

Abstract
Vitiligo is an autoimmune depigment disease results from extensive melanocytes destruction. The destruction of melanocyte is thought to be of multifactorial causation. Genome‐wide associated studies have identified single‐nucleotide polymorphisms in a panel of susceptible loci as risk factors in melanocyte death. But vitiligo onset can't be solely attributed to a susceptive genetic background. Oxidative stress triggered by elevated levels of reactive oxygen species accounts for melanocytic molecular and organelle dysfunction, a minority of melanocyte demise, and melanocyte‐specific antigens exposure. Of note, the self‐responsive immune function directly contributes to the bulk of melanocyte deaths in vitiligo. The aberrantly heightened innate immunity, type‐1‐skewed T helper, and incompetent regulatory T cells tip the balance toward autoreaction and CD8+ cytotoxic T lymphocytes finally execute the killing of melanocytes, possibly alarmed by resident memory T cells. In addition to the well‐established apoptosis and necrosis, we discuss several death modalities like oxeiptosis, ferroptosis, and necroptosis that are probably employed in melanocyte destruction. This review focuses on the various mechanisms of melanocytic death in vitiligo pathogenesis to demonstrate a panorama of that. We hope to provide new insights into vitiligo pathogenesis and treatment strategies by the review.
Keywords: autoimmunity, death, melanocyte, oxidative stress, vitiligo
1. INTRODUCTION
Vitiligo is an autoimmune disease featured as chronic depigmentation and milk‐white lesion in the skin with a prevalence of 0.5% of the general population (Figure 1A). Although not accompanied by disturbing senses like pruritus or pain, vitiligo might negatively affect people's self‐esteem and even cause anxiety or depression, as the disfiguring spots hamper their social interaction. 1 What's worse, the curative effects of existent treatments for vitiligo are not optimistic. Moderately effective therapies like phototherapy (psoralen combined with ultraviolet A, narrowband ultraviolet B [NB‐UVB]), topical treatment (corticosteroid, calcineurin inhibitors), and systemic treatment (corticosteroid) are still practically and financially burdensome. 2 The wide prevalence of this chronic disease and unsatisfying therapeutic effect underscore the need to further explore vitiligo pathogenesis. Pathologic destruction of melanocyte is the major foci in vitiligo onset and development, as depicted in Figure 1B,C, and how to forestall and even reverse the process has always been an emphasis of vitiligo basic research. Although both segmental vitiligo (SV) and nonsegmental vitiligo (NSV) exhibit melanocyte destruction, whether the destruction of melanocytes in SV is attributed to the autoimmune response or inherent cellular abnormalities remains obscure. 3 Considering that NSV is more prevalent 4 with its pathogenesis more thoroughly studied, we discuss mechanisms of melanocyte death within the scope of NSV in the following parts and do not distinguish between “NSV” and “vitiligo”. Recent years have seen unprecedented growth in the understanding of vitiligo pathogenesis,which results from a sizeable set of studies focusing on dissecting the molecular mechanism involved in vitiligo melanocyte destruction. And in this review, we summarize these molecular mechanisms and their interplay in the perspective of melanocyte death in the spectrum of genetic predisposition, oxidative stress‐related subversion, and immunological onslaught. The same causal mechanisms by no means apply to all cases, and different pathogenetic mechanisms might work together, ultimately leading to the same clinical result. We also briefly discuss some death modalities putatively harbored in melanocyte destruction (Figure 2). Besides, the cutting‐edge technologies assisting vitiligo diagnosis as well as current status and future perspectives of therapeutic approaches to prevent melanocytes from death is also included in the hope of providing sparks for future research.
Figure 1.
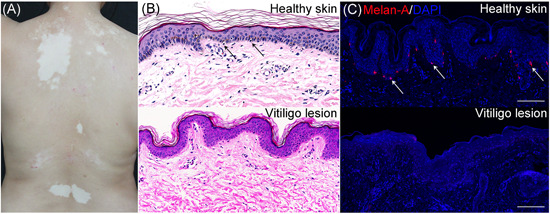
Clinical and pathological manifestation of vitiligo. A, Vitiligo is characterized by chronic depigmentation and milk‐white lesion in the skin. B, Representative images of normal melanocytes in the stratum basale of healthy control and an absence of melanocytes in vitiligo skin. The sections stained with hematoxylin and eosin (×400, black arrowheads in the inset: Melanocytes). C, Healthy skin with normal quantity of melanocytes in contrast to vitiligo lesional skin being devoid of melanocytes on account of melanocyte destruction, as detected by immunofluorescence. Melanocytes in the epidermis were stained with antibodies to Melan‐A (red). Nuclei were counterstained with 4′,6‐diamidino‐2‐phenylindole (blue) (scale bar = 100 μm) (white arrowheads in the inset: Melanocytes) [Color figure can be viewed at wileyonlinelibrary.com]
Figure 2.
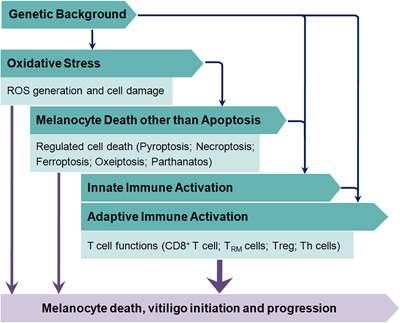
A road map for melanocyte in vitiligo. The figure briefly illustrates the main contents of melanocyte death triggers and the logical relation between them in vitiligo. ROS, reactive oxygen species; Th cells, T‐helper cells; Treg: regulatory T cell; TRM, resident memory T cell [Color figure can be viewed at wileyonlinelibrary.com]
2. GENETIC BACKGROUND
Vitiligo is thought to be of multifactorial causation. Specifically, the concordance of generalized vitiligo in monozygotic twins is approximately 23%. 5 Vitiligo patients’ relatives also suffer an approximately 6‐ to 18‐fold risk for vitiligo than the masses. 5 Additionally, the epidemiological study revealed that single‐nucleotide polymorphisms (SNPs) in multiple risk genes are related to the arising of vitiligo. 6 Concerning all these facts, the genetic background has been emphasized to be a significant endogenous factor in vitiligo pathogenesis. In recent years, extensive genome‐wide associated studies shed light on more than 40 susceptibility loci, including MC1R, TYR, IFIH1, CD44, CD80, GZMB, HLA‐A, XBP1, CAT, and MTHFR. 7 , 8 Of the identified candidate loci, more than half are involved in immunoregulation, T‐cell receptor repertoire, and immune cell‐associated apoptosis, indicating the critical effect of aberrant immune function in vitiligo pathogenesis. 8 The extending list of susceptibility loci might form a scale, to which the results of patients’ gene sequencing could be compared, to evaluate the patients’ “vitiligo risk score". The evaluation might help to optimize vitiligo diagnosis, personalized treatment, and prognosis.
Polymorphisms in TYR and HLA‐A (see below) direct the immune system to target melanocytes. TYR encodes tyrosinase, which is not only an enzyme catalyzing melanogenesis but an autoantigen presented by human leukocyte antigen‐A on the surface of the melanocyte. Tyrosinase epitopes are presented to immature T cells by Langerhans cell to activate an autoimmune response, ultimately induce apoptosis of melanocytes. 9 Intriguingly, the effects of two European‐derived missense variants R402Q (SNP rs1126809) and S192Y (SNP rs1042602) have been solidified in diminishing vitiligo susceptibility by reducing the thermostability of tyrosinase protein, conducing to the decline of tyrosinase epitopes presented, which weakens tyrosinase autoantigen availability. 6 , 10 , 11 HLA‐A encodes the α chain of HLA class I histocompatibility antigen. Jin et al. 12 performed DNA sequence analysis and identified the high‐risk allele HLA‐A*02:01:01:01. 12 The underlying causal mechanism might be an SNP haplotype (rs12206499) spanning transcriptional regulatory region downstream of HLA‐A, which instigates the upregulation of HLA‐A*02:01:01:01 messenger RNA (mRNA), ultimately precipitating autoreactive T‐cell to recognize melanocyte antigens and to destruct melanocytes. 13 Moreover, there exists significant epistasis between HLA‐A SNP rs12206499 and TYR SNP rs1393350, 10 implying the interplay of the two polymorphisms in promoting vitiligo susceptibility.
FAS and FASLG polymorphisms are also critical members of vitiligo SNPs. The Fas–Fas ligand (FasL) system (encoded by FAS and FASLG) controls the final step of cell apoptosis, and functional polymorphisms of the two genes might perturb their transcriptional activities. The FAS polymorphisms (FAS‐1377A>G) and FASLG SNP rs78037977 are reported to be associated with higher vitiligo risk. 8 , 14 Variants at many other loci like CAT (encodes catalase to detoxify hydrogen peroxide [H2O2]), 15 IFIH1 (functions as pattern recognition receptor to activate innate immune response), 16 and GZMB (encodes granzyme B to mediate cytotoxic T cell‐induced apoptosis) 17 also confer increased predisposition to vitiligo onset. Nevertheless, consensus on precise mechanisms whereby most of the candidate genes confer vitiligo risk has not been established, except their epidemiological association with vitiligo susceptibility. After all, the candidate loci merely provide a genetic background, and many other factors such as oxidative stress and derailed immune function must be included to elucidate vitiligo pathogenesis.
3. OXIDATIVE STRESS
Oxidative stress is considered one of the most crucial initiators in vitiligo occurrence, 18 despite consensus on an exact etiology of vitiligo has not been established. Other factors like metabolism possibly engender melanocyte deaths, yet examples backing them up are sparse. 19 Oxidative stress is disturbed redox homeostasis characterized by the imbalance of prooxidants and antioxidants. Oxidative stress in tissue and cell always results from excessive reactive oxygen species (ROS). ROS embody H2O2, hydroxyl radical, hypochlorous acid, and hydroperoxyl radical. 20 In the past several decades, ROS has been proved to be an important second messenger molecule, nevertheless, a high concentration of ROS is implicated in murdering melanocyte in all aspects, including undermining DNA, lipid, protein, and their metabolites structurally and functionally. 21 , 22 Furthermore, ROS‐induced oxidative stress widely instigates aberrant organelle functions, derails metabolism pathways and compromises defensive mechanism against the onslaught of oxidative agents. A growing body of evidence has provided a plausible connection between oxidative stress and deficiency of keratinocyte, melanocyte stem cell, and extracellular microenvironment. 4 , 23 , 24 All the factors, as mentioned above, may help inform our understanding of melanocyte destruction in vitiligo. As for this part, we try to illustrate the panorama of oxidative stress and its association with melanocyte obliteration.
3.1. Source of ROS
The generation of highly enriched ROS to which melanocytes are subject can be attributed to two reasons, excessive formation and inadequate scavenging (Figure 3). Overproduction of ROS is partly triggered by stimuli from the environment, such as ultraviolet (UV) radiation, cytotoxic chemical agents, trauma, pregnancy, stress, and vaccination. 4 , 25 The generation of ROS is an instant intracellular response when the cell is exposed to UV irradiation. 26 Phenols, including monobenzone, hydroquinone, and 4‐tertiary butyl‐phenol, are structurally comparable to melanogenesis substrate tyrosine, thus, capable of covalently binding to tyrosinase, which proceeds to the generation of ROS. 27 , 28 , 29 Excessive ROS might also simultaneously come from melanogenesis and mitochondrial energy metabolism. In the process of melanosynthesis, ROS generation ensues from the conversion of dopa to dopaquinone and dopaquinone to dopachrome. 30 Moreover, melanogenesis is an energy‐consuming procedure, indicating the need for large amounts of adenosine triphosphate (ATP). Biosynthesis of ATP per se is accompanied by the generation of ROS in mitochondria. Mitochondria are suggested to be a dominating source of ROS due to their manipulation of oxidative stress‐related aging and apoptosis. 4 Under cellular stress, electrons escape from the respiratory chain to react with molecular oxygen thus generating superoxide anions and the secondary ROS. 31 In an immortalized perilesional vitiligo melanocyte line PIG3V, suppression on PPARGC1A (encoding peroxisome proliferator‐activated receptor γ coactivator‐1α) expression was attenuated owing to downregulated microRNA (miR)‐211. The expression of PPARGC1A gives rise to the ROS overproduction. 32 Interestingly, keratinocytes appear to transfer H2O2 into neighboring cells including melanocytes, which can be significantly reduced by adding catalase to intercellular media. 33
Figure 3.
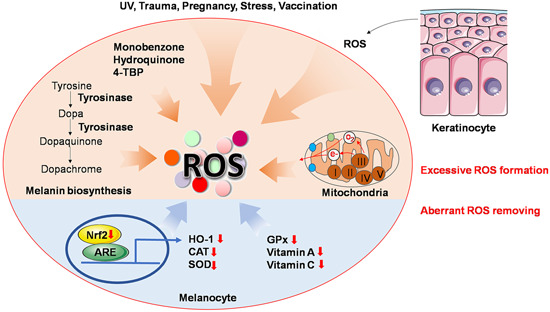
Source of ROS. Accumulation of ROS can be generated by either excessive formation or inadequate scavenging. Environmental stimuli like UV, trauma, pregnancy, stress, and vaccination promote ROS formation. External phenols including monobenzone, hydroquinone and 4‐TBP also give rise to ROS as known. Melanin biosynthesis and energy metabolism in mitochondria physically yield ROS. Neighboring keratinocytes also reportedly transfer ROS to melanocytes. With respect to inadequate scavenging of ROS, downregulated Nrf2 pathway and enzymic or nonenzymic antioxidant agents should be attributable. 4‐TBP, 4‐tertiary butyl‐phenol; ARE, antioxidant response element; CAT, catalase; GPx, glutathione peroxidase; HO‐1, heme oxygenase‐1; Nrf2, nuclear factor E2‐related factor 2; ROS, reactive oxygen species; SOD, superoxide dismutase [Color figure can be viewed at wileyonlinelibrary.com]
Other than excessive formation, aberrant ROS removing mechanisms also account for a plethora of ROS in the epidermis. Downregulated enzymic or nonenzymic defense against oxidants in the epidermis, such as the incapacity of catalase, glutathione peroxidase, and decrease of vitamins A and C underpin the accumulation of ROS. 25 , 34 , 35 Another impaired antioxidant pathway in both melanocyte and keratinocyte, 36 nuclear factor E2‐related factor 2 (Nrf2)‐antioxidant response element (ARE)/heme oxygenase‐1 (HO‐1), also merits mention. Once cellular oxidative stress occurs, the critical transcription factor Nrf2 regulates expression of antioxidant genes by binding to ARE sequence and fuels the transcription of extensive downstream antioxidative enzymes such as HO‐1, catalase, NADH quinone oxidoreductase 1 and superoxide dismutase. 37 , 38 Jian et al. 35 , 39 highlighted that the Nrf2‐ARE pathway deficiency and concomitant antioxidant incapacity confer a persuasive mechanism for melanocytes’ sensitivity towards oxidative stress in vitiligo patients.
3.2. Macromolecules compromised by ROS
Accumulated ROS‐induced oxidative stress in the epidermis of vitiligo patients has been validated. About the impact of oxidative stress on vitiligo occurrence, one plausible hypothesis states that oxidative stress compromises macromolecules in epidermis melanocytes, which precipitate the loss of functional melanocytes. A series of studies have enrolled macromolecule oxidation products such as advanced oxidative protein products, advanced glycosylation end‐products, malondialdehyde, and 8‐hydroxy‐2’‐deoxyguanosine as diameters of evaluating disease progression in vitiligo patients, because levels of these products are directly associated with extension, duration, and disease activity of vitiligo. 40 , 41
Noteworthily, as a result of chronic oxidative stress stimuli, melanocyte DNA undergoes persistent damage. 41 After that, the cell might be orchestrated into cellular senescence featuring irreversible cell proliferation arrest. 42 , 43 The process is governed by a strong cell cycle checkpoint mechanism wherein activation of p53, p16, and p21 are indispensable, 44 and extensive melanocyte cell‐cycle arrest might accentuate the obliteration. Furthermore, ROS‐induced lipid peroxidation might also lead to mitochondrial deterioration, which will be discussed exclusively later. In this context, senescent melanocyte exhibits a special “senescence‐associated secretory phenotype (SASP)” and expresses a multitude of factors including interleukin (IL)‐6, cyclooxygenase, extracellular matrix metalloproteinases (MMPs) and insulin‐like growth factor binding protein. 45 , 46 Part of those secretions might even recruit immune cells to scavenge the “senescent melanocyte,” termed as “senescence‐surveillance” (from antitumor immunity), 47 which is a potential way of melanocyte destruction. It deserves noting that the procedure is a vicious spiral, signals released by the cell via SASP induce senescence of surrounding cells in a paracrine manner, and of themselves in an autocrine manner, thus autonomously enhancing cellular senescence.
3.3. Mitochondrial dysfunction
Mitochondria are not only origin of ROS, but also victims in the flood of oxidative stress (Figure 4). Governing energy supply, aging, and apoptosis of healthy cells, mitochondria are closely linked to the fate of cells, accordingly, mitochondrial impairment is bound to affect melanocyte survival. 4 , 25 , 48 Kang et al. 49 ascertained that melanocyte demise can be interpreted as a transient receptor potential cation channel subfamily M member 2 (TRPM2)‐mediated Ca2+ influx into the cytoplasm and further mitochondrial‐dependent apoptosis. In the upstream, oxidative stress induce the production of TRPM2 by amplifying DNA demethylation. 49 Activated sirtuin 3‐optic atrophy 1 axis is also substantiated to exacerbate mitochondrial damage, which is documented as an indispensable part of oxidative stress‐triggered melanocyte apoptosis. 50 ROS‐associated mitochondrial dysfunction, which is illustrated by mitochondrial outer membrane permeabilization, mitochondrial membrane potential loss, and subsequent apoptosis, have also been reported in other cell lines. 51 , 52 , 53 And the intricate molecular mechanisms of mitochondrial‐associated apoptosis are reviewed in Reference 50.
Figure 4.

Oxidative stress leads to multiple death modalities through various pathways in melanocyte. Excessive ROS hyperactivate the UPR to trigger endoplasmic‐reticulum‐associated melanocytic apoptosis. Translocation of CRT from endoplasmic reticulum under oxidative stress adds to melanocyte immunogenicity. Mitochondria also participate in ROS‐related apoptosis through forming MOMP. Besides, redundant ROS give rise to deficient Nrf2 pathway, generation of HMGB1 and E‐cadherin diminution, which underlie melanocyte apoptosis. Of note, oxidative stress also evokes other modalities of melanocytic death, like necroptosis, necrosis, oxeiptosis, and probably pyroptosis and parthanatos. Some pathways surmised to exist are drawn in dotted lines. Ac‐OPA1, acetylated optic atrophy 1; AIFM1, apoptosis‐inducing factor mitochondrion‐associated 1; ATF6, activating transcription factor 6; CASP1, caspase 1; CHOP, CAAT/enhancer‐binding protein homologous protein; CRT, calreticulin; ER, endoplasmic reticulum; GSDMD, gasdermin D; IRE1α, inositol‐requiring enzyme‐1α; KEAP1, kelch‐like ECH‐associated protein 1; MIF, macrophage migration inhibitory factor; MLKL, mixed lineage kinase domain‐like protein; MOMP, mitochondrial outer membrane permeabilization; mtROS, mitochondrial ROS; Cyto C, cytochrome c; NLRP3, NLR family pyrin domain containing 3; Nrf2, nuclear factor E2‐related factor 2; PARP1, poly(ADP‐ribose) polymerase 1; PERK, PRKR‐like ER kinase; PGAM5, PGAM family member 5; p‐JNK, phosphorylated c‐Jun N‐terminal kinase; RIDD, regulated IRE1‐dependent messenger RNA decay; RIPK1, receptor‐interacting serine/threonine kinase 1; RIPK3, receptor‐interacting serine/threonine kinase 3; SIRT3, sirtuin 3; HMGB1, high‐mobility group box 1; TRPM2, transient receptor potential cation channel subfamily M member 2; UPR, unfolded protein response; ΔΨm, the mitochondrial membrane potential [Color figure can be viewed at wileyonlinelibrary.com]
Aberrant lipid composition and compromised intactness of the respiratory chain in mitochondria are also parts of oxidativestress‐induced impairment. As extremely activated radicals, ROS potently damage biological macromolecules like lipid, reportedly causing lipid peroxidation, less cardiolipin, and more cholesterol in distribution. 54 , 55 Associated with that, the housing of the electron transport chain on lipid is also modified, which is a biochemical basis of mitochondrial impairment. 56 Due to oxidative stress‐derived deficiency in assembly and intrinsic vulnerability to oxidative injury, mitochondrial complex, especially complex I, 32 , 57 exhibits dysfunction and further induces more ROS production thus forming a vicious spiral 58 to destroy melanocyte, and to elevate the sensibility to proapoptotic stimuli. 59
A series of research has been conducted about the effect of several medicines on improving mitochondrial homeostasis. Corresponding results document that baicalein and berberine can restore mitochondrial dysfunction thus promoting melanocyte viability, mainly by decreasing intracellular ROS, downregulating proapoptotic protein levels and facilitating mitochondrial biogenesis. 60 , 61 To conclude, a growing body of evidence has been proving mitochondria as a crux in oxidative stress‐mediated melanocyte obliteration, which is also presenting potential targets for clinical therapy to concur vitiligo.
3.4. Endoplasmic reticulum abnormality
Except being an important site, whereby proteins are synthesized, folded, modified, and transported, the endoplasmic reticulum (ER) is also an organelle sensing cellular stress and regulating the survival or perishment of the cell. Among the various exogenous and endogenous perturbations, like UV irradiation, redox stress, infections and chemical agents, oxidative stress is a vital factor for ER dysfunction in vitiligo (Figure 4). 21 , 62 Morphologically, dilated ER in melanocytes from vitiligo patients was observed, 63 and the effect of ROS on ER dilation can't be excluded. Recently, a series of research substantiates that ER subverted by ROS even induces melanocytes apoptosis. Cellular responses to perturbation like excessive ROS in ER function, termed “ER stress,” is characterized as aberrantly folded protein in the ER lumen, which further triggers unfolded protein response (UPR). Subtoxic UPR is required for restoring normal cellular function, but excessive and prolonged UPR might be a cell death mechanism. 64
UPR comprises three parallel signaling branches: Inositol‐requiring enzyme 1α‐X‐box‐binding protein 1 pathway, activating transcription factor 6 pathway and PRKR‐like ER kinase‐eukaryotic translation initiation factor 2α pathway. Continuously activated by oxidative stress‐induced ER stress, the three branches might work independently or in concert to elicit apoptosis (Figure 4). 65 Highly enriched molecular mechanisms link UPR and apoptosis (like regulated IRE1 dependent decay and phosphorylated Jun amino‐terminal kinase pathways), 65 however, scarcely are there researches to thoroughly substantiate the specific underlying mechanism in vitiligo. Oxidative stress‐triggered Ca2+ disturbance in the ER may also evoke UPR and the final apoptosis. 66 In oxidative stress‐affected melanocyte and keratinocyte, the release of UPR‐regulated cytokines and chemokines (IL‐6, IL‐8, C‐X‐C motif chemokine ligand [CXCL] 12, CC chemokine ligand [CCL] 5) also recruits immune cells and indirectly devotes to melanocyte destruction. 21 , 67 Noteworthily, UPR‐induced autophagy is speculated to stimulate the production of exosomes and soluble inflammatory signals, which are proinflammatory and autoimmunity‐promoting. 62 But autophagy is essentially a protective response in the face of cellular stress, 68 and defective autophagy adds to melanocytic sensitivity towards oxidative stress. 69 For this assessment, we think autophagy is more of an antiapoptotic mechanism, whose deficiency might lead to melanocytic apoptosis in the pathogenesis of vitiligo.
The effect of ROS also extends to other macromolecular in ER. Calreticulin (CRT), a ubiquitous ER protein modulating intracellular Ca2+ homeostasis, is manifested to be positively related to lesional area and duration of vitiligo in patients. The putative mechanism is that oxidative stress propels CRT redistribution from the ER lumen to cell membrane thus elevating the immunogenicity of stressed melanocyte and induces apoptosis (Figure 4). Furthermore, CRT stimulates the generation of proinflammatory cytokines that recruit immunocytes to eliminate melanocytes. 25 , 70 ER is an organelle standing at the crossing of cellular biosynthesis and survival, and is sensitive to various stimuli especially oxidative stress. The central role ER plays in melanocyte destruction of vitiligo pathogenesis can never be overemphasized.
3.5. Keratinocyte‐mediated melanocyte loss
Keratinocyte numerically dominates the cell composition of the epidermis, it also interacts with melanocyte directly. 71 , 72 Cumulative research indicate that keratinocyte is not only an innocent bystander in melanocyte destruction, but a vital nexus in vitiligo pathomechanism. Keratinocyte is a mediator of oxidative stress detriment, secreting cytokines to recruit autoreactive T cells and interfering signal transduction in melanocytes which consequently induce melanocyte destruction. 73 , 74 Intriguingly, Ca2+ and membrane‐associated thioredoxin reductase (TR) synergistically plays a kindred role in melanocytes undermining. Vitiliginous keratinocyte reportedly manifests defective Ca2+ uptake kinetics, which underlies an elevated‐Ca2+ microenvironment, thus TR, a pivotal enzyme modulating free‐radical defense and melanin biosynthesis, is allosterically inhibited and culminates in defense breakdown and melanogenesis inhibition in melanocytes. 75 , 76 Shi et al. 77 substantiated that oxidative stress triggers overexpression of miR‐25 in keratinocytes and melanocytes, a microRNA that epigenetically suppresses the expression of microphthalmia‐associated transcription factor thus leading to a dysfunctional response to oxidative stress due to decreased activity of antioxidative enzymes and impaired transportation of melanosome. Moreover, the elevated level of miR‐25 undermines the paracrine of growth factors like stem cell factor and basic fibroblast growth factor from keratinocytes, both of which support melanocyte melanogenesis and survival. Similar to miR‐25, high‐mobility group box 1 (HMGB1) released from keratinocytes stimulated by oxidative stress may also put melanogenesis and melanocyte survival in peril, mainly by downregulating the activity of melanogenetic molecular like gp100, and initiating the apoptosis signaling, respectively. 78 Parenthetically, HMGB1 translocating from melanocyte nucleus to cytoplasm in oxidative stress could also induce apoptosis through suppressing Nrf2 expression. 79
Apart from the aforementioned factors, keratinocyte‐dependent recruitment of immunocyte under oxidative stress is also implicated in melanocyte destruction. IL‐17 is reported to be upregulated in the skin/serum of vitiligo patients. 80 Excessive IL‐17 promotes IL‐1β secretion from keratinocytes via the ROS–NOD‐like receptors (NLR) family pyrin domain containing 3 (NLRP3)‐caspase pathway, thus coordinating local inflammation and attracts and activates adaptive immunocytes. 58 Stimulated by ROS, secretion of CXCL‐16 and trans‐presentation of IL‐15 in keratinocytes recruits and activates CD8+T cell, respectively, and mediate melanocyte‐specific cytotoxic response. 73 , 81 ROS‐induced release of ATP from keratinocyte might not only stimulate neighboring keratinocytes to generate CXCL9 to chemoattract CD8+T cell, but also trigger caspase‐3‐mediated melanocyte apoptosis. 82 Besides, in the context of ROS, elevated TRPM2 expression gives rise to NLRP3 inflammasome activation. In the downstream, CD8+T cell migration and effector function are both potentiated by IL‐1β/IL‐1R signaling. 83 Hence keratinocyte is decisive for recruiting autoreactive immunocytes to kill melanocytes, as elaborated in the last several emblematic studies.
In vitiligo patients, melanocytes in the basal layer of unaffected zone epidermis are 25% less compared with healthy control, and melanocytes detachment from the basal layer into the upper layer has also been observed, which is termed as “transepidermal loss”. 84 The results described herein concur to delineate a pathogenetic scenario that melanocyte detach from the basal layer was followed by transepidermal migration and eventually, melanocyte death occurs. 85 , 86 Specifically, unlike keratinocyte–keratinocyte adhesion, melanocytes are born poorly attached to surrounding keratinocyte due to less E‐cadherin expressed on surface. 85 Under oxidative stress, elevated Src kinase expression and altered lipid raft arrangement disrupt protein interaction of the β‐catenin with E‐cadherin on melanocyte surface, ultimately weaker melanocyte–keratinocyte adhesion and enhanced melanocyte detachment. 87 , 88 And the shedding of E‐cadherin seems to be precipitated also by type‐1 cytokines like interferon (IFN)‐γ and tumor necrosis factor (TNF)‐α through activated MMP‐9, according to a recent publication. 89 Furthermore, the detachment disrupts fibronectin adhesion‐mediated apoptosis suppression, and migrating to the upper layer of epidermis indicates more onslaught from deleterious stimulations like UV, both of which intensify melanocytes apoptosis and necrosis. 90 , 91 Taken together, oxidative stress amplifies the innate deficiency of melanocyte–keratinocyte adhesion and causes melanocyte transepidermal loss. 92 Keratinocyte‐related mechanisms summarized above might work in parallel or hierarchical fashion to trigger or aggravate melanocyte obliteration in vitiligo pathogenesis (Figure 4).
3.6. Potential therapeutic approaches to ameliorate oxidative stress
Inferring from the factors mentioned above, we think it's high time to find effective therapeutic approaches to ameliorate oxidative stress in vitiligo patients to prevent melanocytes from destruction. NB‐UVB is reported to relieve oxidative stress in vitiligo patients. 93 But the evidence remains to be solidified considering redox‐related indicators in erythrocyte are far from portraying epidermis redox status in this study. Aside from phototherapy, a sizeable set of antioxidants have been validated to protect melanocytes from oxidative stress‐induced mortality in vitro or/and in vivo. Polyphenols are preferred owing to their antioxidant activities. Polyphenols compounds including Ginkgo biloba extracts, apigenin and baicalin have all been documented in vitro to activate the Nrf2 pathway to counteract oxidative stress as yet. 61 , 94 , 95 Quercetin also possesses anti‐inflammatory and antioxidative capacity, albeit not studied in vitiligo treatment. 96 Apart from polyphenols, an array of organics, such as glycyrrhizin, afzelin, 6‐Shogaol, simvastatin, aspirin, also demonstrated their antioxidative ability by targeting Nrf2 pathways. 97 , 98 , 99 , 100 , 101 Vitamin D can ameliorate ROS in melanocytes and restore their viability in oxidative stress through activating Wnt/β‐catenin pathway. 102 Surprisingly, adipose tissue secretome might normalize H2O2‐ and UV‐induced oxidative stress in fibroblasts, despite its relatively weak efficacy in melanocytes. 103 Inorganic substances like molecular hydrogen, palladium and platinum also target Nrf2 pathways in vitro, 104 , 105 but they are still far from therapeutically adopted considering the poor operability of molecular hydrogen and allergenic activity of metals.
Albeit extensively studied and applied, the antioxidants mentioned above are still not recommended as monotherapy in clinical practice. The main reason is the limited efficacy of antioxidants observed in clinical trials due to small numbers of participants and heterogeneity in the design of the trials. Thus, clinical trials with larger amounts of patients and more standardized compatibilities of the antioxidants should be implemented in the future. Moreover, we can conclude that highly enriched studies on counteracting oxidative stress are centered on scavenging the ROS that have been generated. But research on how to retard the excessive generation of ROS in the upstream are still sparse. Future studies might focus on restricting redundant ROS emission in melanocyte metabolism to control oxidative stress forwardly.
For now, innovative strategies for drug delivery, namely, nano‐drug delivery systems, 106 are under development as reviewed in Reference 107. Particles like liposomes, niosomes, microemulsions and nanoparticles carrying drugs permeate into the deeper skin layer and play as the drug repository to slow down the drug release and reduce the frequency of treatments. 107 Several in vitro experiments have been carried out utilizing the nano‐drug delivery system, the preliminary results of which are optimistic. 108 , 109 Additionally, precise drug delivery might also be employed to promote drug accumulation around the target cell and protect melanocyte from death. The affinity between receptors and ligands are utilized in this strategy. Vectors with ligand activity carry drugs that could target melanocyte‐specific receptors like melanocortin‐1 receptor and endothelin receptor. 106 The combination of efficient drug delivery systems and efficacious drugs would upgrade vitiligo therapies to a higher level.
4. OTHER MELANOCYTE DEATH MODALITIES
Concluding from what was mentioned earlier, we find that apoptosis is the most extensively documented way of melanocyte demise, with few melanocytes undergo necrosis. But forms of cell death are not merely restricted to apoptosis and necrosis. Cells may die from accidental cell death (ACD) or regulated cell death (RCD). ACD, like necrosis, is biologically uncontrolled, whereas RCD (apoptosis, necroptosis, pyroptosis, oxeiptosis, ferroptosis, parthanatos, etc.) involves precise signaling cascade and molecular mechanisms, despite rare studies on nonapoptotic melanocyte death in vitiligo etiology. Hence, we introduce several nonapoptotic RCD forms and their possible connection with melanocyte destruction in vitiligo, in hope of providing potential directions for future studies (Figure 4).
4.1. Pyroptosis
Pyroptosis is mechanistically distinct from other forms of cell death with gasdermin D (GSDMD) and caspase‐1/4/5/11 activation as its defining feature. 110 Upon being stimulated by damage‐associated molecular patterns molecules (DAMPs), pathogen‐associated molecular patterns molecules or altered homeostasis, caspases are activated to cleave the downstream GSDMD. GSDMD fragment with membrane pore‐forming activity yields plasma membrane rupture, cytosolic content release, and concurrent cell swelling and lysis. 111 Pyroptotic cell releases a formidable number of cytokines and proteins that may act as “find me” or “eat me” signals. Recently, pyroptosis has been found to participate in target cell death induced by cytotoxic lymphocytes through granzyme‐A‐cleaved gasdermin B. 112 These results raise the interesting possibility in vitiligo, that pyroptosis might partly contribute to ROS‐ or cytotoxic T lymphocytes (CTLs)‐induced melanocyte death and the ensuing immunological outcomes.
4.2. Necroptosis
Necroptosis is a programmed form of necrosis, featured by receptor‐interacting serine/threonine kinase (RIPK) 3‐mixed lineage kinase domain‐like pseudokinase (MLKL) pathway. 113 It can be triggered by various stimuli, for example, activation of toll‐like receptors (e.g., TLR3 and TLR4), 114 death receptors (e.g., Fas), 115 and adhesion receptors. 116 Besides, previous research reported mitochondrial events like the production of mitochondrial ROS and the presence of mitochondrial permeability transition pore might trigger necroptosis. 117 , 118 Our laboratory has disclosed that oxidative stress precipitates melanocyte necroptosis via RIPK1‐RIPK3‐MLKL pathway (article accepted by Journal of Investigative Dermatology) in vitiligo. And further studies should be carried out to elucidate the immune consequence of melanocyte necroptosis.
4.3. Ferroptosis
Ferroptosis is an iron‐ and lipotoxicity‐dependent form of RCD, characterized by dysmorphic small mitochondria with decreased crista and condensed outer membranes in the absence of hallmarks of apoptosis or necroptosis. 107 , 119 A panel of vital factors like inactivation of glutathione peroxidase, 107 p53 elevation, 120 ROS accumulation and lipid peroxidation of membranes in ferroptosis 107 is also shared by aforementioned melanocyte demise. Additionally, ferroptosis is reported to partake in IFN‐γ‐associated cell destruction, and that IFN‐γ could lead to melanocyte death is well‐established in vitiligo pathogenesis. 121 , 122 But whether ferroptosis is a participant in melanocyte destruction is undefined.
4.4. Oxeiptosis
Oxeiptosis is a ROS‐induced noninflammatory RCD. This novel kind of cell death is independent of apoptotic or pyroptotic caspases, necroptosis, autophagy and ferroptosis. 123 Oxeiptosis is mediated by kelch‐like ECH‐associated protein 1 (KEAP1)‐PGAM family member 5 (PGAM5)‐apoptosis‐inducing factor mitochondrion‐associated 1 (AIFM1) pathway. KEAP1 is responsible for sensing and quantification of ROS. In low ROS concentration, KEAP1 activates Nrf2 as a cellular protectant, whereas in high ROS concentration, KEAP1 might release PGAM5 and interact with AIFM1 thus initiating the death signaling. 123 We have found that oxeiptosis existed in the perilesional skin of vitiligo patients and ROS‐treated melanocytes manifested oxeiptosis (unpublished data). We surmise that self‐antigen exposure might be the immunological consequence of melanocyte oxeiptosis, despite oxeiptosis reportedly does not cause inflammatory reaction. 124 The exact effect of melanocyte oxeiptosis on vitiligo pathogenesis remains to be explicated.
4.5. Parthanatos
Parthanatos is a caspase‐independent RCD activated by oxidative stress‐induced DNA damage. 125 In parthanatos, DNA base injured by oxidative stress triggers poly polymerase‐1 (PARP‐1) hyperactivation. Overactivated PARP‐1 synthesizes PAR polymer to elicit the translocation of AIFM1 from the mitochondria to the nucleus, and the AIFM1 recruits macrophage migration inhibitory factor (MIF) to cleave genomic DNA into large fragments. 126 Parthanatos is widely deployed in stroke, myocardial infarction, neurodegenerative diseases, and ROS‐induced injuries. 126 It deserves noting that MIF concentrations were also found to be higher in serum and lesional skin of vitiligo patients. 127 , 128 Thus, whether parthanatos is instrumental in the oxidative stress‐related disease, vitiligo, might be explored in the future. And inhibiting parthanatos to make melanocyte step back from the brink of parthanatotic cell death might be well pursuing.
So far, we have introduced the genetic background of vitiligo, oxidative stress‐induced melanocyte destruction, and other putative nonapoptotic regulated cell death modalities. On one hand, they provide evidence for the death of minority melanocytes in vitiligo. On the other hand, they bring about leakage of dead cell antigens and generation of the inflammatory microenvironment to trigger the immunity against melanocyte or “immunogenic cell death (ICD)” (a term in tumor immunity describing exposure or release of endogenous signaling from tumor cell rendering their death immunogenic). 113 The ICD is the final form of most of the melanocytes which are killed in vitiligo. Thus, we employed a chart to illustrate that the genetic background of vitiligo, oxidative stress‐induced melanocyte destruction, and other putative nonapoptotic regulated cell death modalities that dictate melanocyte demise as well as pave the road for the following immunological consequences, the death of more melanocytes (Figure 5).
Figure 5.
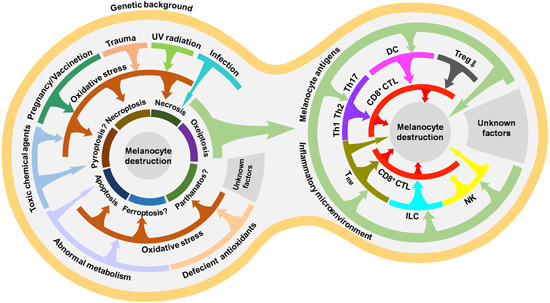
Overview of factors triggering melanocyte death in vitiligo. In the predisposing genetic background, environmental stimuli, aberrant metabolism and deficient antioxidants indirectly cause several possible death modalities of a fraction of melanocytes through ROS, though minor direct death‐triggering pathways exist. Outcomes of these melanocyte deaths, antigens exposure and inflammatory microenvironment, confer the majority of melanocyte demise via immune cells breaching the self‐tolerance. CD8+CTL, CD8+ cytotoxic T lymphocytes; DC, dendritic cell; ILC, innate lymphoid cells; NK, natural killer cells; Th1, T‐helper cell 1; Th17, T‐helper cell 17; Th2, T‐helper cell 2; Treg, regulatory T cell; TRM, resident memory T cell [Color figure can be viewed at wileyonlinelibrary.com]
5. INNATE IMMUNE ACTIVATION
In skin reside cells from nearly every division of the immune system, from macrophages, nature killer (NK) cells, and innate lymphoid cells (ILC) to CTL, regulatory T cells (Treg) and resident memory T (TRM) cells. These cells and associated cytokines/chemokines work in concert to form an immunological barrier and scavenge intrusive pathogens. In pathological conditions, however, the immunological barrier recognizes self‐antigens and destruct target cells, thus leading to autoimmune disease. On the basis of the research published hitherto, aberrantly heightened innate immune might play the role of initiator in the derailed immune function, considering that the immune system activates in a precise spatio‐temporal order (Figure 6). Under stress, DAMPs embodying melanocyte‐specific neoantigens like inducible heat shock protein 70 (HSP70i), HMGB1, S100B, and mitochondria DNA are released into the extracellular microenvironment in the form of cell debris and exosome. These DAMPs carrying danger signals further provoke innate immune activation 28 via pattern recognition receptors (PRR) embracing TLRs, RIG‐I‐like receptors and NLRs. This process might both adversely kill melanocytes and ignite adaptive immune through generating cytokines/chemokines and antigen‐presenting. 129 , 130 Discussion about several critical innate immune subgroups contributing to the process is presented as followed.
Figure 6.
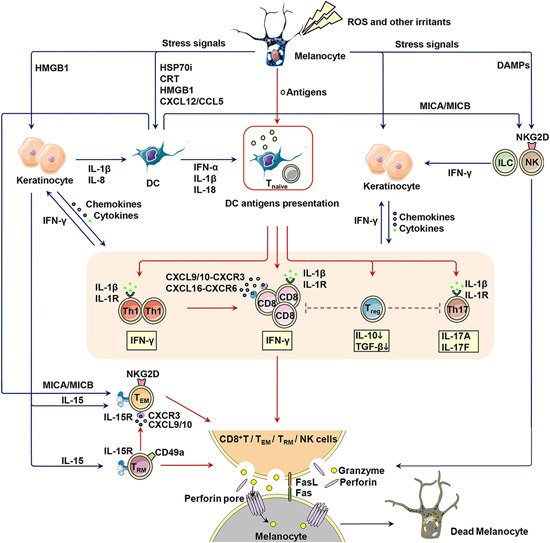
Aberrantly activated immune response in vitiligo. Outlined in the figure are members and their reciprocal relationship of the aberrantly activated innate and adaptive immunity in vitiligo pathogenesis. The interaction between innate immune cells are drawn in blue long‐tail arrow, and the interaction between adaptive immune cells in red. CCL5, CC chemokine ligand 5; CRT, calreticulin; CXCL12, C‐X‐C motif chemokine ligand 12; CXCL16, C‐X‐C motif chemokine ligand 16; CXCL9/10, C‐X‐C motif chemokine ligand 9/10; CXCR3, C‐X‐C motif chemokine receptor 3; CXCR6, C‐X‐C motif chemokine receptor 6; DAMP, damage‐associated molecular patterns molecule; DC, dendritic cell; Fas/FasL, Fas/Fas ligand; HMGB1, high‐mobility group box 1; HSP70i, inducible heat shock protein 70; IFN‐α, interferon‐α; IFN‐γ, interferon‐γ; IL‐10, interleukin‐10; IL‐15, interleukin‐15; IL‐15R, interleukin‐15 receptor; IL‐17A, interleukin‐17A; IL‐17F, interleukin‐17F; IL‐1R, interleukin‐1 receptor; IL‐1β, interleukin‐1β; IL‐8, interleukin‐18; ILC, innate lymphoid cells; MICA/MICB, major histocompatibility complex class I chain‐related protein A and B; NK, nature killer cells; NKG2D, natural killer group 2D; PRR, pattern recognition receptors; ROS, reactive oxygen species; TEM, effector memory T cells; TGF‐β, transforming growth factor‐β; Th1, T‐helper cells; Th17, T‐helper cell 17; TLR, toll‐like receptors; Tnaive, naïve T cells; Treg, regulatory T cells; TRM, resident memory T cells [Color figure can be viewed at wileyonlinelibrary.com]
Once released into the extracellular milieu, DAMPs exert an extensive effect on cell populations in the skin, such as dendritic cell (DC), NK, ILC, keratinocyte, and melanocytes per se. First, DAMPs propel the process of antigen exposure and presenting. Essentially being a ubiquitous cytoprotectant preventing the cell from undergoing apoptosis, 131 HSP70i might enhance the delivery of melanocyte antigens to major histocompatibility complex (MHC) class II expressed on melanocytes, thus potentiating immunogenicity of the stressed melanocyte, as observed in other cell lines. 132 Both HSP70i and HMGB1 bind to PRR on DC to beget its maturation and antigen‐presenting process. 133 , 134 , 135 Second, DAMPs cultivate a proinflammatory milieu to elicit an adaptive immune response. DCs activated by DAMPs release a cascade of cytokines spanning IFN‐α, IL‐1β, and IL‐18 into the extracellular environment. 135 , 136 Stimulated keratinocytes also generate IL‐1β, IL‐8, CXCL9, CXCL10, and CXCL16. 133 , 134 This proinflammatory milieu further introduces activated NK and chemoattracted melanocyte‐specific CD8+T cells to melanocytes and spawns their apoptosis. Intriguingly, of the mentioned cytokines and chemokines, many are reported to be biomarkers for vitiligo like IL‐1β, CXCL9, CXCL10, and CXCL16, which might aid vitiligo diagnosis and prognosis. 81 , 137 , 138 Recent proteomic studies have also proposed a panel of proinflammatory factors like lysophosphatidylcholine, platelet‐activating factor, succinic acid, CXCL4, and CXCL7 to be potential vitiligo biomarkers. 139 There is also an elegant study integrating genomics and proteomics to define a vitiligo diseasome network, and further identified a series of potential biomarkers like CXCL12, fibronectin 1, chloride intracellular channel 1, catenin β1. 140 Albeit these results of proteomic studies might assist vitiligo diagnosis, their mechanical implication in vitiligo pathogenesis remain enigmatic.
Whereas some melanocytes might have died even before the CD8+T cell arrives. HSP70‐exposed TNF related apoptosis‐inducing ligand (TRAIL)+DC are lytic towards TRAIL‐receptor‐expressing melanocytes. 141 , 142 Cells expressing TRAIL are also found to infiltrate the papillary dermis of patients with inflammatory vitiligo. 143 Besides, upon endogenous and exogenous stress, NK, and ILC1 secret IFN‐γ to induce expression of C‐X‐C motif chemokine receptor (CXCR) 3B on melanocyte surface and release of CXCL9, CXCL10, and CXCL11 from keratinocyte and melanocyte. CXCL10 then activates CXCR3B and triggers the apoptosis of melanocytes. 130 This result is solidified by another research illustrating that CXCR3 depleting antibodies are efficacious in preventing depigmentation. 144
Of note, expression of “stress molecules” like MHC class I chain‐related protein A and B (MICA/MICB) also increases on DC. MICA/MICB might further activate natural killer group 2D receptor expressed by NK cells and a subgroup of cytotoxic αβ T cells like effector memory T cells (TEM), which might unleash their effector mechanisms, followed by melanocytes being killed and release of autoantigen from dying melanocytes. 145
Parenthetically, a newly emergent lymphoid subclass called ILC is garnering increased attention. The ILC family is a heterogeneous population of non‐B non‐T lymphocytes encompassing ILC1, ILC2, ILC3, and, possibly, regulatory ILC subclasses. 146 , 147 They share many similarities with T cells and are even suggested to be the innate counterparts of T cell subsets. ILC family manifests great complexity due to their subgroup interconversion and heterogeneity between individuals. 147 Interestingly, their residency in nonlymphoid tissues in steady state and rapid response under stimulation are somehow akin to TRM. 148 And their implication in killing melanocytes still merits more detailed analysis.
Here we can conclude that deranged innate immunity paves the road for the coming of further adaptive immunity by creating a local inflammatory milieu and destructing a minority of melanocyte to release melanocyte‐specific antigens.
A cessation of the abnormal innate immunity is urgently needed as a part of vitiligo management, unfortunately, novel strategies to accomplish the goal are few. Contributions of Mosenson et al. 149 are the mainstay in the development of strategies for innate immunity‐normalization in vitiligo. In 2013, the team switched one amino acid in the sequence of HSP70i through a DNA vaccine, and the mutated HSP70i prevented the activation of DC into inflammatory status thus abrogated the concomitant antigen‐specific T cells accumulation. 149 They successfully reversed the depigmentation in mice with spontaneous vitiligo. In 2018, similar results were acquired in Sinclair swine. 150 And now the team is planning for administering the gene‐based approach in patients, which might induce long‐term repigmentation of vitiligo lesions. As for pharmacotherapy, topically applied vitamin D is reported to reduce the number of DC in the vitiligo skin. 151 Besides, in psoriasis, vitamin D also inhibited the function of DC. 152 But the efficacy of vitamin D in vitiligo treatment still warrants clarification. And the phototherapy, NB‐UVB might also be associated with decreased Langerhans cell distribution in vitiligo epidermis after treatment. 153
Apart from what we have discussed, IL‐27 was also reported to act on DC to suppress the T cell function, thus employing a DNA vaccine to topically overexpress IL‐27 is also therapeutically exploitable. 154 However, members functioning in the innate immune system are not restricted to DC, and pharmacological exploration on regulating NK cell and other cells might also be concerned.
6. ADAPTIVE IMMUNE ACTIVATION
Distinguished from innate immune activation, adaptive immunity is characterized by giving antigen‐specific responses and memorizing antigens. In vitiligo, adaptive immune activation is responsible for killing melanocytes specifically, leaving other cells in the vicinity untouched (Figure 6). It's also a major player in melanocyte destruction in vitiligo. In this part, we mainly talk about T cells rather than B cells, as it remains an open conundrum whether B‐cell‐mediated humoral immunity participates in vitiligo pathogenesis.
6.1. Executioner: CD8+T cell
In cell‐specific autoimmune diseases, such as multiple sclerosis, rheumatoid arthritis, and vitiligo, CD8+CTL has always been underpinned as a fundamental role in disease progression. 155 , 156 The notion that CD8+T cell serves as the last procedure in the process of extensive melanocytes destruction and confers the final and fatal onslaught to melanocytes in the pathogenesis of vitiligo is widely accepted. In fact, the character of CD8+T cell as the executioner in vitiligo was gradually elucidated by a multitude of elegant experiments as follows. Higher serum frequency of melanocyte‐specific CD8+T was observed in vitiligo patients compared to their healthy counterparts. 157 Additionally, their frequency is related to disease severity. 28 Patchy infiltration of T cells was found adjacent to melanocytes, especially in the leading edge of vitiligo depigmentation. 158 , 159 , 160 The proportion of T cells in this area is also CD8+T‐skewed, as manifested by elevated CD8+T cell number and decreased CD4/CD8 ratio. 160 Isolated T cells from biopsies exhibited autoreactivity to a series of melanocyte‐specific antigens, such as melan‐A, gp100 and tyrosinase, and cytotoxicity to autologous melanocytes in vitro. 161 , 162 The depletion of CD8+T cells abrogates melanocyte destruction, however, enrichment for these cells aggravates it. 161 Recently, programmed cell death protein (PD)‐1 and T‐cell immunoglobulin and mucin‐domain containing‐3 molecules, which serve as immune checkpoints to mediate braking of cytotoxic response, are found to be highly expressed on vitiligo CD8+T cells. This is surmised to be a compensation mechanism of the immune system to keep peripheral tolerance towards self‐tissue, in face of hyperactivated CD8+T cells. 163 A study administrating programmed death‐ligand 1 on vitiligo mice successfully reversed the depigmentation also endorses the participation of immune checkpoint in self‐immunoresponse against melanocytes. 164
Mechanically, CD8+T cell mediates melanocyte apoptosis in various ways. Secreting perforin/granzymes and applying Fas–FasL mechanisms are the two most frequently mentioned. 165 But differing views disputed about the contribution of the two mechanisms to melanocyte destruction in vitiligo. Research based on a mouse model with DNA immunization against a xenogeneic form of TRP‐2 revealed that autoimmunity against normal melanocytes required perforin but Fas–FasL was dispensable. 166 Of the other three studies, depending on transgenic mice with tyrosinase‐specific T cell clone, 167 transgenic mice carrying T cells with an HLA‐A2 restricted human tyrosinase reactive T cell receptor (TCR) 168 and mice manifesting melanoma‐induced autoimmune symptoms (vitiligo‐like white coat), 143 respectively, the former two regard perforin as an unnecessary mechanism in melanocyte destruction. And the first and third affirmed the implication of the Fas–FasL system in killing melanocyte. The inconsistency among these research probably results from the heterogeneity of modeling methods. And these vitiligo models are simulating different pathophysiology of distinct genetic background and risk factors. Thus, the four viewpoints might not exclude each other in vitiligo pathogenesis. The contrasting mechanisms employed in transgenic or self‐antigen challenged cytotoxic‐T‐cells‐induced melanocyte destruction also merit further exploration. Moreover, IFN‐γ released by CD8+T cells also dictates the apoptosis of melanocytes in multifaceted ways. First, the IFN‐γ‐CXCL9/CXCL10‐CXCR3 axis further collects more CD8+T cells and promotes their effector function, leading to a severe immune response. 122 , 144 , 169 , 170 Another study established the role of IFN‐γ in increasing the ratio of Treg versus effector T cells. 168 Second, a transient pulse of IFN‐γ may provide a microenvironment of locally high concentrations at which IFN‐γ exerts a cytotoxic effect on melanocytes precisely and efficaciously, leaving other cells in the vicinity uninfluenced. 171 Third, IFN‐γ generated by CD8+T might impair cysteine uptake and trigger ferroptosis in tumor cells as recently reported, 121 whether this mechanism operates in IFN‐γ‐associated melanocyte destruction in vitiligo remains to be clearly delineated.
To conclude, extensive studies having lent credence to the role of executioner played by CD8+T cell in vitiligo, researchers have shifted their interest to explore activation, chemotaxis, suppression, homing, residency, and survival of CD8+CTL, exactly the part we will expound later.
6.2. Alarm sensor and cytotoxic killer: Tissue‐resident memory T cells (TRM) cells
Vitiligo lesions are reversible in the course of the disease. About 50% recurrences take place at the same location within one year after discontinued treatment, 172 this provides some hint as to a memory component of autoimmunity in vitiligo. Memory T cells, especially TRM cells, are proposed to be the culprit underlying this “skin memory disease.” 173
According to the expression of the homing receptors and migratory patterns, memory cells protecting human skin are parsed into four subgroups, which were labeled central memory T cells (TCM), TEM, migratory memory T cells, and TRM. 174 , 175 Canonical surface markers of CD69 (a T cell activation marker), CD103 (integrin αE) and CD49a (α‐subunit of the α1β1 integrin receptor) are frequently utilized to distinguish epidermal TRM from circulating memory cells. In healthy adult human skin, most TRM cells are CD103−CD4+ and reside in the dermis. While CD103+TRM, both CD4+ and CD8+, are enriched in the basal layer epidermis. 176 Skin TRM cells are large in number, but variable among individuals. 177 Of the estimated 2.0 × 1010 resident T cells in the entire skin surface, 178 skin TRM occupies a large fraction, with the proportions vary between 20% and 60%. 175 Assuming a protective role in normal organisms, TRM appears after the resolution of skin inflammation, persists for a long time in tissue, and is endowed with the propensity for more rapid response to invasive pathogen or tumor cells than circulatory memory cells. 172 , 179 , 180 , 181 Aside from eliminating pathogens, when aberrantly activated and sensitized to harmless self‐antigens, nonetheless, TRM might contribute to autoimmune diseases. 182 TRM is also long‐lived. Because the role of CD4+TRM is still ill‐defined in vitiligo, we discuss only CD8+TRM in this part.
Lack of recirculation is a characteristic feature of TRM, and the mechanism for its residency in the skin is being gradually unveiled. TRM residency is a reciprocal interaction between TRM endogenous motivating factors and the microenvironment. TRM has to adapt to local survival cues, tethering within the skin and ignore egress signals. 183 CD103 is the α‐subunit of the α3β7 integrin receptor which enables TRM to anchor to epithelial cells by binding to E‐cadherin. However, binding to E‐cadherin is dispensable for TRM residency, 71 though TRM cells expressing CD103 are more potent in generating cytokines. 174 Sphingosine1‐phosphate receptor 1 is required for lymphocyte egress from peripheral tissues, intriguingly motivated by transcription factor Kruppel‐like factor 2, and CD69 enhances its internalization and degradation thereby maintaining TRM residency in the skin. 184 CD8+TRM in the skin also requires the aryl hydrocarbon receptor for long‐term survival. 185 To facilitate TRM conversion, differentiation, development and residency, tissue also forms appropriate cytokine niches embodying IL‐12, IL‐15, IL‐18, IL‐33, IFN‐β, TNF‐α, and transforming growth factor (TGF)‐β. 184 , 186 , 187 , 188 Among the cytokines, TGF‐β induces expression of CD103, 189 and IL‐15 supports TRM development, maintenance, and survival. 188 Reliance of TRM on IL‐15 is substantiated by several studies, for example, melanocyte‐specific TRM cells cluster in high IL‐15 production sites like hair follicles in the vitiligo skin, 190 and antibody blockade of IL‐15 signaling durably reverses depigmentation. 181
As an alarm sensor, TRM alone is insufficient for inducing and maintaining depigmentation though possessing cytotoxicity. Ensuing melanocyte‐specific antigen stimulation, CD8+TRM secrets IFN‐γ, which proves to be apoptosis‐inducing. 171 CD49a+CD8+TRM are even poised for cytotoxic responses by producing perforin and granzyme B. 176 CD8+TRM residing around hair follicular might also curtail the entry of melanocyte precursors from the follicular reservoir to vitiligo lesions, thus conferring disease flares or repigmentation blockade. 175 But the seminal research by Richmond et al. 191 solidified that TRM must work in concert with recirculating T cells in vitiligo pathogenesis, through depleting recirculating TCM or inhibiting their recruitment to the skin to successfully reverse vitiligo. Vigorously secreted IFN‐γ is also recruiting TCM and B cells to the sites TRM resides in, presumably by upregulating the production of downstream chemokines and vascular cell adhesion molecule 1. 191 , 192 Similarly, CD49a−CD8+TRM also manifests its robust competence in recruiting other immune cells via IL‐17 production. 176
Deranged TRM is a critical component of the overridden natural immunity intertwined in the development of vitiligo. Accordingly, we summarized TRM hallmarks and residency along with their functions in melanocyte destruction in the hope of illuminating novel therapies to tip the balance towards autoimmune suppression or tolerance. Nevertheless, methods to fundamentally deplete TRM from the skin are still far from a definite therapeutic effect. 181 , 193 Considering the effect of TRM in countermining infectious pathogen and tumor cell challenge, an in‐depth understanding of TRM must be obtained before manipulating them in therapeutic approaches.
6.3. Incompetent suppressor: Treg
Tregs contribute to immune homeostasis by suppressing immune activation and maintaining peripheral self‐tolerance. They enforce a negative effect on the activation and expansion of autoreactive CD4+ or CD8+ T cells. 194 , 195 That melanocyte‐specific CD8+ T cells can be detected in healthy people without vitiligo suggests autoimmune kept in check, but in vitiligo, the checkpoint is invalid. 196 The sparse Treg found in vitiligo skin is also indicative of autoreactive CD8+ T cells' unopposed action. 197
The transcription factor Forkhead box P3 (FoxP3) remains the most reliable Treg marker. Apart from FoxP3, Tregs are also known to highly express CC chemokine receptor (CCR) 4 and CCR7, both of which mediating Tregs homing to tissues together with their ligands CCL21 and CCL22. 194 CD25, cytotoxic T lymphocyte‐associated antigen‐4, PD‐1, and inducible T‐cell costimulator are also expressed on Treg surface as suppressive molecules. 198 Treg employs mainly two mechanisms in suppressive function, namely, cytokine secretion (TGF‐β1 and IL‐10) and cell–cell contact. Also, Tregs reportedly restrain melanocyte‐specific CD8+ T cells by inducing their anergy and dampening their proliferation and cytokine production (IFN‐γ, TNF‐α, and IL‐2). 199 Tregs‐orchestrated downregulation of costimulatory molecules CD80 and CD86 on antigen‐presenting cells contributes to the process of suppression as well.
Extensive reports have delineated the landscape of disequilibrium between hyperactivated autoreactive CD8+ T cells and incompetent suppressive Tregs, with some displaying contradictory results. But research having effectively found the underlying cause of the incapable immunosuppression are few. Numbers and proportion of serum and skin Tregs are found to be significantly decreased in a series of reports 195 , 200 , 201 when comparing vitiligo patients with healthy controls. Intriguingly, some studies demonstrated unaltered or even elevated numbers of Treg in vitiligo skin. 196 , 202 , 203 Discrepancies between studies about counts of Tregs might be attributed to (1) each sampling reflects merely a cross‐section of a developing disease progression, thus the panorama of changing Treg counts can't be obtained; (2) many other activated CD4+ cells also express FoxP3 and FoxP3 antibodies employed in the experiment can also bind to them 204 ; (3) racial and age factors. 194 Several studies illustrated the notably decreased levels of TGF‐β 195 , 201 , 205 and IL‐10 205 , 206 in the serum of vitiligo patients in contrast to controls. FoxP3 expression in serum and lesional skin, 201 FoxP3 mRNA expression in lesional and perilesional skin declined in vitiligo patients. 207 The two points indicate functional deficiencies in Tregs derived from vitiligo patients, though only a few studies tried to find the underlying pathomechanism. Klarquist et al. 196 attributed sparse Tregs in vitiligo skin to reduced expression of CCL22‐induced Treg homing failure. Zhang et al. 195 ascribed Treg incapability to a diminution of functional modulator HO‐1, and applying HO‐1 agonist restored immunoregulatory Treg function probably by increasing IL‐10 and latency‐associated peptide expression. 195 A subset of Treg maintaining in tissue for relatively long periods termed memory Tregs (mTregs) is also found to be dysfunctional in psoriasis. And the contribution of mTregs to vitiligo pathogenesis remains to be elucidated in the future. 208
Tregs deficiency‐induced unrestricted autoimmune response culminates in melanocyte death in vitiligo pathogenesis. Therefore, the analysis above might contribute to the clarification of mechanisms underlying incompetent Tregs and melanocyte destruction.
6.4. Probable booster: T helper cells
Early studies found that CD4+ T helper (Th) cells infiltrating the perilesional skin are type‐1‐skewed, 209 , 210 with their proportion reaching approximately 95%. 178 This leads to enhanced production of inflammatory cytokines like IFN‐γ and TNF‐α. 211 Moreover, keratinocytes respond to Th1 cytokines by eliciting the migration of Th1 and to Th2 cytokines by recruiting both Th1 and Th2 cells. 212 This imbalanced sensitivity to Th1‐ and Th2‐ cytokines might partly explain the Th1‐ polarization in vitiligo skin. CD4+ T cells are also involved in melanocyte loss by destroying MHC class II expressing cells via Fas–FasL interaction, and melanocytes exactly express both MHC class II and Fas. 213 The participation of Th2 in vitiligo pathogenesis has not been validated, though Th2‐produced IL‐4 can inhibit melanogenesis directly in vitro. 214
Another subgroup of T‐helper cells, Th17, together with its cytokine IL‐17 is implicated in the etiology of several autoimmune diseases, psoriasis, rheumatoid arthritis, multiple sclerosis, and primary Sjogren's syndrome included. 215 And its role remains to be further defined in vitiligo though progress has been obtained. Th17 has been found to infiltrate the vitiliginous skin. 129 They diffusely infiltrate the upper dermis, hence Th17 cells putatively act on melanocytes through cytokines secretion instead of direct interaction. 129 A wealth of studies has illustrated that levels of serum Th17, 216 , 217 serum IL‐17, 80 , 216 , 218 , 219 lesional tissue IL‐17 mRNA, 80 , 220 perilesional skin IL‐17A and IL‐17A receptor 129 are all relatively higher in vitiligo patients. That NB‐UVB light treatment could decrease lesional and perilesional IL‐17 expression accompanied by a reduction in Vitiligo Area Scoring Index (VASI) reiterates Th17‐participation in vitiligo. 207 Singh et al. 215 reviewed that serum IL‐17 levels are positively correlated with the extent of body area involvement, VASI and Vitiligo Disease Activity. Accordingly, Th17 and IL‐17 might participate in the pathomechanism of vitiligo given the evidence enumerated above.
Mechanistically, the capacity for melanogenesis and antiapoptosis could be dampened by IL‐17 in melanocyte in vitro. 129 It should be highlighted that Zhou et al. 58 demonstrated IL‐17‐stimulated melanocytes experience apoptosis because of mitochondrial dysfunction and ROS accumulation. This result directly connects IL‐17 to melanocyte demise. Besides, in a study of adoptive cell transfer therapy in melanoma, the authors illustrated that Th17‐polarized CD4+ cells confer antitumor efficacy and concomitant autoimmune vitiligo, despite the efficacy is surprisingly IFN‐γ‐dependent. 221 Studies of Speeckaert et al. 222 , 223 provide insights into a new paradigm of Th17 in vitiligo. They showed that Th17.1 cells instead of Th17 cells are increased in vitiligo. As a subset of Th17 cells, Th17.1 cells secret both IL‐17 and IFN‐γ and gradually differentiate into nonclassical Th1 cells. And there is a delicate Th17/Th17.1/Th1 balance which is altered significantly in the disease progress.
Although a series of publications have underpinned the putative role of Th17 in melanocyte destruction, outcomes of therapy pertinent to Th17 still could not meet people's anticipation. For example, secukinumab (IL‐17A inhibitor) application leads to new depigmentation onset in patients rather than halt vitiligo development in the clinical trial. 223 This indicates that the effect of Th17 should be properly understood, especially in the context of complex plasticity of Th17 encountering different environmental triggers. 224 And deeper insights into immune mechanisms might instigate an ongoing discussion about the role of Th17 in vitiligo.
6.5. Possible interventions to retrieve adaptive immune anomalies
Immunosuppressive therapies have always been the mainstay of vitiligo treatment, which is echoed by the profound effect of immunology anomaly on melanocyte destruction. For now, the recommended immunosuppressive interventions include topical steroids, calcineurin inhibitors (tacrolimus and pimecrolimus), and oral pulse steroids. But these interventions are just moderately effective and financially burdensome. Fortunately, researchers have been translating basic research into target therapies. And in this part, we will discuss state‐of‐the‐art strategies for targeted immunomodulation in vitiligo.
As previously mentioned, IFN‐γ‐CXCL9/CXCL10‐CXCR3 axis is indispensable in the killing of melanocytes by the CD8+ T cells, and compromising the axis was effective in halting vitiligo progression and facilitating repigmentation. 122 , 144 , 225 Mediated by Janus kinase (JAK) 1 and 2, in this axis, keratinocyte senses IFN‐γ and generates CXCL9/CXCL10. 122 Rosmarin et al. 226 focus on the research of JAK inhibitors in vitiligo treatment, which is of high clinical relevance and therapeutic significance. Their prospective, randomized, vehicle‐controlled phase‐2 trial revealed that JAK inhibitor ruxolitinib was effective as monotherapy in vitiligo treatment, without treatment‐related serious adverse events reported. 226 However, this therapeutic option must be sustained to protect from disease relapses. That JAK inhibitors might just abrogate chemotactic approach of cytotoxic cells instead of dislodging the TRM, which recruits cytotoxic cells to the skin, might explain the failure in generating durable treatment responses. 193
Referring to TRM, it might be a culprit manipulating vitiligo relapses. Their continuous recruitment of autoreactive cytotoxic T cells from circulation by secreting IFN‐γ should be targeted to realize durably vitiligo reverse. Richmond et al. 181 has successfully depleted TRM from vitiligo lesional skin through long‐term administration of the anti‐CD122 antibody. CD122 is a subunit of the heterotrimeric IL‐15 receptor, thus, CD122 blockade might impair the residency of TRM in the skin by nullifying the support from IL‐15. 181 Besides, TRM exhibits unique metabolic panorama compared to effector T cells and other memory T cells. TRM relies on exogenous free fatty acid (FFA) uptake to maintain its residency in the skin. Accordingly, it might be promising to intercept the critical metabolism pathway to deplete TRM, for example, trimetazidine, which blocks mitochondrial β‐oxidation of FFA to decrease the longevity of TRM in the skin. 227 , 228
Treg is endowed with the immunosuppressive properties, hence it is always acclaimed as “living drugs.” In therapeutic strategies for vitiligo, “quantitively” or “qualitatively” potentiating Treg might be an optimistic option due to its endogeneity. First, Treg expansion is an efficient strategy. IL‐2 is fundamental for the differentiation, survival, and function of Treg. 229 Moreover, Treg expresses the high‐affinity IL‐2 receptor to collect IL‐2 from the microenvironment. Thus IL‐2 therapy is speculated to be efficacious in Treg expansion. Low‐dose IL‐2 treatment has exhibited efficacy in alopecia areata, 230 systemic lupus erythematosus, 231 type 1 diabetes, 232 and hepatitis virus C‐induced vasculitis. 233 Besides, TNF receptor 2 (TNFR2) is highly expressed on Treg compared to other T cells, and using specific antibodies or agonists to stimulate TNFR2 is reported to efficiently expand natural Tregs in graft‐versus‐host disease. 234 Furthermore, targeting the Notch‐1 signaling pathway reportedly enhance Treg populations and suppressive function, 235 which is pharmacologically exploitable. Another possible option is adoptive Treg therapy, which is harvesting Treg from the patient's circulation to expand them in vitro, and transfer the expanded Tregs back to the patient. But the scarcity of Tregs in the blood and slow rate of expansion in vitro are the major constraints for the clinical application. 236 Alternative to Treg expansion, elevating the recruitment of Treg to the epidermis through gene‐gun‐treatment‐induced CCL22 overexpression also delayed vitiligo progression. 237
Second, “qualitatively” edited Treg has been much heralded in recent years. Antigen‐specific Tregs accumulate in targeted tissue and function 1,000– to 10,000‐ fold higher than the nonspecific “bystander” Tregs. It is, therefore, tempting to speculate investing a pool of antigen‐specific Treg into one treatment. Approaches like genetically engineered Treg cells expressing chimeric antigen receptor (CAR) or artificial TCR enable targeting specific antigens of interest. With the help of CAR and TCR technology, abundant antigen‐specific Tregs can be acquired through in vitro expansion. 238 Apart from adding specificity to Tregs, T cells with specificity can be transformed into Tregs. The effector T cells could portray the immunosuppression of Treg after lentiviral‐based overexpression of FoxP3. 239 But the transduced Treg is at the risk of instability that may still function as effector T cells. Additionally, mesenchymal stem cell is being developed to exert their broad immunosuppressive capacity on autoimmune diseases. 240 Limitations still exist, however, considering the current processes of Treg manufacturing are costly and labor‐intensive. Multiple obstacles must be circumvented before these technologies are applied in vitiligo, in light of the low‐priority of vitiligo in global health despite its impact on patients’ psychological health.
An objective assessment method of therapeutic efficacy is necessary in vitiligo management, in addition to standard photographing protocol 241 and helpful instruments (Wood's lamp, dermoscopy, and in vivo confocal microscopy). To meet the clinical needs, several studies employed imaging analysis to evaluate the changes of the depigmentation area, but restricted by factors like large depigmentation, curved body surface and complex repigmentation patterns. 242 , 243 , 244 , 245 A recent publication introduced a delicate method to generate VASI automatically, through superpixel‐generated computerized digital image analysis of clinical photographs with ImageJ. 246 Another study proposed a protocol for calculating the true depigmentation area which could serve as a “gold standard” for validation of other digital tools. 247 The combination of these two methods might facilitate both vitiligo diagnosis and therapeutic efficacy assessment.
7. CONCLUSIONS
In the context of genetic vulnerability, ROS is a crucial player in the initiation of vitiligo. ROS contribute to the melanocyte destruction in the starting stage from many aspects, like DNA damage, lipid peroxidation, mitochondrial dysfunction and endoplasmic reticulum stress. Besides, investigations into the death modalities like necroptosis, pyroptosis, ferroptosis, oxeiptosis, and parthanatos should extend from tumor research to melanocyte death studies in vitiligo to discover new vitiligo therapies. Derailed immunity is a central factor in melanocyte destruction in vitiligo. Innate immunity paves the road for adaptive immunity response through generating inflammatory microenvironment and antigen‐presenting. In the adaptive immunity, Treg fails to suppress the autoreactive immunity, TRM works in concert with CD8+CTL to kill melanocytes with support of Th. Besides, measures to potentiate the suppressive capability of Treg or scavenge dysfunctional TRM specifically to rebuild the self‐tolerance in vitiligo remain further development.
From environmental stimulus to ROS‐induced damage and autoimmunity, the program of killing melanocytes is activated in a roughly hierarchical fashion in vitiligo pathogenesis, as depicted in Figure 5. The figure might help to better understand the mechanisms of vitiligo onset and development, and the blank representing unknown factors underlying the melanocyte destruction needs to be fulfilled by further studies.
ACKNOWLEDGMENT
This study has received funding from the National Natural Science Foundation of China (nos. 81930087, 91742201, and 81625020).
Chen J, Li S, Li C. Mechanisms of Melanocyte Death in Vitiligo. Med Res Rev. 2021;41:1138–1166. 10.1002/med.21754
Contributor Information
Shuli Li, Email: lishli@fmmu.edu.cn.
Chunying Li, Email: lichying@fmmu.edu.cn.
REFERENCES
- 1. Dabas G, Vinay K, Parsad D, Kumar A, Kumaran MS. Psychological disturbances in patients with pigmentary disorders: a cross‐sectional study. J Eur Acad Dermatol Venereol. 2020;34(2):392–399. [DOI] [PubMed] [Google Scholar]
- 2. Frisoli ML, Essien K, Harris JE. Vitiligo: mechanisms of pathogenesis and treatment. Annu Rev Immunol. 2020;38:621–648. [DOI] [PubMed] [Google Scholar]
- 3. van Geel N, Speeckaert R. Segmental vitiligo. Dermatol Clin. 2017;35(2):145–150. [DOI] [PubMed] [Google Scholar]
- 4. Picardo M, Dell'Anna ML, Ezzedine K, et al. Vitiligo. Nat Rev Dis Primers. 2015;1:15011. [DOI] [PubMed] [Google Scholar]
- 5. Alkhateeb A, Fain PR, Thody A, Bennett DC, Spritz RA. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16(3):208–214. [DOI] [PubMed] [Google Scholar]
- 6. Spritz RA, Andersen GH. Genetics of vitiligo. Dermatol Clin. 2017;35(2):245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin Y, Birlea SA, Fain PR, et al. Genome‐wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44(6):676–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jin Y, Andersen G, Yorgov D, et al. Genome‐wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat Genet. 2016;48(11):1418–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spritz RA. Modern vitiligo genetics sheds new light on an ancient disease. J Dermatol. 2013;40(5):310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin Y, Birlea SA, Fain PR, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med. 2010;362(18):1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spritz RA. Six decades of vitiligo genetics: genome‐wide studies provide insights into autoimmune pathogenesis. J Invest Dermatol. 2012;132(2):268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jin Y, Ferrara T, Gowan K, et al. Next‐generation DNA re‐sequencing identifies common variants of TYR and HLA‐A that modulate the risk of generalized vitiligo via antigen presentation. J Invest Dermatol. 2012;132(6):1730–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hayashi M, Jin Y, Yorgov D, et al. Autoimmune vitiligo is associated with gain‐of‐function by a transcriptional regulator that elevates expression of HLA‐A*02:01 in vivo. Proc Natl Acad Sci USA. 2016;113(5):1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li M, Sun D, Li C, et al. Functional polymorphisms of the FAS gene associated with risk of vitiligo in Chinese populations: a case‐control analysis. J Invest Dermatol. 2008;128(12):2820–2824. [DOI] [PubMed] [Google Scholar]
- 15. Mansuri MS, Jadeja SD, Singh M, Laddha NC, Dwivedi M, Begum R. The catalase gene promoter and 5'‐untranslated region variants lead to altered gene expression and enzyme activity in vitiligo. Br J Dermatol. 2017;177(6):1590–1600. [DOI] [PubMed] [Google Scholar]
- 16. Jin Y, Andersen GHL, Santorico SA, Spritz RA. Multiple functional variants of IFIH1, a gene involved in triggering innate immune responses, protect against vitiligo. J Invest Dermatol. 2017;137(2):522–524. [DOI] [PubMed] [Google Scholar]
- 17. Xu M, Liu Y, Liu Y, et al. Genetic polymorphisms of GZMB and vitiligo: a genetic association study based on Chinese Han population. Sci Rep. 2018;8(1):13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Di Dalmazi G, Hirshberg J, Lyle D, Freij JB, Caturegli P. Reactive oxygen species in organ‐specific autoimmunity. Auto Immun Highlights. 2016;7(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen J, Zhuang T, Chen J, et al. Homocysteine induces melanocytes apoptosis via PERK‐eIF2alpha‐CHOP pathway in vitiligo. Clin Sci (Lond). 2020;134(10):1127–1141. [DOI] [PubMed] [Google Scholar]
- 20. Wang X, Hai C. Novel insights into redox system and the mechanism of redox regulation. Mol Biol Rep. 2016;43(7):607–628. [DOI] [PubMed] [Google Scholar]
- 21. Manga P, Elbuluk N, Orlow SJ. Recent advances in understanding vitiligo. F1000Res. 2016;5(F1000 Faculty Rev):2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sastry KS, Naeem H, Mokrab Y, Chouchane AI. RNA‐seq reveals dysregulation of novel melanocyte genes upon oxidative stress: implications in vitiligo pathogenesis. Oxid Med Cell Longev. 2019;2019:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y, Zhang CF, Rossiter H, et al. Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J Invest Dermatol. 2013;133(6):1629–1637. [DOI] [PubMed] [Google Scholar]
- 24. Speeckaert R, Dugardin J, Lambert J, et al. Critical appraisal of the oxidative stress pathway in vitiligo: a systematic review and meta‐analysis. J Eur Acad Dermatol Venereol. 2018;32(7):1089–1098. [DOI] [PubMed] [Google Scholar]
- 25. Xie H, Zhou F, Liu L, et al. Vitiligo: How do oxidative stress‐induced autoantigens trigger autoimmunity? J Dermatol Sci. 2016;81(1):3–9. [DOI] [PubMed] [Google Scholar]
- 26. Hofer S, Stonig M, Wally V, et al. Contradictory effects of chemical filters in UV/ROS‐stressed human keratinocyte and fibroblast cells. ALTEX. 2019;36(2):231–244. [DOI] [PubMed] [Google Scholar]
- 27. van den Boorn JG, Picavet DI, van Swieten PF, et al. Skin‐depigmenting agent monobenzone induces potent T‐cell autoimmunity toward pigmented cells by tyrosinase haptenation and melanosome autophagy. J Invest Dermatol. 2011;131(6):1240–1251. [DOI] [PubMed] [Google Scholar]
- 28. Richmond JM, Frisoli ML, Harris JE. Innate immune mechanisms in vitiligo: danger from within. Curr Opin Immunol. 2013;25(6):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frisoli ML, Harris JE. Vitiligo: mechanistic insights lead to novel treatments. J Allergy Clin Immunol. 2017;140(3):654–662. [DOI] [PubMed] [Google Scholar]
- 30. Denat L, Kadekaro AL, Marrot L, Leachman SA, Abdel‐Malek ZA. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol. 2014;134(6):1512–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. [DOI] [PubMed] [Google Scholar]
- 32. Sahoo A, Lee B, Boniface K, et al. MicroRNA‐211 regulates oxidative phosphorylation and energy metabolism in human vitiligo. J Invest Dermatol. 2017;137(9):1965–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ostankovitch M, Robila V, Engelhard VH. Regulated folding of tyrosinase in the endoplasmic reticulum demonstrates that misfolded full‐length proteins are efficient substrates for class I processing and presentation. J Immunol. 2005;174(5):2544–2551. [DOI] [PubMed] [Google Scholar]
- 34. Khan R, Satyam A, Gupta S, Sharma VK, Sharma A. Circulatory levels of antioxidants and lipid peroxidation in Indian patients with generalized and localized vitiligo. Arch Dermatol Res. 2009;301(10):731–737. [DOI] [PubMed] [Google Scholar]
- 35. Jian Z, Li K, Song P, et al. Impaired activation of the Nrf2‐ARE signaling pathway undermines H2O2‐induced oxidative stress response: a possible mechanism for melanocyte degeneration in vitiligo. J Invest Dermatol. 2014;134(8):2221–2230. [DOI] [PubMed] [Google Scholar]
- 36. Kim H, Park CS, Lee AY. Reduced Nrf2 activation in PI3K phosphorylation‐impaired vitiliginous keratinocytes increases susceptibility to ROS‐generating chemical‐induced apoptosis. Environ Toxicol. 2017;32(12):2481–2491. [DOI] [PubMed] [Google Scholar]
- 37. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1‐Nrf2‐ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. [DOI] [PubMed] [Google Scholar]
- 38. Kobayashi M, Yamamoto M. Nrf2‐Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. [DOI] [PubMed] [Google Scholar]
- 39. Jian Z, Li K, Liu L, et al. Heme oxygenase‐1 protects human melanocytes from H2O2‐induced oxidative stress via the Nrf2‐ARE pathway. J Invest Dermatol. 2011;131(7):1420–1427. [DOI] [PubMed] [Google Scholar]
- 40. Vaccaro M, Bagnato G, Cristani M, et al. Oxidation products are increased in patients affected by non‐segmental generalized vitiligo. Arch Dermatol Res. 2017;309(6):485–490. [DOI] [PubMed] [Google Scholar]
- 41. Mitra S, de Sarkar S, Pradhan A, et al. Levels of oxidative damage and proinflammatory cytokines are enhanced in patients with active vitiligo. Free Radic Res. 2017;51(11–12):986–994. [DOI] [PubMed] [Google Scholar]
- 42. Watanabe S, Kawamoto S, Ohtani N, Hara E. Impact of senescence‐associated secretory phenotype and its potential as a therapeutic target for senescence‐associated diseases. Cancer Sci. 2017;108(4):563–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gluck S, Ablasser A. Innate immunosensing of DNA in cellular senescence. Curr Opin Immunol. 2019;56:31–36. [DOI] [PubMed] [Google Scholar]
- 44. Chandler H, Peters G. Stressing the cell cycle in senescence and aging. Curr Opin Cell Biol. 2013;25(6):765–771. [DOI] [PubMed] [Google Scholar]
- 45. Ohtani N. Deciphering the mechanism for induction of senescence‐associated secretory phenotype (SASP) and its role in aging and cancer development. J Biochem. 2019;166:289–295. [DOI] [PubMed] [Google Scholar]
- 46. Bellei B, Pitisci A, Ottaviani M, et al. Vitiligo: a possible model of degenerative diseases. PLOS One. 2013;8(3):e59782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre‐malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. [DOI] [PubMed] [Google Scholar]
- 48. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. [DOI] [PubMed] [Google Scholar]
- 49. Kang P, Zhang W, Chen X, et al. TRPM2 mediates mitochondria‐dependent apoptosis of melanocytes under oxidative stress. Free Radic Biol Med. 2018;126:259–268. [DOI] [PubMed] [Google Scholar]
- 50. Yi X, Guo W, Shi Q, et al. SIRT3‐dependent mitochondrial dynamics remodeling contributes to oxidative stress‐induced melanocyte degeneration in vitiligo. Theranostics. 2019;9(6):1614–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gogvadze V, Norberg E, Orrenius S, Zhivotovsky B. Involvement of Ca2+ and ROS in alpha‐tocopheryl succinate‐induced mitochondrial permeabilization. Int J Cancer. 2010;127(8):1823–1832. [DOI] [PubMed] [Google Scholar]
- 52. Sharaf MS, Stevens D, Kamunde C. Zinc and calcium alter the relationship between mitochondrial respiration, ROS and membrane potential in rainbow trout (Oncorhynchus mykiss) liver mitochondria. Aquat Toxicol. 2017;189:170–183. [DOI] [PubMed] [Google Scholar]
- 53. Wang Q, Huang L, Yue J. Oxidative stress activates the TRPM2‐Ca(2+)‐CaMKII‐ROS signaling loop to induce cell death in cancer cells. Biochim Biophys Acta, Mol Cell Res. 2017;1864(6):957–967. [DOI] [PubMed] [Google Scholar]
- 54. Dell'Anna ML, Ottaviani M, Albanesi V, et al. Membrane lipid alterations as a possible basis for melanocyte degeneration in vitiligo. J Invest Dermatol. 2007;127(5):1226–1233. [DOI] [PubMed] [Google Scholar]
- 55. Dell'Anna ML, Ottaviani M, Bellei B, et al. Membrane lipid defects are responsible for the generation of reactive oxygen species in peripheral blood mononuclear cells from vitiligo patients. J Cell Physiol. 2010;223(1):187–193. [DOI] [PubMed] [Google Scholar]
- 56. Dell'Anna ML, Ottaviani M, Kovacs D, et al. Energetic mitochondrial failing in vitiligo and possible rescue by cardiolipin. Sci Rep. 2017;7(1):13663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dell'Anna ML, Urbanelli S, Mastrofrancesco A, et al. Alterations of mitochondria in peripheral blood mononuclear cells of vitiligo patients. Pigment Cell Res. 2003;16(5):553–559. [DOI] [PubMed] [Google Scholar]
- 58. Zhou J, An X, Dong J, et al. IL‐17 induces cellular stress microenvironment of melanocytes to promote autophagic cell apoptosis in vitiligo. FASEB J. 2018;32(9):4899–4916. [DOI] [PubMed] [Google Scholar]
- 59. Wang Y, Li S, Li C. Perspectives of new advances in the pathogenesis of vitiligo: from oxidative stress to autoimmunity. Med Sci Monit. 2019;25:1017–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jiang W, Li S, Chen X, et al. Berberine protects immortalized line of human melanocytes from H2O2‐induced oxidative stress via activation of Nrf2 and Mitf signaling pathway. J Dermatol Sci. 2019;94(1):236–243. [DOI] [PubMed] [Google Scholar]
- 61. Ma J, Li S, Zhu L, et al. Baicalein protects human vitiligo melanocytes from oxidative stress through activation of NF‐E2‐related factor2 (Nrf2) signaling pathway. Free Radic Biol Med. 2018;129:492–503. [DOI] [PubMed] [Google Scholar]
- 62. Harris JE. Cellular stress and innate inflammation in organ‐specific autoimmunity: lessons learned from vitiligo. Immunol Rev. 2016;269(1):11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Boissy RE, Liu YY, Medrano EE, Nordlund JJ. Structural aberration of the rough endoplasmic reticulum and melanosome compartmentalization in long‐term cultures of melanocytes from vitiligo patients. J Invest Dermatol. 1991;97(3):395–404. [DOI] [PubMed] [Google Scholar]
- 64. Park K, Lee SE, Shin KO, Uchida Y. Insights into the role of endoplasmic reticulum stress in skin function and associated diseases. FEBS J. 2019;286(2):413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13(3):184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Carreras‐Sureda A, Pihan P, Hetz C. Calcium signaling at the endoplasmic reticulum: fine‐tuning stress responses. Cell Calcium. 2018;70:24–31. [DOI] [PubMed] [Google Scholar]
- 67. Rezk AF, Kemp DM, El‐Domyati M, et al. Misbalanced CXCL12 and CCL5 chemotactic signals in vitiligo onset and progression. J Invest Dermatol. 2017;137(5):1126–1134. [DOI] [PubMed] [Google Scholar]
- 68. Marino G, Niso‐Santano M, Baehrecke EH, Kroemer G. Self‐consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. He Y, Li S, Zhang W, et al. Dysregulated autophagy increased melanocyte sensitivity to H2O2‐induced oxidative stress in vitiligo. Sci Rep. 2017;7(1):42394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Y, Liu L, Jin L, et al. Oxidative stress‐induced calreticulin expression and translocation: new insights into the destruction of melanocytes. J Invest Dermatol. 2014;134(1):183–191. [DOI] [PubMed] [Google Scholar]
- 71. Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9(10):679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Delmas V, Larue L. Molecular and cellular basis of depigmentation in vitiligo patients. Exp Dermatol. 2019;28(6):662–666. [DOI] [PubMed] [Google Scholar]
- 73. Chen X, Guo W, Chang Y, et al. Oxidative stress‐induced IL‐15 trans‐presentation in keratinocytes contributes to CD8(+) T cells activation via JAK‐STAT pathway in vitiligo. Free Radic Biol Med. 2019;139:80–91. [DOI] [PubMed] [Google Scholar]
- 74. Qiao Z, Wang X, Xiang L, Zhang C. Dysfunction of autophagy: a possible mechanism involved in the pathogenesis of vitiligo by breaking the redox balance of melanocytes. Oxid Med Cell Longev. 2016;2016:3401570–3401577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schallreuter KU, Pittelkow MP. Defective calcium uptake in keratinocyte cell cultures from vitiliginous skin. Arch Dermatol Res. 1988;280(3):137–139. [DOI] [PubMed] [Google Scholar]
- 76. Schallreuter KU, Wood JM. Thioredoxin reductase—its role in epidermal redox status. J Photochem Photobiol B. 2001;64(2–3):179–184. [DOI] [PubMed] [Google Scholar]
- 77. Shi Q, Zhang W, Guo S, et al. Oxidative stress‐induced overexpression of miR‐25: the mechanism underlying the degeneration of melanocytes in vitiligo. Cell Death Differ. 2016;23(3):496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kim JY, Lee EJ, Seo J, Oh SH. Impact of high‐mobility group box 1 on melanocytic survival and its involvement in the pathogenesis of vitiligo. Br J Dermatol. 2017;176(6):1558–1568. [DOI] [PubMed] [Google Scholar]
- 79. Mou K, Liu W, Miao Y, Cao F, Li P. HMGB1 deficiency reduces H2O2‐induced oxidative damage in human melanocytes via the Nrf2 pathway. J Cell Mol Med. 2018;22(12):6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bassiouny DA, Shaker O. Role of interleukin‐17 in the pathogenesis of vitiligo. Clin Exp Dermatol. 2011;36(3):292–297. [DOI] [PubMed] [Google Scholar]
- 81. Li S, Zhu G, Yang Y, et al. Oxidative stress drives CD8(+) T‐cell skin trafficking in patients with vitiligo through CXCL16 upregulation by activating the unfolded protein response in keratinocytes. J Allergy Clin Immunol. 2017;140(1):177‐189 e179. [DOI] [PubMed] [Google Scholar]
- 82. Ahn Y, Seo J, Lee EJ, et al. ATP‐P2X7‐induced inflammasome activation contributes to melanocyte death and CD8(+) T‐cell trafficking to the skin in vitiligo. J Invest Dermatol. 2020;140:1794–1804. [DOI] [PubMed] [Google Scholar]
- 83. Li S, Kang P, Zhang W, et al. Activated NLR family pyrin domain containing 3 (NLRP3) inflammasome in keratinocytes promotes cutaneous T‐cell response in patients with vitiligo. J Allergy Clin Immunol. 2020;145(2):632–645. [DOI] [PubMed] [Google Scholar]
- 84. Wagner RY, Luciani F, Cario‐André M, et al. Altered E‐cadherin levels and distribution in melanocytes precede clinical manifestations of vitiligo. J Invest Dermatol. 2015;135(7):1810–1819. [DOI] [PubMed] [Google Scholar]
- 85. Kumar R, Parsad D. Melanocytorrhagy and apoptosis in vitiligo: connecting jigsaw pieces. Indian J Dermatol Venereol Leprol. 2012;78(1):19–23. [DOI] [PubMed] [Google Scholar]
- 86. Kumar R, Parsad D, Kanwar AJ. Role of apoptosis and melanocytorrhagy: a comparative study of melanocyte adhesion in stable and unstable vitiligo. Br J Dermatol. 2011;164(1):187–191. [DOI] [PubMed] [Google Scholar]
- 87. Nelson WJ, Nusse R. Convergence of Wnt, beta‐catenin, and cadherin pathways. Science. 2004;303(5663):1483–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chan HL, Chou HC, Duran M, et al. Major role of epidermal growth factor receptor and Src kinases in promoting oxidative stress‐dependent loss of adhesion and apoptosis in epithelial cells. J Biol Chem. 2010;285(7):4307–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Boukhedouni N, Martins C, Darrigade AS, et al. Type‐1 cytokines regulate matrix metalloprotease‐9 production and E‐cadherin disruption to promote melanocyte loss in vitiligo. JCI Insight. 2020;5(11):e133772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gauthier Y, Cario Andre M, Taieb A. A critical appraisal of vitiligo etiologic theories. Is melanocyte loss a melanocytorrhagy? Pigment Cell Res. 2003;16(4):322–332. [DOI] [PubMed] [Google Scholar]
- 91. Gauthier Y, Cario‐Andre M, Lepreux S, Pain C, Taieb A. Melanocyte detachment after skin friction in non lesional skin of patients with generalized vitiligo. Br J Dermatol. 2003;148(1):95–101. [DOI] [PubMed] [Google Scholar]
- 92. Cario‐Andre M, Pain C, Gauthier Y, Taieb A. The melanocytorrhagic hypothesis of vitiligo tested on pigmented, stressed, reconstructed epidermis. Pigment Cell Res. 2007;20(5):385–393. [DOI] [PubMed] [Google Scholar]
- 93. Karsli N, Akcali C, Ozgoztasi O, Kirtak N, Inaloz S. Role of oxidative stress in the pathogenesis of vitiligo with special emphasis on the antioxidant action of narrowband ultraviolet B phototherapy. J Int Med Res. 2014;42(3):799–805. [DOI] [PubMed] [Google Scholar]
- 94. Zhang S, Yi X, Su X, et al. Ginkgo biloba extract protects human melanocytes from H2O2‐induced oxidative stress by activating Nrf2. J Cell Mol Med. 2019;23(8):5193–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhang B, Wang J, Zhao G, et al. Apigenin protects human melanocytes against oxidative damage by activation of the Nrf2 pathway. Cell Stress Chaperones. 2020;25(2):277–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gianfaldoni S, Tchernev G, Lotti J, et al. Unconventional treatments for vitiligo: are they (un) satisfactory? Open Access Maced J Med Sci. 2018;6(1):170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Mou K, Pan W, Han D, et al. Glycyrrhizin protects human melanocytes from H2O2‐induced oxidative damage via the Nrf2dependent induction of HO1. Int J Mol Med. 2019;44(1):253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Jung E, Kim JH, Kim MO, Lee SY, Lee J. Melanocyte‐protective effect of afzelin is mediated by the Nrf2‐ARE signalling pathway via GSK‐3beta inactivation. Exp Dermatol. 2017;26(9):764–770. [DOI] [PubMed] [Google Scholar]
- 99. Yang L, Yang F, Teng L, Katayama I. 6‐Shogaol protects human melanocytes against oxidative stress through activation of the Nrf2‐antioxidant response element signaling pathway. Int J Mol Sci. 2020;21:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Chang Y, Li S, Guo W, et al. Simvastatin protects human melanocytes from H2O2‐induced oxidative stress by activating Nrf2. J Invest Dermatol. 2017;137(6):1286–1296. [DOI] [PubMed] [Google Scholar]
- 101. Jian Z, Tang L, Yi X, et al. Aspirin induces Nrf2‐mediated transcriptional activation of haem oxygenase‐1 in protection of human melanocytes from H2O2‐induced oxidative stress. J Cell Mol Med. 2016;20(7):1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Tang L, Fang W, Lin J, Li J, Wu W, Xu J. Vitamin D protects human melanocytes against oxidative damage by activation of Wnt/beta‐catenin signaling. Lab Invest. 2018;98(12):1527–1537. [DOI] [PubMed] [Google Scholar]
- 103. Bellei B, Papaccio F, Filoni A, et al. Extracellular fraction of adipose tissue as an innovative regenerative approach for vitiligo treatment. Exp Dermatol. 2019;28(6):695–703. [DOI] [PubMed] [Google Scholar]
- 104. Fang W, Tang L, Wang G, et al. Molecular hydrogen protects human melanocytes from oxidative stress by activating Nrf2 signaling. J Invest Dermatol. 2020;140:2230–2241. [DOI] [PubMed] [Google Scholar]
- 105. Tsuji G, Hashimoto‐Hachiya A, Takemura M, Kanemaru T, Ichihashi M, Furue M. Palladium and platinum nanoparticles activate AHR and NRF2 in human keratinocytes‐implications in vitiligo therapy. J Invest Dermatol. 2017;137(7):1582–1586. [DOI] [PubMed] [Google Scholar]
- 106. Sun MC, Xu XL, Lou XF, Du YZ. Recent progress and future directions: the nano‐drug delivery system for the treatment of vitiligo. Int J Nanomedicine. 2020;15:3267–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Doppalapudi S, Mahira S, Khan W. Development and in vitro assessment of psoralen and resveratrol co‐loaded ultradeformable liposomes for the treatment of vitiligo. J Photochem Photobiol B. 2017;174:44–57. [DOI] [PubMed] [Google Scholar]
- 109. Mir‐Palomo S, Nácher A, Díez‐Sales O, et al. Inhibition of skin inflammation by baicalin ultradeformable vesicles. Int J Pharm. 2016;511(1):23–29. [DOI] [PubMed] [Google Scholar]
- 110. Shi J, Gao W, Shao F. Pyroptosis: gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42(4):245–254. [DOI] [PubMed] [Google Scholar]
- 111. Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–157. [DOI] [PubMed] [Google Scholar]
- 112. Zhou Z, He H, Wang K, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368:eaaz7548. [DOI] [PubMed] [Google Scholar]
- 113. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. He S, Liang Y, Shao F, Wang X. Toll‐like receptors activate programmed necrosis in macrophages through a receptor‐interacting kinase‐3‐mediated pathway. Proc Natl Acad Sci USA. 2011;108(50):20054–20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Holler N, Zaru R, Micheau O, Thome M, Tschopp J. Fas triggers an alternative, caspase‐8‐independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2001;1(6):489–495. [DOI] [PubMed] [Google Scholar]
- 116. Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM‐CSF priming. J Immunol. 2016;197(10):4090–4100. [DOI] [PubMed] [Google Scholar]
- 117. Berghe TV, Vanlangenakker N, Parthoens E, Deckers W, Vandenabeele P. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2009;17(6):922–930. [DOI] [PubMed] [Google Scholar]
- 118. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148(1–2):228–243. [DOI] [PubMed] [Google Scholar]
- 119. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron‐dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53‐mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang W, Green M, Choi JE, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rashighi M, Agarwal P, Richmond JM, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. 2014;6(223):223ra223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Holze C, Michaudel C, Mackowiak C, et al. Oxeiptosis, a ROS‐induced caspase‐independent apoptosis‐like cell‐death pathway. Nat Immunol. 2018;19(2):130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Scaturro P, Pichlmair A. Oxeiptosis: a discreet way to respond to radicals. Curr Opin Immunol. 2019;56:37–43. [DOI] [PubMed] [Google Scholar]
- 125. David KK, Andrabi SA, Dawson TM, Dawson VL. Parthanatos, a messenger of death. Front Biosci (Landmark Ed). 2009;14(3):1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang Y, An R, Umanah GK, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP‐ribose) polymerase‐1. Science. 2016;354(6308):aad6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Serarslan G, Yönden Z, Söğüt S, Savaş N, Çelik E, Arpaci A. Macrophage migration inhibitory factor in patients with vitiligo and relationship between duration and clinical type of disease. Clin Exp Dermatol. 2010;35(5):487–490. [DOI] [PubMed] [Google Scholar]
- 128. Ma L, Xue HB, Guan XH, et al. Relationship of macrophage migration inhibitory factor levels in PBMCs, lesional skin and serum with disease severity and activity in vitiligo vulgaris. Braz J Med Biol Res. 2013;46(5):460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kotobuki Y, Tanemura A, Yang L, et al. Dysregulation of melanocyte function by Th17‐related cytokines: significance of Th17 cell infiltration in autoimmune vitiligo vulgaris. Pigment Cell Melanoma Res. 2012;25(2):219–230. [DOI] [PubMed] [Google Scholar]
- 130. Tulic MK, Cavazza E, Cheli Y, et al. Innate lymphocyte‐induced CXCR3B‐mediated melanocyte apoptosis is a potential initiator of T‐cell autoreactivity in vitiligo. Nat Commun. 2019;10(1):2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Denman CJ, McCracken J, Hariharan V, et al. HSP70i accelerates depigmentation in a mouse model of autoimmune vitiligo. J Invest Dermatol. 2008;128(8):2041–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Srivastava P. Interaction of heat shock proteins with peptides and antigen presenting cells: chaperoning of the innate and adaptive immune responses. Annu Rev Immunol. 2002;20:395–425. [DOI] [PubMed] [Google Scholar]
- 133. Cui T, Zhang W, Li S, et al. Oxidative stress‐induced HMGB1 Release from melanocytes: a paracrine mechanism underlying the cutaneous inflammation in vitiligo. J Invest Dermatol. 2019;139(10):2174–2184. [DOI] [PubMed] [Google Scholar]
- 134. Bertolotti A, Boniface K, Vergier B, et al. Type I interferon signature in the initiation of the immune response in vitiligo. Pigment Cell Melanoma Res. 2014;27(3):398–407. [DOI] [PubMed] [Google Scholar]
- 135. Jacquemin C, Rambert J, Guillet S, et al. Heat shock protein 70 potentiates interferon alpha production by plasmacytoid dendritic cells: relevance for cutaneous lupus and vitiligo pathogenesis. Br J Dermatol. 2017;177(5):1367–1375. [DOI] [PubMed] [Google Scholar]
- 136. Speeckaert R, van Geel N. Vitiligo: an update on pathophysiology and treatment options. Am J Clin Dermatol. 2017;18(6):733–744. [DOI] [PubMed] [Google Scholar]
- 137. Marie J, Kovacs D, Pain C, et al. Inflammasome activation and vitiligo/nonsegmental vitiligo progression. Br J Dermatol. 2014;170(4):816–823. [DOI] [PubMed] [Google Scholar]
- 138. Speeckaert R, Speeckaert M, de Schepper S, van Geel N. Biomarkers of disease activity in vitiligo: a systematic review. Autoimmun Rev. 2017;16(9):937–945. [DOI] [PubMed] [Google Scholar]
- 139. Liang L, Li Y, Tian X, Zhou J, Zhong L. Comprehensive lipidomic, metabolomic and proteomic profiling reveals the role of immune system in vitiligo. Clin Exp Dermatol. 2019;44(7):e216–e223. [DOI] [PubMed] [Google Scholar]
- 140. Malhotra AG, Singh S, Jha M, Pandey KM. A parametric targetability evaluation approach for vitiligo proteome extracted through integration of gene ontologies and protein interaction topologies. IEEE/ACM Trans Comput Biol Bioinform. 2019;16(6):1830–1842. [DOI] [PubMed] [Google Scholar]
- 141. Kroll TM, Bommiasamy H, Boissy RE, et al. 4‐Tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell‐mediated killing: relevance to vitiligo. J Invest Dermatol. 2005;124(4):798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Edgunlu T, Solak Tekin N, Ozel Turkcu U, Karakas‐Celik S, Urhan‐Kucuk M, Tekin L. Evaluation of serum trail level and DR4 gene variants as biomarkers for vitiligo patients. J Eur Acad Dermatol Venereol. 2016;30(10):e97–e98. [DOI] [PubMed] [Google Scholar]
- 143. Jimbo H, Nagai H, Fujiwara S, Shimoura N, Nishigori C. Fas‐FasL interaction in cytotoxic T cell‐mediated vitiligo: the role of lesional expression of tumor necrosis factor‐alpha and interferon‐gamma in Fas‐mediated melanocyte apoptosis. Exp Dermatol. 2020;29(1):61–70. [DOI] [PubMed] [Google Scholar]
- 144. Richmond JM, Masterjohn E, Chu R, Tedstone J, Youd ME, Harris JE. CXCR3 depleting antibodies prevent and reverse vitiligo in mice. J Invest Dermatol. 2017;137(4):982–985. [DOI] [PubMed] [Google Scholar]
- 145. Jacquemin C, Martins C, Lucchese F, et al. NKG2D defines a subset of skin effector memory CD8 T cells with proinflammatory functions in vitiligo. J Invest Dermatol. 2019;140(6):1143–1153. [DOI] [PubMed] [Google Scholar]
- 146. Wang S, Xia P, Chen Y, et al. Regulatory innate lymphoid cells control innate intestinal inflammation. Cell. 2017;171(1):201–216. [DOI] [PubMed] [Google Scholar]
- 147. Colonna M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. 2018;48(6):1104–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bando JK, Colonna M. Innate lymphoid cell function in the context of adaptive immunity. Nat Immunol. 2016;17(7):783–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Mosenson JA, Zloza A, Nieland JD, et al. Mutant HSP70 reverses autoimmune depigmentation in vitiligo. Sci Transl Med. 2013;5(174):174ra128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Henning SW, Fernandez MF, Mahon JP, et al. HSP70iQ435A‐encoding DNA repigments vitiligo lesions in Sinclair swine. J Invest Dermatol. 2018;138(12):2531–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Gorman S, Judge MA, Hart PH. Immune‐modifying properties of topical vitamin D: focus on dendritic cells and T cells. J Steroid Biochem Mol Biol. 2010;121(1–2):247–249. [DOI] [PubMed] [Google Scholar]
- 152. Karthaus N, van Spriel AB, Looman MWG, et al. Vitamin D controls murine and human plasmacytoid dendritic cell function. J Invest Dermatol. 2014;134(5):1255–1264. [DOI] [PubMed] [Google Scholar]
- 153. Prignano F, Ricceri F, Bianchi B, et al. Dendritic cells: ultrastructural and immunophenotypical changes upon NB‐UVB in vitiligo skin. Arch Dermatol Res. 2011;303(4):231–238. [DOI] [PubMed] [Google Scholar]
- 154. Mascanfroni ID, Yeste A, Vieira SM, et al. IL‐27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013;14(10):1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Carvalheiro H, da Silva JA, Souto‐Carneiro MM. Potential roles for CD8(+) T cells in rheumatoid arthritis. Autoimmun Rev. 2013;12(3):401–409. [DOI] [PubMed] [Google Scholar]
- 156. Sabatino JJ, Jr. , Wilson MR, Calabresi PA, Hauser SL, Schneck JP, Zamvil SS. Anti‐CD20 therapy depletes activated myelin‐specific CD8(+) T cells in multiple sclerosis. Proc Natl Acad Sci USA. 2019;116(51):25800–25807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Ogg GS, Rod Dunbar P, Romero P, Chen JL, Cerundolo V. High frequency of skin‐homing melanocyte‐specific cytotoxic T lymphocytes in autoimmune vitiligo. J Exp Med. 1998;188(6):1203–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lili Y, Yi W, Ji Y, Yue S, Weimin S, Ming L. Global activation of CD8+ cytotoxic T lymphocytes correlates with an impairment in regulatory T cells in patients with generalized vitiligo. PLOS One. 2012;7(5):e37513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Westerhof W, d'Ischia M. Vitiligo puzzle: the pieces fall in place. Pigment Cell Res. 2007;20(5):345–359. [DOI] [PubMed] [Google Scholar]
- 160. van den Wijngaard R, Wankowicz‐Kalinska A, Le Poole C, Tigges B, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+T cells at the perilesional site. Lab Invest. 2000;80(8):1299–1309. [DOI] [PubMed] [Google Scholar]
- 161. van den Boorn JG, Konijnenberg D, Dellemijn TAM, et al. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. 2009;129(9):2220–2232. [DOI] [PubMed] [Google Scholar]
- 162. Mandelcorn‐Monson RL, Shear NH, Yau E, et al. Cytotoxic T lymphocyte reactivity to gp100, MelanA/MART‐1, and tyrosinase, in HLA‐A2‐positive vitiligo patients. J Invest Dermatol. 2003;121(3):550–556. [DOI] [PubMed] [Google Scholar]
- 163. Rahimi A, Hossein‐Nataj H, Hajheydari Z, et al. Expression analysis of PD‐1 and Tim‐3 immune checkpoint receptors in patients with vitiligo; positive association with disease activity. Exp Dermatol. 2019;28(6):674–681. [DOI] [PubMed] [Google Scholar]
- 164. Miao X, Xu R, Fan B, et al. PD‐L1 reverses depigmentation in Pmel‐1 vitiligo mice by increasing the abundance of Tregs in the skin. Sci Rep. 2018;8(1):1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Deng Q, Luo Y, Chang C, Wu H, Ding Y, Xiao R. The emerging epigenetic role of CD8+T cells in autoimmune diseases: a systematic review. Front Immunol. 2019;10:856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Bowne WB, Srinivasan R, Wolchok JD, et al. Coupling and uncoupling of tumor immunity and autoimmunity. J Exp Med. 1999;190(11):1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase‐specific TCR transgenic mice. J Immunol. 2010;184(4):1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Chatterjee S, Eby JM, Al‐Khami AA, et al. A quantitative increase in regulatory T cells controls development of vitiligo. J Invest Dermatol. 2014;134(5):1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Rashighi M, Harris JE. Vitiligo pathogenesis and emerging treatments. Dermatol Clin. 2017;35(2):257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Richmond JM, Bangari DS, Essien KI, et al. Keratinocyte‐derived chemokines orchestrate T‐cell positioning in the epidermis during vitiligo and may serve as biomarkers of disease. J Invest Dermatol. 2017;137(2):350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Yang L, Wei Y, Sun Y, et al. Interferon‐gamma inhibits melanogenesis and induces apoptosis in melanocytes: a pivotal role of CD8+ cytotoxic T lymphocytes in vitiligo. Acta Derm Venereol. 2015;95(6):664–670. [DOI] [PubMed] [Google Scholar]
- 172. Boniface K, Jacquemin C, Darrigade AS, et al. Vitiligo skin is imprinted with resident memory CD8 T cells expressing CXCR3. J Invest Dermatol. 2018;138(2):355–364. [DOI] [PubMed] [Google Scholar]
- 173. Boniface K, Seneschal J. Vitiligo as a skin memory disease: the need for early intervention with immunomodulating agents and a maintenance therapy to target resident memory T cells. Exp Dermatol. 2019;28(6):656–661. [DOI] [PubMed] [Google Scholar]
- 174. Watanabe R, Gehad A, Yang C, et al. Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med. 2015;7(279):279ra239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Willemsen M, Linkute R, Luiten RM, Matos TR. Skin‐resident memory T cells as a potential new therapeutic target in vitiligo and melanoma. Pigment Cell Melanoma Res. 2019;32(5):612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Cheuk S, Schlums H, Gallais Sérézal I, et al. CD49a expression defines tissue‐resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity. 2017;46(2):287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Riding RL, Harris JE. The role of memory CD8(+) T cells in vitiligo. J Immunol. 2019;203(1):11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Clark RA, Chong B, Mirchandani N, et al. The vast majority of CLA+T cells are resident in normal skin. J Immunol. 2006;176(7):4431–4439. [DOI] [PubMed] [Google Scholar]
- 179. Sasson SC, Gordon CL, Christo SN, Klenerman P, Mackay LK. Local heroes or villains: tissue‐resident memory T cells in human health and disease. Cell Mol Immunol. 2020;17(2):113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Kabashima K, Honda T, Ginhoux F, Egawa G. The immunological anatomy of the skin. Nat Rev Immunol. 2019;19(1):19–30. [DOI] [PubMed] [Google Scholar]
- 181. Richmond JM, Strassner JP, Zapata L, et al. Antibody blockade of IL‐15 signaling has the potential to durably reverse vitiligo. Sci Transl Med. 2018;10:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. 2015;7(269):269rv261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Masopust D, Schenkel JM. The integration of T cell migration, differentiation and function. Nat Rev Immunol. 2013;13(5):309–320. [DOI] [PubMed] [Google Scholar]
- 184. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. 2013;14(12):1285–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zaid A, Mackay LK, Rahimpour A, et al. Persistence of skin‐resident memory T cells within an epidermal niche. Proc Natl Acad Sci USA. 2014;111(14):5307–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Mackay LK, Rahimpour A, Ma JZ, et al. The developmental pathway for CD103(+)CD8+ tissue‐resident memory T cells of skin. Nat Immunol. 2013;14(12):1294–1301. [DOI] [PubMed] [Google Scholar]
- 187. Bergsbaken T, Bevan MJ, Fink PJ. Local inflammatory cues regulate differentiation and persistence of CD8(+) tissue‐resident memory T cells. Cell Rep. 2017;19(1):114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Mackay LK, Wynne‐Jones E, Freestone D, et al. T‐box transcription factors combine with the cytokines TGF‐beta and IL‐15 to control tissue‐resident memory T cell fate. Immunity. 2015;43(6):1101–1111. [DOI] [PubMed] [Google Scholar]
- 189. El‐Asady R, Yuan R, Liu K, et al. TGF‐{beta}‐dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft‐versus‐host disease. J Exp Med. 2005;201(10):1647–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Malik BT, Byrne KT, Vella JL, et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci Immunol. 2017;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Richmond JM, Strassner JP, Rashighi M, et al. Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J Invest Dermatol. 2019;139(4):769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. 2014;346(6205):98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Azzolino V, Zapata L, Jr. , Garg M, et al. Jak inhibitors reverse vitiligo in mice but do not deplete skin resident memory T cells. J Invest Dermatol. 2020. 10.1016/j.jid.2020.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Ujiie H. Regulatory T cells in autoimmune skin diseases. Exp Dermatol. 2019;28(6):642–646. [DOI] [PubMed] [Google Scholar]
- 195. Zhang Q, Cui T, Chang Y, et al. HO‐1 regulates the function of Treg: association with the immune intolerance in vitiligo. J Cell Mol Med. 2018;22(9):4335–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Klarquist J, Denman CJ, Hernandez C, et al. Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res. 2010;23(2):276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Le Poole IC, Mehrotra S. Replenishing regulatory T cells to halt depigmentation in vitiligo. J Investig Dermatol Symp Proc. 2017;18(2):S38–S45. [DOI] [PubMed] [Google Scholar]
- 198. Cabral J, Hanley SA, Gerlach JQ, et al. Distinctive surface glycosylation patterns associated with mouse and human CD4(+) regulatory T cells and their suppressive function. Front Immunol. 2017;8:987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Maeda Y, Nishikawa H, Sugiyama D, et al. Detection of self‐reactive CD8(+) T cells with an anergic phenotype in healthy individuals. Science. 2014;346(6216):1536–1540. [DOI] [PubMed] [Google Scholar]
- 200. Hegab DS, Attia MA. Decreased circulating T regulatory cells in Egyptian patients with nonsegmental vitiligo: correlation with disease activity. Dermatol Res Pract. 2015;2015:145409–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Tembhre MK, Parihar AS, Sharma VK, Sharma A, Chattopadhyay P, Gupta S. Alteration in regulatory T cells and programmed cell death 1‐expressing regulatory T cells in active generalized vitiligo and their clinical correlation. Br J Dermatol. 2015;172(4):940–950. [DOI] [PubMed] [Google Scholar]
- 202. Terras S, Gambichler T, Moritz RK, Altmeyer P, Lambert J. Immunohistochemical analysis of FOXP3+ regulatory T cells in healthy human skin and autoimmune dermatoses. Int J Dermatol. 2014;53(3):294–299. [DOI] [PubMed] [Google Scholar]
- 203. Zhou L, Li K, Shi YL, et al. Systemic analyses of immunophenotypes of peripheral T cells in non‐segmental vitiligo: implication of defective natural killer T cells. Pigment Cell Melanoma Res. 2012;25(5):602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Das D, Akhtar S, Kurra S, Gupta S, Sharma A. Emerging role of immune cell network in autoimmune skin disorders: an update on pemphigus, vitiligo and psoriasis. Cytokine Growth Factor Rev. 2019;45:35–44. [DOI] [PubMed] [Google Scholar]
- 205. Tembhre MK, Sharma VK, Sharma A, Chattopadhyay P, Gupta S. T helper and regulatory T cell cytokine profile in active, stable and narrow band ultraviolet B‐treated generalized vitiligo. Clin Chim Acta. 2013;424:27–32. [DOI] [PubMed] [Google Scholar]
- 206. Kidir M, Karabulut AA, Ercin ME, Atasoy P. Regulatory T‐cell cytokines in patients with nonsegmental vitiligo. Int J Dermatol. 2017;56(5):581–588. [DOI] [PubMed] [Google Scholar]
- 207. Hegazy RA, Fawzy MM, Gawdat HI, Samir N, Rashed LA. T helper 17 and Tregs: a novel proposed mechanism for NB‐UVB in vitiligo. Exp Dermatol. 2014;23(4):283–286. [DOI] [PubMed] [Google Scholar]
- 208. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, et al. Memory regulatory T cells reside in human skin. J Clin Invest. 2014;124(3):1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Czarnowicki T, He H, Leonard A, et al. Blood endotyping distinguishes the profile of vitiligo from that of other inflammatory and autoimmune skin diseases. J Allergy Clin Immunol. 2019;143(6):2095–2107. [DOI] [PubMed] [Google Scholar]
- 210. Sabat R, Wolk K, Loyal L, Docke WD, Ghoreschi K. T cell pathology in skin inflammation. Semin Immunopathol. 2019;41(3):359–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Wańkowicz‐Kalińska A, van den Wijngaard RMJGJ, Tigges BJ, et al. Immunopolarization of CD4+ and CD8+ T cells to Type‐1‐like is associated with melanocyte loss in human vitiligo. Lab Invest. 2003;83(5):683–695. [DOI] [PubMed] [Google Scholar]
- 212. Albanesi C, Scarponi C, Sebastiani S, et al. A cytokine‐to‐chemokine axis between T lymphocytes and keratinocytes can favor Th1 cell accumulation in chronic inflammatory skin diseases. J Leukoc Biol. 2001;70(4):617–623. [PubMed] [Google Scholar]
- 213. Lambe T, Leung JCH, Bouriez‐Jones T, et al. CD4 T cell‐dependent autoimmunity against a melanocyte neoantigen induces spontaneous vitiligo and depends upon Fas–Fas ligand interactions. J Immunol. 2006;177(5):3055–3062. [DOI] [PubMed] [Google Scholar]
- 214. Choi H, Choi H, Han J, et al. IL‐4 inhibits the melanogenesis of normal human melanocytes through the JAK2‐STAT6 signaling pathway. J Invest Dermatol. 2013;133(2):528–536. [DOI] [PubMed] [Google Scholar]
- 215. Singh RK, Lee KM, Vujkovic‐Cvijin I, et al. The role of IL‐17 in vitiligo: a review. Autoimmun Rev. 2016;15(4):397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Zhou L, Shi YL, Li K, et al. Increased circulating Th17 cells and elevated serum levels of TGF‐beta and IL‐21 are correlated with human non‐segmental vitiligo development. Pigment Cell Melanoma Res. 2015;28(3):324–329. [DOI] [PubMed] [Google Scholar]
- 217. Zhen Y, Yao L, Zhong S, Song Y, Cui Y, Li S. Enhanced Th1 and Th17 responses in peripheral blood in active non‐segmental vitiligo. Arch Dermatol Res. 2016;308(10):703–710. [DOI] [PubMed] [Google Scholar]
- 218. Khan R, Gupta S, Sharma A. Circulatory levels of T‐cell cytokines (interleukin [IL]‐2, IL‐4, IL‐17, and transforming growth factor‐beta) in patients with vitiligo. J Am Acad Dermatol. 2012;66(3):510–511. [DOI] [PubMed] [Google Scholar]
- 219. Elela MA, Hegazy RA, Fawzy MM, Rashed LA, Rasheed H. Interleukin 17, interleukin 22 and FoxP3 expression in tissue and serum of non‐segmental vitiligo: a case‐controlled study on eighty‐four patients. Eur J Dermatol. 2013;23(3):350–355. [DOI] [PubMed] [Google Scholar]
- 220. Wang CQF, Cruz‐Inigo AE, Fuentes‐Duculan J, et al. Th17 cells and activated dendritic cells are increased in vitiligo lesions. PLOS One. 2011;6(4):e18907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Muranski P, Boni A, Antony PA, et al. Tumor‐specific Th17‐polarized cells eradicate large established melanoma. Blood. 2008;112(2):362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Speeckaert R, Lambert J, van Geel N. Learning from success and failure: biologics for non‐approved skin diseases. Front Immunol. 2019;10:1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Speeckaert R, Mylle S, van Geel N. IL‐17A is not a treatment target in progressive vitiligo. Pigment Cell Melanoma Res. 2019;32(6):842–847. [DOI] [PubMed] [Google Scholar]
- 224. Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. 2017;17(9):535–544. [DOI] [PubMed] [Google Scholar]
- 225. Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN‐gamma for autoreactive CD8(+) T‐cell accumulation in the skin. J Invest Dermatol. 2012;132(7):1869–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Rosmarin D, Pandya AG, Lebwohl M, et al. Ruxolitinib cream for treatment of vitiligo: a randomised, controlled, phase 2 trial. Lancet. 2020;396(10244):110–120. [DOI] [PubMed] [Google Scholar]
- 227. Pan Y, Tian T, Park CO, et al. Survival of tissue‐resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543(7644):252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Pan Y, Kupper TS. Metabolic reprogramming and longevity of tissue‐resident memory T cells. Front Immunol. 2018;9:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Liao W, Lin JX, Leonard WJ. Interleukin‐2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38(1):13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Castela E, Le Duff F, Butori C, et al. Effects of low‐dose recombinant interleukin 2 to promote T‐regulatory cells in alopecia areata. JAMA Dermatol. 2014;150(7):748–751. [DOI] [PubMed] [Google Scholar]
- 231. He J, Zhang X, Wei Y, et al. Low‐dose interleukin‐2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med. 2016;22(9):991–993. [DOI] [PubMed] [Google Scholar]
- 232. Hartemann A, Bensimon G, Payan CA, et al. Low‐dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double‐blind, placebo‐controlled trial. Lancet Diabetes Endocrinol. 2013;1(4):295–305. [DOI] [PubMed] [Google Scholar]
- 233. Saadoun D, Rosenzwajg M, Joly F, et al. Regulatory T‐cell responses to low‐dose interleukin‐2 in HCV‐induced vasculitis. N Engl J Med. 2011;365(22):2067–2077. [DOI] [PubMed] [Google Scholar]
- 234. Zou H, Li R, Hu H, Hu Y, Chen X. Modulation of regulatory T cell activity by TNF receptor type ii‐targeting pharmacological agents. Front Immunol. 2018;9:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Magee CN, Murakami N, Borges TJ, et al. Notch‐1 inhibition promotes immune regulation in transplantation via regulatory T cell‐dependent mechanisms. Circulation. 2019;140(10):846–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T cells and human disease. Annu Rev Immunol. 2020;38:541–566. [DOI] [PubMed] [Google Scholar]
- 237. Eby JM, Kang HK, Tully ST, et al. CCL22 to Activate Treg migration and suppress depigmentation in vitiligo. J Invest Dermatol. 2015;135(6):1574–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Raffin C, Vo LT, Bluestone JA. Treg cell‐based therapies: challenges and perspectives. Nat Rev Immunol. 2020;20(3):158–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Passerini L, Mel ER, Sartirana C, et al. CD4(+) T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med. 2013;5(215):215ra174. [DOI] [PubMed] [Google Scholar]
- 240. Cras A, Farge D, Carmoi T, Lataillade JJ, Wang DD, Sun L. Update on mesenchymal stem cell‐based therapy in lupus and scleroderma. Arthritis Res Ther. 2015;17:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. van Geel N, Hamzavi I, Kohli I, et al. Standardizing serial photography for assessing and monitoring vitiligo: a core set of international recommendations for essential clinical and technical specifications. J Am Acad Dermatol. 2019.83(6):1639–1646. [DOI] [PubMed] [Google Scholar]
- 242. Linthorst Homan MW, Wolkerstorfer A, Sprangers MA, van der Veen JP. Digital image analysis vs. clinical assessment to evaluate repigmentation after punch grafting in vitiligo. J Eur Acad Dermatol Venereol. 2013;27(2):e235–e238. [DOI] [PubMed] [Google Scholar]
- 243. Nugroho H, Ahmad Fadzil MH, Shamsudin N, Hussein SH. Computerised image analysis of vitiligo lesion: evaluation using manually defined lesion areas. Skin Res Technol. 2013;19(1):e72–e77. [DOI] [PubMed] [Google Scholar]
- 244. Sheth VM, Rithe R, Pandya AG, Chandrakasan A. A pilot study to determine vitiligo target size using a computer‐based image analysis program. J Am Acad Dermatol. 2015;73(2):342–345. [DOI] [PubMed] [Google Scholar]
- 245. Mikhael NW, Sabry HH, El‐Refaey AM, Salem RM, El‐Gendy MF, Farid SA. Clinimetric analysis of recently applied quantitative tools in evaluation of vitiligo treatment. Indian J Dermatol Venereol Leprol. 2019;85(5):466–474. [DOI] [PubMed] [Google Scholar]
- 246. Toh JJH, Bhoi S, Tan VWD, et al. Automated scoring of vitiligo using superpixel‐generated computerized digital image analysis of clinical photographs: a novel and consistent way to score vitiligo. Br J Dermatol. 2018;179(1):220–221. [DOI] [PubMed] [Google Scholar]
- 247. van Geel N, Vandendriessche D, Vandersichel E, et al. Reference method for digital surface measurement of target lesions in vitiligo: a comparative analysis. Br J Dermatol. 2019;180(5):1198–1205. [DOI] [PubMed] [Google Scholar]