

Abstract
Background
The ataxin‐2 (ATXN2) gene contains a cytosine‐adenine‐guanine repeat sequence ranging from 13 to 31 repeats, but when surpassing certain thresholds causes neurodegeneration. Genetic alterations in ATXN2 other than pathological cytosine adenine guanine (CAG) repeats are unknown.
Methods/Results
We have identified a 9–base pair duplication in the 2‐gene ATXN2 sense/antisense region. The duplication was found in a Swedish family with spinocerebellar ataxia 3 with parkinsonism, conferring a deviated age at onset unexplained by the concomitant presence of ATXN2 intermediate alleles. Similarly, C9ORF72 amyotrophic lateral sclerosis cases bearing the same duplication had earlier age at onset than those with C9ORF72 and ATXN2 intermediate alleles. No effect was evident in Parkinson's disease (PD) cases without known PD gene mutations.
Conclusions
We describe the first genetic alteration other than the known intermediate‐range CAG repeats in ATXN2. This 9–base pair duplication may act as an additional hit among carriers of pathological nucleotide expansions in ATXN3 and C9ORF72 with ATXN2 intermediate. © 2020 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society.
Keywords: ATXN2, SCA3, Parkinson's disease, C9ORF72, gene modifier, genotype–phenotype correlations
The ataxin‐2 gene (ATXN2) contains a cytosine‐adenine‐guanine (CAG) repeat sequence in exon 1. The normal length of CAG repeats ranges from 13 to 31 repeats. Intermediate repeats between 27 and 31 CAG repeats are associated with neurological diseases, and expansions beyond 34 CAG repeats cause spinocerebellar ataxia 2 (SCA2). 1 , 2 , 3 , 4
Genetic alterations of ATXN2 other than pathological CAG repeats are unknown. We identified a 9–base pair (bp) duplication in the ATXN2 promoter/exon 1 region lowering age at onset (AO) for spinocerebellar ataxia 3/Machado Joseph disease (SCA3/MJD) and C9ORF72‐amyotrophic lateral sclerosis (ALS).
1. Material and Methods
Details on Material and Methods are presented as supplementary material (Figs. 1A and S1, Tables S1 and S2).
FIG. 1.
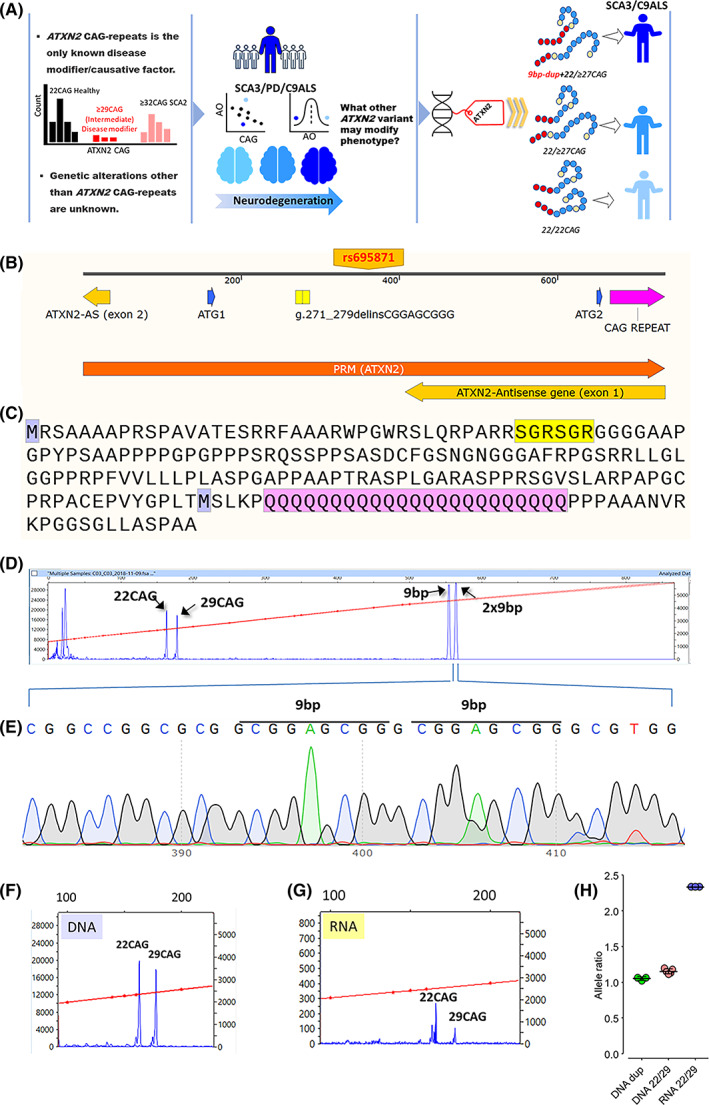
Unique 9‐bp duplication in the 2‐gene region ATXN2‐sense/antisense. (A) Schematic representation of the background of the study and the main results. We examined a total of 323 DNA samples from different patients with neurodegenerative diseases (SCAs, PD, and C9ORF72‐ALS) and 823 DNA samples from controls from the United States and Sweden. DNA samples, demographics, and clinical data were obtained from the Coriell Institute for Medical Research and Karolinska University Hospital. In addition to the CAG repeats, DNA was examined for other ATXN2 genetic alterations potentially contributing as disease modifiers of SCAs, PD, and C9ORF72‐ALS. Age at disease onset and clinical rating scales were used as phenotype markers for determining genotype–phenotype relationship. Figure S1 and Tables S1, S2 show the extended flow of the investigation, as well as the general methods applied for each cohort. (B) Map for promoter/exon 1 of ataxin‐2 gene including the CAG repeat and the relative positions for some markers close to the 9‐bp duplication. Transcription start sites are also indicated with blue arrows and the encoded region from the first putative start site and the position for the rs695871. (C) The encoded fragment of the ataxin‐2 is included in the map. The 9‐bp duplication encodes the duplicated motif SGR, located in the intrinsically disordered region of ataxin‐2. In navy blue are the 2 methionines, and shadowed in pink is the polyQ tract. (D) Capillary electrophoresis of both ATXN2 CAG repeat and the 9‐bp duplication in the index case (II‐1). (E) Representative electropherograms of the 9‐bp duplication in the ATXN2 gene in reverse direction. (F–H) PCR fragment size analysis of cDNA showing that the mutant mRNA allele with the duplication is expressed 2.3‐fold more than the intermediate allele of 29 CAG repeats in the index case. Data are shown as the average of triplicate samples, and error bars denote SD. [Color figure can be viewed at wileyonlinelibrary.com]
2. Results
2.1. Duplication of 9 bp in a 2‐Gene ATXN2 Sense and Antisense Region
We identified a 9‐bp duplication, c.109_117delinsCGGAGCGGG, ref seq. NM_002973, located in the 2‐gene ATXN2 sense/antisense region. The 9‐bp duplication causes the reiteration of the SGR motif. For ATXN2‐S, the 9‐bp duplication is in the promoter/exon 1 region, and for the natural antisense, ATXN2‐AS, ~60 bp toward the end of intron 1 (Fig. 1A,B).
2.2. Duplication of 9 bp in ATXN2 in Different Cohorts
The 9‐bp duplication was present in 1 of 28 Swedish SCA3/MJD cases (1 of 28, 3.57%; Fig. 1C–G), 2 C9ORF72‐ALS (2 of 70, 2.86%), and 4 patients with Parkinson's disease (4 of 198, 2%). Moreover, this duplication was absent in 10 SCA patients from the Coriell Cell repository. Four of 823 controls (0.48%) had the 9‐bp duplication. This variant was absent in the following databases: gnomAD, ExAC, SNPdB, and the1000 Genome Project.
2.3. Parkinsonian Phenotype in Swedish SCA3 Family With the Duplication
As shown in the family pedigree in Figure 2, both cases had full penetrant ATXN3 alleles (I‐1 = 65 CAG repeats, II‐1 = 69 CAG repeats). By history, the paternal grandmother was also affected. SCA3 manifested in the father (I‐1) with insidious resting tremor, reduced arm swing and facial expression at age 50, which was responsive to l‐dopa and thus diagnosed as Parkinson's disease (PD). Motor fluctuations were evident in the course of the disease. Later, cerebellar signs became evident, motivating an investigation for SCA3/MJD. This patient died of pulmonary complications at age 72 years. Neuropathological examination demonstrated loss of pigmented neurons in the substantia nigra and ubiquitin‐immunoreactive inclusions. Neuronal intranuclear polyQ‐positive inclusions were found in the pontine nuclei, whereas loss of Purkinje cells in the cerebellar cortex was mild (Fig. 2C–F and supplements).
FIG. 2.

Clinical information, genetics, and neuropathology of Swedish SCA3 parkinsonian family with both intermediate CAG repeats and novel 9‐bp duplication. (A) Pedigree of the SCA3 family with the 9‐bp duplication. (B) Genotype and phenotype of individuals involved in this familiar study. (C) Neuropathology of case I‐1, indicating moderate loss of pigmented neurons in the substantia nigra and also being positive for ubiquitin and p62. (D) Three arrows indicate 3 ubiquitin‐positive intranuclear inclusions in a pigmented neuron in the substantia nigra. (E) Numerous neurons in the pons contained intranuclear polyQ‐positive inclusions. (F) Some loss of Purkinje cells in the cerebellar cortex was noticed. (G) CT scan for the index case. (H) [123I]FP‐CIT SPECT in patient II‐1 displaying significantly reduced binding to dopamine transporter. (I) [123I] FP‐CIT SPECT image from a healthy control. (J) Generalized estimated equation analysis of the AO‐CAG relationship in the Swedish cohort highlighting individuals I‐1 (father) and II‐1 (daughter). Red curve is for the full SCA3 cohort, blue curve is when excluding the 9‐bp duplication carrier (II‐1), and black is when both the 9‐bp duplication carrier (II‐1) and I‐1 with an intermediate ATXN2 CAG are excluded. Model effects are presented in the inset box. The index case II‐1 deviates from the expected AO, as shown in the residual analysis in (K) and in different models (Table S4). (L) The same effect of lowering disease onset is also found in the 2 C9ORF72‐ALS cases carrying the 9‐bp duplication. In the box‐and‐whiskers plots, there are 3 groups: ALS with C9ORF72 mutation only (C9), ALS with C9ORF72 plus intermediate ATXN2 CAG (ATXN2), and the 2 C9ORF72‐ALS carriers with the 9‐bp duplication (DUP). Box‐and‐whisker plots represent median and the 25%–75% interquartile range as well as the 5th–95th percentiles. Note that there is no overlap between the ATXN2 and DUP groups. Purple points represent values outside the 5th–95th percentiles. [Color figure can be viewed at wileyonlinelibrary.com]
Case II‐1 is the index case in this family. This 49‐year‐old woman presented with insidious balance impairment, slurred speech, and disturbed coordination starting at age 30. At age 45, her examination revealed ataxia signs, bradykinesia, and reduced arm swing. A CT scan demonstrated cerebellar atrophy, particularly in the vermis, but less atrophy was evident in the mesencephalon, pons, and cerebellar peduncles (Fig. 2G). Recent assessment of the dopamine transporter with [123I]FP‐CIT SPECT demonstrated markedly reduced binding in both putamina and to a lesser degree in the caudate compared with controls (Fig. 2H,I). Treatment with l‐dopa was initially beneficial; later, motor fluctuations appeared motivating add‐on medication with amantadine, which reduced her fluctuations. SARA score was 10 at age 45 and increased to 20 at age 49.
2.4. Segregation Analysis in the SCA3‐Parkinsonian Family
The SCA3 mutation elongated from 65 to 69 CAG repeats in the index case when transmitted from the deceased father. In addition, we found cosegregation of the SCA3 mutation with the A‐allele of rs1048755 located in exon 8 of ATXN3 (Fig. S2).
As shown in Figure 2, the index case inherited the ATXN2 intermediate allele of 29 CAG repeats from her father. However, the 9‐bp duplication came from her healthy 78‐year‐old mother (I‐2), who harbors the ATXN2 genotype 22/22 CAG repeats. The mother also transmitted one of these 22 CAG repeats and the duplication to one of her offspring, II‐2, who also inherited the father's ATXN2 intermediate allele. From this, we conclude that the novel 9‐bp duplication in II‐1 is in cis with 1 maternal 22‐CAG allele and in trans with the paternal ATXN2 29‐CAG‐repeat allele. The cosegregation to the G variant of rs695871 located 200 bp downstream of the duplication supported this segregation. Interestingly, Digital Droplet PCR analysis using rs695871 confirmed the inclusion of the duplication in the main ATXN2 transcript and gene expression occurring from transcription start site 1 (TSS1; Fig. S3A–C).
There were no differences in ATXN3 allelic expression (Fig. S2A–D). However, PCR fragment analysis of cDNA from case II‐1 demonstrated that the ATXN2 22CAG allele, in cis with the duplication, has a 2.3‐fold‐higher expression than its accompanying 29‐CAG allele (Fig. 1E–G). Moreover, bidirectional ATXN2‐S/AS gene expression analysis showed higher expression associated with the 9‐bp duplication (Fig. S3A–C). The 22/29CAG ratio was ~1 in the genomic DNA for the duplication (not shown) and the different allelic expression was not due to ATXN2‐S/AS epigenetic gene methylation (data not shown).
2.5. Duplication of 9 bp, Genetic Modifiers, and Age at Onset at SCA3
Interestingly, both cases had not only SCA3 but also clear parkinsonism and lacked mutations in genes commonly associated with PD (Supplementary Information).
The meiotic instability of only 4 CAG repeats (65 to 69 CAG) does not explain the striking genetic anticipation of 20 years in this parent–daughter pair. Moreover, our index case has a much earlier AO than expected according to the AO‐CAG using the generalized estimating equation for linear regression model (R 2 = 0.48, P < 0.0001; R 2 = 0.53, P < 0.0002 excluding the 9 bp‐dup case; R 2 = 0.55, P < 0.0006 excluding this parent offspring pair) for this Swedish cohort. According to this model, the expected AO for our case was 44.75 years, so our index case has an anticipation of ~15 years for ataxia onset (AOobs/AOexp = 0.67) and in other predictive models 5 (Fig. 2J,K, Table S4).
Both patients carried otherwise normal ATXN3 alleles of 12 and 20 CAG repeats in the normal allele, which lack a modifying effect. 6 Somatic mosaicism for the ATXN3 CAG repeats in blood was not a source for phenotype variability either (Mosaicism Index, 3.00 ± 0.51 vs 3.04 ± 0.13, not significant). For the 2 single‐nucleotide polymorphisms, rs910369 and rs709930, located in ATXN3 3′‐UTR, the A allele was previously reported to decrease AO for SCA3, 7 but this was not confirmed in our cohort. Neither did rs7969300 in ATXN2 decrease AO (not shown).
The ε2‐ApoE allele accounts for earlier SCA3 AO, but our cases were ε3ε3 without modifying effect. 8 Other potential phenotype modifiers were not present in ATN1, HTT, TBP, CACNA1A, and C9ORF72 (Table S5).
Because all the above‐mentioned potential modifiers are excluded, it is suggested that the earlier AO in our index case is explained by the combined effect of the overexpressed 9‐bp duplication allele in trans with the intermediate 29‐CAG allele.
2.6. Duplication of 9 bp and Modulatory Effect in C9ORF72‐ALS and PD
The lowering effect on AO was also found in 2 C9ORF72‐ALS cases carrying the 9‐bp duplication, called c94 (ATXN2, 22/28 CAG, upper‐limb onset; ALS Functional Rating Scale, 41; AO, 45 years; survival, 43 months) and c67 (ATXN2, 22/22 CAG, bulbar onset; ALS Functional Rating Scale , 32; AO, 52 years). Despite that both cases have methylated C9ORF72 promoter (25%–55%), which is neuroprotective, they developed ALS 10.7 years earlier (median, 48.5 years; interquartile range [IQR], 25%–75%, 45.0–52.0 years) than those bearing both C9ORF72 plus ATXN2CAG ≥ 27CAG (median, 59.0 years; IQR, 56.5–62.0 years), and compared with the C9ORF72 mutation, only their AO tended to be lower (median, 57.0 years; IQR, 52.0–64.0 years); see Figure 2L and supplementary information. Patient c94, with ATXN2 = 22/28 CAG, is a mosaic for 2 different CAA‐repeat interruption patterns within the 22‐CAG allele associated with the 9‐bp duplication. Interestingly, his AO of 45 years is below the 5th percentile (47 years) of the whole C9ORF72‐ALS cohort. One configuration had 3CAA and the other only 2CAA motifs. The sequences are (7CAG–2CAA–4CAG–1CAA–8CAG) and (8CAG–1CAA–4CAG–1CAA–8CAG), respectively (Fig. S5), and this may have effects on RNA folding (Fig. S6).
All PD patients harboring the 9‐bp duplication lack mutations in known PD genes but did not have clear phenotypic differences to other PD patients without the 9‐bp duplication and with or without intermediate ATXN2 alleles (data not shown).
2.7. Gene Expression Analysis in C9ORF72‐ALS and PD
Similar to the SCA3 family, we detected the rs695871 using DDPCR and confirmed gene expression starting from the TSS1 in the C9ORF72‐ALS and PD cases with the 9‐bp‐duplication (Fig. S3D,E). We also studied bidirectional qPCR gene expression (ATXN2‐S/AS) and found that the ATXN2‐S transcript levels were significantly higher (MWU, 15; P < 0.001) in 9‐bp duplication carriers (Fig. S3E and supplemental information).
3. Discussion
We have identified a novel 9‐bp duplication located in the dygenic ATXN2‐S/AS region 9 and demonstrated its expression within the ATXN2‐S transcript. Previously, only 1 group has examined genetic alterations other than the CAG/CAA repeats in ATXN2, but they found no variants in the same region that we investigated 10 and concluded that CAG repeats are the unique cause for the parkinsonian SCA2 phenotype. Unlike previous studies (see supplementary discussion 11 , 12 , 13 ), only ascribing the effect to the CAG expansion, we found that the 9‐bp duplication acted in trans accompanying the ATXN2 intermediate allele in C9ORF72‐ALS (28 CAG) and in SCA3 (29 CAG). Therefore, our finding is novel and adds a new level of complexity for ATXN2‐related diseases.
The duplication influenced AO for SCA3 and C9ORF72‐ALS. Both ATXN3 and C9ORF72 genes are sensitive, when mutated, to ATXN2 functions. 14 , 15 For instance, unexpanded ataxin‐2 has been found in intranuclear inclusions of SCA3 brains, and a meta‐analysis confirmed ATXN2 intermediate alleles as the strongest modulators for earlier AO in SCA3. 5 , 16 For C9ORF72, coexpression of ATXN2 30Q combined with lack of C9ORF72 increases neuronal toxicity. 17 In addition, ATXN2 intermediate CAG‐repeat lengths constitute a susceptibility factor to develop motor neuron diseases among C9ORF72 mutation carriers in which other modifiers, that is, NIPA1, SMN1, and SMN2, were ruled out. 18
We did not observe modifying effects on AO of the 9‐bp duplication in the studied PD cohort; however, none of them had any known underlying PD mutations. Of interest, PINK and Parkin genes (both involved in PD) are affected by gain/loss of ataxin‐2 function. 19 , 20 Therefore, the potential modifier role merits studies in cohorts with familial PD cases. The presence of parkinsonism in 2 of our SCA3 patients supports a wide modifier role for intermediate alleles, as noted in Frontemporal Dementia and in atypical parkinsonism. 11 , 12
Our results are in line with recent observations supporting that, beyond poly‐Q tract per se, perturbations of normal aspects of ATXN2 function and its expression have implications for neurodegeneration. Interestingly, therapeutic silencing of ATXN2 increased survival and improved motor function in ALS and SCA2 mouse models. 21 , 22 Our findings warrant further studies in larger SCA2, SCA3, C9ORF72‐FTD/ALS, and familial PD cohorts.
Financial Disclosures
P.S. received honoraria from AbbVie. Other authors did not received royalties, funding, interests, grants, patents, or any commercial benefits related to this work in the last 12 months.
Author Roles
Research project: J.M.L.M., M.P., I.N., P.S.; conception: J.M.L.M., M.P., P.S.; organization: J.M.L.M., M.P., P.S.; execution: J.M.L.M., M.P., I.N.; statistical analysis: J.M.L.M.; design: J.M.L.M., M.P., P.S.; Manuscript preparation: J.M.L.M., M.P., P.S.; writing of the first draft: J.M.L.M.; review and critique: J.M.L.M., M.P., I.N., P.S..
Supporting information
Appendix S1. Supporting Information
Acknowledgments
The authors are extremely grateful to the patients and their relatives participating in this study. The authors are also grateful to the SCA network association in Sweden and to the Follin Foundation. We thank Dr. Mohsen Karimi for fruitful discussions and Ms. Lovisa Brodin for preparing PBMCs from PD patients and Jesper Eisfeldt, PhD, for helping with GEE models performed in R.
Relevant conflicts of interest/financial disclosures: Authors have nothing to declare.
Funding agencies: Parkinson Fonden 2019 and ALF support from the Stockholm City Council, Region Stockholm, P. S. is a Wallenberg Clinical Scholar.
Contributor Information
Jose Miguel Laffita‐Mesa, Email: per.svenningsson@ki.se, Email: jose.laffita@ki.se.
Per Svenningsson, Email: per.svenningsson@ki.se.
References
- 1. Pulst SM, Nechiporuk A, Nechiporuk T, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 1996;14(3):269–276. [DOI] [PubMed] [Google Scholar]
- 2. Imbert G, Saudou F, Yvert G, et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 1996;14(3):285–291. [DOI] [PubMed] [Google Scholar]
- 3. Sanpei K, Takano H, Igarashi S, et al. Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 1996;14(3):277–284. [DOI] [PubMed] [Google Scholar]
- 4. Laffita‐Mesa JM, Velázquez‐Pérez LC, Santos Falcón N, et al. Unexpanded and intermediate CAG polymorphisms at the SCA2 locus (ATXN2) in the Cuban population: evidence about the origin of expanded SCA2 alleles. Eur J Hum Genet 2012;20(1):41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. De mattos EP, Leotti VB, Soong BW, et al. Age at onset prediction in spinocerebellar ataxia type 3 changes according to population of origin. Eur J Neurol 2019;26(1):113–120. [DOI] [PubMed] [Google Scholar]
- 6. Tezenas du montcel S, Durr A, Bauer P, et al. Modulation of the age at onset in spinocerebellar ataxia by CAG tracts in various genes. Brain 2014;137(9):2444–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Long Z, Chen Z, Wang C, et al. Two novel SNPs in ATXN3 3’ UTR may decrease age at onset of SCA3/MJD in Chinese patients. PLoS One 2015;10(2):e0117488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bettencourt C, Raposo M, Kazachkova N, et al. The APOE ε2 allele increases the risk of earlier age at onset in Machado‐Joseph disease. Arch Neurol 2011;68(12):1580–1583. [DOI] [PubMed] [Google Scholar]
- 9. Li PP, Sun X, Xia G, et al. ATXN2‐AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann Neurol 2016;80(4):600–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang C, Xu Y, Feng X, et al. Linkage analysis and whole‐exome sequencing exclude extra mutations responsible for the Parkinsonian phenotype of spinocerebellar ataxia‐2. Neurobiol Aging 2015;36(1):545.e1–e7. [DOI] [PubMed] [Google Scholar]
- 11. Rubino E, Mancini C, Boschi S, et al. ATXN2 intermediate repeat expansions influence the clinical phenotype in frontotemporal dementia. Neurobiol Aging 2019;73:231.e7–231.e9. [DOI] [PubMed] [Google Scholar]
- 12. Fournier C, Anquetil V, Camuzat A, et al. Interrupted CAG expansions in ATXN2 gene expand the genetic spectrum of frontotemporal dementias. Acta Neuropathol Commun 2018;6(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tojima M, Murakami G, Hikawa R, et al. Homozygous 31 trinucleotide repeats in the SCA2 allele are pathogenic for cerebellar ataxia. Neurol Genet 2018;4(6):e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nóbrega C, Carmo‐silva S, Albuquerque D, et al. Re‐establishing ataxin‐2 downregulates translation of mutant ataxin‐3 and alleviates Machado‐Joseph disease. Brain 2015;138(Pt 12):3537–3554. [DOI] [PubMed] [Google Scholar]
- 15. Ciura S, Sellier C, Campanari ML, Charlet‐berguerand N, Kabashi E. The most prevalent genetic cause of ALS‐FTD, C9orf72 synergizes the toxicity of ATXN2 intermediate polyglutamine repeats through the autophagy pathway. Autophagy 2016;12(8):1406–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Uchihara T, Fujigasaki H, Koyano S, Nakamura A, Yagishita S, Iwabuchi K. Non‐expanded polyglutamine proteins in intranuclear inclusions of hereditary ataxias‐triple‐labeling immunofluorescence study. Acta Neuropathol 2001;102(2):149–152. [DOI] [PubMed] [Google Scholar]
- 17. Sellier C, Campanari M‐L, Corbier CJ, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin‐2 to induce motor neuron dysfunction and cell death. EMBO J 2016;35(12):1276–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Blitterswijk M, Mullen B, Heckman MG, et al. Ataxin‐2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol Aging 2014;35(10):2421.e13–e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huynh DP, Nguyen DT, Pulst‐korenberg JB, Brice A, Pulst SM. Parkin is an E3 ubiquitin‐ligase for normal and mutant ataxin‐2 and prevents ataxin‐2‐induced cell death. Exp Neurol 2007;203(2):531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sen NE, Drost J, Gispert S, et al. Search for SCA2 blood RNA biomarkers highlights Ataxin‐2 as strong modifier of the mitochondrial factor PINK1 levels. Neurobiol Dis 2016;96:115–126. [DOI] [PubMed] [Google Scholar]
- 21. Elden AC, Kim HJ, Hart MP, et al. Ataxin‐2 intermediate‐length polyglutamine expansions are associated with increased risk for ALS. Nature 2010;466(7310):1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee T, Li YR, Chesi A, et al. Evaluating the prevalence of polyglutamine repeat expansions in amyotrophic lateral sclerosis. Neurology 2011;76(24):2062–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information