

Abstract
Background
Many trials of amyloid‐modulating agents fail to improve cognitive outcome in Alzheimer's disease despite substantial reduction of amyloid β levels.
Methods
We applied a mechanism‐based Quantitative Systems Pharmacology model exploring the pharmacodynamic interactions of apolipoprotein E (APOE), Catechol ‐O ‐methyl Transferase (COMTVal158Met), and 5‐HT transporter (5‐HTTLPR) rs25531 genotypes and aducanumab.
Results
The model predicts large clinical variability. Anticipated placebo differences on Alzheimer's Disease Assessment Scale (ADAS)‐COG in the aducanumab ENGAGE and EMERGE ranged from 0.77 worsening to 1.56 points improvement, depending on the genotype‐comedication combination. 5‐HTTLPR L/L subjects are found to be the most resilient. Virtual patient simulations suggest improvements over placebo between 4% and 20% at the 10 mg/kg dose, depending on the imbalance of the 5‐HTTLPR genotype and exposure. In the Phase II PRIME trial, maximal anticipated placebo difference at 10 mg/kg ranges from 0.3 worsening to 5.3 points improvement.
Discussion
These virtual patient simulations, once validated against clinical data, could lead to better informed future clinical trial designs.
Keywords: aducanumab, genotype, medication, pharmacodynamic effect, responder profile
1. INTRODUCTION
Amyloid‐modulating trials, despite robust effects on reducing levels of amyloid β (Aβ) have been disappointing in Alzheimer's disease (AD), leading some to question the amyloid hypothesis. 1 , 2 It has been proposed that the treatment came too late in the disease, and that they would be more effective for healthy elderly with specific risk factors. 3 Other problems include levels of target engagement, although so far all trials with β‐secretase enzyme inhibition (BACE‐I) and γ‐secretase inhibitors (GSIs) that had very robust target engagement actually worsened cognitive outcome. 2 Possible reasons include (1) a toxic off‐target effect of BACE‐I and GSI; (2) a more complex non‐linear biology for Aβ, with beneficial effects for shorter peptides at low doses 4 , 5 that might lead to a “sweet spot” of amyloid reduction; (3) differential impact of Aβ baseline and rate of accumulation on cognitive outcomes; and (4) the pharmacodynamic effect of comedications and genotypes on the dose‐response of amyloid‐modulating agents.
To address the last two issues, we applied the novel technology of Quantitative Systems Pharmacology (QSP) to a virtual patient simulation of clinical trials with aducanumab, a monoclonal antibody that lowers aggregated forms of Aβ. QSP is an advanced computer model that integrates the biology of different Aβ peptides on action potential firing of neuronal circuits, in this case a model for cognitive performance. 4 , 6 In general, QSP, especially for central nervous system (CNS) disorders increasingly becomes more appreciated as a tool with academia, funding organizations, industry, and regulatory agencies to address the large clinical trial failure rate. 7
It is suspected that the large variability in clinical responses is due partly to the pharmacodynamic interactions of comedications and genotypes on the dose‐response of a new investigative drug, in addition to baseline and natural progression of amyloid levels. By explicitly modeling the neurophysiological impact of certain genotypes from clinical imaging observations, a QSP model, in principle, can estimate the actual impact of these interactions. The platform has demonstrated its predictive validity in a prospective prediction of an unexpected clinical outcome for a novel pro‐cognitive target in AD. 8
Here we focus on clinical trials of aducanumab, 9 a monoclonal antibody against aggregated forms of Aβ 10 with cognitive benefits in a small Phase II trial at 3 and 10 mg/kg, but not at 6 mg/kg. 11 The large Phase III EMERGE and ENGAGE trial was halted for futility in March 2019, but subsequent analysis of further subjects found a signal in one of the trials. By implementing the clinical trial design and the pharmacodynamic interactions with genotypes and comedications, we aim to generate hypotheses about the lack of dose‐response in the Phase II trial and the different responses in the Phase III trials.
We focus on COMTVal158Met rs4680, 12 5‐HTTLPR rs25531 s/L, 13 and APOE, as they are common variants that affect cognitive state and their effects on dopamine and serotonin dynamics (important for cognition) and on amyloid physiology have been documented.
The effect of the 5‐HTTLPR genotype in AD has not been studied in the clinical setting; however, in schizophrenia an association between the 5‐HTTLPR genotype and the risk for schizophrenia was found in a South Indian population, 14 but not in a Japanese population. 15 Recent studies suggest the presence of a tri‐allelic impact with an additional G‐A mutation in the L‐form of the promotor 16 with the A‐form, but not with the G‐form; enhancing the L‐phenotype on 5‐HTT transporter expression. In patients with major depression, response to antidepressants is strongly modulated by the 5‐HTTLPR rs25531 genotype. 17 In principle, subjects with the 5‐HTTLPR L/L genotype, who have lower basal serotonin levels, while not affecting Aβ dynamics could perform better on cognitive readouts, probably due to the lower 5‐HT3 and 5‐HT6 activation levels that improve neuronal firing and network stability. 5‐HT6 antagonism has been shown to improve cognition in preclinical models 18 but the effect in clinical AD trials has been modest. 19 Of interest, the 5‐HTTLPR L/L genotype is over‐represented in obsessive compulsive disorder 16 and in aggression associated with AD. 20 We speculate that this could be due to the fact that the 5‐HTTLPR L/L allele overstabilizes representations in the cortical network at the expense of flexibility. 21
RESEARCH IN CONTEXT
Systematic review: This study uses an advanced computer model of neuronal human brain circuits relevant to cognition in combination with experimentally documented effects of amyloid β (Aβ) peptides. The model is based on domain expertise and has been calibrated previously for clinical cognitive scales.
Interpretation: Implementing the clinical trial design of aducanumab using virtual patients with different genotypes, medications, and Aβ loads, the model generates testable hypotheses on the differential outcomes between Phase II and III and identifies a possible responder genotype.
Future directions: If these predictions can be validated with actual clinical data, this platform allows for evaluation of the interactions between comedications, genotypes, and amyloid status for improving new trial designs in Alzheimer's disease.
The catechol‐O‐methyl transferase (COMT) gene product catalyzes the transfer of a methyl group from S‐adenosylmethionine to catecholamines such as dopamine and norepinephrine, which is a necessary step in the metabolism of these endogenous neurotransmitter, especially in the human cortex. The homozygote Met/Met form is more thermolabile; therefore is associated with lower activity of the enzyme 22 and higher ambient dopamine and norepinephrine levels. Given the impact of dopamine on cognitive performance, 23 it is of interest to study the impact of this genotype on functional changes associated with Aβ changes. Of interest this genotype was modestly associated with AD plus psychosis in female patients, whereas no association was found with male AD patients. 24 For both genotypes, positron emission tomography (PET) tracer imaging studies in unmedicated healthy volunteers have documented the impact on the dynamics of the respective neurotransmitters. 25 , 26
It should be strongly emphasized that this study is a hypothesis‐generating project to better understand the outcomes of clinical trials and that final validation of these predictions needs to be performed by comparing actual clinical trial outcomes. Nevertheless the platform has shown prospective validation in a number of clinical trials in Neurology 8 and Psychiatry. 27 , 28
2. METHODS
2.1. Calibrated model for ADAS‐COG readout
The calibrated QSP model for cognition in AD has been described extensively before. 6 , 29 Basically, the model consists of a biophysically realistic network of 80 prefrontal cortex pyramidal glutamatergic and 40 γ‐aminobutyric acid (GABA)ergic interneurons, with the effects of dopaminergic, serotonergic, noradrenergic, and cholinergic modulation (see also Supplementary Information S3) and is based on the stability of a memory trace within a working memory paradigm. The model has been calibrated using 28 different drug‐dose–duration interventions with acetylcholinesterase inhibitors (AChE‐I) and 5‐HT6 antagonists. 6
2.2. Impact of Aβ load on cognitive outcome
In previous work on this QSP platform 4 we showed that in order to explain three different clinical data sets on cognition, the following properties of the amyloid peptide needed to be included, based on preclinical work in cortical slices: 5
The biological effect of the short form (Aβ40) is neurostimulatory at low concentrations but reduces glutamatergic neurotransmission at higher concentrations,
the long form (Aβ42) dose‐dependently reduces glutamatergic neurotransmission and
both forms dose‐dependently reduce alpha7 nicotinic neurotransmission.
We generated look‐up tables covering glutamatergic transmission on the N‐methyl‐d‐aspartate receptor (NMDA‐R) in combination with nicotinic neurotransmission at the alpha7 nicotinic acetylcholine receptor (nAChR). These tables are then converted to look‐up tables with Aβ40 and Aβ42 levels (for a maximum of 17 units) using the dose‐response relationship on glutamatergic and nicotinic neurotransmission. This results in 54 different conditions of three genotypes with and without AChE‐I and for different trial durations (corresponding to patients at the start of the trial and after 52 and 104 weeks).
Previous model simulations 4 suggest a level of 3 units for amyloid positivity threshold based on PET imaging and a natural increase in both Aβ40 and Aβ42 amyloid levels of 1 unit over 12 weeks. We also simulated fast progressors (1 unit/10 weeks) and slow progressors (1 unit/16 weeks).
2.3. Effect of aducanumab on Aβ changes
In the Phase III study, patients were gradually uptitrated to either a low dose (6 mg/kg) or a high dose (10 mg/kg), mostly to mitigate ARIA side‐effects. We simulate a slow titration schedule, with patients at 1 mg/kg for the first 8 weeks, 3 mg/kg for the next 16 weeks, 6 mg/kg for the next 20 weeks, and 10 mg/kg after week 44 for the high dose arm or they continued on 6 mg/kg for the low dose.
The Phase II trial did not include any titration, and included 1, 3, 6, and 10 mg/kg for 52 weeks.
2.4. Implementation of AChE‐I
The receptor model has been described in detail before 30 , 31 (see Supplementary information S1) and simulates the competition between neurotransmitters, drugs, and tracer molecules at the postsynaptic receptor, for example, a cholinergic synapse under natural in vivo firing conditions.
Target engagement of donepezil, an AChE‐inhibitor with a Ki of 20 nM, 33 is derived from imaging studies with 11C‐PMP, 34 corresponding to brain AChE‐inhibition levels of 35% at 10 mg. 34 , 35 The subsequent changes in ACh half‐life affects activation levels of muscarinic and nicotinic receptors, leading to corresponding modifications in glutamate and GABA (see Supplementary Information Table S2 for biological references).
2.5. Implementation of genotypes
We study all possible combinations of the following genotypes: COMTVal158Met, 5‐HTTLPR rs25531, and APOE (all together 27 cases). The genotypes are MM, MV, and VV for COMTVal158Met; LL, Ls, and ss for 5‐HTTLPR rs25531; and APOE44, APOE4X, and APOEXX, where X = 2,3 for APOE.
The same receptor competition model can be used to determine the pharmacodynamic effect of genotypes. To reproduce experimental findings that the COMTVal158Met genotype affects the displacement of the dopamine 1 receptor (D1R) PET radiotracer NNC‐112 in healthy unmedicated volunteers, 25 the synaptic half‐life of dopamine in the COMTVV case was adjusted to 100 ms, 130 ms in the COMTMV, and 160 ms in the COMTMM case. Similarly, the displacement of the 5‐HT4 PET tracer [11C]SB207145 is dependent on the 5‐HTTLPR s/l isoform, 26 resulting in a half‐life of 55 ms for the LL case, 75 ms for the Ls case, and 100 ms for the ss case.
We implemented the APOE genotype using different synapse densities with APOE44, a 20% lower, and APOEXX, a 20% higher synapse density compared to APOE4X genotype. 36 , 37 , 38 The effect of APOE on Aβ clearance 40 is implemented as a 10% decrease in the naturalistic amyloid accumulation for APOEXX and a 10% increase for APOE44, compared to the APOE4X genotype.
We assume a Hardy‐Weinberg distribution for all genotypes, 41 except for APOE, where the allele frequency of the ε4 allele increases from 0.16 in controls to 0.40 in AD. 42
2.6. Virtual patient trial
We sample the genotype combinations using the appropriate distributions described above by creating a cumulative distribution function for the 54 possible combinations and using a random number generator for a unique signature of each virtual patient. Baseline amyloid level and amyloid accumulation rates are sampled from Gaussian distributions with defined average value and variance. This procedure creates a unique profile of changes in Aβ40 and Aβ42 for each patient, which is then allocated to placebo, low‐dose, or high‐dose active treatment arm in the ratio of 1:1:1.
Using the look‐up tables described earlier, we generate a unique trajectory of glutamatergic and nicotinic changes for each virtual patient resulting in anticipated ADAS‐COG changes from their own baseline at 52 and 104 weeks.
3. RESULTS
Figure 1 shows the concept of the virtual patient platform. In many cases, the antibody‐mediated reduction in oligomeric Aβ levels over time cannot be determined experimentally but in principle can be estimated from amyloid imaging using biophysically realistic aggregation dynamics models (see further in discussion). We consider the process of aducanumab‐mediated removal of oligomeric and aggregated amyloid forms as competitive with the natural processes of amyloid synthesis and aggregation into oligomeric forms and finally plaques. For this study, however, we considered this parameter as an independent variable and tested different reductions (40% to 80%) in the natural oligomeric Aβ increase at the highest dose of 10 mg/kg.
FIGURE 1.
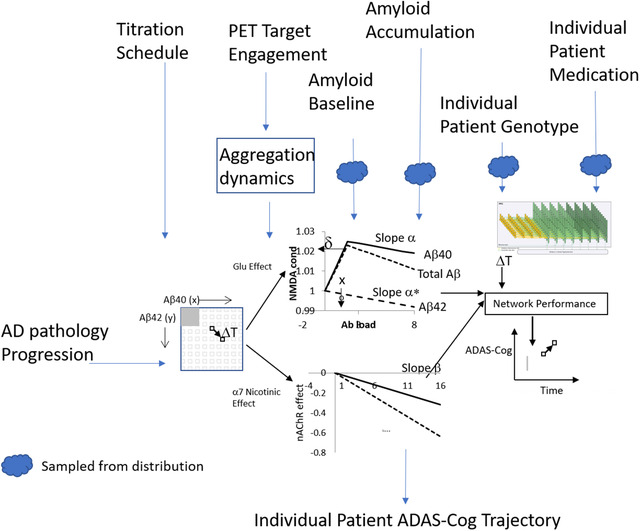
Schematic representation of the virtual patient platform. The core computer model consists of a QSP model that simulates the effect of actual Aβ loads on glutamatergic and nicotinic neurotransmission in an ADAS‐Cog calibrated neuronal cortical network. The input is defined as the number of patients, average baseline amyloid level and variance, their average rate of amyloid accumulation and variance, pharmacodynamic effect of amyloid agents on levels of both oligomeric Aβ40 and Aβ42, specific clinical trial design, fraction of patients on AChE‐I, and genotype distribution (in this example only APOE, COMTVal158Met and 5‐HTTLPR as an example).Changes in Aβ oligomeric load can be calculated from natural history in the placebo arm and pharmacodynamic effects of therapeutic interventions. The output is an individualized cognitive ADAS‐Cog trajectory for that specific patient
3.1. Effect of genotypes and procholinergic medication on cognitive trajectory in placebo patients
We first studied the impact of the different genotype and AChE‐I combinations in the placebo condition. For different values of baseline amyloid and accumulation rates, the simulations show that (1) the baseline ADAS‐COG ranges from 20.48 to 22.85, with an average of 21.28; (2) ADAS‐COG at 52 weeks ranges from 26.40 to 33.20, with an average of 30.50; and (3) ADAS‐COG at 104 weeks ranges from 31.12 to 34.26, with an average of 32.66. This translates into average placebo changes of 9.23 points at 52 weeks (range 4.28 to 12.12) and 11.38 points (range 9.06 to 13.17) at 104 weeks, thus suggesting that genotypes and medications can interact with amyloid physiology to generate a large variability in clinical response.
3.2. Effect of genotypes and procholinergic medication on efficacy of aducanumab in phase III titration study
Next, we introduced the effect of aducanumab on Aβ accumulation using the Phase III slow titration schedule and calculated the improvement over placebo for each of the 54 different configurations and various amyloid baseline and accumulation rates.
Figure 2 shows that the anticipated improvement of high‐dose aducanumab over placebo depends on amyloid baseline values and accumulation rate at 52 and 104 weeks, and is around 1 point on ADAS‐Cog in an “ideally randomized” patient population where the different configurations are weighted according to their incidence.
FIGURE 2.
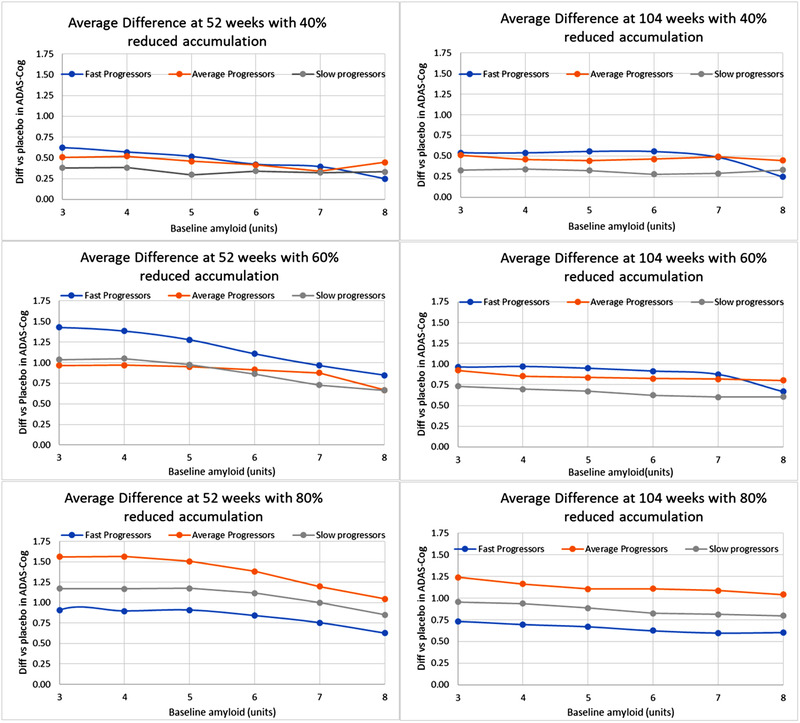
Effect of baseline amyloid and rate of amyloid accumulation on changes in ADAS‐Cog for aducanumab and placebo in ADAS‐Cog averaged over the 54 genotypes and comedications. Shown is the difference in changes versus baseline between aducanumab and placebo (positive outcome favors aducanumab). Note that the average changes in ADAS‐Cog from baseline are around 7 and 10.5 points for placebo at 52 and 104 weeks, respectively. The titration schedule of the Phase III ENGAGE and EMERGE trial at 10 mg/kg at 52 weeks (left) and 104 weeks (right) is used for different reductions of oligomeric amyloid accumulation rate by aducanumab (40%‐80%). Higher reductions increase the cognitive improvement over placebo to a maximum of 1.6 points for fast progressors and relatively low baseline (3‐4 units, around the threshold for amyloid PET positivity). Note that the clinical trial included patients with Aβ positivity (baseline >3)
The data show a complex relationship between improvement and reduction in amyloid accumulation rate, with the 60% better than the 40% and almost equal to the 80%. Of interest at this high exposure, at very low amyloid baseline (below the positivity threshold) the antibody becomes worse than placebo. From here on we will show simulations for the highest exposure (ie, a decrease of 80% vs naturalistic progression), except where noted.
To identify responders, we rank ordered all outcomes for the 54 different combinations and looked at the distribution of genotypes on the top 25%. Figure 3 suggests that the 5‐HTTLPR LL genotype is significantly over‐represented in the aducanumab responders.
FIGURE 3.
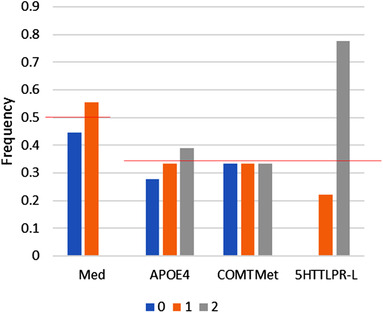
Responder analysis for aducanumab. Frequency of alleles in top 30% of responder combinations. When rank ordering the different genotype‐medication combinations for their greatest improvement over placebo for both 52 and 104 weeks trial duration, subjects with the 5‐HTTLPR LL genotype are highly represented in the top 30%. Red lines correspond to the expected random distribution
3.3. Effect of genotypes and procholinergic medication on efficacy of aducanumab in phase II dose‐finding study
For the small Phase II study PRIME we tested the effect of acute single dosing in the platform with the appropriate distribution over all combinations. At the highest dose (10 mg/kg), average improvement was 2.36 points. Of interest, when eliminating APOE44 carriers the effect at 10 mg/kg is on average about 0.04 points smaller.
When taking into account the small numbers of the actual Phase II trial (n = 20‐30), variability can be much greater. The outcomes for the best and worst case scenarios were derived by comparing the average outcome for the top half of the active arm versus bottom half of placebo and vice versa. For the highest dose of 10 mg/kg, the average value suggests a best‐case outcome of 5.3 points (a 59% improvement) improvement at 52 weeks. Conversely for the worst‐case scenario, a 3.2‐point worsening (a 39% deterioration) for the 1 mg/kg dose is observed. The low number of patients leads to large variability in outcomes and can partially explain the lack of dose‐response in the PRIME trial. Figure 4 shows the effect of all doses on the difference with placebo. Table 1 summarizes the results of the simulation for the different trial designs.
FIGURE 4.
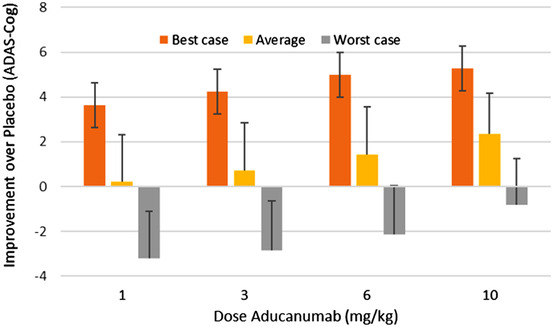
Best, worse, and average scenario for the improvement over placebo after 52 weeks in the PRIME Phase II trial for four different doses for fast progressors with a baseline amyloid level of 3 units (around the threshold for amyloid positivity as explained in the text). Shown is the difference in changes versus baseline between aducanumab and placebo (positive outcome favors aducanumab). Because of the low numbers in the trial, differences between the active arm and placebo can vary greatly depending on the genotype and medication distribution. A maximal improvement of 5 points on the ADAS‐Cog can be achieved with the 10 mg/kg dose, whereas the worst‐case scenario will result in an over 3 point worsening for the 1 mg/kg dose
TABLE 1.
Average difference between aducanumab and placebo changes versus baseline (positive outcome favors aducanumab) and range of effects (in points on ADAS‐Cog scales) for all possible scenarios with aducanumab (different amyloid base‐line values and Aβ rate accumulations over the 54 different configurations when weighted according to the Hardy‐Weinberg distribution for COMT and 5‐HTTLPR and with observed frequencies for APOE in AD patients)
| Trial | Difference with placebo in ADAS‐Cog at 52 weeks | Difference with placebo in ADAS‐Cog at 104 weeks |
|---|---|---|
| ENGAGE/EMERGE titration (40% reduction) | 0.48 (range 0.30‐0.61) | 0.48 (range 0.13‐0.60) |
| ENGAGE/EMERGE titration (60% reduction) | 0.87 (range 0.59‐1.11) | 0.82 (range 0.68‐1.07) |
| ENGAGE/EMERGE titration (80% reduction) | 0.82 (range 0.72‐1.13) | 0.86 (range 0.68‐1.24) |
| ENGAGE/EMERGE fast titration | 0.72 (range 0.61‐0.90) | 0.59 (range 0.24‐0.70) |
| ENGAGE/EMERGE with APOE44 switch | N/A | 0.35 (range 0.02‐0.48) |
| PRIME (10 mg/kg) – 40% reduction | 1.04 (range −0.3 to 4.29) | N/A |
| PRIME (10 mg/kg) – 60% reduction | 1.96 (range −0.3 to 5.15) | N/A |
| PRIME (10 mg/kg) – 80% reduction | 2.16 (range 0.3 to 5.39) | N/A |
| PRIME (6 mg/kg) range 40% to 80% reduction | 0.70 (range −1.4 to 2.87) | N/A |
| PRIME (3 mg/kg) range 40% to 80% reduction | 0.30 (range −1.8 to 2.42) | N/A |
| PRIME (1 mg/kg) range 40% to 80% reduction | 0.12 (range −2.0 to 2.25) | N/A |
ENGAGE/EMERGE are Phase III trials and PRIME is a Phase II trial.
3.4. Virtual patient trials
A series of 100 virtual patient trials of 1200 subjects in the Phase III trial leads to an average improvement at 52 weeks of about 12% or 1.23 points (range 1.11‐1.45) over placebo for the high dose. At 104 weeks the improvement over placebo is about 10% or 1.18 points (range 1.10‐1.28). Standard deviations are around 2 points for the 52‐week outcome and 1 point for the 104‐week outcome. The variability at the individual patient level is substantial, with a range of 8 points at 52 weeks and 5 points at 104 weeks, with somewhat smaller ranges for placebo subjects (7 points at 52 weeks and 4.5 points at 104 weeks).
Figure 5 shows the relationship between baseline functional performance on ADAS‐Cog and changes in ADAS‐Cog after 52 weeks. Patients with worse baseline (higher number of errors) deteriorate less, likely because of a more restricted dynamic range. Of interest subjects with the 5‐HTTLPR LL genotype but not APOEXX are more resilient (have a smaller deterioration for the same baseline) to increases in amyloid load. This is in line with the observation that this genotype is over‐represented in the aducanumab responder population.
FIGURE 5.
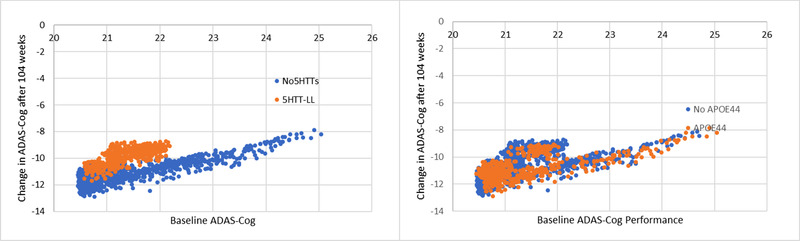
Virtual patient trial outcome after 52 weeks for changes in ADAS‐Cog as a function of baseline ADAS‐Cog. The genotypes/medications are distributed according to their incidence in the population, and both baseline and amyloid accumulation are sampled from a Gaussian distribution around the average value (here a baseline level of 6 units) and an amyloid accumulation of 1 unit/12 weeks. (Left) Subjects in brown are carriers of the 5‐HTTTLPR LL genotype. As expected, the worsening in ADAS‐Cog with amyloid accumulation decreases as the baseline ADAS‐Cog gets worse. However, 5‐HTTLPR LL carriers are more resilient to functional cognitive worsening for a similar amount of amyloid accumulation. (Right) There is little difference in the distribution of responders with (brown) or without (blue) the APOE44 homozygote genotype
3.5. Lowering dose during the trial for a specific group of patients
We then studied the impact of lowering the aducanumab dose of 10 mg/kg to 6 mg/kg at week 52 for APOE44 homozygotes for a cognitive readout at 104 weeks. The dose lowering leads to an average decrease in efficacy of 0.15 points on ADAS‐Cog (range 0.05‐0.24), depending upon the baseline amyloid level and for the fast progressors. This difference further reduces an already modest response.
3.6. Effect of imbalanced genotype distribution on virtual patient trials
Finally, we calculated the effect of an imbalance in responder genotypes (ie, the 5‐HTTLPR L/L) between high‐dose aducanumab and placebo. Figure 6 shows the relative improvement over placebo for a 1200 patient trial as a function of the degree of imbalance. For the highest exposure levels and a shift of 40 responder subjects (of 400) between active arm and placebo, the simulated improvements over placebo can reach the 20% range (1.8 points on ADAS‐Cog). For lower exposure and more equilibrated distribution, improvements are around 5%.
FIGURE 6.
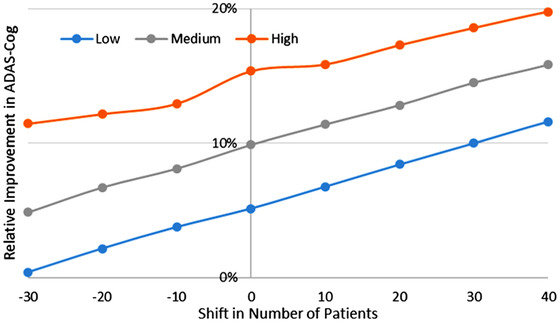
Anticipated improvement over placebo at 52 weeks for a virtual patient trial (n = 1500) with an average baseline of 3 units and an amyloid accumulation of 10 weeks/unit, both sampled from a distribution with 50% variance. Shown is the difference in changes versus baseline between aducanumab and placebo (positive outcome favors aducanumab).The figure suggests that the highest exposure (80% inhibition of oligomer formation) in combination with an imbalance of only 40 responder subjects (of 400 possible subjects) for the 5‐HTTLPR L/L genotype can lead to an improvement over placebo of close to 20% at 52 weeks. Conversely, low exposure with an unfavorable distribution only leads to a 2%‐3% improvement over placebo
4. DISCUSSION
This study uses a novel computer‐based approach using virtual patients to perform a post hoc analysis of aducanumab, focusing on the pharmacodynamic interaction with genotypes and medications. The simulations suggest a substantial impact of genotypes and medication status on the cognitive trajectory of individual patients, which might explain part of the variability in clinical outcomes.
This mechanism‐based model, 4 constrained by clinical data, 42 , 43 , 44 allows the identification of biological principles driving the complex relationship between functional effect and aducanumab‐mediated changes in amyloid accumulation. The dose‐dependent difference in cognitive changes compared to the changes in the placebo arm saturates between 60% and 80% reduction in oligomeric concentration with a complex dependence on baseline amyloid and natural rate of amyloid accumulation.
The major hypothesis generated by the model is the prediction that the 5‐HTTLPR genotype, in particular the LL genotype which is associated with a higher SERT expression 26 and lower baseline 5‐HT levels, is resilient against amyloid accumulation. The effect is likely a consequence of lower 5‐HT3 receptor activation that affects GABAergic tone 46 and of reduced 5‐HT6 activation that leads to indirect changes in cholinergic, 47 glutamatergic, and dopaminergic neurotransmitter systems. 48 Clinically these effects lead to a stabilization of excitation dynamics and improved cognition in schizophrenia patients 49 and in Alzheimer's patients. 19 , 49 Of interest, a clinical study with citalopram, a 5‐HTT blocker that increases ambient 5‐HT levels for addressing agitation in AD patients, resulted in cognitive worsening. 51 We acknowledge that other genotypes that affect 5‐HTT expression or in other pathways that we didn't explicitly model might also play a role.
Although APOE44 carriers usually start out at lower functional baseline scores, their cognitive deterioration over time is not different from non‐APOE4 carriers, in line with clinical observations that APOE most importantly drives age at onset, but not necessarily cognitive deterioration after diagnosis. 52 Furthermore, no significant pharmacodynamic interaction of COMTVal158Met genotype was observed, suggesting that dopamine levels are not modulating cognitive changes after Aβ intervention.
The simulations show a clear effect of the titration schedule on the aducanumab clinical outcome as opposed to a non–titration‐based multiple‐dose study. Without titration, improvement over placebo can reach about 2 points at the highest dose and exposure in a large patient trial when all genotypes and medications are contributing equally. This is the same range of effects for AChE‐I such as donepezil, galantamine, and rivastigmine. 52 , 53 In a much smaller patient sample, as in the PRIME study, maximal improvement can be higher when a majority of responder profiles are in the active arm and non‐responder profiles are in the placebo arm. Therefore, a trial with small patient numbers can yield widely different outcomes, depending on the distribution of genotypes and medications over treatment arms. It is not inconceivable that the patients who could tolerate the highest aducanumab dose were the ones with the largest responses, maybe carrying a 5‐HTTLPR LL genotype. This could explain the lack of dose‐response in the small Phase II PRIME trial. 11
In contrast, in the Phase III ENGAGE and EMERGE trial with a progressive titration schedule over 44 weeks, total exposure is smaller and the differences with placebo are modest (about half the size of AChE‐I) with substantial variability; such values are not likely to be detected statistically. It is of interest to note that the magnitude of this outcome is in the range of the reported differences between solanezumab and placebo at 78 weeks. 55 The simulations also suggested that switching all APOE44 subjects to a lower dose at 52 weeks would shave off only an undetectable 0.15 points on an already modest outcome. Overall, the data suggest that any amyloid‐related intervention fundamentally has limited effects on cognitive readouts (exposure‐dependent but reaching only about 1.25 point on ADAS‐Cog).
However, imbalances between genotypes, especially for responders in the different treatment arms, can substantially affect the anticipated outcomes. Even with the numbers of subjects in the two, Phase III trials, EMERGE and ENGAGE, the simulations show that a 20% improvement can be achieved for a relatively modest imbalance of 40 responder subjects (of 400) between placebo and active arm.
There are a number of important limitations to the model. The model assumes a complete lack of direct neurotoxicity of amyloid peptides in the AD brain. The QSP model already indirectly assumes a linear loss of synapses and neurons over time, 6 possibly triggered by many other processes (which we do not explicitly model) such as neuroinflammation and tau pathology. Region‐of‐interest imaging in cognitively normal subjects show indeed differences in hypometabolism (in line with effects on glutamate transmission) but not atrophy in Aβ+ versus Aβ− subjects. 56
The APOE genotype has a pleotropic phenotype, including effects on microglia and astrocyte biology, 57 but we limit ourselves to an effect on synapse densities and amyloid clearance. The current QSP model does not include any biological processes related to non‐neuronal cells, but it is conceivable to introduce the effect of secreted cytokines such as TNFα on voltage‐gated ion channels. 58
Levels of target engagement, that is, how much aducanumab reduces oligomeric amyloid concentrations, can only be derived indirectly from imaging studies on plaque density that show an almost complete clearance after 52 weeks of 10 mg/kg aducanumab treatment. 11 To derive estimates of oligomeric amyloid levels in the living AD brain, one has to rely on extrapolations based on models of aggregation kinetics. 58 , 59 These models suggest that even with complete clearance of aggregated plaques, substantial soluble small‐order oligomeric amyloid peptides would remain, probably due to breakdown of plaques in smaller aggregates and/or reduced interaction of oligomeric peptides to a lower number of already formed plaques (see for instance Figure 2 from 60 ). Moreover, studies with solanezumab suggest a change in “soluble” CSF Aβ40 in the 30% and CSF Aβ42 in the 60% range compared to placebo. 61 Our simulations suggest that higher reduction of amyloid accumulation increases the functional improvement in a non‐linear way that saturates at around 1.2 points on the ADAS‐Cog scale for the Phase III titration schedule.
The effect of Aβ peptides is limited to glutamate and nicotinic neurotransmission; other targets have been proposed, such as Kv4.2 and Kv4.3 channels, 62 upregulation of the 5‐HT1AR, 63 or Ca‐dysregulation, 64 which all could affect the electrophysiological properties of the neuronal circuits. Because there are other K+ channels active in cortical neuron that are not affected by Aβ, we believe these effects might be more modulatory in nature with a more limited impact on the outcomes. In addition, the upregulation of 5‐HT1AR, activation of which has been linked to improved cognitive outcome, 64 , 65 is specific for the short Aβ40 form, providing even more evidence for a neurostimulatory effect. We plan to include these in future iterations of the platform.
A major limitation of the current QSP model is the absence of tau pathology, microglia and astrocyte involvement, and vascular pathology. The current calibrated version of the QSP platform with ADAS‐Cog readout 6 assumes already a calibrated parameter for progressive synapse and neuronal loss as a consequence of non–amyloid‐related neuropathological processes, however, without implementing a lot of detail. Subsequent iterations of the platform can elaborate these processes in detail and will certainly be necessary to support the development of specific disease‐modifying interventions. Although this is an important issue, we would argue that the trial duration (1‐2 years) simulated here may be too short to have substantial changes in these pathologies that often take many years to develop and that therefore the major process is a change in Aβ. Imaging studies have indeed demonstrated an average hippocampal volume loss of 0.6 mm3/year and a maximal cortical thinning rate of 0.07 mm/year with a Mini‐Mental State Exam (MMSE) score of 21. 67 We fully acknowledge that for modeling long‐term prevention studies, this type of slowly progressing pathology certainly needs to be included. For instance, tau pathology could be implemented through its effect on action potential properties that affect synchronization of neuronal circuit activity. 68
The platform in its current form intends to quantitatively estimate the impact of the complex non‐linear nature of amyloid biology that could possibly explain the unexpected dissociation between target exposure and clinical functional outcome. It might further allow optimization of titration schedules for cognitive performance while mitigating side‐effects such as ARIA, 69 the level of exposure and corresponding reduction of amyloid accumulation, and the selection of specific genotype populations. In principle, the platform can be extended to include other CNS‐active medications, which are often used in clinical practice, 70 allowing more Real‐world Experience to be incorporated at the trial design stage. Many of these concepts can be applied to other similar amyloid‐modulating agents, and this mechanism‐based QSP modeling platform provides a framework to improve the design of future clinical trials.
However, for the ultimate validation of this QSP model, the predictions need to be verified against the actual analyses of the clinical trial.
CONFLICT OF INTEREST
At the time of the study, the authors were employees of In Silico Biosciences. HG is now with Certara‐QSP.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
AS and HG are employees of In Silico Biosciences, a company providing QSP services to the medical community in CNS disorders. AS and HG developed the model and HG ran the experiments and wrote the paper.
Geerts H, Spiros A. Learning from amyloid trials in Alzheimer's disease. A virtual patient analysis using a quantitative systems pharmacology approach. Alzheimer's Dement. 2020;16:862–872. 10.1002/alz.12082
REFERENCES
- 1. Foroutan N, Hopkins RB, Tarride JE, Florez ID, Levine M. Safety and efficacy of active and passive immunotherapy in mild‐to‐moderate Alzheimer's disease: A systematic review and network meta‐analysis. Clin Invest Med. 2019;42:E53‐E65. [DOI] [PubMed] [Google Scholar]
- 2. Penninkilampi R, Brothers HM, Eslick GD. Safety and Efficacy of Anti‐Amyloid‐beta immunotherapy in Alzheimer's disease: a systematic review and meta‐analysis. J Neuroimmune Pharmacol. 2017;12:194‐203. [DOI] [PubMed] [Google Scholar]
- 3. Aisen PS, Siemers E, Michelson D, et al. What have we learned from expedition III and EPOCH trials? Perspective of the CTAD task force. J Prev Alzheimers Dis. 2018;5:171‐174. [DOI] [PubMed] [Google Scholar]
- 4. Geerts H, Spiros A, Roberts P. Impact of amyloid‐beta changes on cognitive outcomes in Alzheimer's disease: analysis of clinical trials using a quantitative systems pharmacology model. Alzheimers Res Ther. 2018;10:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y, Zhou TH, Zhi Z, Barakat A, Hlatky L, Querfurth H. Multiple effects of beta‐amyloid on single excitatory synaptic connections in the PFC. Front Cell Neurosci. 2013;7:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts PD, Spiros A, Geerts H. Simulations of symptomatic treatments for Alzheimer's disease: computational analysis of pathology and mechanisms of drug action. Alzheimers Res Ther. 2012;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Geerts H, Wikswo J, van der Graaf PH, et al. Quantitative systems pharmacology for neuroscience drug discovery and development: current status, opportunities, and challenges. CPT Pharmacometrics Syst Pharmacol. 2020;9(1):5‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nicholas T, Duvvuri S, Leurent C, et al. Systems pharmacology modeling in neuroscience: Prediction and outcome of PF‐04995274, a 5‐HT4 partial agonist, in a clinical scopolamine impairment trial. Advances in Alzheimer's Disease. 2013;2:83‐98. [Google Scholar]
- 9. Budd Haeberlein S, O'Gorman J, Chiao P, et al. Clinical development of aducanumab, an anti‐abeta human monoclonal antibody being investigated for the treatment of early Alzheimer's disease. J Prev Alzheimers Dis. 2017;4:255‐263. [DOI] [PubMed] [Google Scholar]
- 10. Arndt JW, Qian F, Smith BA, et al. Structural and kinetic basis for the selectivity of aducanumab for aggregated forms of amyloid‐beta. Sci Rep. 2018;8:6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50‐56. [DOI] [PubMed] [Google Scholar]
- 12. Lachman HM, Morrow B, Shprintzen R, et al. Association of codon 108/158 catechol‐O‐methyltransferase gene polymorphism with the psychiatric manifestations of velo‐cardio‐facial syndrome. Am J Med Genet. 1996;67:468‐472. [DOI] [PubMed] [Google Scholar]
- 13. Heils A, Teufel A, Petri S, et al. Functional promoter and polyadenylation site mapping of the human serotonin (5‐HT) transporter gene. J Neural Transm: Gen Sect. 1995;102:247‐254. [DOI] [PubMed] [Google Scholar]
- 14. Vijayan NN, Iwayama Y, Koshy LV, et al. Evidence of association of serotonin transporter gene polymorphisms with schizophrenia in a South Indian population. J Hum Genet. 2009;54:538‐542. [DOI] [PubMed] [Google Scholar]
- 15. Ikeda M, Iwata N, Suzuki T, et al. No association of serotonin transporter gene (SLC6A4) with schizophrenia and bipolar disorder in Japanese patients: association analysis based on linkage disequilibrium. J Neural Transm. 2006;113:899‐905. [DOI] [PubMed] [Google Scholar]
- 16. Hu XZ, Lipsky RH, Zhu G, et al. Serotonin transporter promoter gain‐of‐function genotypes are linked to obsessive‐compulsive disorder. Am J Hum Genet. 2006;78:815‐826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Staeker J, Leucht S, Laika B, Steimer W. Polymorphisms in serotonergic pathways influence the outcome of antidepressant therapy in psychiatric inpatients. Genet Test Mol Biomarkers. 2014;18:20‐31. [DOI] [PubMed] [Google Scholar]
- 18. de Jong IEM, Mork A. Antagonism of the 5‐HT6 receptor ‐ Preclinical rationale for the treatment of Alzheimer's disease. Neuropharmacology. 2017;125:50‐63. [DOI] [PubMed] [Google Scholar]
- 19. Maher‐Edwards G, Zvartau‐Hind M, Hunter AJ, et al. Double‐blind, controlled phase II study of a 5‐HT6 receptor antagonist, SB‐742457, in Alzheimer's disease. Curr Alzheimer Res. 2010;7:374‐385. [DOI] [PubMed] [Google Scholar]
- 20. Sukonick DL, Pollock BG, Sweet RA, et al. The 5‐HTTPR*S/*L polymorphism and aggressive behavior in Alzheimer disease. Arch Neurol. 2001;58:1425‐1428. [DOI] [PubMed] [Google Scholar]
- 21. Mekern VN, Sjoerds Z, Hommel B. How metacontrol biases and adaptivity impact performance in cognitive search tasks. Cognition. 2019;182:251‐259. [DOI] [PubMed] [Google Scholar]
- 22. Weinshilboum RM, Raymond FA. Inheritance of low erythrocyte catechol‐o‐methyltransferase activity in man. Am J Hum Genet. 1977;29:125‐135. [PMC free article] [PubMed] [Google Scholar]
- 23. Leijenaar JF, Groeneveld GJ, Klaassen ES, et al. Methylphenidate and galantamine in patients with vascular cognitive impairment‐the proof‐of‐principle study STREAM‐VCI. Alzheimers Res Ther. 2020;12:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sweet RA, Devlin B, Pollock BG, et al. Catechol‐O‐methyltransferase haplotypes are associated with psychosis in Alzheimer disease. Mol Psychiatry. 2005;10:1026‐1036. [DOI] [PubMed] [Google Scholar]
- 25. Slifstein M, Kolachana B, Simpson EH, et al. COMT genotype predicts cortical‐limbic D1 receptor availability measured with [11C]NNC112 and PET. Mol Psychiatry. 2008;13:821‐827. [DOI] [PubMed] [Google Scholar]
- 26. Fisher PM, Holst KK, Mc Mahon B, et al. 5‐HTTLPR status predictive of neocortical 5‐HT4 binding assessed with [(11)C]SB207145 PET in humans. NeuroImage. 2012;62:130‐136. [DOI] [PubMed] [Google Scholar]
- 27. Liu J, Ogden A, Comery TA, Spiros A, Roberts P, Geerts H. Prediction of efficacy of vabicaserin, a 5‐HT2C agonist, for the treatment of schizophrenia using a quantitative systems pharmacology model. CPT Pharmacometrics Syst Pharmacol. 2014;3:e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Geerts H, Spiros A, Roberts P, Twyman R, Alphs L, Grace AA. Blinded prospective evaluation of computer‐based mechanistic schizophrenia disease model for predicting drug response. PLoS ONE. 2012;7:e49732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nicholas T, Duvvuri Sridhar, Leurent Claire, et al. Systems pharmacology modeling in neuroscience: prediction and outcome of PF‐04995274, a 5HT4 partial agonist, in a clinical scopolamine impairment trial. Advances in Alzheimer's Disease. 2013;2:83‐98. [Google Scholar]
- 30. Spiros A, Carr R, Geerts H. Not all partial dopamine D(2) receptor agonists are the same in treating schizophrenia. Exploring the effects of bifeprunox and aripiprazole using a computer model of a primate striatal dopaminergic synapse. Neuropsychiatr Dis Treat. 2010;6:589‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spiros A, Geerts H. A quantitative way to estimate clinical off‐target effects for human membrane brain targets in CNS research and development. J Exp Pharmacol. 2012;4:53‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woodruff‐Pak DS, Lander C, Geerts H. Nicotinic cholinergic modulation: galantamine as a prototype. CNS Drug Rev. 2002;8:405‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Geerts H, Roberts P, Spiros A. Assessing the synergy between cholinomimetics and memantine as augmentation therapy in cognitive impairment in schizophrenia. A virtual human patient trial using quantitative systems pharmacology. Front Pharmacol. 2015;6:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Darreh‐Shori T, Kadir A, Almkvist O, et al. Inhibition of acetylcholinesterase in CSF versus brain assessed by 11C‐PMP PET in AD patients treated with galantamine. Neurobiol Aging. 2008;29:168‐184. [DOI] [PubMed] [Google Scholar]
- 35. Shinotoh H, Aotsuka A, Fukushi K, et al. Effect of donepezil on brain acetylcholinesterase activity in patients with AD measured by PET. Neurology. 2001;56:408‐410. [DOI] [PubMed] [Google Scholar]
- 36. Kim J, Yoon H, Basak J. Apolipoprotein E in synaptic plasticity and Alzheimer's disease: potential cellular and molecular mechanisms. Mol Cells. 2014;37:767‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chung WS, Verghese PB, Chakraborty C, et al. Novel allele‐dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A. 2016;113:10186‐10191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dumanis SB, Tesoriero JA, Babus LW, et al. ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J Neurosci. 2009;29:15317‐15322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Castellano JM, Kim J, Stewart FR, et al. Human apoE isoforms differentially regulate brain amyloid‐beta peptide clearance. Sci Transl Med. 2011;3:89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lesch KP, Bengel D, Heils A, et al. Association of anxiety‐related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527‐1531. [DOI] [PubMed] [Google Scholar]
- 41. Roses AD, Saunders AM, Alberts MA, et al. Apolipoprotein E E4 allele and risk of dementia. JAMA. 1995;273:374‐375; author reply 5‐6. [PubMed] [Google Scholar]
- 42. Lim YY, Maruff P, Schindler R, et al. Disruption of cholinergic neurotransmission exacerbates Abeta‐related cognitive impairment in preclinical Alzheimer's disease. Neurobiol Aging. 2015;36:2709‐2715. [DOI] [PubMed] [Google Scholar]
- 43. Doraiswamy PM, Sperling RA, Johnson K, et al. Florbetapir F 18 amyloid PET and 36‐month cognitive decline: a prospective multicenter study. Molecular Psychiatry. 2014;19:1044‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Samtani MN, Xu SX, Russu A, et al. Alzheimer's disease assessment scale‐cognitive 11‐item progression model in mild‐to‐moderate Alzheimer's disease trials of bapineuzumab. Alzheimers Dement (N Y). 2015;1:157‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Puig MV. In vivo excitation of GABA interneurons in the medial prefrontal cortex through 5‐HT3 receptors. Cerebral Cortex. 2004;14:1365‐1375. [DOI] [PubMed] [Google Scholar]
- 46. Marcos B, Gil‐Bea FJ, Hirst WD, Garcia‐Alloza M, Ramirez MJ. Lack of localization of 5‐HT6 receptors on cholinergic neurons: implication of multiple neurotransmitter systems in 5‐HT6 receptor‐mediated acetylcholine release. Eur J Neurosci. 2006;24:1299‐1306. [DOI] [PubMed] [Google Scholar]
- 47. Lacroix LP, Dawson LA, Hagan JJ, Heidbreder CA. 5‐HT6 receptor antagonist SB‐271046 enhances extracellular levels of monoamines in the rat medial prefrontal cortex. Synapse. 2004;51:158‐164. [DOI] [PubMed] [Google Scholar]
- 48. Akhondzadeh S, Mohammadi N, Noroozian M, et al. Added ondansetron for stable schizophrenia: a double blind, placebo controlled trial. Schizophr Res. 2009;107:206‐212. [DOI] [PubMed] [Google Scholar]
- 49. Maher‐Edwards G, Dixon R, Hunter J, et al. SB‐742457 and donepezil in Alzheimer disease: a randomized, placebo‐controlled study. Int J Geriatr Psychiatry. 2011;26(5):536‐544. [DOI] [PubMed] [Google Scholar]
- 50. Porsteinsson AP, Drye LT, Pollock BG, et al. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized clinical trial. JAMA. 2014;311:682‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aerssens J, Raeymaekers P, Lilienfeld S, Geerts H, Konings F, Parys W. APOE genotype: no influence on galantamine treatment efficacy nor on rate of decline in Alzheimer's disease. Dement Geriatr Cogn Disord. 2001;12:69‐77. [DOI] [PubMed] [Google Scholar]
- 52. Rogers SL, Friedhoff LT. The efficacy and safety of donepezil in patients with Alzheimer's disease: results of a US Multicentre, Randomized, Double‐Blind, Placebo‐Controlled Trial. The Donepezil Study Group. Dementia. 1996;7:293‐303. [DOI] [PubMed] [Google Scholar]
- 53. Tariot PN, Solomon PR, Morris JC, Kershaw P, Lilienfeld S, Ding C. A 5‐month, randomized, placebo‐controlled trial of galantamine in AD. The Galantamine USA‐10 Study Group. Neurology. 2000;54:2269‐2276. [DOI] [PubMed] [Google Scholar]
- 54. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370:311‐321. [DOI] [PubMed] [Google Scholar]
- 55. Kljajevic V, Grothe MJ, Ewers M, Teipel S. Distinct pattern of hypometabolism and atrophy in preclinical and predementia Alzheimer's disease. Neurobiol Aging. 2014;35:1973‐1981. [DOI] [PubMed] [Google Scholar]
- 56. Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer's disease. Front Aging Neurosci. 2019;11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sydow A, Hochgrafe K, Konen S, et al. Age‐dependent neuroinflammation and cognitive decline in a novel Ala152Thr‐Tau transgenic mouse model of PSP and AD. Acta Neuropathol Commun. 2016;4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Proctor CJ, Boche D, Gray DA, Nicoll JA. Investigating interventions in Alzheimer's disease with computer simulation models. PLoS ONE. 2013;8:e73631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Proctor CJ, Macdonald C, Milner JM, Rowan AD, Cawston TE. A computer simulation approach to assessing therapeutic intervention points for the prevention of cytokine‐induced cartilage breakdown. Arthritis Rheumatol. 2014;66:979‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Willis BA, Sundell K, Lachno DR, et al. Central pharmacodynamic activity of solanezumab in mild Alzheimer's disease dementia. Alzheimers Dement (N Y). 2018;4:652‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Plant LD, Webster NJ, Boyle JP, et al. Amyloid beta peptide as a physiological modulator of neuronal ‘A’‐type K+ current. Neurobiol Aging. 2006;27:1673‐1683. [DOI] [PubMed] [Google Scholar]
- 62. Verdurand M, Chauveau F, Daoust A, et al. Differential effects of amyloid‐beta 1‐40 and 1‐42 fibrils on 5‐HT1A serotonin receptors in rat brain. Neurobiol Aging. 2016;40:11‐21. [DOI] [PubMed] [Google Scholar]
- 63. Lazzari C, Kipanyula MJ, Agostini M, Pozzan T, Fasolato C. Abeta42 oligomers selectively disrupt neuronal calcium release. Neurobiol Aging. 2015;36:877‐885. [DOI] [PubMed] [Google Scholar]
- 64. He P, Ouyang X, Zhou S, et al. A novel melatonin agonist Neu‐P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer' disease. Horm Behav. 2013;64:1‐7. [DOI] [PubMed] [Google Scholar]
- 65. Citrome L, Stensbol TB, Maeda K. The preclinical profile of brexpiprazole: what is its clinical relevance for the treatment of psychiatric disorders? Expert Rev Neurother. 2015;15:1219‐1229. [DOI] [PubMed] [Google Scholar]
- 66. Sabuncu MR, Desikan RS, Sepulcre J, et al. The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011;68:1040‐1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hill E, Karikari TK, Moffat KG, Richardson MJE, Wall MJ. Introduction of tau oligomers into cortical neurons alters action potential dynamics and disrupts synaptic transmission and plasticity. eNeuro. 2019;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sperling RA, Jack CR, Jr , Black SE, et al. Amyloid‐related imaging abnormalities in amyloid‐modifying therapeutic trials: recommendations from the Alzheimer's Association Research Roundtable Workgroup. Alzheimers Dement. 2011;7:367‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lau DT, Mercaldo ND, Shega JW, Rademaker A, Weintraub S. Functional decline associated with polypharmacy and potentially inappropriate medications in community‐dwelling older adults with dementia. Am J Alzheimers Dis Other Demen. 2011;26:606‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information