

Abstract
Introduction
MAPT H1 haplotype is implicated as a risk factor for neurodegenerative diseases including Alzheimer's disease (AD).
Methods
Using Alzheimer's Disease Genetics Consortium (ADGC) genome‐wide association study (GWAS) data (n = 18,841), we conducted a MAPT H1/H2 haplotype–stratified association to discover MAPT haplotype–specific AD risk loci.
Results
We identified 11 loci—5 in H2‐non‐carriers and 6 in H2‐carriers—although none of the MAPT haplotype–specific associations achieved genome‐wide significance. The most significant H2 non‐carrier–specific association was with a NECTIN2 intronic (P = 1.33E‐07) variant, and that for H2 carriers was near NKX6‐1 (P = 1.99E‐06). The GABRG2 locus had the strongest epistasis with MAPT H1/H2 variant rs8070723 (P = 3.91E‐06). Eight of the 12 genes at these loci had transcriptome‐wide significant differential expression in AD versus control temporal cortex (q < 0.05). Six genes were members of the brain transcriptional co‐expression network implicated in “synaptic transmission” (P = 9.85E‐59), which is also enriched for neuronal genes (P = 1.0E‐164), including MAPT.
Discussion
This stratified GWAS identified loci that may confer AD risk in a MAPT haplotype–specific manner. This approach may preferentially enrich for neuronal genes implicated in synaptic transmission.
Keywords: Alzheimer's disease, case‐control studies, co‐expression networks, differential gene expression, haplotype‐stratified genome‐wide association, MAPT
1. INTRODUCTION
Tauopathies, a class of neurodegenerative disorders, are characterized by neurofibrillary tangles (NFTs) in the brain due to pathological aggregation of hyperphosphorylated microtubule‐associated protein tau (MAPT), encoded by the MAPT gene on chromosome 17q21.3. Tau tangles are present in the brains of patients with progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), Pick disease, dementia pugilistica, frontotemporal dementia, and parkinsonism linked to chromosome‐17 (FTDP‐17 or frontotemporal lobar degeneration with tau pathology (FTLD‐tau)), and other neurodegenerative diseases, including Alzheimer's disease (AD), the most prevalent tauopathy and cause of dementia. 1 In addition to senile plaques composed primarily of extracellular amyloid beta (Aβ), the presence of NFTs is a hallmark of AD pathology.
MAPT variants have been implicated in the etiology and pathogenesis of multiple neurodegenerative diseases. The discovery of multiple MAPT mutations in FTDP‐17 provided some of the first evidence that changes in tau alone could cause neurodegenerative disease. The FTDP‐17 splice‐site mutations within MAPT demonstrated that an imbalance in the ratio of 3R and 4R tau isoforms is sufficient to cause disease. 2 , 3 , 4 Further association studies revealed that the locus can be divided into two major haplotypes: H1 and H2. MAPT falls within the largest known block of linkage disequilibrium (LD) in the human genome, spanning ≈1.8 Mb. There is a 900 kb inversion of the H2 haplotype with respect to the H1 haplotype, covering a region encompassing several genes, including MAPT, IMP5, CRHR1, and NSF. The inversion results in a reduced recombination between the inverted H2 and non‐inverted H1 haplotypes.
The common MAPT haplotype H1 shows robust association with risk for the primary tauopathies PSP 5 and CBD, 6 as well as Parkinson disease (PD), which is not considered as a tauopathy. 7 MAPT H1 haplotype–tagging single‐nucleotide polymorphisms (SNPs) were identified among the top PSP 8 and PD genome‐wide association study (GWAS) 9 signals. In addition, MAPT H1 haplotype shows considerable variation 10 , 11 and leads to H1‐ subhaplotypes, where H1c, has been implicated in the risk of PSP, CBD, AD, and PD. 12 , 13 MAPT H2 haplotype has been associated with reduced risk for several neurodegenerative disorders. 14 , 15
Although MAPT is a compelling candidate for neurodegenerative disease susceptibility, evidence of association of AD with the MAPT H1 and H2 haplotypes have produced equivocal results. 12 , 16 , 17 This may in part be due to limited sample sizes, and therefore limited power for most MAPT haplotype association studies in AD. In a large study from Genetic and Environmental Risk for Alzheimer's Disease (GERAD1) consortium, 18 the MAPT H2 haplotype–tagging variant was found to have association with reduced AD risk. In a study of >20,000 individuals from Mayo Clinic and the Alzheimer's Disease Genetics Consortium (ADGC), we identified associations with both reduced AD risk and reduced brain MAPT levels with the H2 haplotype. 14 In addition, a recent meta‐analysis pooling 39 studies in AD again demonstrated association of reduced AD risk with the MAPT H2 haplotype. 15
HIGHLIGHTS
Microtubule‐associated protein tau gene (MAPT) H1 and H2 carriers have discordant Alzheimer's disease (AD) risk loci, most of which are novel.
Many of the genes at these loci are differentially expressed in AD brains.
The MAPT haplotype–stratified approach identified genes in synaptic transmission networks.
RESEARCH IN CONTEXT
Systemic review: Comprehensive review of the literature shows that the microtubule‐associated protein tau gene (MAPT) is a strong candidate for neurodegenerative disease susceptibility. The MAPT H2 haplotype is associated with lower Alzheimer's disease (AD) risk in large cohorts and lower brain MAPT levels.
Interpretation: We hypothesized that AD risk variants exhibit MAPT haplotype–dependent association. Through haplotype‐stratified association analyses using data from the Alzheimer's Disease Genetics Consortium (ADGC) on 18,841 participants, we identified 11 loci with MAPT H1– or H2–specific AD risk association. Eight genes at these loci had significant differential expression in AD versus control brains. Six genes were members of the neuronal‐enriched brain transcriptional co‐expression network implicated in synaptic transmission.
Future directions: Replication of MAPT haplotype–stratified associations should be sought in larger cohorts. Candidate genes from this study should be evaluated for the presence of functional variants that may influence tau‐related outcomes. Emerging larger cohorts with multiomics data and generation of more complex model systems will enable these studies.
In the current study, we sought to further elucidate the role of MAPT H1 and H2 haplotypes in AD susceptibility by leveraging the genome‐wide genotype data available from the sizable ADGC case‐control series. Using haplotype‐stratified analyses, we tested the hypothesis that AD risk variants exhibit MAPT haplotype–dependent association and may therefore potentially identify novel AD risk variants with implications for functional pathways. Analysis of a stratum with a more homogeneous AD risk profile with respect to MAPT H1/H2 haplotype may help uncover loci that have differential influence on AD risk in a MAPT context‐specific manner. For example, given the association of MAPT H2 with lower brain MAPT levels, it is plausible that those loci with MAPT H2–specific associations harbor genes that influence neurodegeneration via pathways that are not dependent on elevated tau. In contrast, AD risk associations in H2 non‐carriers may enrich for loci that confer risk in a tau level‐dependent fashion.
Our approach herein is akin to pursuing GWAS in an apolipoprotein E gene (APOE)–stratified fashion. 19 Although MAPT haplotypes tested to date in the literature clearly have smaller effect sizes than that of APOE genotypes for AD risk, it is nonetheless worthwhile to pursue this MAPT haplotype–stratified analysis not only because of its potential to identify novel loci but also because of the plethora of data implicating tau in AD in functional studies. 20 In this study, we evaluated known International Genomics of Alzheimer's Project (IGAP) AD 21 risk loci in a MAPT haplotype–stratified analysis, which did not reveal evidence of MAPT haplotype–specific associations. We also identified novel AD risk loci with association only in MAPT H2 carriers (six loci) or H2‐non‐carriers (five loci). We characterize genes near both the known and the new loci for their expression levels and co‐expression networks in a brain transcriptome data set of AD and control temporal cortex. 22 , 23 Our findings, which require replication in larger cohorts, suggest that MAPT haplotype–stratified GWAS may identify novel loci, and that genes at these loci are expressed predominantly within neuron‐enriched networks implicated in synaptic transmission.
2. METHODS
2.1. Study populations
The ADGC data were used for this study. Subjects available through the ADGC have been described previously and are available through ftps from the UPENN server (alois.med.upenn.edu). 24 , 25 , 26 , 27 The data set included all the covariates required for the analysis and all actual and imputed genotypes. Post–quality control (post‐QC) data for both the actual and imputed genotypes and designations for all the sub‐cohorts included in the ADGC data were obtained. The demographics detailing each cohort and stratified group are described in Table S1. The cohort for the expression analysis was the Mayo Clinic RNAseq data set. 22 Detailed methods are provided in Supplementary Methods.
2.2. AD risk association analysis
Variants were evaluated for association with AD using multivariable logistic regression implemented in PLINK. 28 Both joint (full data set of 21 cohorts analyzed jointly, adjusting for cohort) and meta‐(separate cohorts) analyses were performed. For the meta‐analysis, a random effects method was adopted due to presence of heterogeneity, I 2 > 25. 29 An additive model for the minor alleles determined in the unstratified data set was applied with the covariates age, sex, and PC1‐3 (principal components 1‐3) used throughout all models. A second model using the additional APOE covariate in the joint and meta‐analyses was also evaluated. Two IGAP loci variants rs4147929 and rs9331896 were filtered out of the original data set due to the QC procedures described previously. 27 They were evaluated separately for the joint analyses using the same method above. Meta analyses could not be performed for rs4147929 and rs9331896 due to their absence from the original data set. To generate forest plots for the variants of interest, meta‐analysis was performed in R30 with the Metafor package 31 using the random effects method with DerSimonian Laird estimator for the variance between studies/cohorts. To determine the joint effect of the tested SNPs and MAPT haplotypes on AD risk, we also performed a bivariate analysis, described in Supplementary Methods.
2.3. Epistasis analysis
SNP–SNP interactions of epistasis between each of the 3,067,502 SNPs and the H2 tagging variant rs8070723‐G were conducted. Two models were evaluated for the H2 tagging variant, a carrier model (H1H1 and H1H2+H2H2) and a dosage model (H1H1, H1H2, and H2H2). The analysis was performed by creating a distance matrix in PLINK between each SNP and rs8070723‐G. Two general linear models (with SNPx rs8070723‐G interaction and without interaction) were executed using age, sex, ADGC cohort, and PC1‐3 as covariates followed by an analysis of variance (ANOVA) to assess the significance between the models using the chi‐square method as implemented in R.
2.4. Gene expression analyses
Differential gene expression and co‐expression network analyses were conducted as previously published. 23 , 32 For each gene, multiple linear regression was performed in which normalized gene expression was the dependent variable, diagnosis (AD vs control) was the independent variable of primary interest and sex, flowcell, age at death, RNA integrity number (RIN), and center from which the samples were obtained were the covariates. Weighted Gene Co‐Expression Network Analysis (WGCNA) was utilized to identify brain co‐expression networks and test their associations with AD as we reported previously. 23
2.5. Visuals
The figures were generated using the lattice 33 and metafor packages in R and Inkscape (www.inkscape.org).
3. RESULTS
3.1. MAPT haplotype–specific association analysis at known AD risk loci
Using genome‐wide genotype data from 21 cohorts within ADGC, we tested the hypothesis that AD risk variants exhibit MAPT haplotype–specific association. Following QC measures, approximately 3 million variants with a minor allele frequency (MAF) ≥0.02, and all index variants identified by the IGAP consortium 21 were retained for analysis and evaluated for MAPT haplotype–specific association. MAPT H2 haplotype tagging allele rs8070723‐G was used to stratify study participants into H2 carriers (H1H2+H2H2: 3631 cases, 3729 controls) and H2 non‐carriers (H1H1: 5958 cases, 5523 controls). The demographics of the cohorts of the H2 carriers and non‐carriers are described in Table S1.
GWAS analyses with AD were performed using joint and meta‐analyses. There was no evidence of population stratification based on the quantile‐quantile plots (QQ plots) (Figure S1) and the genomic inflation factors of 1.04, 1.04, and 1.01 for the unstratified, H2 non‐carrier, and H2 carrier joint analyses, respectively. The joint and meta‐analyses yielded similar results with respect to genomic inflation. Likewise, the addition of APOE as a covariate did not significantly alter the results. We adopted the joint analysis approach without APOE covariate as the primary model.
We first evaluated the previously reported IGAP 21 AD risk variants to determine if they exhibit MAPT haplotype–specific association. As expected, the unstratified analysis results were similar to those reported in the IGAP study, albeit with reduced significance due to the smaller cohort size (Figure S2, Table 1). IGAP index variants had similar direction of AD risk in both the H2 non‐carrier and H2 carrier analyses. To determine whether any of these variants had a significantly different effect on AD risk based on the MAPT haplotype, we performed epistasis analysis with the MAPT H1/H2 haplotype tagging variant. Only two IGAP variants, rs10948363 (CD2AP) and rs1476679 (ZCWPW1/PILRB), showed a trend of epistasis (uncorrected P < 0.05) with the MAPT H1/H2 haplotype–tagging variant (Table 1); however, the odds ratios (ORs) for both variants were in the same direction with overlapping 95% confidence intervals (CIs). In summary, we found no strong evidence of MAPT haplotype–specific association for the reported IGAP AD risk SNPs.
TABLE 1.
IGAP loci associations stratified by H2 non‐carriers and H2 carriers
| Unstratified f | H2 non‐carriers f | H2 carriers f | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP a | CHR c | Position c | Closest gene d | Major/Minor c | MAF e | Nj g | ORJ (95% CI) g | P J g | NM h | ORm h | PM h | Nj g | ORJ (95% CI) g | PJ g | NM h | ORm h | PM h | Nj g | ORJ (95% CI) g | PJ g | NM h | ORm h | PM h | Epistatis P i |
| rs6656401 | 1 | 207692049 | CR1 | G/A | 0.183 | 17833 | 1.15 (1.08‐1.21) | 2.37E‐06 | 21 | 1.15 | 1.37E‐06 | 10854 | 1.13 (1.05‐1.21) | 9.82E‐04 | 21 | 1.14 | 7.48E‐04 | 6979 | 1.17 (1.07‐1.28) | 7.45E‐04 | 21 | 1.17 | 1.36E‐03 | 5.37E‐01 |
| rs6733839 | 2 | 127892810 | BIN1;CYP27C1 | C/T | 0.394 | 14091 | 1.23 (1.17‐1.3) | 4.39E‐16 | 21 | 1.23 | 1.89E‐10 | 8627 | 1.23 (1.15‐1.31) | 2.64E‐10 | 21 | 1.23 | 1.25E‐09 | 5464 | 1.24 (1.14‐1.35) | 3.43E‐07 | 21 | 1.27 | 6.23E‐08 | 8.38E‐01 |
| rs35349669 | 2 | 234068476 | INPP5D | C/T | 0.482 | 17273 | 1.04(1‐1.09) | 7.35E‐02 | 21 | 1.04 | 7.39E‐02 | 10535 | 1.04 (0.99‐1.11) | 1.37E‐01 | 21 | 1.05 | 9.74E‐02 | 6738 | 1.03 (0.96‐1.11) | 3.69E‐01 | 21 | 1.02 | 5.38E‐01 | 8.82E‐01 |
| rs190982 | 5 | 88223420 | MEF2C‐AS1 | A/G | 0.384 | 16317 | 0.94 (0.9‐0.98) | 9.25E‐03 | 21 | 0.93 | 5.87E‐03 | 9911 | 0.93 (0.88‐0.99) | 2.53E‐02 | 21 | 0.92 | 1.03E‐02 | 6406 | 0.95 (0.88‐1.02) | 1.78E‐01 | 21 | 0.96 | 2.87E‐01 | 7.69E‐01 |
| rs9271192 | 6 | 32578530 | HLA‐DRB1;HLA‐DQA1 | A/C | 0.272 | 18208 | 1.1 (1.05‐1.15) | 1.51E‐04 | 21 | 1.10 | 2.37E‐03 | 11080 | 1.14 (1.07‐1.21) | 7.56E‐05 | 21 | 1.14 | 1.85E‐03 | 7128 | 1.04 (0.96‐1.13) | 2.92E‐01 | 21 | 1.05 | 2.73E‐01 | 1.02E‐01 |
| rs10948363 | 6 | 47487762 | CD2AP | A/G | 0.274 | 18827 | 1.09 (1.04‐1.14) | 5.18E‐04 | 21 | 1.09 | 5.22E‐04 | 11476 | 1.05 (0.98‐1.11) | 1.56E‐01 | 21 | 1.05 | 1.47E‐01 | 7351 | 1.16 (1.07‐1.25) | 1.60E‐04 | 21 | 1.16 | 6.81E‐04 | 3.47E‐02 |
| rs2718058 | 7 | 37841534 | GPR141;NME8 | A/G | 0.364 | 18336 | 0.93 (0.89‐0.98) | 2.81E‐03 | 21 | 0.94 | 7.69E‐02 | 11182 | 0.94 (0.89‐1) | 4.77E‐02 | 21 | 0.97 | 3.96E‐01 | 7154 | 0.92 (0.86‐0.99) | 2.78E‐02 | 21 | 0.92 | 2.50E‐02 | 6.38E‐01 |
| rs1476679 | 7 | 100004446 | ZCWPW1/PILRB | T/C | 0.280 | 18617 | 0.92 (0.88‐0.97) | 1.22E‐03 | 21 | 0.92 | 1.73E‐03 | 11346 | 0.88 (0.83‐0.94) | 7.06E‐05 | 21 | 0.88 | 8.06E‐05 | 7271 | 0.99 (0.92‐1.07) | 7.82E‐01 | 21 | 0.98 | 7.21E‐01 | 2.20E‐02 |
| rs11771145 | 7 | 143110762 | EPHA1‐AS1 | G/A | 0.334 | 17291 | 0.93 (0.89‐0.97) | 1.97E‐03 | 21 | 0.93 | 4.22E‐03 | 10525 | 0.93 (0.88‐0.99) | 1.85E‐02 | 21 | 0.93 | 2.87E‐02 | 6766 | 0.93 (0.86‐1) | 4.27E‐02 | 21 | 0.93 | 6.38E‐02 | 8.76E‐01 |
| rs28834970 | 8 | 27195121 | PTK2B | T/C | 0.363 | 18538 | 1.13 (1.08‐1.18) | 1.68E‐07 | 21 | 1.13 | 1.64E‐07 | 11305 | 1.14 (1.07‐1.2) | 1.48E‐05 | 21 | 1.14 | 1.67E‐04 | 7233 | 1.12 (1.04‐1.2) | 3.15E‐03 | 21 | 1.12 | 3.42E‐03 | 6.97E‐01 |
| rs9331896 b | 8 | 27467686 | CLU | T/C | 0.377 | 16160 | 0.90 (0.86‐0.95) | 2.45E‐05 | NA | NA | NA | 9842 | 0.91 (0.85‐0.96) | 1.36E‐03 | NA | NA | NA | 6318 | 0.90 (0.84‐0.98) | 9.41E‐03 | NA | NA | NA | 9.87E‐01 |
| rs10838725 | 11 | 47557871 | CELF1 | T/C | 0.316 | 18596 | 1.04 (0.99‐1.09) | 1.25E‐01 | 21 | 1.04 | 9.93E‐02 | 11334 | 1.04 (0.98‐1.11) | 1.63E‐01 | 21 | 1.04 | 2.03E‐01 | 7262 | 1.03 (0.96‐1.11) | 4.20E‐01 | 21 | 1.04 | 2.99E‐01 | 7.52E‐01 |
| rs983392 | 11 | 59923508 | MS4A2;MS4A6A | A/G | 0.398 | 18095 | 0.87 (0.84‐0.91) | 2.44E‐09 | 21 | 0.87 | 5.14E‐09 | 11017 | 0.87 (0.83‐0.93) | 3.61E‐06 | 21 | 0.87 | 3.55E‐06 | 7078 | 0.87 (0.81‐0.93) | 1.11E‐04 | 21 | 0.86 | 4.95E‐03 | 9.28E‐01 |
| rs10792832 | 11 | 85867875 | PICALM;EED | G/A | 0.355 | 18651 | 0.88 (0.84‐0.92) | 7.90E‐09 | 21 | 0.88 | 9.90E‐09 | 11368 | 0.89 (0.84‐0.94) | 3.32E‐05 | 21 | 0.89 | 7.71E‐05 | 7283 | 0.86 (0.8‐0.93) | 5.00E‐05 | 21 | 0.85 | 4.90E‐05 | 5.56E‐01 |
| rs11218343 | 11 | 121435587 | SORL1 | T/C | 0.039 | 18816 | 0.76 (0.68‐0.84) | 7.44E‐07 | 21 | 0.75 | 9.94E‐07 | 11465 | 0.8 (0.69‐0.92) | 1.46E‐03 | 21 | 0.80 | 2.20E‐03 | 7351 | 0.69 (0.58‐0.83) | 5.43E‐05 | 18 | 0.69 | 1.34E‐04 | 2.26E‐01 |
| rs17125944 | 14 | 53400629 | FERMT2 | T/C | 0.093 | 18809 | 1.12 (1.04‐1.21) | 2.75E‐03 | 21 | 1.13 | 2.59E‐02 | 11459 | 1.11 (1.01‐1.22) | 3.68E‐02 | 21 | 1.09 | 1.84E‐01 | 7350 | 1.15 (1.02‐1.29) | 2.38E‐02 | 21 | 1.15 | 2.32E‐02 | 5.89E‐01 |
| rs10498633 | 14 | 92926952 | SLC24A4 | G/T | 0.217 | 18720 | 0.92 (0.88‐0.97) | 2.27E‐03 | 21 | 0.93 | 5.80E‐03 | 11409 | 0.9 (0.84‐0.96) | 1.45E‐03 | 21 | 0.90 | 2.50E‐03 | 7311 | 0.96 (0.88‐1.04) | 3.27E‐01 | 21 | 0.97 | 5.18E‐01 | 2.21E‐01 |
| rs8093731 | 18 | 29088958 | DSG2 | C/T | 0.010 | 18683 | 0.94 (0.76‐1.16) | 5.49E‐01 | 20 | 1.01 | 9.39E‐01 | 11389 | 0.9 (0.69‐1.18) | 4.62E‐01 | 18 | 0.84 | 2.92E‐01 | 7294 | 0.99 (0.72‐1.38) | 9.70E‐01 | 15 | 1.14 | 6.31E‐01 | 6.37E‐01 |
| rs4147929 b | 19 | 1063443 | ABCA7 | G/A | 0.171 | 16429 | 1.14 (1.07‐1.21) | 4.60E‐05 | NA | NA | NA | 10002 | 1.09 (1.01‐1.18) | 2.97E‐02 | NA | NA | NA | 6427 | 1.21 (1.09‐1.33) | 1.61E‐04 | NA | NA | NA | 1.34E‐01 |
| rs3865444 | 19 | 51727962 | CD33 | C/A | 0.303 | 18707 | 0.91 (0.87‐0.96) | 1.78E‐04 | 21 | 0.91 | 1.70E‐04 | 11395 | 0.92 (0.87‐0.98) | 8.80E‐03 | 21 | 0.92 | 1.14E‐02 | 7312 | 0.9 (0.84‐0.97) | 6.84E‐03 | 21 | 0.89 | 3.35E‐03 | 6.27E‐01 |
| rs7274581 | 20 | 55018260 | CASS4 | T/C | 0.084 | 18599 | 0.87 (0.8‐0.94) | 3.21E‐04 | 21 | 0.86 | 2.92E‐04 | 11333 | 0.88 (0.8‐0.98) | 1.71E‐02 | 21 | 0.88 | 2.01E‐02 | 7266 | 0.84 (0.74‐0.95) | 5.15E‐03 | 21 | 0.84 | 5.66E‐03 | 4.88E‐01 |
IGAP variants determined in the publication Lambert et al.21
SNPs rs4147929 and rs9331896 were filtered out of the original analysis due to the QC procedures in Boehme et al.27 They were evaluated separately for the joint analysis.
Chromosome, position, alleles from the February 2009 (GRCh37/hg19) build.
Nearest gene(s) located within ±100 kb of the IGAP SNP.
MAF calculated in PLINK derived from the unstratified genotypes.
Unstratified contains all subjects from the 21 ADGC cohorts used for analysis. The H2 tagging variant rs8070723 was used to stratify study participants into H2 non‐carriers (H1H1) and H2 carriers (H1H2+H2H2).
Joint analysis performed with logistic regression in PLINK using as covariates age, sex, cohort, and PC1‐3. Rs4147929 is missing in the GSK cohort. N, number of subjects analyzed; OR, odds ratio for AD risk for the minor allele; 95% CI, 95% confidence interval, P‐value.
Meta‐analysis performed with logistic regression analysis in PLINK using the random effects model, and using the covariates age, sex, and PC1‐3. Meta‐analysis could not be performed for rs4147929 and rs9331896. N, number of cohorts analyzed; OR, odds ratio, P‐value.
Epistasis analyses performed in R. All cohorts were combined and stratified into two groups by the H2 tagging variant rs8070723: H2 non‐carriers (H1H1) and H2 carriers (H1H2+H2H2). Results from the carrier model are shown.
3.2. Genome‐wide MAPT haplotype–specific AD risk association analysis
To identify any additional AD risk variants with MAPT haplotype–specific association, we evaluated the genome‐wide results for the unstratified, H2 non‐carrier and H2 carrier groups (Figure 1). We tested for significance of MAPT haplotype–specific associations by genome‐wide epistasis analysis with rs8070723 (Table 2). We defined loci with MAPT haplotype–specific AD risk associations as being discordant. To be classified as discordant, the following criteria had to be met: Discordant locus (1) has AD risk association P value of < 1E‐05 in one of the stratified analysis, but statistically insignificant in the other one (P > 5E‐02); (2) has nominally significant epistasis interaction with rs8070723 (P < 0.05).
FIGURE 1.
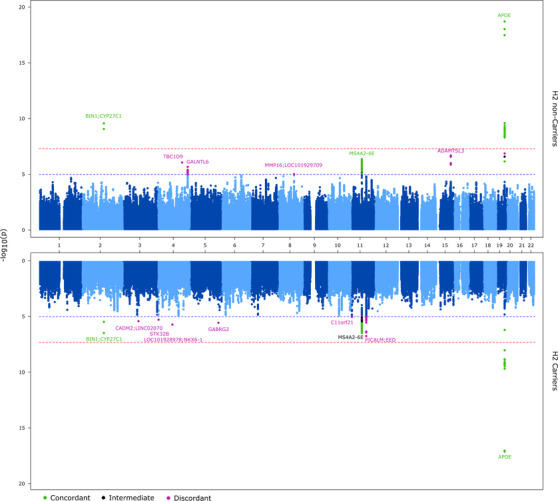
Miami plot of MAPT haplotype–stratified association results: P values from the joint association analyses are shown. APOE was not included as a covariate in these analyses. Top: H2 non‐carriers. Bottom: H2 carriers. The threshold for genome‐wide significance (P < 5E10‐8) is indicated by the red line and the threshold for trending significance (p < 1E‐5) is indicated by the blue line. Loci with P < 1E‐05 are annotated as follows: dark green, concordant (P < 1E‐05 in both data sets with epistatic P > 0.05); dark purple, discordant (P < 1E‐05 in one data set only, with epistatic P < 0.05); black, intermediate (P < 1E‐05 in one data set only with epistatic P > 0.05)
TABLE 2.
MAPT‐haplotype specific associations
| Unstratified d | H2 non‐carriers d | H2 carriers d | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNPa | CHR b | Position b | Closest gene c | Major/Ref allele b | Minor/Alt allele b | Nj e | ORJ (95% CI) e | Pj e | Nm f | ORm f | Pm f | Nj e | ORJ (95% CI) e | Pj e | Nm f | ORm f | Pm f | Nj e | ORJ (95% CI) e | Pj e | Nm f | ORm f | Pm f | Epistatis P g |
| Discordant loci with P ≤ 1E‐5 in the H2 non‐carriers a.1 | ||||||||||||||||||||||||
| rs4555724 | 4 | 141658697 | TBC1D9 | T | C | 18797 | 0.91 (0.87‐0.95) | 3.64E‐05 | 21 | 0.92 | 3.19E‐03 | 11454 | 0.87 (0.82‐0.92) | 8.64E‐07 | 21 | 0.87 | 1.11E‐05 | 7343 | 0.98 (0.92‐1.06) | 6.60E‐01 | 21 | 0.99 | 6.95E‐01 | 6.78E‐03 |
| rs28705797 | 4 | 173902942 | GALNTL6 | A | C | 18759 | 1.13 (1.06‐1.2) | 1.12E‐04 | 21 | 1.13 | 1.97E‐04 | 11424 | 1.21 (1.12‐1.32) | 2.10E‐06 | 21 | 1.21 | 4.73E‐06 | 7335 | 1.02 (0.92‐1.13) | 6.96E‐01 | 21 | 1.00 | 9.37E‐01 | 7.96E‐03 |
| rs1685555 | 8 | 89483894 | MMP16;LOC101929709 | A | G | 18637 | 0.79 (0.69‐0.9) | 5.13E‐04 | 21 | 0.79 | 1.08E‐02 | 11371 | 0.68 (0.57‐0.81) | 9.49E‐06 | 20 | 0.67 | 3.08E‐04 | 7266 | 1 (0.81‐1.24) | 9.79E‐01 | 20 | 1.02 | 8.78E‐01 | 4.98E‐03 |
| rs4354897 | 15 | 84644971 | ADAMTSL3 | T | C | 18707 | 0.91 (0.87‐0.96) | 2.90E‐04 | 21 | 0.91 | 5.72E‐03 | 11397 | 0.84 (0.79‐0.9) | 2.04E‐07 | 21 | 0.84 | 1.22E‐06 | 7310 | 1.03 (0.95‐1.12) | 4.87E‐01 | 21 | 1.02 | 7.13E‐01 | 1.22E‐04 |
| rs11665676 | 19 | 45378719 | NECTIN2;TOMM40:APOE | C | T | 18622 | 0.77 (0.7‐0.85) | 6.16E‐07 | 21 | 0.76 | 2.40E‐05 | 11355 | 0.7 (0.62‐0.8) | 1.33E‐07 | 21 | 0.69 | 9.76E‐07 | 7267 | 0.89 (0.75‐1.05) | 1.63E‐01 | 21 | 0.89 | 1.70E‐01 | 3.45E‐02 |
| Discordant loci with P ≤ 1E‐5 in the H2 carriers a.2 | ||||||||||||||||||||||||
| rs7356060 | 3 | 86238011 | CADM2;LINC02070 | A | T | 18424 | 1.13 (1.05‐1.21) | 4.39E‐04 | 21 | 1.12 | 7.63E‐04 | 11231 | 1.04 (0.95‐1.13) | 4.28E‐01 | 21 | 1.03 | 5.77E‐01 | 7193 | 1.29 (1.16‐1.43) | 3.99E‐06 | 21 | 1.29 | 7.64E‐06 | 1.67E‐03 |
| rs348732 | 4 | 85250583 | LOC101928978;NKX6‐1 | C | A | 18605 | 0.91 (0.88‐0.96) | 5.82E‐05 | 21 | 0.91 | 5.32E‐05 | 11342 | 0.96 (0.91‐1.02) | 1.47E‐01 | 21 | 0.96 | 1.46E‐01 | 7263 | 0.84 (0.79‐0.91) | 1.99E‐06 | 21 | 0.83 | 4.49E‐07 | 6.17E‐03 |
| rs1838973 | 4 | 5157262 | STK32B | G | A | 18586 | 1.26 (1.14‐1.4) | 1.40E‐05 | 21 | 1.26 | 2.19E‐05 | 11332 | 1.14 (1‐1.31) | 5.43E‐02 | 21 | 1.14 | 6.95E‐02 | 7254 | 1.46 (1.24‐1.72) | 5.46E‐06 | 20 | 1.56 | 1.27E‐05 | 2.28E‐02 |
| rs55712126 | 5 | 161528378 | GABRG2 | T | G | 18703 | 1.12 (0.99‐1.26) | 6.85E‐02 | 21 | 1.09 | 1.66E‐01 | 11412 | 0.9 (0.78‐1.05) | 1.85E‐01 | 20 | 0.89 | 1.36E‐01 | 7291 | 1.62 (1.33‐1.99) | 2.88E‐06 | 21 | 1.60 | 9.85E‐06 | 3.91E‐06 |
| rs77007065 | 11 | 2319730 | C11orf21 | G | A | 18743 | 0.93 (0.84‐1.03) | 1.79E‐01 | 21 | 0.94 | 3.07E‐01 | 11426 | 1.12 (0.99‐1.28) | 7.88E‐02 | 21 | 1.12 | 8.86E‐02 | 7317 | 0.68 (0.58‐0.81) | 9.78E‐06 | 20 | 0.70 | 8.35E‐05 | 5.62E‐06 |
| rs140869727 | 11 | 85751041 | PICALM;EED | G | A | 18516 | 1.11 (1.06‐1.17) | 5.00E‐05 | 21 | 1.12 | 3.79E‐04 | 11283 | 1.05 (0.98‐1.12) | 1.57E‐01 | 21 | 1.05 | 3.05E‐01 | 7233 | 1.22 (1.12‐1.33) | 3.00E‐06 | 21 | 1.23 | 2.38E‐06 | 4.51E‐03 |
This table shows the loci with AD risk associations of P ≤1E‐5 in only H2 non‐carriers (a.1) or H2 carriers (a.2). The H2 tagging variant rs8070723 was used to stratify study participants into H2 non‐carriers (H1H1) and H2 carriers (H1H2+H2H2). The table shows the top loci from the LD‐based clumping implemented in PLINK using an r2 threshold of 0.2 within 1000kb.
Loci with P ≤ 1E‐5 in H2 non‐carriers and P ≥ 0.05 in the in H2 carriers.
Loci with P ≤ 1E‐5 in H2 carriers and P ≥ 0.05 in the in H2 non‐carriers. Rs140869727 at the PICALM locus was clumped with rs17159904, but rs17159904 did not meet the P ≥ 0.05 threshold in the H2 non‐carriers. Therefore, the result from the former is shown.
Chromosome, position, alleles from the February 2009 (GRCh37/hg19) build.
Nearest gene(s) located within ±100 kb of the top SNP.
Unstratified contains all subjects from the 21 ADGC cohorts used for analysis.
Joint analysis performed with logistic regression in PLINK using as covariates age, sex, cohort and PC1‐3. N, number of subjects analyzed; OR, odds ratio; 95% CI, 95% confidence interval, P‐value.
Meta‐analysis of logistic regression analysis in PLINK using the random effects model, and using the covariates age, sex and PC1‐3. N, number of cohorts analyzed; OR, odds ratio, P‐value.
Epistasis analyses performed in R. All cohorts were combined and stratified into two groups by the H2 tagging variant rs8070723: H2 non‐carriers (H1H1) and H2 carriers (H1H2+H2H2). Results from the carrier model are shown.
We identified five loci in the H2 non‐carriers and six in the H2 carriers with discordant MAPT haplotype–specific AD risk associations (Figure 1, Table 2). These loci (nearest genes at loci) are as follows: In the H2 non‐carriers: chr4 (TBC1D9), chr4 (GALNTL6), chr8 (MMP16;LOC101929709), chr15 (ADAMTSL3), and chr19 (NECTIN2;TOMM40;APOE); and in the H2 carriers: chr3 (CADM2;LINC02070), chr4 (STK32B), chr 4 (LOC101928978;NKX6‐1), chr5 (GABRG2), chr11 (C11orf21), chr11 (PICALM;EED). None of these loci reached genome‐wide significance, although they had a stronger association in their relevant MAPT haplotype–stratified groups than in the combined unstratified group, despite the smaller sample size of the former. Forest plots of the discordant loci and their meta‐analysis results are shown in Figure S3.
We checked the regional association plots of the discordant loci to determine whether any of them represented known IGAP AD risk loci (Figure 2). All but two of the discordant loci are novel, which is not surprising because the most significant associations detected by IGAP are likely to be enriched for concordant loci. The two discordant loci that are also known AD risk loci are NECTIN2;TOMM40;APOE and PICALM;EED, which have differentially greater significance in the MAPT H2 non‐carriers and H2 carriers, respectively. We further evaluated these two loci to determine the extent to which the discordant associations are influenced by the known index variants.
FIGURE 2.
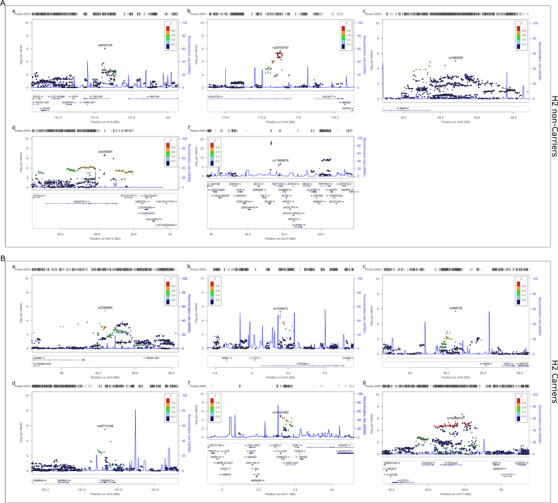
Regional association plots of discordant MAPT haplotype–stratified association results: The figures are shown for the 11 loci depicted in Table 2 and reflect the results of haplotype‐stratified joint association analyses without the APOE covariate. Discordant loci results with significance in the (A) H2 non‐carriers or (B) H2 carriers.
For the NECTIN2;TOMM40;APOE locus, we determined that the minor T allele of rs11665676 is more enriched in APOE ε4–negative than in APOE ε4–positive participants, with allele frequencies of 0.06 versus 0.03, respectively. The frequency of rs11665676‐T in participants with the APOE ε2/ε2; ε2/ε3; ε2/ε4; ε3/ε3; ε3/ε4; and ε4/ε4 backgrounds is 0; 0.037; 0.006; 0.065; 0.035; and 0.006, respectively, which demonstrates the enrichment of this allele, particularly in APOE ε3. When we repeated the analysis adjusting for APOE ε4 dosage, the AD risk association of rs11665676 in MAPT H2 non‐carriers was abolished (APOE‐unadjusted OR = 0.7 and P = 1.33E‐07; APOE‐adjusted OR = 0.93 and P = 0.28), which is not surprising given the strong linkage disequilibrium (LD) of this variant with those that define APOE ε2/ε3/ε4 (rs429358 and rs7412). APOE ε4 dosage association with AD risk did not reveal differences between the MAPT H2 non‐carriers (OR = 1.35, P = 3.28E‐264) and H2 carriers (OR = 1.42, P = 2.80E‐190). The 95% CI for APOE ε4 AD risk ORs were overlapping between these two stratified groups, and there was no evidence of epistasis interaction between APOE ε4 dose and MAPT H1/H2 haplotype. Collectively, our findings suggest that although there are no discordant associations for APOE ε4 dose per se based on MAPT H1/H2 status, rs11665676‐T may be tagging a subtype of APOE, which confers greater protection in MAPT H2 non‐carriers.
The PICALM locus index IGAP SNP rs10792832 did not have any evidence of differential MAPT haplotype–stratified association (Table 1, epistasis P value > 0.05). In contrast, the discordant variant rs140869727 that resides in an intron of PICALM has epistasis (P = 4.51E‐03) with AD risk association in the MAPT H2 carriers (OR = 1.22, P = 3.0E‐06, Table 2). The linkage disequilibrium r 2 value for these SNPs is 0.15 in the unstratified and both stratified cohorts, with D' = 0.99. These results support a model where the rarer and discordant rs140869727‐A may be tagging a PICALM variant, which confers a greater risk of AD in MAPT H2 carriers.
Of the discordant loci with significance in the H2 non‐carriers, the four novel ones had essentially no overlap in their 95% CIs with the H2‐carrier results (Table 2). The level of significance for joint analyses in the H2 non‐carriers ranged between P = 2.04E‐7 (ADAMTSL3) and P = 9.49E‐6 (MMP16;LOC101929709). For these discordant variants that are significant in the H2 non‐carriers, there was no evidence of association in the H2 carriers (ORs ≈1 and P = 0.5‐1.0). Similarly, the five discordant novel loci with significance in the H2 carriers had P = 1.99E‐6 (LOC101928978;NKX6‐1) to P = 9.78E‐6 (C11orf21), whereas in the H2 non‐carriers, these variants had ORs at ≈1 with essentially non‐overlapping 95% CIs and P = 0.054 to 0.4.
By definition, all discordant loci had nominally significant epistasis P values, although none reached genome‐wide significance (Table 2). Considering the 21 IGAP and 11 discordant loci evaluated, and applying a study‐wide epistasis P ‐value of 1.52E‐3 (Bonferroni P = 0.05/33), there was one discordant SNP with MAPT H2 non‐carrier–specific association and two discordant SNPs in the MAPT H2‐carrier group. The SNP with the smallest epistasis P value and MAPT H2 non‐carrier–specific association is rs4354897 on chromosome 15 (Table 2), an intronic variant within ADAMTSL3 (Figure 2). Among the discordant loci, this is the second most significant variant (P = 2.04E‐07) after the chromosome 19 APOE locus variant. The minor allele of ADAMTSL3 rs4354897 is associated with a lower risk of AD (OR = 0.84) in MAPT H2 non‐carriers.
The two MAPT H2 carrier–specific variants with study‐wide significant epistasis were rs55712126 on chromosome 5, an intronic variant in GABRG2; and rs77007065 on chromosome 11, which is intronic for C11orf21 and also 2 kb upstream of TSPAN32 (Figure 2). GABRG2 rs55712126‐G and C11orf21 rs77007065‐A are associated with higher (OR = 1.62, P = 2.88E‐06) and lower AD risk (OR = 0.68, P = 9.78E‐06), respectively, in MAPT H2 carriers (Table 2).
To determine the joint effect of the discordant SNPs and MAPT haplotypes on AD risk, we performed a bivariate analysis (Table S2). The MAPT H2 non‐carriers with the SNP major homozygote genotypes were designated as the reference. We tested the AD risk association of each SNP genotype in the MAPT H2‐carrier or H2 non‐carrier background against this reference. The bivariate analysis results are consistent with their corresponding MAPT haplotype–specific associations and depict the joint effect of each SNP genotype and the MAPT haplotype on AD risk.
3.3. Brain expression analyses of MAPT‐stratified AD risk association loci genes
We characterized the brain expression patterns of the genes at the discordant MAPT‐stratified association loci (Table 2) using the temporal cortex (TCX) RNAseq transcriptome data from Mayo Clinic. 22 , 23 , 32 Of the 17 genes at the 11 discordant loci, 12 were present in this data set (Table S3). We evaluated these genes for differential expression (DE) between neuropathologic AD and control TCX RNAseq data. In addition, we determined the brain gene co‐expression networks, 34 which harbor these genes and annotated these networks for their enriched gene ontology (GO) biological processes 35 and brain cell types, as described previously. 23 , 32 Eight of the 12 genes evaluated had significantly different expression in AD versus control TCX (Table S3). The genes with transcriptome‐wide significant differential expression (q<0.05) were GALNTL6, TBC1D9, TOMM40, APOE, PVRL2, ADAMTSL3, GABRG2, and PICALM, with q values ranging between 1.99E‐02 and 3.29E‐06.
Of interest, six of these genes reside in a co‐expression network module (TCX1) that is enriched for both neuronal cell types and “synaptic transmission” GO process (GO:0007268). The “synaptic transmission” module itself is also associated with AD (P = 4.70E‐03). Four of the (GALNTL6, TOMM40, TBC1D9, MMP16) “synaptic transmission” module genes had MAPT H2 non‐carrier, and the other two (CADM2, GABRG2) had H2 carrier–specific AD risk association (Table 2). The “synaptic transmission” module and all but one discordant gene in this module were lower in AD TCX, as would be expected from neuronal loss observed in AD brains in this region. The discordant loci genes GALNTL6, TBC1D9, and GABRG2 had high module membership levels >0.80, suggesting strong correlations with the rest of the network. Notably, MAPT also resides in the ``synaptic transmission” module: TCX1. Of the IGAP loci genes, PTK2B, EPDR1, and CELF1 also reside within TCX1.
Of the other differentially expressed genes, two were from modules that had cell type enrichment: NECTIN2 (PVRL2) belonged to the module enriched for “defense response” (GO:0006952) and microglia. Both the NECTIN2 (PVRL2) gene (differential expression = DE q = 9.93E‐04) and its module (DE P = 4.19E‐06) had significant differential expression in the AD versus control brains (Table S3). The other was APOE (DE q = 9.42E‐04), which resided in the module (DE P = 1.12E‐04) enriched for “carboxylic acid catabolic process” (GO:0046395), and both astrocytes and endothelia. These genes reside at the same chromosome 19 locus. Both of these modules and genes were higher in the AD TCX, which may again be expected based on microglial and astrocytic population increases observed in brain regions affected with AD neuropathology. Finally, two genes, ADAMTSL3 (DE q = 1.53E‐03) and PICALM (DE q = 7.72E‐03), which reside at MAPT H2 non‐carrier and H2 carrier–specific loci, respectively, are both significantly higher in AD TCX and belong to modules enriched for “regulation of transcription, DNA‐templated” (GO:0006355) (DE P = 3.71E‐02).
We performed the same analyses also for the known AD risk loci genes (Table S3). Eight of the 17 IGAP loci genes with brain expression data had significant differential expression, both at the gene (q < 0.05) and module levels (P < 0.05). Two modules enriched for “immune response” (GO:0006955) and “synaptic transmission” (GO:0007268) genes had the highest number of IGAP risk loci genes. Three genes (HLA‐DRB1, INPP5D, MS4A6A) were in the microglial gene–enriched “immune response” module, as we have shown previously 36 ; and three others (CELF1, EPDR1, PTK2B) were in the neuronal gene enriched “synaptic transmission” module. We noted that there were IGAP risk loci genes within oligodendrocyte (BIN1, ZCWPW1), astrocyte/endothelia (CLU, FERMT2), and endothelia gene–enriched modules (CASS4). In summary, half of the discordant MAPT‐stratified loci genes were from neuronal modules, whereas the IGAP AD risk loci genes had similar representation across network modules that were enriched for any of the five brain cell types.
To determine whether any of the MAPT haplotype‐specific AD risk SNPs influenced brain expression levels of MAPT or the “synaptic transmission” co‐expression module TCX1, which also harbors MAPT, we performed expression quantitative trait loci (eQTL) and module QTL (modQTL) analyses, respectively, as described previously. 14 , 32 , 37 , 38 None of the MAPT haplotype–specific AD risk SNPs had significant associations with temporal cortex MAPT levels or the “synaptic transmission” module eigengene (data not shown). We conclude that these MAPT haplotype‐specific loci are not likely to confer AD risk through their influence on brain gene expression of MAPT or synaptic transmission network genes.
4. DISCUSSION
Despite significant progress in identifying genetic risk factors and the increased understanding in Alzheimer's disease (AD) etiology, the ability to develop effective preventions or cures continues to remain elusive. Novel approaches to analyzing available multiscale genomic and phenotypic data will provide further insights into the complexity of AD and provide mechanisms to foster the development of precision medicine.
In this study we sought to evaluate available genome data by performing a stratified analytic approach. Stratified methods based on sex 39 , 40 , 41 and APOE 19 have been reported previously and have shown background‐dependent associations with AD. Due to the implication of MAPT in both AD neuropathology 1 , 7 , 20 and risk, 14 , 15 , 18 we performed MAPT haplotype–stratified association analyses in the genotype data from the ADGC to test the hypothesis that AD risk variants may exhibit MAPT haplotype‐dependent association. We tested previously identified AD risk loci 21 to determine whether they have differential associations in a MAPT haplotype context–dependent manner. We also extended this analysis genome‐wide to determine if this approach may identify novel AD risk variants.
We found that the index AD risk variants reported previously had similar directions of associations in both the MAPT H2 non‐carrier and H2 carrier analyses. Epistasis analysis with these and the MAPT H1/H2 haplotype tagging variants revealed no evidence of differential association (P > 0.05) for all but two AD risk loci. Even though CD2AP‐rs10948363 and ZCWPW1/PILRB‐rs1476679 had nominally significant epistasis (P = 0.035 and 0.022, respectively), the estimated effects of these variants were largely overlapping in the MAPT H2 carriers and non‐carriers. These findings are not surprising given that the loci that rise to significance in the overall GWAS are likely to have a more consistent effect across stratified groups.
In contrast, stratified analysis may uncover novel loci with group‐specific associations that may be missed in the combined cohort. Although we did not identify any MAPT haplotype–specific associations at genome‐wide significance in this study, we uncovered 11 discordant loci that had association at P < 1E–05 in one stratum (five in MAPT H2 non‐carriers and six in MAPT H2‐carriers), no association (P > 0.05) in the other stratum and evidence of epistasis (P < 0.05) with the MAPT H1/H2 tagging variant rs8070723. The most significant MAPT–haplotype–specific association was observed for chromosome 19 variant rs11665676 at the NECTIN2;TOMM40;APOE locus. The minor T allele of this variant was associated with a lower AD risk (OR = 0.7, P = 1.33E‐07) only in the MAPT H2 non‐carriers (ie, those with MAPT H1/H1 haplotype). It is important to note that although there was no evidence of MAPT haplotype–specific associations for APOE ε4 dose in our study per se, rs11665676‐T is enriched in APOE ε3 carriers.
These findings suggest the following model: In the presence of the strong effect conferred by APOE ε4, the presence of MAPT H1 versus H2 haplotype does not make a significant difference with respect to AD risk. Consequently, there is no MAPT haplotype–specific associations for APOE ε4 dose. However, rs11665676‐T, which is enriched in APOE ε3 carriers, may be marking a variant of APOE that confers greater protection in those who are MAPT H2 non‐carriers. We and others previously showed that MAPT H2 haplotype is associated with a lower risk of AD. 14 , 15 , 18 The preferential protection of rs11665676‐T in MAPT H2 non‐carriers may be due to the fact that in the presence of the protective MAPT H2 haplotype, any further protection conferred by this variant may be negligible. This may explain the lack of association of rs11665676‐T with lower AD risk in MAPT H2‐carriers.
The discordant rs11665676 variant resides within an intron of NECTIN2 (aka PVRL2), which is within a LD region with BCAM and in proximity to the TOMM40‐APOE‐APOC1 locus. 42 It has been shown previously that the LD structure of the polymorphisms across these five genes displayed heterogeneity between AD and control individuals, suggesting that the genes within this region in addition to APOE may play a role in AD risk. 42 , 43 Indeed, a highly polymorphic variant of TOMM40 (poly‐T variant) was found to associate with AD risk and its endophenotypes independent of APOE in some studies. 44 Given the complexity of this region on chromosome 19, including LD across multiple genes, plentiful polymorphisms, and the strong APOE ε2/ε3/ε4 effect on AD risk, alternative approaches focused on haplotype analysis of this region are proposed to uncover novel variants that influence AD independent of APOE. 45 Our analysis of stratifying samples according to specific genotypic/haplotypic backgrounds provides another approach in the discovery of polymorphisms that may influence AD risk under a specific genomic context. Our approach identified a polymorphism in NECTIN2 (PVRL2), which is enriched in APOE ε3 carriers and which has differential protective association in MAPT H2 non‐carriers. This finding suggests a biological link between NECTIN2 and/or APOE with MAPT.
In a previous APOE‐stratified analysis, 19 a variant in the MAPT region, rs2732703‐G, which is more common in H2 carriers, was found to confer greater protection from AD in APOE ε4 negative individuals. This finding is different and independent of our report, and suggests that variability at the MAPT locus influences APOE association with AD risk, whereas our results indicate that variability at the APOE locus has distinct AD risk association on different MAPT haplotype backgrounds. Both findings support the notion of heterogeneity at both APOE and MAPT haplotypic regions, which may modify AD risk associations depending on the combinations of variants harbored. Understanding the full set of functional variants at these important loci, their genetic/biological interactions, and their collective effects on AD risk and its endophenotypes is necessary to successfully practice precision medicine in the future.
Whether the NECTIN2 (aka PVRL2) rs11665676 variant signifies association with this gene per se or marks another variant within APOE remains to be established. NECTIN2 (nectin, cell adhesion molecule 2), also known as poliovirus receptor‐related 2 (and formerly as herpesvirus entry mediator B, HVEB), encodes a plasma membrane glycoprotein that has been implicated in a multitude of central nervous system (CNS) functions. 43 NECTIN2 is involved in adherens junction, which is important to maintain blood‐brain barrier and to prevent the spread of viral infections. In our brain expression data 22 , 23 , 32 analyzed herein, we determined NECTIN2 to be significantly elevated in AD TCX, and to reside in a co‐expression module enriched for “defense response” GO biological process and microglia‐enriched genes. These findings support a role for this gene in innate immune pathways. Our findings along with prior association of another NECTIN2 variant (rs6859) with AD risk in African Americans independent of APOE, 46 merit further evaluation of this gene as a plausible AD gene.
In addition to the NECTIN2 variant at the APOE locus, MAPT‐stratified analysis revealed one other discordant association in a known AD risk locus, which was PICALM intronic SNP rs140869727 that revealed increased risk in MAPT H2‐carriers. The minor A allele of rs140869727 has frequency (MAF) of 0.17 and is rarer than the PICALM locus index IGAP SNP rs10792832, which has a MAF of 0.36. The latter did not have differential MAPT haplotype–stratified association, whereas rs140869727 had epistasis (P = 4.51E‐03). We concluded that the discordant rarer SNP may be tagging a PICALM variant, which confers greater risk of AD in MAPT H2 carriers. PICALM was found to associate with both 3R and 4R tau inclusions in AD and primary tauopathies, and soluble PICALM levels were inversely correlated with phosphotau, 47 suggesting a biological link between this protein involved in clathrin‐mediated endocytosis and tau.
We identified nine discordant loci that were not previously identified in AD risk GWAS, including the largest recent studies. 48 , 49 The four novel H2 non‐carrier–specific associations were near TBC1D9 (chr4), GALNTL6 (chr 4), MMP16;LOC101929709 (chr8), and ADAMTSL3 (chr15). Of these, ADAMTSL3 locus had the strongest AD risk association (P = 2.04E‐7), where the minor allele of intronic SNP rs4354897 conferred protection (OR = 0.84, CI = 0.79 to 0.9), only in MAPT H2 non‐carriers, but not in H2 carriers (epistasis P = 1.22E–04). ADAMTSL3 encodes a glycoprotein that localizes to the extracellular matrix, belongs to a family of metalloproteases, and is proposed to be a candidate gene for schizophrenia, with proposed function in synaptogenesis. 50 Of interest, another H2 non‐carrier–specific association locus resides near a different matrix metalloproteinase encoding gene, MMP16. Matrix metalloproteases have been implicated in AD and other neurodegenerative diseases through their roles in Aß degradation, inflammatory processes, and processing of neurodegenerative proteins including tau. 51 Given this, metalloproteases have been proposed as potential therapeutic targets in AD and other neurodegenerative diseases. The other two genes at MAPT H2 non‐carrier–specific AD risk loci have been identified previously in vascular and/or neuropsychiatric genetic studies. GALNTL6 has been associated with lipid metabolism, 52 body mass index, 53 and hypertension. In addition, a separate SNP in GALNTL6 was associated with AD at age of onset, although it lost its significance after correcting for the APOE. 54 TBC1D9 is a brain‐expressed gene encoding a protein with Rab3A‐GAP activity. There are no reports linking this gene to AD to date. Recently, a de novo and potentially pathogenic TBC1D9 missense variant was identified in sporadic Attention‐Deficit/ Hyperactivity Disorder (ADHD). 55
Five novel loci showed AD risk association only in the H2 carriers, namely, CADM2;LINC02070 on chromosome 3, STK32B on chromosome 4, LOC101928978;NKX6‐1 on chromosome 4, GABRG2 on chromosome 5, and C11orf21 on chromosome 11. Of these, GABRG2 locus has the strongest AD risk association (P = 2.88E‐06) and evidence of epistasis with MAPT H1/H2 locus (3.91E‐06). GABRG2 encodes the γ2 subunit of the pentameric γ‐aminobutyric acid receptor A (GABAA) ligand‐gated ion channels that bind the major inhibitory neurotransmitter in mammalian brains, GABA. Previously, missense, nonsense, frameshift, splice‐site, and deletion mutations within GABRG2 were associated with simple febrile seizures and genetic epilepsy syndromes through different mechanisms leading to reduced channel levels and/or function. 56 GABRG2 levels were found to be reduced in iPSC‐derived neurons and brains from MAPT p.R406W carriers, mouse models of tauopathy, 57 and in the Mayo Clinic brain RNAseq data 22 from patients with the primary tauopathy PSP compared with controls, in both TCX and cerebellum. In our study, we also evaluated the Mayo Clinic brain RNAseq data and determined lower levels of GABRG2 in TCX (q = 1.99E‐02), but not in the cerebellum (data not shown) in AD compared with controls. Collectively, these findings suggest that tauopathies could lead to lower expression of the inhibitory channel proteins, including GABRG2, possibly through loss of these neurons in affected brain areas. This could in turn lead to excitatory/inhibitory imbalance, culminating in enhanced Aß production and ultimately further neuronal loss. 58 Our findings suggest that GABRG2 variants increase AD risk preferentially in MAPT H2 carriers, who are expected to have lower brain MAPT levels and greater protection against AD. 14 Hence, risk conferred by other pathways, such as disruption of GABAergic signaling, may be more important for and detectable in this lower MAPT risk group.
The intronic variant rs7356060 that discordantly confers risk in MAPT H2 carriers (OR = 1.29, CI = 1.16 to 1.43, P = 3.99E‐06) marks another interesting candidate CADM2, which was also identified as a candidate gene in a GWAS of cognitive function, specifically executive function and processing speed. 59 CADM2 encodes cell adhesion molecule 2 and is also known as synaptic cell adhesion molecule 2 (SYNCAM2) and nectin‐like protein 3 (NECL3). That the MAPT‐stratified analysis led to the discovery of a nectin (NECTIN2 on chromosome 19) and a nectin‐like protein (CADM2 = NECL3 = SYNACM2) as candidates is noteworthy. CADM2 was also identified as a locus for habitual physical activity, along with APOE, 60 and was also suggested as a gene that may link obesity with psychiatric traits. 61
The three other candidate genes at the AD risk loci identified in MAPT H2‐carriers—C11orf21, STK32B, and NKX6‐1—were also implicated in CNS diseases or function. C11orf21 has an intronic variant rs77007065‐A, which confers AD protection in MAPT H2 carriers (0.68, CI = 0.58 to 0.81, P = 9.78E‐06) and is one of the most discordant SNPs (epistasis P = 5.62E‐06). This variant is also upstream of TSPAN32, which together with C11orf21 resides in a region of differential methylation in autistic brain samples. 62 STK32B is a serine/threonine kinase and resides at a locus previously identified in a GWAS for essential tremor. 63 The promoter region of this gene is differentially methylated in blood samples from adolescents with generalized anxiety disorder. 64 Finally, NKX6‐1, which is a transcription factor, was found to be involved in midbrain dopaminergic neuron differentiation, 65 in addition to its role in the differentiation of pancreatic ß islet cells. 66 Whether these are the genes that harbor functional variants that influence AD risk in a MAPT haplotype–dependent manner and their biological interaction with tau‐related pathways remains to be established.
In our study, we also performed a systematic evaluation of all of the candidate genes at the discordant AD risk loci for their expression in AD versus control temporal cortex (TCX), 22 , 23 , 32 their membership in brain gene co‐expression networks identified in these samples, and annotation of these networks for their enriched biological processes and CNS cell types. For these analyses, we utilized the Mayo Clinic Brain RNAseq data generated by our group, and implemented approaches as previously described. 22 , 23 , 32 We also analyzed the candidate genes at the known IGAP AD risk loci 21 in the same fashion. Eight of 12 discordant loci genes and eight of 17 IGAP loci genes were differentially expressed in AD versus control TCX with transcriptome‐wide significance (q < 0.05). Half of the discordant loci genes (GALNTL6, TOMM40, TBC1D9, GABRG2, MMP16, CADM2) were members of the co‐expression network that was enriched in “synaptic transmission” GO biological process. This network had also a significantly higher representation of neuron‐enriched genes. In comparison to the discordant loci genes, the known IGAP AD risk loci genes had a lower representation of “synaptic transmission” membership, with 3 (PTK2B, EPDR1, CELF1) of 17 genes that were assessed in the transcriptome data. The published IGAP loci genes had membership within a variety of networks with broader enrichment of GO processes and cell types. These included “axon ensheathment”/oligodendrocyte (BIN1, ZCWPW1); “immune response”/microglia (HLA‐DRB1, INPP5D, MS4A6A); “carboxylic acid catabolic process”/astrocyte and endothelia (CLU; FERMT2); and “vasculature development”/endothelia (CASS4). Neither GWAS associations nor co‐expression network and differential gene expression analyses per se definitively identifies the disease risk genes. Nevertheless, the concurrent presence of GWAS candidate genes within networks that are enriched in processes known to be perturbed in the disease process (such as “immune response,” “synaptic transmission,” “axon ensheathment”) provides further strength for the candidacy of these genes and information about the pathways with which they are likely to be involved. The presence of half of the discordant loci in “synaptic transmission” networks suggests that the MAPT haplotype stratified approach may be preferentially identifying neuronal genes that are involved in this crucial process in a MAPT haplotype–dependent manner. This finding is congruent with known and proposed roles of tau in synaptic transmission or its disruption in AD. 67 In comparison, the un‐stratified GWAS appears to uncover genes that pertain to a wider spectrum of pathways and cellular processes that may be due to the lack of the dependency on MAPT haplotype context.
Because the transcriptome data was obtained in bulk brain tissue in a region affected with AD neuropathology, the observed transcriptional differences between AD and controls may reflect cell population changes. 22 Despite this caveat, we and others have successfully utilized bulk brain transcriptome data to identify transcriptional networks that associate with neurodegenerative diseases and their endophenotypes. 23 , 32 , 42 , 43 , 45 Many of these networks are enriched in pathways and genes that have been implicated previously in these diseases through independent data including genetic associations. 32 , 36 , 42 This suggests that integrative analysis of transcriptional networks and disease association data can provide cross‐validation for the genes. This approach also provides transcriptional context for the candidate genes discovered from disease GWAS as demonstrated here.
In summary, we performed a MAPT H1/H2 haplotype–stratified association in the ADGC GWAS data and identified 11 loci with evidence of association in one stratum (P < 1.0E‐05), no association in the other stratum (P > 0.05), and epistasis (P < 0.05) with the MAPT H1/H2 haplotype–tagging variant rs8070723. With the exception of a NECTIN2 variant at the NECTIN2;TOMM40;APOE locus and a rare variant in PICALM, these are novel loci that have not been reported previously. Half of the candidate genes at these loci reside within a co‐expression network enriched in neuronal genes and implicated in “synaptic transmission.” These findings contrast with those from the known IGAP loci, where we did not find evidence of MAPT H1/H2 haplotype–stratified association and where the candidate genes are members of co‐expression networks that represent a broader range of cellular and biological process enrichment.
There are several limitations to our study. Notwithstanding their novelty, the MAPT H1/H2 haplotype–stratified association results should be interpreted with caution due to falling short of genome‐wide significance (P < 5.0E‐08), as they may represent false‐positive findings. It will be important to apply this approach in larger available GWAS data and seek confirmation. Given that MAPT H2 haplotype is rarer, our MAPT H2 carriers were smaller in size (n = 7360) than MAPT H2 non‐carriers (n = 11,481). This may explain the presence of two loci that approached genome‐wide significance in the MAPT H2 non‐carriers, whereas the strongest association remained at P = 1.99E‐06 in the MAPT H2 carriers. We also acknowledge that our MAPT H1/H2 haplotype definition was based on the tagging variant rs8070723 and that the H1 haplotype, which has considerable variation, 10 , 11 can be divided into additional sub‐haplotypes. Future studies utilizing whole genome sequencing (WGS) can enable more accurate assignment of haplotypes, although sub‐haplotypic stratification would require even greater sample sizes. We discovered that many of the candidate genes at the discordant AD risk loci are differentially expressed in AD TCX and reside in the “synaptic transmission” co‐expression network, which also harbors MAPT. Despite their intriguing biological implications, it is possible that these congruent genomic and transcriptomic findings are coincidental. Definitive determination of biological interactions between the discordant loci genes with MAPT requires detailed studies in model systems, which is beyond the scope of this work. Our findings provide testable hypotheses for such functional studies. Finally, our brain transcriptome data are driven from bulk tissue, where the gene expression findings may simply reflect cell population changes and where biologically important differential expression results in rarer cell types may be obscured. It will be important to evaluate brain cell–type specific expression patterns of the genes nominated in this study in the single‐nucleus and single‐cell transcriptome data from AD and control brains, once sizable data sets become available.
Our study represents an alternative approach in leveraging available GWAS data for discovery of loci and genes that may confer AD risk in a MAPT context–dependent manner. Integrative utilization of independent genomic and transcriptomic data provide cross‐validation for our findings. The candidate genes that emerge from this study should be evaluated for the presence of functional variants that may influence tau‐related outcomes in model systems or human cohorts. Emerging larger cohorts with multi‐omics data and generation of more complex model systems should enable these future studies.
ADGC Affiliations
1Department of Neurology, Johns Hopkins University, Baltimore, Maryland, 2Department of Neurology, University of Michigan, Ann Arbor, Michigan, 3Geriatric Research, Education and Clinical Center (GRECC), VA Ann Arbor Healthcare System (VAAAHS), Ann Arbor, Michigan, 4Michigan Alzheimer Disease Center, Ann Arbor, Michigan, 5Department of Neurology, University of California Los Angeles, Los Angeles, California, 6Department of Psychiatry, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania, 7Geriatric Research, Education and Clinical Center (GRECC), University of Wisconsin, Madison, Wisconsin, 8Department of Medicine, University of Wisconsin, Madison, Wisconsin, 9Wisconsin Alzheimer's Institute, Madison, Wisconsin, 10Department of Medicine (Genetics Program), Boston University, Boston, Massachusetts, 11Department of Pharmacology and Neuroscience, University of North Texas Health Science Center, Fort Worth, Texas, 12Department of Human Genetics, University of Pittsburgh, Pittsburgh, Pennsylvania, 13Department of Neurological Sciences, Rush University Medical Center, Chicago, Illinois, 14Department of Behavioral Sciences, Rush University Medical Center, Chicago, Illinois, 15Civin Laboratory for Neuropathology, Banner Sun Health Research Institute, Phoenix, Arizona, 16Departments of Psychiatry, Neurology, and Psychology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania, 17The John P. Hussman Institute for Human Genomics, University of Miami, Miami, Florida, 18Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami, Miami, Florida, 19National Alzheimer's Coordinating Center, University of Washington, Seattle, Washington, 20Rush Alzheimer's Disease Center, Rush University Medical Center, Chicago, Illinois, 21Department of Pathology, Northwestern University Feinberg School of Medicine, Chicago, Illinois, 22Cognitive Neurology and Alzheimer's Disease Center, Northwestern University Feinberg School of Medicine, Chicago, Illinois, 23Department of Neurology, University of Washington, Seattle, Washington, 24VA Puget Sound Health Care System/GRECC, Seattle, Washington, 25Department of Epidemiology, Harvard School of Public Health, Boston, Massachusetts, 26Department of Psychiatry, Massachusetts General Hospital/Harvard Medical School, Boston, Massachusetts, 27Department of Neurology, Mayo Clinic, Rochester, Minnesota, 28Swedish Medical Center, Seattle, Washington, 29Department of Neurology, University of California San Francisco, San Francisco, California, 30Department of Medicine, Duke University, Durham, North Carolina, 31Department of Neuroscience, Mount Sinai School of Medicine, New York, New York, 32Department of Psychiatry, Mount Sinai School of Medicine, New York, New York, 33Departments of Genetics and Genomic Sciences, Mount Sinai School of Medicine, New York, New York, 34Department of Pathology and Immunology, Washington University, St. Louis, Missouri, 35Department of Pathology and Laboratory Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, Pennsylvania, 36USF Health Byrd Alzheimer's Institute, University of South Florida, Tampa, Florida, 37Fred Hutchinson Cancer Research Center, Seattle, Washington, 38Department of Psychiatry and Behavioral Sciences, Miller School of Medicine, University of Miami, Miami, Florida, 39Department of Pathology, University of Alabama at Birmingham, Birmingham, Alabama, 40Department of Neurology, University of Southern California, Los Angeles, California, 41Department of Neurology, University of Alabama at Birmingham, Birmingham, Alabama, 42Neurogenomics Division, Translational Genomics Research Institute, Phoenix, Arizona, 43Department of Medicine, University of Washington, Seattle, Washington,44Department of Neurology, University of California Irvine, Irvine, California, 45Department of Psychiatry and Hope Center Program on Protein Aggregation and Neurodegeneration, Washington University School of Medicine, St. Louis, Missouri, 46Program in Translational NeuroPsychiatric Genomics, Institute for the Neurosciences, Department of Neurology & Psychiatry, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts, 47Program in Medical and Population Genetics, Broad Institute, Cambridge, Massachusetts, 48Department of Neurology, University of California Davis, Sacramento, California, 49University of Virginia School of Medicine, Charlottesville, Virginia, 50Institute for Memory Impairments and Neurological Disorders, University of California Irvine, Irvine, California, 51Wien Center for Alzheimer's Disease and Memory Disorders, Mount Sinai Medical Center, Miami Beach, Florida, 52Rush Institute for Healthy Aging, Department of Internal Medicine, Rush University Medical Center, Chicago, Illinois, 53Department of Medical and Molecular Genetics, Indiana University, Indianapolis, Indiana, 54Department of Neurology, Indiana University, Indianapolis, Indiana, 55Department of Psychiatry, New York University, New York, New York, 56C.S. Kubik Laboratory for Neuropathology, Massachusetts General Hospital, Charlestown, Massachusetts, 57Department of Neurosciences, University of California San Diego, La Jolla, California, 58Department of Psychiatry, University of Pittsburgh, Pittsburgh, Pennsylvania, 59Department of Pathology and Laboratory Medicine, Emory University, Atlanta, Georgia, 60Emory Alzheimer's Disease Center, Emory University, Atlanta, Georgia, 61Neurogenetics Program, University of California Los Angeles, Los Angeles, California, 62Department of Pathology and Laboratory Medicine, Indiana University, Indianapolis, Indiana, 63Department of Neurology, Emory University, Atlanta, Georgia, 64Division of Genetics, Department of Medicine and Partners Center for Personalized Genetic Medicine, Brigham and Women's Hospital and Harvard Medical School, Boston, Massachusetts, 65Department of Neurology, Massachusetts General Hospital/Harvard Medical School, Boston, Massachusetts, 66Center for Applied Genomics, Children's Hospital of Philadelphia, Philadelphia, Pennsylvania, 67Department of Pathology (Neuropathology), University of Pittsburgh, Pittsburgh, Pennsylvania, 68Institute of Neurology, University College London, Queen Square, London,69Sanders‐Brown Center on Aging, Department of Molecular and Biomedical Pharmacology, University of Kentucky, Lexington, Kentucky, 70Taub Institute on Alzheimer's Disease and the Aging Brain, Department of Neurology, Columbia University, New York, New York, 71Department of Pathology, Duke University, Durham, North Carolina, 72Department of Genome Sciences, University of Washington, Seattle, Washington, 73Department of Medicine (Medical Genetics), University of Washington, Seattle, Washington, 74Sanders‐Brown Center on Aging, Department Neurology, University of Kentucky, Lexington, Kentucky, 75Department of Pathology and Laboratory Medicine, University of California Davis, Sacramento, California, 76Department of Biostatistics, Boston University, Boston, Massachusetts, 77Department of Ophthalmology, Boston University, Boston, Massachusetts, 78University of Pittsburgh Alzheimer's Disease Research Center, Pittsburgh, Pennsylvania, 79Department of Neurology, Oregon Health & Science University, Portland, Oregon, 80Department of Neurology, Portland Veterans Affairs Medical Center, Portland, Oregon, 81Department of Pathology and Laboratory Medicine, University of California Irvine, Irvine, California, 82Department of Neurology, Boston University, Boston, Massachusetts, 83Department of Pathology, Boston University, Boston, Massachusetts, 84Department of Neuropsychology, University of California San Francisco, San Francisco, California, 85Department of Molecular & Medical Genetics, Oregon Health & Science University, Portland, Oregon, 86Department of Epidemiology, University of Washington, Seattle, Washington, 87Department of Neurobiology and Behavior, University of California Irvine, Irvine, California, 88Group Health Research Institute, Group Health, Seattle, Washington, 89Department of Pathology, University of Washington, Seattle, Washington, 90Department of Psychiatry and Behavioral Sciences, University of Washington School of Medicine, Seattle, Washington, 91Department of Pathology, University of Michigan, Ann Arbor, Michigan, 92Department of Psychiatry, Johns Hopkins University, Baltimore, Maryland, 93Department of Preventive Medicine, University of Southern California, Los Angeles, California, 94Department of Medicine ‐ Pulmonary, New York University, New York, New York, 95Department of Neurology, University of Miami, Miami, Florida, 96Department of Pathology, University of California San Diego, La Jolla, California, 97School of Nursing Northwest Research Group on Aging, University of Washington, Seattle, Washington, 98Department of Neurology, Northwestern University Feinberg School of Medicine, Chicago, Illinois, 99Department of Pathology, University of Southern California, Los Angeles, California, 100Department of Neurology, Washington University, St. Louis, Missouri, 101Arizona Alzheimer's Consortium, Phoenix, Arizona, 102Department of Psychiatry, University of Arizona, Phoenix, Arizona, 103Banner Alzheimer's Institute, Phoenix, Arizona, 104Alzheimer's Disease Center, New York University, New York, New York, 105Gertrude H. Sergievsky Center, Columbia University, New York, New York, 106Department of Neurology, Columbia University, New York, New York, 107Tanz Centre for Research in Neurodegenerative Disease, University of Toronto, Toronto, Ontario, Canada, 108Department of Neurology, University of Texas Southwestern, Dallas, Texas, 109Department of Radiology and Imaging Sciences, Indiana University, Indianapolis, Indiana, 110Department of Pathology (Neuropathology), Rush University Medical Center, Chicago, Illinois, 111Department of Psychiatry, University of Southern California, Los Angeles, California, 112Cambridge Institute for Medical Research and Department of Clinical Neurosciences, University of Cambridge, Cambridge, 113Center for Human Genetics and Research, Department of Molecular Physiology and Biophysics, Vanderbilt University, Nashville, Tennessee, 114Department of Pathology, Johns Hopkins University, Baltimore, Maryland, 115Sanders‐Brown Center on Aging, Department of Anatomy and Neurobiology, University of Kentucky, Lexington, Kentucky, 116Department of Pathology & Laboratory Medicine, University of California Los Angeles, Los Angeles, California, 117Taub Institute on Alzheimer's Disease and the Aging Brain, Department of Pathology, Columbia University, New York, New York, 118Department of Psychiatry, Northwestern University Feinberg School of Medicine, Chicago, Illinois, 119Department of Psychiatry & Behavioral Sciences, Duke University, Durham, North Carolina, 120Department of Pathology, Oregon Health & Science University, Portland, Oregon, 121Evelyn F. McKnight Brain Institute, Department of Neurology, Miller School of Medicine, University of Miami, Miami, Florida.
ADGC Co‐Authors
Marilyn S. Albert1, Roger L. Albin2‐4, Liana G. Apostolova5, Steven E. Arnold6, Sanjay Asthana7‐9, Craig S. Atwood7,9, Clinton T. Baldwin10, Robert Barber11, Michael M. Barmada12, Lisa L. Barnes13,14, Thomas G. Beach15, James T. Becker16, Gary W. Beecham17,18, Duane Beekly19, David A. Bennett13,20, Eileen H. Bigio21,22, Thomas D. Bird23,24, Deborah Blacker25,26, Bradley F. Boeve27, James D. Bowen28, Adam Boxer29, James R. Burke30, Joseph D. Buxbaum31‐33, Nigel J. Cairns34, Laura B. Cantwell35, Chuanhai Cao36, Chris S. Carlson37, Cynthia M. Carlsson8, Regina M. Carney38, Steven L. Carroll39, Helena C. Chui40, David G. Clark41, Jason Corneveaux42, David H. Cribbs44, Elizabeth A. Crocco38, Carlos Cruchaga45, Philip L. De Jager46,47, Charles DeCarli48, Steven T. DeKosky49, F. Yesim Demirci12, Malcolm Dick50, Ranjan Duara51, Denis Evans52, Kelley M. Faber53, Kenneth B. Fallon39, Martin R. Farlow54, Steven Ferris55, Tatiana M. Foroud53, Matthew P. Frosch56, Douglas R. Galasko57, Mary Ganguli58, Marla Gearing59,60, Daniel H. Geschwind61, Bernardino Ghetti62, John R. Gilbert17,18, Jonathan D. Glass63, Alison M. Goate45, Robert C. Green64, John H. Growdon65, Hakon Hakonarson66, Ronald L. Hamilton67, Kara L. Hamilton‐Nelson17, John Hardy68, Lindy E. Harrell41, Elizabeth Head69, Lawrence S. Honig70, Matthew J. Huentelman42, Christine M. Hulette71, Bradley T. Hyman65, Gail P. Jarvik72,73, Gregory A. Jicha74, Lee‐Way Jin75, Gyungah Jun10,76,77, M. Ilyas Kamboh12,78, Anna Karydas29, Jeffrey A. Kaye79,80, Ronald Kim81, Edward H. Koo57, Neil W. Kowall82,83, Joel H. Kramer84, Patricia Kramer79,85, Walter A. Kukull86, Brian W. Kunkle17, Frank M. LaFerla87, James J. Lah63, Eric B. Larson43,88, James B. Leverenz89, Allan I. Levey63, Ge Li90, Andrew P. Lieberman91, Chiao‐Feng Lin35, Oscar L. Lopez78, Kathryn L. Lunetta76, Constantine G. Lyketsos92, Wendy J. Mack93, Daniel C. Marson41, Eden R. Martin17,18, Frank Martiniuk94, Deborah C. Mash95, Eliezer Masliah57,96, Wayne C. McCormick43, Susan M. McCurry97, Andrew N. McDavid37, Ann C. McKee82,83, Marsel Mesulam22,98, Bruce L. Miller29, Carol A. Miller99, Joshua W. Miller75, Thomas J. Montine89, John C. Morris34,100, Jill R. Murrell53,62, Amanda J. Myers38, Adam C. Naj35, John M. Olichney48, Amanda Partch35, Henry L. Paulson2, William Perry17, Elaine Peskind90, Aimee Pierce44, Wayne W. Poon50, Huntington Potter36, Joseph F. Quinn79, Ashok Raj36, Murray Raskind90, Eric M. Reiman42,101‐103, Barry Reisberg55,104, Christiane Reitz70,105,106, John M. Ringman5, Erik D. Roberson41, Ekaterina Rogaeva107, Howard J. Rosen29, Roger N. Rosenberg108, Mark A. Sager8, Mary Sano32, Andrew J. Saykin53,109, Julie A. Schneider13,110, Lon S. Schneider40,111, William W. Seeley29, Amanda G. Smith36, Joshua A. Sonnen89, Salvatore Spina62, Peter St George‐Hyslop107,112, Robert A. Stern82, Rudolph E. Tanzi65, Tricia A. Thornton‐Wells113, John Q. Trojanowski35, Juan C. Troncoso114, Debby W. Tsuang24,90, Otto Valladares35, Vivianna M. Van Deerlin35, Linda J. Van Eldik115, Badri N. Vardarajan70,105,106, Harry V. Vinters5,116, Jean Paul Vonsattel117, Li‐San Wang35, Sandra Weintraub22,118, Kathleen A. Welsh‐Bohmer30,119, Jennifer Williamson70, Sarah Wishnek17, Randall L. Woltjer120, Clinton B. Wright121, Chang‐En Yu43, Lei Yu13.
ADGC Declarations of Interest
T.D.B. received licensing fees from and is on the speaker's bureau of Athena Diagnostics, Inc. M.R.F. receives research funding from BristolMyersSquibb Company, Danone Research, Elan Pharmaceuticals, Inc., Eli Lilly and Company, Novartis Pharmaceuticals Corporation, OctaPharma AG, Pfizer Inc., and Sonexa Therapeutics, Inc.; Receives honoraria as scientific consultant from Accera, Inc., Astellas Pharma US Inc., Baxter, Bayer Pharmaceuticals Corporation, BristolMyersSquibb, Eisai Medical Research, Inc., GE Healthcare, Medavante, Medivation, Inc., Merck & Co., Inc., Novartis Pharmaceuticals Corp., Pfizer, Inc., Prana Biotechnology Ltd., QR Pharma., Inc., The sanofi‐aventis Group, and Toyama Chemical Co., Ltd.; and is speaker for Eisai Medical Research, Inc., Forest Laboratories, Pfizer Inc. and Novartis Pharmaceuticals Corporation. A.M.G. has research funding from AstraZeneca, Pfizer and Genentech, and has received remuneration for giving talks at Pfizer and Genentech. R.C.P. is on the Safety Monitory Committee of Pfizer, Inc. (Wyeth) and a consultant to the Safety Monitoring Committee at Janssen Alzheimer's Immunotherapy Program (Elan), to Elan Pharmaceuticals, and to GE Healthcare. R.E.T. is a consultant to Eisai, Japan in the area of Alzheimer's genetics and a shareholder in, and consultant to Pathway Genomics, Inc, San Diego, CA.
Supporting information
Figure S1. QQ plots. Joint analysis results’ QQ plots and genomic inflation factors are shown for (A) unstratified, (B) H2 non‐carrier, and (C) H2 carrier analyses. All analyses included cohorts, age, sex, PC1‐3, and APOE as covariates. QQ plots display observed versus expected P ‐values given the number of statistical tests performed for each SNP. The number of SNPs shown was LD pruned for visualization. A red diagonal represents the expected distribution. Points to the left of the diagonal represent associations that are more significant than expected. Genomic inflation (λ) estimates were obtained for each data set.
Figure S2. Manhattan plot of unstratified joint association analysis. P values from the analytic model without APOE as a covariate is shown. The threshold for genome‐wide significance (P < 5E10‐8) is indicated by the red line and the threshold for trending significance (p < 1E‐5) is indicated by the blue line. Loci with p < 1E‐5 are annotated as mustard yellow.
Figure S3. Forest plot of discordant loci. Results are shown for the discordant loci depicted in Table 2 for joint association analysis excluding APOE. (A) Discordant loci with P ≤ 1E‐5 in H2 non‐carriers. (B) Discordant loci with P ≤ 1E‐5 in H2 carriers. Point size of odds ratio is weighted by the N of each group. Chromosome, nearest gene name(s), and most significant variant at each of the discordant loci are shown at the top of the figures. ADGC cohort names are shown on the left; and their corresponding odds ratios (ORs) and 95% confidence intervals (95% CIs) are shown to the right of the figures. Meta‐analysis OR and 95% CI results of the discordant variants are shown on the bottom of each figure.
ACKNOWLEDGMENTS
We thank the patients and their families for their participation, without which these studies would not have been possible.
This work was supported by National Institute on Aging [RF AG051504; U01 AG046139; R01 AG061796 to NET]; and the National Institute of Neurological Disorders and Stroke [R01 NS080820 to NET].
For samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona and utilized in the brain expression studies: The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05‐901, and 1001 to the Arizona Parkinson's Disease Consortium), and the Michael J. Fox Foundation for Parkinson's Research.
DECLARATIONS OF INTEREST
None.
ADGC ACKNOWLEDGMENTS
The National Institutes of Health, National Institute on Aging (NIH‐NIA) supported this work through the following grants: ADGC, U01 AG032984, RC2 AG036528; NACC, U01 AG016976; NCRAD, U24 AG021886; NIA LOAD, U24 AG026395, U24 AG026390; Banner Sun Health Research Institute P30 AG019610; Boston University, P30 AG013846, U01 AG10483, R01 CA129769, R01 MH080295, R01 AG017173, R01 AG025259, R01AG33193; Columbia University, P50 AG008702, R37 AG015473; Duke University, P30 AG028377, AG05128; Emory University, AG025688; Group Health Research Institute, UO1 AG06781, UO1 HG004610; Indiana University, P30 AG10133; Johns Hopkins University, P50 AG005146, R01 AG020688; Massachusetts General Hospital, P50 AG005134; Mayo Clinic, P50 AG0016574, U01 AG006786; Mount Sinai School of Medicine, P50 AG005138, P01 AG002219; New York University, P30 AG08051, MO1RR00096, UL1 RR029893, 5R01AG012101, 5R01AG022374, 5R01AG013616, 1RC2AG036502, 1R01AG035137; Northwestern University, P30 AG013854; Oregon Health & Science University, P30 AG008017, R01 AG026916; Rush University, P30 AG010161, R01 AG019085, R01 AG15819, R01 AG17917, R01 AG30146; TGen, R01 NS059873; University of Alabama at Birmingham, P50 AG016582, UL1RR02777; University of Arizona, R01 AG031581; University of California, Davis, P30 AG010129; University of California, Irvine, P50 AG016573, P50, P50 AG016575, P50 AG016576, P50 AG016577; University of California, Los Angeles, P50 AG016570; University of California, San Diego, P50 AG005131; University of California, San Francisco, P50 AG023501, P01 AG019724; University of Kentucky, P30 AG028383, AG05144; University of Michigan, P50 AG008671; University of Pennsylvania, P30 AG010124; University of Pittsburgh, P50 AG005133, AG030653; University of Southern California, P50 AG005142; University of Texas Southwestern, P30 AG012300; University of Miami, R01 AG027944, AG010491, AG027944, AG021547, AG019757; University of Washington, P50 AG005136; Vanderbilt University, R01 AG019085; and Washington University, P50 AG005681, P01 AG03991. The Kathleen Price Bryan Brain Bank at Duke University Medical Center is funded by NINDS grant # NS39764, NIMH MH60451 and by Glaxo Smith Kline. Genotyping of the TGEN2 cohort was supported by Kronos Science. The TGen series was also funded by NIA grant AG034504 to AJM, The Banner Alzheimer's Foundation, The Johnnie B. Byrd Sr. Alzheimer's Institute, the Medical Research Council, and the state of Arizona and also includes samples from the following sites: Newcastle Brain Tissue Resource (funding via the Medical Research Council, local NHS trusts and Newcastle University), MRC London Brain Bank for Neurodegenerative Diseases (funding via the Medical Research Council), South West Dementia Brain Bank (funding via numerous sources including the Higher Education Funding Council for England [HEFCE], Alzheimer's Research Trust [ART], BRACE as well as North Bristol NHS Trust Research and Innovation Department and DeNDRoN), The Netherlands Brain Bank (funding via numerous sources including Stichting MS Research, Brain Net Europe, Hersenstichting Nederland Breinbrekend Werk, International Parkinson Fonds, Internationale Stichting Alzheimer Onderzoek), Institut de Neuropatologia, Servei Anatomia Patologica, Universitat de Barcelona. ADNI funding for ADNI is through the Northern California Institute for Research and Education by grants from Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol‐Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman‐La Roche, Schering‐Plough, Synarc, Inc., Alzheimer's Association, Alzheimer's Drug Discovery Foundation, the Dana Foundation, and by the National Institute of Biomedical Imaging and Bioengineering and NIA grants U01 AG024904, RC2 AG036535, K01 AG030514. We thank Drs. D. Stephen Snyder and Marilyn Miller from NIA who are ex‐officio ADGC members. Support was also from the Alzheimer's Association (LAF, IIRG‐08‐89720; MP‐V, IIRG‐05‐14147) and the US Department of Veterans Affairs Administration, Office of Research and Development, Biomedical Laboratory Research Program. P.S.G.‐H. is supported by Wellcome Trust, Howard Hughes Medical Institute, and the Canadian Institute of Health Research.
Strickland SL, Reddy JS, Allen M, et al. MAPT haplotype–stratified GWAS reveals differential association for AD risk variants. Alzheimer's Dement. 2020;16:983–1002. 10.1002/alz.12099
Samantha L. Strickland and Joseph S. Reddy contributed equally to this study.
REFERENCES
- 1. Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609‐622. [DOI] [PubMed] [Google Scholar]
- 2. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature. 1998;393:702‐705. [DOI] [PubMed] [Google Scholar]
- 3. Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737‐7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. D'Souza I, Poorkaj P, Hong M, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism‐chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999;96:5598‐5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dickson DW, Rademakers R, Hutton ML. Progressive supranuclear palsy: pathology and genetics. Brain Pathol. 2007;17:74‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kouri N, Ross OA, Dombroski B, et al. Genome‐wide association study of corticobasal degeneration identifies risk variants shared with progressive supranuclear palsy. Nat Commun. 2015;6:7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vandrovcova J, Anaya F, Kay V, Lees A, Hardy J, de Silva R. Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res. 2010;7:726‐734. [DOI] [PubMed] [Google Scholar]
- 8. Hoglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699‐6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Simon‐Sanchez J, Schulte C, Bras JM, et al. Genome‐wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pastor P, Ezquerra M, Perez JC, et al. Novel haplotypes in 17q21 are associated with progressive supranuclear palsy. Ann Neurol. 2004;56:249‐258. [DOI] [PubMed] [Google Scholar]
- 11. Pittman AM, Myers AJ, Duckworth J, et al. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet. 2004;13:1267‐1274. [DOI] [PubMed] [Google Scholar]
- 12. Myers AJ, Kaleem M, Marlowe L, et al. The H1c haplotype at the MAPT locus is associated with Alzheimer's disease. Hum Mol Genet. 2005;14:2399‐2404. [DOI] [PubMed] [Google Scholar]
- 13. Pittman AM, Myers AJ, Abou‐Sleiman P, et al. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42:837‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Allen M, Kachadoorian M, Quicksall Z, et al. Association of MAPT haplotypes with Alzheimer's disease risk and MAPT brain gene expression levels. Alzheimers Res Ther. 2014;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang CC, Zhu JX, Wan Y, et al. Meta‐analysis of the association between variants in MAPT and neurodegenerative diseases. Oncotarget. 2017;8:44994‐45007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baker M, Graff‐Radford D, Wavrant DeVrieze F, et al. No association between TAU haplotype and Alzheimer's disease in population or clinic based series or in familial disease. Neurosci Lett. 2000;285:147‐149. [DOI] [PubMed] [Google Scholar]
- 17. Bullido MJ, Aldudo J, Frank A, Coria F, Avila J, Valdivieso F. A polymorphism in the tau gene associated with risk for Alzheimer's disease. Neurosci Lett. 2000;278:49‐52. [DOI] [PubMed] [Google Scholar]
- 18. Gerrish A, Russo G, Richards A, et al. The role of variation at AbetaPP, PSEN1, PSEN2, and MAPT in late onset Alzheimer's disease. J Alzheimers Dis. 2012;28:377‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jun G, Ibrahim‐Verbaas CA, Vronskaya M, et al. A novel Alzheimer disease locus located near the gene encoding tau protein. Mol Psychiatry. 2016;21:108‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke‐Iqbal I. Mechanisms of tau‐induced neurodegeneration. Acta Neuropathol (Berl). 2009;118:53‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allen M, Carrasquillo MM, Funk C, et al. Human whole genome genotype and transcriptome data for Alzheimer's and other neurodegenerative diseases. Sci Data. 2016;3:160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allen M, Wang X, Burgess JD, et al. Conserved brain myelination networks are altered in Alzheimer's and other neurodegenerative diseases. Alzheimers Dement. 2018;14:352‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet. 2011;43:436‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Naj AC, Jun G, Reitz C, et al. Effects of multiple genetic loci on age at onset in late‐onset Alzheimer disease: a genome‐wide association study. JAMA Neurol. 2014;71:1394‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sims R, van der Lee SJ, Naj AC, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat Genet. 2017;49:1373‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boehme KL, Mukherjee S, Crane PK, Kauwe JSK. ADGC 1000 Genomes combined data workflow (electronic document).
- 28. Purcell S, Neale B, Todd‐Brown K, et al. PLINK: a tool set for whole‐genome association and population‐based linkage analyses. Am J Hum Genet. 2007;81:559‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ. 2003;327:557‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Team RDC . R: A Language and Environment for Statistical Computing. Vienna, Austria. 2008.
- 31. Viechtbauer W. Conducting meta‐analyses in R with the metafor Package. J Stat Softw. 2010;36:1548‐7660. [Google Scholar]
- 32. Allen M, Wang X, Serie DJ, et al. Divergent brain gene expression patterns associate with distinct cell‐specific tau neuropathology traits in progressive supranuclear palsy. Acta Neuropathol (Berl). 2018;136:709‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sarkar D. Lattice: Multivariate Data Visualization with R. New York: Springer; 2008. [Google Scholar]
- 34. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ekins S, Nikolsky Y, Bugrim A, Kirillov E, Nikolskaya T. Pathway mapping tools for analysis of high content data. Methods Mol Biol. 2007;356:319‐350. [DOI] [PubMed] [Google Scholar]
- 36. Conway OJ, Carrasquillo MM, Wang X, et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol Neurodegener. 2018;13:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carrasquillo MM, Allen M, Burgess JD, et al. A candidate regulatory variant at the TREM gene cluster associates with decreased Alzheimer's disease risk and increased TREML1 and TREM2 brain gene expression. Alzheimers Dement. 2017;13:663‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zou F, Chai HS, Younkin CS, et al. Brain expression genome‐wide association study (eGWAS) identifies human disease‐associated variants. PLoS Genet. 2012;8:e1002707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liesinger AM, Graff‐Radford NR, Duara R, et al. Sex and age interact to determine clinicopathologic differences in Alzheimer's disease. Acta Neuropathol. 2018;136(6):873‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ferrari R, Dawoodi S, Raju M, et al. Androgen receptor gene and sex‐specific Alzheimer's disease. Neurobiol Aging. 2013;34 2077e19‐2077e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ronquillo JG, Baer MR, Lester WT. Sex‐specific patterns and differences in dementia and Alzheimer's disease using informatics approaches. J Women Aging. 2016;28:403‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kulminski AM, Huang J, Wang J, He L, Loika Y, Culminskaya I. Apolipoprotein E region molecular signatures of Alzheimer's disease. Aging Cell. 2018;17:e12779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yashin AI, Fang F, Kovtun M, et al. Hidden heterogeneity in Alzheimer's disease: Insights from genetic association studies and other analyses. Exp Gerontol. 2018;107:148‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chiba‐Falek O, Gottschalk WK, Lutz MW. The effects of the TOMM40 poly‐T alleles on Alzheimer's disease phenotypes. Alzheimers Dement. 2018;14:692‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lutz MW, Crenshaw D, Welsh‐Bohmer KA, Burns DK, Roses AD. New Genetic Approaches to AD: Lessons from APOE‐TOMM40 Phylogenetics. Curr Neurol Neurosci Rep. 2016;16:48. [DOI] [PubMed] [Google Scholar]
- 46. Logue MW, Schu M, Vardarajan BN, et al. A comprehensive genetic association study of Alzheimer disease in African Americans. Arch Neurol. 2011;68:1569‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ando K, Tomimura K, Sazdovitch V, et al. Level of PICALM, a key component of clathrin‐mediated endocytosis, is correlated with levels of phosphotau and autophagy‐related proteins and is associated with tau inclusions in AD, PSP and Pick disease. Neurobiol Dis. 2016;94:32‐43. [DOI] [PubMed] [Google Scholar]
- 48. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dow DJ, Huxley‐Jones J, Hall JM, et al. ADAMTSL3 as a candidate gene for schizophrenia: gene sequencing and ultra‐high density association analysis by imputation. Schizophr Res. 2011;127:28‐34. [DOI] [PubMed] [Google Scholar]
- 51. Rivera S, Garcia‐Gonzalez L, Khrestchatisky M, Baranger K. Metalloproteinases and their tissue inhibitors in Alzheimer's disease and other neurodegenerative disorders. Cell Mol Life Sci. 2019;76:3167‐3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kathiresan S, Manning AK, Demissie S, et al. A genome‐wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fox CS, Heard‐Costa N, Cupples LA, Dupuis J, Vasan RS, Atwood LD. Genome‐wide association to body mass index and waist circumference: the Framingham Heart Study 100K project. BMC Med Genet. 2007;8(Suppl 1):S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Szigeti K, Kellermayer B, Lentini JM, et al. Ordered subset analysis of copy number variation association with age at onset of Alzheimer's disease. J Alzheimers Dis. 2014;41:1063‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim DS, Burt AA, Ranchalis JE, et al. Sequencing of sporadic Attention‐Deficit Hyperactivity Disorder (ADHD) identifies novel and potentially pathogenic de novo variants and excludes overlap with genes associated with autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet. 2017;174:381‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kang JQ, Macdonald RL. Molecular Pathogenic Basis for GABRG2 Mutations Associated With a Spectrum of Epilepsy Syndromes, From Generalized Absence Epilepsy to Dravet Syndrome. JAMA Neurol. 2016;73:1009‐1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jiang S, Wen N, Li Z, et al. Integrative system biology analyses of CRISPR‐edited iPSC‐derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry. 2018;8:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Calvo‐Flores Guzman B, Vinnakota C, Govindpani K, Waldvogel HJ, Faull RLM, Kwakowsky A. The GABAergic system as a therapeutic target for Alzheimer's disease. J Neurochem. 2018;146:649‐669. [DOI] [PubMed] [Google Scholar]
- 59. Ibrahim‐Verbaas CA, Bressler J, Debette S, et al. GWAS for executive function and processing speed suggests involvement of the CADM2 gene. Mol Psychiatry. 2016;21:189‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Klimentidis YC, Raichlen DA, Bea J, et al. Genome‐wide association study of habitual physical activity in over 377,000 UK Biobank participants identifies multiple variants including CADM2 and APOE. Int J Obes (Lond). 2018;42:1161‐1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Morris J, Bailey MES, Baldassarre D, et al. Genetic variation in CADM2 as a link between psychological traits and obesity. Sci Rep. 2019;9:7339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Nardone S, Sams DS, Reuveni E, et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry. 2014;4:e433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Muller SH, Girard SL, Hopfner F, et al. Genome‐wide association study in essential tremor identifies three new loci. Brain. 2016;139:3163‐3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ciuculete DM, Bostrom AE, Tuunainen AK, et al. Changes in methylation within the STK32B promoter are associated with an increased risk for generalized anxiety disorder in adolescents. J Psychiatr Res. 2018;102:44‐51. [DOI] [PubMed] [Google Scholar]
- 65. Prakash N, Wurst W. A Wnt signal regulates stem cell fate and differentiation in vivo. Neurodegener Dis. 2007;4:333‐338. [DOI] [PubMed] [Google Scholar]
- 66. Hunter CS, Stein RW. Evidence for Loss in Identity, De‐Differentiation, and Trans‐Differentiation of Islet beta‐Cells in Type 2 Diabetes. Front Genet. 2017;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tai HC, Wang BY, Serrano‐Pozo A, Frosch MP, Spires‐Jones TL, Hyman BT. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer's disease. Acta Neuropathol Commun. 2014;2:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sotiropoulos I, Galas MC, Silva JM, et al. Atypical, non‐standard functions of the microtubule associated Tau protein. Acta Neuropathol Commun. 2017;5:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. QQ plots. Joint analysis results’ QQ plots and genomic inflation factors are shown for (A) unstratified, (B) H2 non‐carrier, and (C) H2 carrier analyses. All analyses included cohorts, age, sex, PC1‐3, and APOE as covariates. QQ plots display observed versus expected P ‐values given the number of statistical tests performed for each SNP. The number of SNPs shown was LD pruned for visualization. A red diagonal represents the expected distribution. Points to the left of the diagonal represent associations that are more significant than expected. Genomic inflation (λ) estimates were obtained for each data set.
Figure S2. Manhattan plot of unstratified joint association analysis. P values from the analytic model without APOE as a covariate is shown. The threshold for genome‐wide significance (P < 5E10‐8) is indicated by the red line and the threshold for trending significance (p < 1E‐5) is indicated by the blue line. Loci with p < 1E‐5 are annotated as mustard yellow.
Figure S3. Forest plot of discordant loci. Results are shown for the discordant loci depicted in Table 2 for joint association analysis excluding APOE. (A) Discordant loci with P ≤ 1E‐5 in H2 non‐carriers. (B) Discordant loci with P ≤ 1E‐5 in H2 carriers. Point size of odds ratio is weighted by the N of each group. Chromosome, nearest gene name(s), and most significant variant at each of the discordant loci are shown at the top of the figures. ADGC cohort names are shown on the left; and their corresponding odds ratios (ORs) and 95% confidence intervals (95% CIs) are shown to the right of the figures. Meta‐analysis OR and 95% CI results of the discordant variants are shown on the bottom of each figure.