fehsenfeld FC: Analysis of the isoprene chemistry observed during the New England Air Quality Study (NEAQS) 2002 intensive experiment, J. Geophys. Res, 111, D23S12, 10.1029/2006JD007570, 2006. [DOI] [Google Scholar]
- Roiger A, Aufmhoff H, Stock P, Arnold F, and Schlager H: An aircraft-borne chemical ionization - ion trap mass spectrometer (CI-ITMS) for fast PAN and PPN measurements, Atmos. Meas. Tech, 4, 173–188, 10.5194/amt-4-173-2011, 2011. [DOI] [Google Scholar]
- Sander SP, Abbatt J, Barker JR, Burkholder JB, Friedl RR, Golden DM, Huie RE, Kolb CE, Kurylo MJ, Moortgat GK, Orkin VL, and Wine PH: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 17, JPL Publication 10–6, Jet Propulsion Laboratory, Pasedena, http://jpldataeval.jpl.nasa.gov (last access: 3 March 2014), 2011. [Google Scholar]
- Saunders SM, Jenkin ME, Derwent RG, and Pilling MJ: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys, 3, 161–180, 10.5194/acp-3-161-2003, 2003. [DOI] [Google Scholar]
- Sauvage B, Martin RV, van Donkelaar A, Liu X, Chance K, L., J., Palmer PI, Wu S, and Fu TM: Remote sensed and in situ constraints on processes affecting tropical tropospheric ozone, Atmos. Chem. Phys, 7, 815–838, 10.5194/acp-7-815-2007, 2007. [DOI] [Google Scholar]
- Shepson PB, Bottenheim JW, Hastie DR, and Venkatram A: Determination of the relative ozone and PAN deposition velocities at night, Geophys. Res. Lett, 19, 1121–1124, 10.1029/92gl01118, 1992. [DOI] [Google Scholar]
- Sillman S and Samson PJ: Impact of temperature on oxidant photochemistry in urban, polluted rural and remote environments, J. Geophys. Res, 100, 11497–11508, 10.1029/94jd02146, 1995. [DOI] [Google Scholar]
- Singh HB: Reactive nitrogen in the troposphere: chemistry and transport of NOx and PAN, Environ. Sci. Tech, 21, 320–327, 10.1021/es00158a001, 1987. [DOI] [PubMed] [Google Scholar]
- Singh HB and Hanst PL: Peroxyacetyl nitrate (PAN) in the unpolluted atmosphere: An important reservoir for nitrogen oxides, Geophys. Res. Lett, 8, 941–944, 10.1029/GL008i008p00941, 1981. [DOI] [Google Scholar]
- Singh HB, Condon E, Vedder J, O’Hara D, Ridley BA, Gandrud BW, Shetter JD, Salas LJ, Huebert B, H¸bler G, Carroll MA, Albritton DL, Davis DD, Bradshaw JD, Sandholm ST, Rodgers MO, Beck SM, Gregory GL, and LeBel PJ: Peroxyacetyl Nitrate Measurements During CITE 2: Atmospheric Distribution and Precursor Relationships, J. Geophys. Res, 95, 10163–10178, 10.1029/JD095iD07p10163, 1990a. [DOI] [Google Scholar]
- Singh HB, Herlth D, O’Hara D, Salas L, Torres AL, Gregory GL, Sachse GW, and Kasting JF: Atmospheric Peroxyacetyl Nitrate Measurements Over the Brazilian Amazon Basin During the Wet Season: Relationships With Nitrogen Oxides and Ozone, J. Geophys. Res, 95, 16945–16954, 10.1029/JD095iD10p16945, 1990b. [DOI] [Google Scholar]
- Singh HB, Herlth D, O’Hara D, Zahnle K, Bradshaw JD, Sandholm ST, Talbot R, Crutzen PJ, and Kanakidou M: Relationship of Peroxyacetyl Nitrate to Active and Total Odd Nitrogen at Northern High Latitudes: Influence of Reservoir Species on NOx and O3, J. Geophys. Res, 97, 16523–16530, 10.1029/91jd00890, 1992. [DOI] [PubMed] [Google Scholar]
- Singh HB, Herlth D, O’Hara D, Zahnle K, Bradshaw JD, Sandholm ST, Talbot R, Gregory GL, Sachse GW, Blake DR, and Wofsy SC: Summertime distribution of PAN and other reactive nitrogen species in the northern high-latitude atmosphere of eastern Canada, J. Geophys. Res, 99, 1821–1835, 10.1029/93JD00946, 1994. [DOI] [Google Scholar]
- Singh HB, Kanakidou M, Crutzen PJ, and Jacob DJ: High concentrations and photochemical fate of oxygenated hydrocarbons in the global troposphere, Nature, 378, 50–54, 1995. [Google Scholar]
- Singh HB, Herlth D, Kolyer R, Chatfield R, Viezee W, Salas LJ, Chen Y, Bradshaw JD, Sandholm ST, Talbot R, Gregory GL, Anderson B, Sachse GW, Browell E, Bachmeier AS, Blake DR, Heikes B, Jacob D, and Fuelberg HE: Impact of biomass burning emissions on the composition of the South Atlantic troposphere: Reactive nitrogen and ozone, J. Geophys. Res, 101, 24203–24219, 10.1029/96JD01018, 1996a. [DOI] [Google Scholar]
- Singh HB, Herlth D, Kolyer R, Salas L, Bradshaw JD, Sandholm ST, Davis DD, Crawford J, Kondo Y, Koike M, Talbot R, Gregory GL, Sachse GW, Browell E, Blake DR, Rowland FS, Newell R, Merrill J, Heikes B, Liu SC, Crutzen PJ, and Kanakidou M: Reactive nitrogen and ozone over the western Pacific: Distribution, partitioning, and sources, J. Geophys. Res, 101, 1793–1808, 10.1029/95JD01029, 1996b. [DOI] [Google Scholar]
- Singh HB, Viezee W, Chen Y, Thakur AN, Kondo Y, Talbot RW, Gregory GL, Sachse GW, Blake DR, Bradshaw JD, Wang Y, and Jacob DJ: Latitudinal distribution of reactive nitrogen in the free troposphere over the Pacific Ocean in late winter/early spring, J. Geophys. Res, 103, 28237–28246, 10.1029/98JD01891, 1998. [DOI] [Google Scholar]
- Singh HB, Brune WH, Crawford JH, Jacob DJ, and Russell PB: Overview of the summer 2004 Intercontinental Chemical Transport Experiment-North America (INTEX-A), J. Geophys. Res, 111, D24S01, 10.1029/2006jd007905, 2006. [DOI] [Google Scholar]
- Singh HB, Salas L, Herlth D, Kolyer R, Czech E, Avery M, Crawford JH, Pierce RB, Sachse GW, Blake DR, Cohen RC, Bertram TH, Perring A, Wooldridge PJ, Dibb J, Huey G, Hudman RC, Turquety S, Emmons LK, Flocke F, Tang Y, Carmichael GR, and Horowitz LW: Reactive nitrogen distribution and partitioning in the North American troposphere and lowermost stratosphere, J. Geophys. Res, 112, D12S04, 10.1029/2006JD007664, 2007. [DOI] [Google Scholar]
- Singh HB, Brune WH, Crawford JH, Flocke F, and Jacob DJ: Chemistry and transport of pollution over the Gulf of Mexico and the Pacific: spring 2006 INTEX-B campaign overview and first results, Atmos. Chem. Phys, 9, 2301–2318, 10.5194/acp-9-2301-2009, 2009. [DOI] [Google Scholar]
- Slusher DL, Huey LG, Tanner DJ, Flocke FM, and Roberts JM: A thermal dissociation chemical ionization mass spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide, J. Geophys. Res, 109, D19315, 10.1029/2004JD004670, 2004. [DOI] [Google Scholar]
- Smyth SB, Sandholm ST, Bradshaw JD, Talbot RW, Blake DR, Blake NJ, Rowland FS, Singh HB, Gregory GL, Anderson BE, Sachse GW, Collins JE, and Bachmeier AS: Factors influencing the upper free tropospheric distribution of reactive nitrogen over the South Atlantic during the TRACE A experiment, J. Geophys. Res, 101, 24165–24186, 10.1029/96jd00224, 1996. [DOI] [Google Scholar]
- Stewart DJ, Taylor CM, Reeves CE, and McQuaid JB: Biogenic nitrogen oxide emissions from soils: impact on NOx and ozone over west Africa during AMMA (African Monsoon Multidisciplinary Analysis): observational study, Atmos. Chem. Phys, 8, 2285–2297, 10.5194/acp-8-2285-2008, 2008. [DOI] [Google Scholar]
- Stone D, Evans MJ, Walker HM, Ingham T, Vaughan S, Ouyang B, Kennedy OJ, McLeod MW, Jones RL, Hopkins J, Punjabi S, Lidster R, Hamilton JF, Lee JD, Lewis AC, Carpenter LJ, Forster G, Oram DE, Reeves CE, Bauguitte S, Morgan W, Coe H, Aruffo E, Dari-Salisburgo C, Giammaria F, Di Carlo P, and Heard DE: Radical chemistry at night: comparisons between observed and modelled HOx, NO3 and N2O5 during the RONOCO project, Atmos. Chem. Phys. Discuss, 13, 9519–9566, 10.5194/acpd-13-9519-2013, 2013. [DOI] [Google Scholar]
- Stroud C, Madronich S, Atlas E, Cantrell C, Fried A, Wert B, Ridley B, Eisele F, Mauldin L, Shetter R, Lefer B, Flocke F, Weinheimer A, Coffey M, Heikes B, Talbot R, and Blake D: Photochemistry in the Arctic Free Troposphere: Ozone Budget and Its Dependence on Nitrogen Oxides and the Production Rate of Free Radicals, J.of Atmos. Chem, 47, 107–138, 10.1023/B:JOCH.0000021026.71906.e1, 2004. [DOI] [Google Scholar]
- Sudo K, Takahashi M, and Akimoto H: CHASER: A global chemical model of the troposphere 2. Model results and evaluation, J. Geophys. Res, 107, 4586, 10.1029/2001jd001114, 2002. [DOI] [Google Scholar]
- Talbot R, Dibb J, Scheuer E, Seid G, Russo R, Sandholm S, Tan D, Singh H, Blake D, Blake N, Atlas E, Sachse G, Jordan C, and Avery M: Reactive nitrogen in Asian continental outflow over the western Pacific: Results from the NASA Transport and Chemical Evolution over the Pacific (TRACE-P) airborne mission, J. Geophys. Res, 108, 8803, 10.1029/2002jd003129, 2003. [DOI] [Google Scholar]
- Talbot RW, Dibb JE, Scheuer EM, Kondo Y, Koike M, Singh HB, Salas LB, Fukui Y, Ballenthin JO, Meads RF, Miller TM, Hunton DE, Viggiano AA, Blake DR, Blake NJ, Atlas E, Flocke F, Jacob DJ, and Jaegle L: Reactive nitrogen budget during the NASA SONEX Mission, Geophys. Res. Lett, 26, 3057–3060, 10.1029/1999GL900589, 1999. [DOI] [Google Scholar]
- Talbot RW, Dibb JE, Scheuer EM, Bradshaw JD, Sandholm ST, Singh HB, Blake DR, Blake NJ, Atlas E, and Flocke F: Tropospheric reactive odd nitrogen over the South Pacific in austral springtime, J. Geophys. Res, 105, 6681–6694, 10.1029/1999JD901114, 2000. [DOI] [Google Scholar]
- Talukdar RK, Burkholder JB, Schmoltner A-M, Roberts JM, Wilson RR, and Ravishankara AR: Investigation of the loss processes for peroxyacetyl nitrate in the atmosphere: UV photolysis and reaction with OH, J. Geophys. Res, 100, 14163–14173, 10.1029/95jd00545, 1995. [DOI] [Google Scholar]
- Tanimoto H, Wild O, Kato S, Furutani H, Makide Y, Komazaki Y, Hashimoto S, Tanaka S, and Akimoto H: Seasonal cycles of ozone and oxidized nitrogen species in northeast Asia 2. A model analysis of the roles of chemistry and transport, J. Geophys. Res, 107, 4706, 10.1029/2001jd001497, 2002. [DOI] [Google Scholar]
- Tereszchuk KA, Moore DP, Harrison JJ, Boone CD, Park M, Remedios JJ, Randel WJ, and Bernath PF: Observations of peroxyacetyl nitrate (PAN) in the upper troposphere by the Atmospheric Chemistry Experiment-Fourier Transform Spectrometer (ACE-FTS), Atmos. Chem. Phys, 13, 5601–5613, 10.5194/acp-13-5601-2013, 2013. [DOI] [Google Scholar]
- Thakur AN, Singh HB, Mariani P, Chen Y, Wang Y, Jacob DJ, Brasseur G, Müller JF, and Lawrence M: Distribution of reactive nitrogen species in the remote free troposphere: data and model comparisons, Atmos. Environ, 33, 1403–1422, 10.1016/s1352-2310(98)00281-7, 1999. [DOI] [Google Scholar]
- Thornton JA, Jaegle L, and McNeill VF: Assessing known pathways for HO2 loss in aqueous atmospheric aerosols: Regional and global impacts on tropospheric oxidants, J. Geophys. Res, 113, 10.1029/2007JD009236, 2008. [DOI] [Google Scholar]
- Tie X, Zhang R, Brasseur G, Emmons L, and Lei W: Effects of lightning on reactive nitrogen and nitrogen reservoir species in the troposphere, J. Geophys. Res, 106, 3167–3178, 10.1029/2000jd900565, 2001. [DOI] [Google Scholar]
- Turquety S, Logan JA, Jacob DJ, Hudman RC, Leung FY, Heald CL, Yantosca RM, Wu S, Emmons LK, Edwards DP, and Sachse GW: Inventory of boreal fire emissions for North America in 2004: Importance of peat burning and pyroconvective injection, J. Geophys. Res, 112, D12S03, 10.1029/2006JD007281, 2007. [DOI] [Google Scholar]
- Tyndall GS, Cox RA, Granier C, Lesclaux R, Moortgat GK, Pilling MJ, Ravishankara AR, and Wallington TJ: Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res, 106, 12157–12182, 10.1029/2000jd900746, 2001. [DOI] [Google Scholar]
- Val Martin M, Honrath RE, Owen RC, and Li QB: Seasonal variation of nitrogen oxides in the central North Atlantic lower free troposphere, J. Geophys. Res, 113, D17307, 10.1029/2007jd009688, 2008. [DOI] [Google Scholar]
- Val Martin M, Logan JA, Kahn RA, Leung FY, Nelson DL, and Diner DJ: Smoke injection heights from fires in North America: analysis of 5 years of satellite observations, Atmos. Chem. Phys, 10, 1491–1510, 10.5194/acp-10-1491-2010, 2010. [DOI] [Google Scholar]
- van der A RJ, Eskes HJ, Boersma KF, van Noije TPC, Van Roozendael M, De Smedt I, Peters DHMU, and Meijer EW: Trends, seasonal variability and dominant NOx source derived from a ten year record of NO2 measured from space, J. Geophys. Res, 113, D04302, 10.1029/2007jd009021, 2008. [DOI] [Google Scholar]
- van der Werf GR, Randerson JT, Giglio L, Collatz GJ, Mu M, Kasibhatla PS, Morton DC, DeFries RS, Jin Y, and van Leeuwen TT: Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997–2009), Atmos. Chem. Phys, 10, 11707–11735, 10.5194/acp-10-11707-2010, 2010. [DOI] [Google Scholar]
- van Donkelaar A, Martin RV, Leaitch WR, Macdonald AM, Walker TW, Streets DG, Zhang Q, Dunlea EJ, Jimenez JL, Dibb JE, Huey LG, Weber R, and Andreae MO: Analysis of aircraft and satellite measurements from the Intercontinental Chemical Transport Experiment (INTEX-B) to quantify long-range transport of East Asian sulfur to Canada, Atmos. Chem. Phys, 8, 2999–3014, 10.5194/acp-8-2999-2008, 2008. [DOI] [Google Scholar]
- van het Bolscher M, Pereira J, Spessa A, Dalsoren S, van Nojie T, and Szopa S: REanalysis of the TROpospheric chemical composition over the past 40 years: A long-term global modeling study of tropospheric chemistry, Max Plank Inst. for Meteorology, Hamburg, Germany, 77, 2008. [Google Scholar]
- Vestreng V, and Klein H: Emission data reported to UNECE/EMEP: Quality assurance and trend analysis & presentation of WebDab, Norwegian Meteorological Institute, Oslo, Norway, 2002. [Google Scholar]
- Villalta PW, Lovejoy ER, and Hanson DR: Reaction probability of peroxyacetyl radical on aqueous surfaces, Geophys. Res. Lett, 23, 1765–1768, 10.1029/96GL01286, 1996. [DOI] [Google Scholar]
- Vivchar A: Wildfires in Russia in 2000–2008: estimates of burnt areas using the satellite MODIS MCD45 data, Remote Sens. Lett, 2, 81–90, 10.1080/01431161.2010.499138, 2010. [DOI] [Google Scholar]
- von Kuhlmann R, Lawrence MG, Crutzen PJ, and Rasch PJ: A model for studies of tropospheric ozone and nonmethane hydrocarbons: Model evaluation of ozone-related species, J. Geophys. Res, 108, 4729, 10.1029/2002jd003348, 2003. [DOI] [Google Scholar]
- von Kuhlmann R, Lawrence MG, Pöschl U, and Crutzen PJ: Sensitivities in global scale modeling of isoprene, Atmos. Chem. Phys, 4, 1–17, 10.5194/acp-4-1-2004, 2004. [DOI] [Google Scholar]
- Walker TW, Jones DBA, Parrington M, Henze DK, Murray LT, Bottenheim JW, Anlauf K, Worden JR, Bowman KW, Shim C, Singh K, Kopacz M, Tarasick DW, Davies J, von der Gathen P, Thompson AM, and Carouge CC: Impacts of midlatitude precursor emissions and local photochemistry on ozone abundances in the Arctic, J. Geophys. Res, 117, D01305, 10.1029/2011JD016370, 2012. [DOI] [Google Scholar]
- Wallington TJ, Andino JM, Ball JC, and Japar SM: Fourier transform infrared studies of the reaction of Cl atoms with PAN, PPN, CH3OOH, HCOOH, CH3COCH3 and CH3COC2H5 at 295 ± 2 K, J. Atmos. Chem, 10, 301–313, 10.1007/bf00053865, 1990. [DOI] [Google Scholar]
- Wang S, Wu D, Wang X-M, Fung JC-H, and Yu JZ: Relative contributions of secondary organic aerosol formation from toluene, xylenes, isoprene, and monoterpenes in Hong Kong and Guangzhou in the Pearl River Delta, China: an emission-based box modeling study, J. Geophys. Res, 118, 507–519, 10.1029/2012JD017985, 2013. [DOI] [Google Scholar]
- Wang X. m., Sheng G. y., Fu J-M, Chan C. y., Lee S-C, Chan LY, and Wang Z. s.: Urban roadside aromatic hydrocarbons in three cities of the Pearl River Delta, People’s Republic of China, Atmos. Environ, 36, 5141–5148, 10.1016/S1352-2310(02)00640-4, 2002. [DOI] [Google Scholar]
- Wang YX, McElroy MB, Jacob DJ, and Yantosca RM: A nested grid formulation for chemical transport over Asia: Applications to CO, Journal of Geophysical Research: Atmospheres, 109, D22307, 10.1029/2004JD005237, 2004. [DOI] [Google Scholar]
- Wang Y, Jacob DJ, and Logan JA: Global simulation of tropospheric O3-NOx -hydrocarbon chemistry: 3. Origin of tropospheric ozone and effects of nonmethane hydrocarbons, J. Geophys. Res, 103, 10757–10767, 10.1029/98JD00156, 1998a. [DOI] [Google Scholar]
- Wang Y, Logan JA, and Jacob DJ: Global simulation of tropospheric O3-NOx-hydrocarbon chemistry 2. Model evaluation and global ozone budget, J. Geophys. Res, 103, 10727–10755, 10.1029/98jd00157, 1998b. [DOI] [Google Scholar]
- Wang Y, Ridley B, Fried A, Cantrell C, Davis D, Chen G, Snow J, Heikes B, Talbot R, Dibb J, Flocke F, Weinheimer A, Blake N, Blake D, Shetter R, Lefer B, Atlas E, Coffey M, Walega J, and Wert B: Springtime photochemistry at northern mid and high latitudes, J. Geophys. Res, 108, 8358, 10.1029/2002jd002227, 2003. [DOI] [Google Scholar]
- Warneke C, Holzinger R, Hansel A, Jordan A, Lindinger W, Pöschl U, Williams J, Hoor P, Fischer H, Crutzen PJ, Scheeren HA, and Lelieveld J: Isoprene and Its Oxidation Products Methyl Vinyl Ketone, Methacrolein, and Isoprene Related Peroxides Measured Online over the Tropical Rain Forest of Surinam in March 1998, J. Atmos. Chem, 38, 167–185, 10.1023/a:1006326802432, 2001. [DOI] [Google Scholar]
- Warneke C, Bahreini R, Brioude J, Brock CA, de Gouw JA, Fahey DW, Froyd KD, Holloway JS, Middlebrook A, Miller L, Montzka S, Murphy DM, Peischl J, Ryerson TB, Schwarz JP, Spackman JR, and Veres P: Biomass burning in Siberia and Kazakhstan as an important source for haze over the Alaskan Arctic in April 2008, Geophys. Res. Lett, 36, L02813, 10.1029/2008GL036194, 2009. [DOI] [Google Scholar]
- Warneke C, Froyd KD, Brioude J, Bahreini R, Brock CA, Cozic J, de Gouw JA, Fahey DW, Ferrare R, Holloway JS, Middlebrook AM, Miller L, Montzka S, Schwarz JP, Sodemann H, Spackman JR, and Stohl A: An important contribution to springtime Arctic aerosol from biomass burning in Russia, Geophys. Res. Lett, 37, L01801, 10.1029/2009gl041816, 2010. [DOI] [Google Scholar]
- Warneke C, Roberts JM, Veres P, Gilman J, Kuster WC, Burling I, Yokelson R, and de Gouw JA: VOC identification and inter-comparison from laboratory biomass burning using PTR-MS and PIT-MS, Int. J. Mass Spectrom, 303, 6–14, 10.1016/j.ijms.2010.12.002, 2011. [DOI] [Google Scholar]
- Wesely ML: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ, 23, 1293–1304, 10.1016/0004-6981(89)90153-4, 1989. [DOI] [Google Scholar]
- Whalley LK, Lewis AC, McQuaid JB, Purvis RM, Lee JD, Stemmler K, Zellweger C, and Ridgeon P: Two high-speed, portable GC systems designed for the measurement of non-methane hydrocarbons and PAN: Results from the Jungfraujoch High Altitude Observatory, J. Environ. Mon, 6, 234–241, 2004. [DOI] [PubMed] [Google Scholar]
- Wiedinmyer C, Akagi SK, Yokelson RJ, Emmons LK, AlSaadi JA, Orlando JJ, and Soja AJ: The Fire INventory from NCAR (FINN): a high resolution global model to estimate the emissions from open burning, Geosci. Model Dev, 4, 625–641, 10.5194/gmd-4-625-2011, 2011. [DOI] [Google Scholar]
- Wiegele A, Glatthor N, Höpfner M, Grabowski U, Kellmann S, Linden A, Stiller G, and von Clarmann T: Global distributions of C2H6, C2H2, HCN, and PAN retrieved from MIPAS reduced spectral resolution measurements, Atmos. Meas. Tech, 5, 723–734, 10.5194/amt-5-723-2012, 2012. [DOI] [Google Scholar]
- Williams JE, van Velthoven PFJ, and Brenninkmeijer CAM: Quantifying the uncertainty in simulating global tropospheric composition due to the variability in global emission estimates of Biogenic Volatile Organic Compounds, Atmos. Chem. Phys, 13, 2857–2891, 10.5194/acp-13-2857-2013, 2013. [DOI] [Google Scholar]
- Wolfe GM, Thornton JA, McNeill VF, Jaffe DA, Reidmiller D, Chand D, Smith J, Swartzendruber P, Flocke F, and Zheng W: Influence of trans-Pacific pollution transport on acyl peroxy nitrate abundances and speciation at Mount Bachelor Observatory during INTEX-B, Atmos. Chem. Phys, 7, 5309–5325, 10.5194/acp-7-5309-2007, 2007. [DOI] [Google Scholar]
- Worthy DEJ, Trivett NBA, Hopper JF, and Bottenheim JW: Analysis of long-range transport events at Alert, Northwest Territories, during the Polar Sunrise Experiment, J. Geophys. Res, 99, 25329–25344, 1994. [Google Scholar]
- Xiao Y, Logan JA, Jacob DJ, Hudman RC, Yantosca R, and Blake DR: Global budget of ethane and regional constraints on U.S. sources, J. Geophys. Res, 113, D21306, 10.1029/2007jd009415, 2008. [DOI] [Google Scholar]
- Xue LK, Wang T, Zhang JM, Zhang XC, Deliger, Poon CN, Ding AJ, Zhou XH, Wu WS, Tang J, Zhang QZ,and Wang WX: Source of surface ozone and reactive nitrogen speciation at Mount Waliguan in western China: New insights from the 2006 summer study, J. Geophys. Res, 116, D07306, 10.1029/2010jd014735, 2011. [DOI] [Google Scholar]
- Yienger JJ and Levy H II: Empirical model of global soilbiogenic NOx emissions, J. Geophys. Res, 100, 11447–11464, 10.1029/95jd00370, 1995. [DOI] [Google Scholar]
- Zanis P, Monks PS, Green TJ, Schuepbach E, Carpenter LJ, Mills GP, Rickard AR, Brough N, and Penkett SA: Seasonal variation of peroxy radicals in the lower free troposphere based on observations from the FREE Tropospheric EXperiments in the Swiss Alps, Geophys. Res. Lett, 30, 1497, 10.1029/2003GL017122, 2003. [DOI] [Google Scholar]
- Zanis P, Ganser A, Zellweger C, Henne S, Steinbacher M, and Staehelin J: Seasonal variability of measured ozone production efficiencies in the lower free troposphere of Central Europe, Atmos. Chem. Phys, 7, 223–236, 10.5194/acp-7-223-2007, 2007. [DOI] [Google Scholar]
- Zellweger C, Ammann M, Buchmann B, Hofer P, Lugauer M, Rüttimann R, Streit N, Weingartner E, and Baltensperger U: Summertime NOy speciation at the Jungfraujoch, 3580 m above sea level, Switzerland, J. Geophys. Res, 105, 6655–6667, 10.1029/1999jd901126, 2000. [DOI] [Google Scholar]
- Zhang J, Wang T, Chameides WL, Cardelino C, Kwok J, Blake DR, Ding A, and So KL: Ozone production and hydrocarbon reactivity in Hong Kong, Southern China, Atmos. Chem. Phys, 7, 557–573, 10.5194/acp-7-557-2007, 2007a. [DOI] [Google Scholar]
- Zhang L, Jacob DJ, Boersma KF, Jaffe DA, Olson JR, Bowman KW, Worden JR, Thompson AM, Avery MA, Cohen RC, Dibb JE, Flock FM, Fuelberg HE, Huey LG, McMillan WW, Singh HB, and Weinheimer AJ: Transpacific transport of ozone pollution and the effect of recent Asian emission increases on air quality in North America: an integrated analysis using satellite, aircraft, ozonesonde, and surface observations, Atmos. Chem. Phys, 8, 6117–6136, 10.5194/acp-8-6117-2008, 2008. [DOI] [Google Scholar]
- Zhang Q, Streets DG, He K, Wang Y, Richter A, Burrows JP, Uno I, Jang CJ, Chen D, Yao Z, and Lei Y: NOx emission trends for China, 1995–2004: The view from the ground and the view from space, J. Geophys. Res, 112, D22306, 10.1029/2007jd008684, 2007b. [DOI] [Google Scholar]
- Zhang Q, Streets DG, Carmichael GR, He KB, Huo H, Kannari A, Klimont Z, Park IS, Reddy S, Fu JS, Chen D, Duan L, Lei Y, Wang LT, and Yao ZL: Asian emissions in 2006 for the NASA INTEX-B mission, Atmos. Chem. Phys, 9, 5131–5153, 10.5194/acp-9-5131-2009, 2009. [DOI] [Google Scholar]
- Zheng W, Flocke FM, Tyndall GS, Swanson A, Orlando JJ, Roberts JM, Huey LG, and Tanner DJ: Characterization of a thermal decomposition chemical ionization mass spectrometer for the measurement of peroxy acyl nitrates (PANs) in the atmosphere, Atmos. Chem. Phys, 11, 6529–6547, 10.5194/acp-11-6529-2011, 2011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.