

Abstract
The etiology of the common, sporadic form of Alzheimer's disease (sAD) is unknown. We hypothesize that tau pathology within select projection neurons with susceptible microenvironments can initiate sAD. This postulate rests on extensive data demonstrating that in human brains tau pathology appears about a decade before the formation of Aβ plaques (Aβps), especially targeting glutamate projection neurons in the association cortex. Data from aging rhesus monkeys show abnormal tau phosphorylation within vulnerable neurons, associated with calcium dysregulation. Abnormally phosphorylated tau (pTau) on microtubules traps APP‐containing endosomes, which can increase Aβ production. As Aβ oligomers increase abnormal phosphorylation of tau, this would drive vicious cycles leading to sAD pathology over a long lifespan, with genetic and environmental factors that may accelerate pathological events. This hypothesis could be testable in the aged monkey association cortex that naturally expresses characteristics capable of promoting and sustaining abnormal tau phosphorylation and Aβ production.
Keywords: β‐amyloid, abnormally phosphorylated tau, association cortex, calcium, rhesus monkey, sporadic Alzheimer's disease, tau seeding
1. OBJECTIVE
Similarities between the autosomal dominant and sporadic forms of Alzheimer's disease presume a shared etiology, but evidence indicates that sporadic Alzheimer's disease (sAD) may have differing origins. We propose that tau pathology can be a key initiating factor in sAD and arises prior to the appearance of amyloid beta plaques (Aβps). This hypothesis rests on extensive neuropathological analyses of human brains across the lifespan and corroborating findings from non‐human primate models, where the earliest stages of tau pathology can be captured.
The etiology of the common, sporadic form of Alzheimer's disease (sAD) is not known
We hypothesize that tau pathology can initiate sAD in vulnerable neurons
Neuropathological data show that tau precedes amyloid beta (Aβ) deposition by ∼10 years
Monkey data show phosphorylated tau (pTau) trafficking early in association cortex
pTau traps amyloid precursor protein‐containing endosomes, which may increase Aβ and drive vicious cycles
2. BACKGROUND
2.1. Historical evolution
The amyloid hypothesis has dominated the AD field and is based largely on the persuasive genetics of autosomal dominant AD and corroborative data from genetic mouse models, positing that Aβ accumulation initiates and drives the tau pathology, which disables and ultimately kills affected neurons. 1 This linear relationship, with Aβ as the trigger and tau the bullet, is compellingly simple and is likely true for some genetic forms of AD. However, there are important findings that disagree with this hypothesis, 2 including extensive neuropathological data from sAD brains, 3 as well as the limited success of anti‐amyloid treatments in sAD, 4 and thus there is now a search for additional etiological factors and therapeutic targets. 4
2.2. Rationale
Tau pathology accumulates intracellularly, as hyperphosphorylated tau fibrillates into the paired helical filaments that form neurofibrillary tangles, ultimately killing the neuron. In contrast, Aβps accumulate extracellularly from the aggregation and fibrillation of Aβ peptides that are released from neurons into the extracellular space.
Analyses of large numbers of human brains across the lifespan show that tau pathology begins about a decade before formation of Aβps (Figure 1). 3 The discovery of a woman with a rare, combined PS1/Christchurch apoE3 mutation, who did not develop dementia despite extensive formation of Aβps—but little tau pathology—reinforces the growing notion that abnormal tau may be a key etiological factor. 5 In addition, tau pathology, but not Aβ, correlates with progressive gray matter loss 6 and cognitive impairment. 7 Importantly, tau pathology exhibits a highly selective pattern and sequence of disease progression (Figure 2). 8
FIGURE 1.
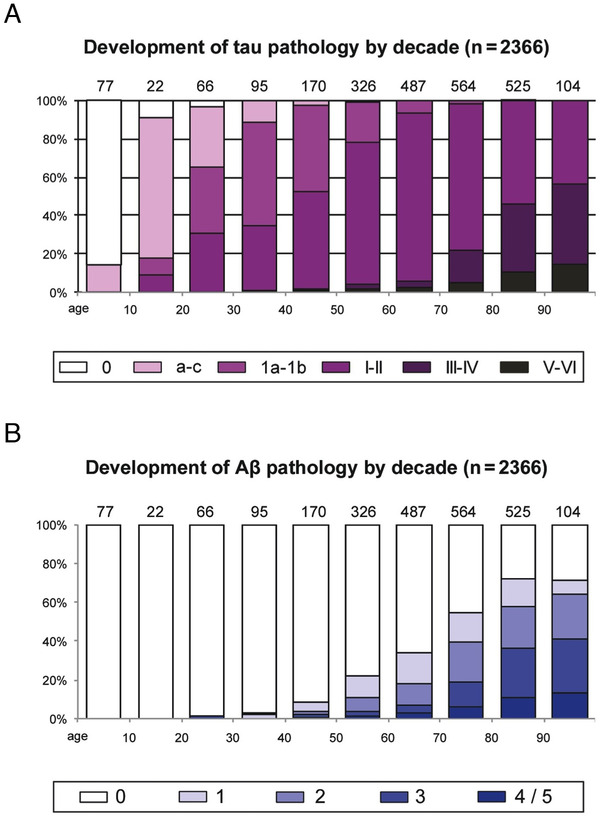
(A) The frequency of fibrillar tau pathology in the human brain across the lifespan. White columns indicate an absence of AT8‐positive abnormal tau. Color‐coded key and columns in violet shading show the relevant frequency of cases at all stages of AT8‐positive tau lesions. The number of cases in each decade appears directly above the columns. (B) The frequency of fibrillar amyloid beta (Aβ) pathology in the human brain across the lifespan. Columns in blue shading indicate the relevant frequency of individuals at various Aβ plaque phases. The number of cases in each decade also appears above each column. Note that tau pathology arises much earlier than Aβ plaques. Adapted with permission from 3
FIGURE 2.
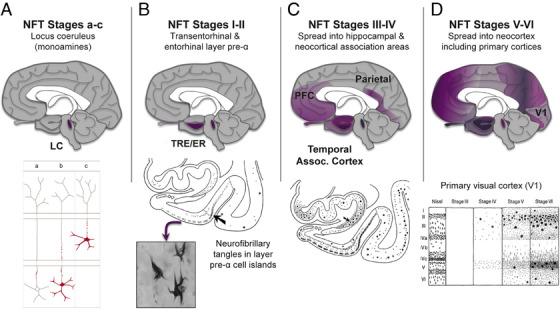
The sequence of tau pathology in the human brain. (A) Tau pathology is first seen in the subcortical nuclei that project widely to the cerebral cortex, such as the noradrenergic locus coeruleus (LC, subcortical stages a‐c). However, these cells do not degenerate until later stages (eg, neurofibrillary tangle [NFT] stages III/IV). Abnormal tau in the LC is shown in red in the bottom portion of panel A. Notably, tau pathology (stage a) begins in the proximal axon and is followed by lesions filling the somatodendritic domain of coeruleus neurons (stage b). Next, tau‐positive axons and/or nerve cells develop in other non‐thalamic subcortical nuclei with diffuse cortical projections (stage c). (B) Tau pathology is initially seen in the cerebral cortex in glutamatergic neurons of the transentorhinal region (TRE) and then entorhinal region (ER) cortices (NFT stages I‐II, arrow in the lower portion of panel B points to the border between the two). Initially, the tau pathology is confined predominantly to layer pre‐α (inset shows neurofibrillary tangles in pre‐α). (C) Tau pathology then develops in the deeper layers of TRE and ER, in the CA1 sector of the Ammon's horn (arrow in the lower portion of panel C points to the prosubiculum) and in the adjoining temporal neocortex (NFT stages III‐IV). (D) Tau pathology finally spreads to the secondary and then primary visual, auditory, somatosensory, and somatomotor cortices at the latest stages (NFT stages V‐VI). The lower portion of panel D shows, for example, the progression of pathology in the primary visual cortex, V1, in greater detail. Based on 3 and 9 with permission
In human brains from 1 to 100 years, AT8‐labeled tau pathology is first evident in brainstem nuclei projecting widely to the cerebral cortex, such as the noradrenergic locus coeruleus (Figure 2A), where it can be seen as early as childhood 3 , 9 However, these neurons do not generate until far later in the course of sAD. In contrast, tau pathology in the cerebral cortex usually begins in the transentorhinal region (TRE) and then in upper cellular layers of the entorhinal region (ER) at a very early age (Figure 2B) and causes rapid degeneration. 9 Tau pathology then extends to deeper layers of the ER and arises in the interconnected hippocampal formation and association cortices (Figure 2C), leaving primary sensory and motor fields untouched until late‐stage disease (Figure 2D). Within the cerebral cortex, tau pathology particularly afflicts highly interconnected, glutamatergic projection neurons, 10 leaving γ–aminobutric acid (GABA)ergic interneurons largely intact. 3 In contrast, extraneuronal Aβps are usually first deposited in the association neocortex of the temporal lobe, and then more generally in cortex. 8 With disease progression, Aβps often extend to subcortical structures, for example striatum and thalamus (reviewed in 3 ).
Importantly, although tau pathology in the TRE/ER is frequent and occurs in persons considered cognitively “normal,” 11 it is not benign, as it ultimately destroys affected neurons and is likely the provenance of more widespread tau pathology in late‐stage disease. Our current cognitive assessments are remarkably crude, reflecting the wide variation in baseline cognitive abilities between subjects, and thus require large declines in cognitive performance to discern impairments. More sophisticated, within‐subjects longitudinal comparisons, using relational memory tasks that challenge TRE/ER function 12 may provide a more in‐depth view of the consequences of early tau pathology.
The sequence of pathological events evident from neuropathology is seemingly at odds with those from cerebrospinal fluid (CSF) assays, where a rise in Aβ is seen before a rise in abnormally pTau. 13 However, this may be an artifact of differential access to the CSF. For instance, Aβ is extruded into the extracellular space, where it can be readily captured in CSF, while abnormal tau is sequestered inside neurons or trafficked between neurons in endosomes, and therefore may not be widely exposed to the CSF until neuronal death causes membrane breakdown. Moreover, the beginning of abnormal tau phosphorylation may not be evident in CSF due to dephosphorylation by ubiquitous phosphatases. New, more sensitive CSF assays of abnormal tau are in progress 14 and may provide data more consistent with early changes in tau phosphorylation.
3. UPDATED HYPOTHESIS
We propose that cortical tau pathology naturally arises in select and extensively interconnected glutamatergic projection neurons 10 with unique molecular characteristics 15 that render them especially vulnerable to abnormal tau phosphorylation and seeding. We propose that pTau can in turn accelerate Aβ cleavage from amyloid precursor protein (APP) 16 and that a range of additional factors can determine the speed and extent of this degenerative cascade.
RESEARCH IN CONTEXT
Systematic review: We hypothesize that tau pathology can initiate sporadic Alzheimer's disease (sAD).
Interpretation: Extensive evaluation of human neuropathological data show that tau pathology begins about a decade before the appearance of amyloid beta (Aβ) plaques, indicating that Aβ is unlikely to be the initial cause of sAD. Data from aging humans and rhesus monkeys show that abnormal tau phosphorylation naturally arises early in the transentorhinal/entorhinal regions, associated with signs of calcium dysregulation and transneuronal propagation of tau seeds, and later develops within select pyramidal cells in the association neocortex with dysregulated cyclic adenosine monophosphate‐calcium signaling. Although Aβ is known to increase abnormal tau phosphorylation, pathological tau may also drive Aβ cleavage by trapping amyloid precursor protein‐containing endosomes in dendrites, propelling a vicious cycle of tau and amyloid pathology over a long lifetime.
Future directions: Focus on reducing early tau pathology may augment therapeutic approaches to treating sAD.
3.1. Early experimental or observational data
Brains of aging rhesus monkeys help illuminate the origins and selectivity of tau pathology, as they can be examined with minimal post‐mortem interval, for example, in perfusion‐fixed tissue, capturing pTau in its earliest forms that are degraded in human postmortem tissue. 16 , 17 Aging rhesus monkeys show the same qualitative pattern and sequence of tau pathology as humans, but do not reach the widespread pathology seen in longer‐lived humans. 16 As described below, analyses of human and non‐human primate brains indicate that tau pathology arises locally within vulnerable neurons, and spreads via seeding between neurons in a network of highly interconnected, glutamatergic projection neurons with permissive intracellular environments. Data from non‐human primate brains also indicate mechanisms by which pTau can increase the generation of Aβ, 16 thus driving vicious cycles.
3.1.1. Local generation of tau pathology in vulnerable, glutamatergic projection neurons in association cortex
Tau normally functions to regulate microtubules 18 and is concentrated in axons, but is also present in dendrites. 17 , 19 , 20 In both humans and monkeys, tau pathology begins within distal dendrites and then spreads to the soma. 16 , 17 , 20 , 21 , 22 ImmunoEM analysis of rhesus monkey cortex by Paspalas 16 shows pTau accumulating in dendrites of entorhinal layer pre‐α cellular islands in young adulthood, with subsequent hyperphosphorylation with advancing age, consonant with the time course in humans. 20 Tau is phosphorylated by a large number of kinases, including phosphorylation by protein kinase A (PKA) at serine 214 (pS214Tau). This is an important early step, as it causes tau to detach from dendritic microtubules 23 and aggregate on microtubules and on the calcium‐storing smooth endoplasmic reticulum (SER) in dendrites, especially under glutamate synapses. 16 The appearance of pS214Tau is associated with evidence of calcium dysregulation from the SER at the same location. 16 pS214Tau then becomes hyperphosphorylated at additional sites, with a distinctive three‐dimensional conformation 24 labeled by the AT8 antibody. 16 The immunoEM reveals AT8‐labeled fibrils in dendrites, which eventually invade the soma, 16 similar to the situation in humans. 20
A similar pattern and progression of tau pathology are seen in the rhesus monkey dorsolateral prefrontal cortex (dlPFC) at a later age, but not in primary visual cortex, 17 consistent with the sequence of pathology seen in human brains. 9 Thus, there is extensive pS214Tau accumulation near glutamate synapses and on dendritic microtubules and the calcium‐containing SER (Figure 3B). 17 These data suggest that cortical glutamate synapses, which expand in number over brain evolution, serve as an “engine of pathology,” which over a long lifespan can create the degenerative pattern seen in sAD. 15 This hypothesis is consistent with the increasing degree of tau pathology seen across primate species, corresponding with increasing numbers of cortical‐cortical glutamatergic connections on spines and increasing lifespan: macaques < chimpanzees < humans. 15 , 25
FIGURE 3.
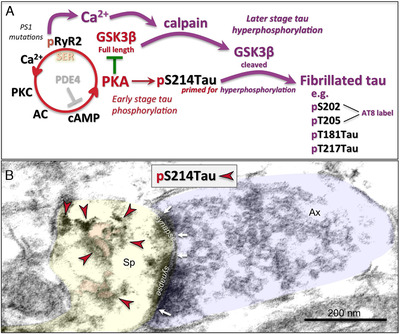
The possible role of calcium dysregulation in tau pathology. (A) Schematic illustration of potential molecular signaling events contributing to early‐ and later‐stage abnormal tau phosphorylation. Many glutamate synapses on spines in the dorsolateral prefrontal cortex (dlPFC) express the molecular machinery to magnify calcium signaling near the synapse, where cyclic adeonosine monophosphate (cAMP)‐ protein kinase A (PKA) signaling increases calcium release from the smooth endoplasmic reticulum (SER) through calcium channel ryanodine receptors (RyR2), a process held in check by phosphodiesterases (PDE4), which catabolizes cAMP. 17 However, PDE4 is lost with advancing age, 17 leading to increased PKA activity. This includes increased PKA phosphorylation of ryanodine receptors (pS2808RyR2), which causes calcium leakage into the cytosol, 16 similar to presenilin mutations in autosomal dominant AD. 28 Increased PKA activity also phosphorylates tau at S214, 17 priming tau for hyperphosphorylation by the kinase, GSK3β, 38 a key factor in driving sporadic Altzheimer's disease (sAD) tau pathology. Full length GSK3β is inhibited by PKA, which may hold this process in check when cytosolic calcium levels are normal. However, when there are sufficiently high levels of calcium to activate the protease calpain, this may “switch” the system into later stage, tau hyperphosphorylation, as calpain can cleave GSK3β at the N‐terminus, removing its inhibition by PKA. 48 Disinhibition of GSK3β would initiate hyperphosphorylation of pS214Tau at key sites for fibrillation, 38 including those labeled by the AT8 antibody currently used to diagnose AD, and pT181Tau and pT217Tau, which have potential as novel, in vivo biomarkers. 49 , 50 Disinhibited GSK3β signaling can also degrade PDE4s, 51 which would further dysregulate calcium and PKA signaling and tau phosphorylation. Thus, the activation of calpain may be a major step in activating advanced pathological events, consistent with it heralding the rise of fibrillated tau pathology in the sAD brain. 30 , 31 However, even early‐stage tau phosphorylation at S214 may have pathological consequences, as its aggregation on microtubules interferes with endosomal trafficking, which may contribute to the production of Aβ (see Figure 5 below). (B) An example of a glutamatergic‐like synapse on a spine in the aged dlPFC, where immunolabeling for PKA‐phosphorylated tau (pS214Tau; indicated by the red arrowheads) is expressed on and near the SER (pseudocolored in pink). The spine (Sp) is pseudocolored in yellow; the axon terminal (Ax) is pseudocolored in blue; the synapse is perforated and is delineated by white arrows; the scale bar indicates 200 nm. Note that pS214Tau labeling in aged dlPFC was first reported in 17
What factors render neurons susceptible versus resilient to tau pathology, 26 and why are the most vulnerable neurons concentrated in the locus coeruleus and neocortical association fields? A decades‐old hypothesis posits that elevated calcium may be a key culprit. 27 This idea was reinforced by the discovery that presenilin mutations cause calcium leak from the SER into the cytosol, 28 which may explain why these mutations create especially aggressive disease progression. More recently, calcium dysregulation has been documented in the brains of patients with sAD, 29 where activated calpain heralds the rise of fibrillated tau pathology. 30 , 31
Data from both animal and human studies suggest that neurons susceptible to tau pathology express signs of elevated cytosolic calcium, for example, due to stress or head injury, 32 and/or when there is age‐related loss of regulatory proteins that control calcium release 17 or bind calcium in the cytosol (eg, calbindin). 33 Indeed, the gradient of calbindin expression in pyramidal cells in the young adult cortex directly correlates with the progression of tau pathology in the aged cortex, with lowest levels in primary sensory cortex and increasing levels in higher association cortices. 34 These data suggest that pyramidal cells that utilize high levels of calcium for normal functioning will be particularly vulnerable if/when calbindin is lost with age. Pyramidal cells in the dlPFC, but not primary visual cortex, express the cyclic adeonosine monophosphate (cAMP)‐signaling machinery to magnify calcium signaling next to glutamate synapses. 35 High levels of calcium signaling may also be needed for normative function in the locus coeruleus and ER. These vulnerable neurons also have increased calcium signaling during stress exposure. 32 , 36 , 37 Hence, these neurons would be particularly vulnerable to tau pathology with advancing age, if regulatory factors are lost. 15 Conversely, resilient neurons appear to have lower calcium requirements or retain factors that regulate calcium. It is noteworthy that cerebellar Purkinje cells and cortical GABAergic interneurons, which are spared in sAD, express exceptionally high levels of calcium‐binding proteins that are retained in sAD and may contribute to their resilience. 33 Thus, levels of cytosolic calcium may be one key factor that determines a vulnerable microenvironment for abnormal tau phosphorylation.
Figure 3 illustrates the molecular events by which calcium dysregulation can lead to early‐stage tau phosphorylation, priming tau for subsequent hyperphosphorylation by disinhibited GSK3β at key diagnostic sites (S202, T205, T181, T217), 38 and creating the fibrils that ultimately form neurofibrillary tangles. It is likely that the balance in phosphorylation versus dephosphorylation would determine the speed and extent of tau pathology, and that this would be influenced by a large number of genetic and environmental factors. Many of the risk factors for sAD also increase calcium dysregulation and/or GSK3β signaling, including head injury, 39 severe emotional distress, 40 and insulin insensitivity. 41 Thus, calcium dysregulation within glutamatergic neurons in association cortex is likely an important factor in initiating and propelling tau pathology. 28
3.1.2. Phosphorylated tau “seeding” within neuronal networks
Compelling data exist that pTau can traffic between neurons to “seed” in higher cortical networks subserving cognition (Figure 4). This idea was first proposed by primate neuroanatomists in the 1980s, who recognized that the pattern of tau pathology was consistent with the pattern of cortical connections (reviewed in 15 ). More detailed evaluation of tau pathology further bolstered this concept, 3 , 20 and mouse AD models have also found evidence of pTau trafficking. 42 Explicit testing of this hypothesis in post‐mortem human tissue shows that seeding from ER is more effective, and can occur at an earlier stage, than seeding from the locus coeruleus (Figure 4A), suggesting the ER may provide much of the early engine of tau propagation. 43 High resolution immunoEM of rhesus monkey cortex allows visualization of pS214Tau trafficking between neurons in association cortex, 16 , 17 for example pS214Tau trafficking between neurons within omega bodies in entorhinal layer pre‐α in a young adult monkey (Figure 4B; 16 ). 17 Notably, pTau trafficking was only seen near excitatory, but not inhibitory synapses, 16 , 17 consistent with tau afflicting glutamatergic, but not GABAergic neurons. 3 Trafficked pTau may only “infect” neurons with permissive intracellular environments, for example high cytosolic calcium. 15 Altogether, these data from human and non‐human primate brains indicate how tau pathology can spread over a lifetime, starting early and eventually compromising the integrity of higher cortical networks. 20
FIGURE 4.
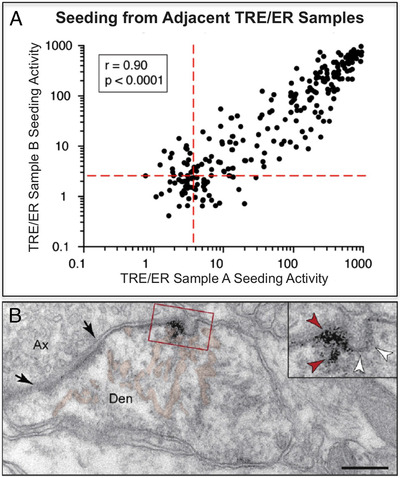
Propagation of tau pathology in vulnerable cortical circuits. (A) Figure reproduced with permission from (42) on network propagation (“seeding”) that starts in the transentorhinal region (TRE) and entorhinal region (ER). Seeding of abnormal tau was most effective at earliest time points from these cortical regions. The generation of large amounts of pTau in TRE/ER early in the aging process may arise from early calcium dysregulation in these circuits, as seen in aging monkey. 16 (B) A Paspalas 16 immunoEM image capturing pS214Tau trafficking (red arrowheads in inset) between excitatory neurons in layer pre‐α of the middle‐aged rhesus monkey ER (from 16 with permission). Black arrows point to the synaptic membrane; white arrowheads indicate an omega‐shaped profile on the plasma membrane. The smooth endoplasmic reticulum (SER) is pseudocolored in pink. Abbreviations: Ax, axon; Den, dendrite. Scale bar = 200 nm
3.1.3. Tau phosphorylation may drive Aβ generation, creating a vicious cycle
The distribution pattern of Aβps in human brains suggests that Aβ is produced and released from the axon terminals of neurons already afflicted with tau pathology. 3 , 44 Although the pattern and sequence of Aβps are less organized than those of tau, Aβps generally arise in brain regions that receive inputs from tau‐afflicted neurons. 13
Data from aging rhesus monkeys indicate a mechanism by which pTau may drive Aβ production within neurons, similar to how genetic alterations in retromer (eg, SORL1) signaling may increase the risk of sAD by causing “endosomal traffic jams,” increasing the time APP spends in endosomes where it is cleaved to Aβ. 45 The immunoEM data 16 , 17 show that aggregated pTau on microtubules similarly causes “endosomal traffic jams” that may drive Aβ production (Figure 5A,B). Endosomes normally traffic on microtubules, but this process is disrupted when pS214Tau aggregates on dendritic microtubules “trap” endosomes, for example in aged dlPFC (Figure 5D‐E), or middle‐aged ER. 16 Similarly, fibrils of hyperphosphorylated tau “trap” endosomes containing APP. 16 Hence, accumulated pTau, like altered retromer signaling, could magnify Aβ production. This important hypothesis should be tested with additional approaches; however, this will be challenging, as it requires the use of neurons that naturally generate phosphorylated tau and Aβ without genetic modifications. However, the immunoEM data are a definitive demonstration at the ultrastructural level of pTau trapping endosomes, providing strong support for a mechanism whereby phosphorylated tau can exacerbate Aβ cleavage. Phosphorylated tau may also aggravate Aβ production in axons through the disruption of microtubules, which increases the accumulation of APP and BACE. 46 Thus, tau pathology can set up a vicious cycle, where pTau increases Aβ production, and Aβ oligomers then drive more tau phosphorylation (Figure 6), a process that can build over a long lifetime. In short, AD pathology could initiate from multiple starting points: in autosomal dominant AD, for example, due to APP duplications, pathology would start with Aβ generation which would subsequently increase the phosphorylation of tau, while in sAD, pathology would start from changes in cell signaling that dysregulate calcium and drive tau phosphorylation, which would then exacerbate Aβ production. The engagement of vicious cycles would ultimately create a similar phenotype, even though they may have differing origins (Figure 6). Anti‐tau therapies may help to interrupt the vicious cycle and reduce pathology. These data help explain the unexpected finding in mouse models that anti‐tau therapies reduce Aβps as well as tau pathology. 47
FIGURE 5.
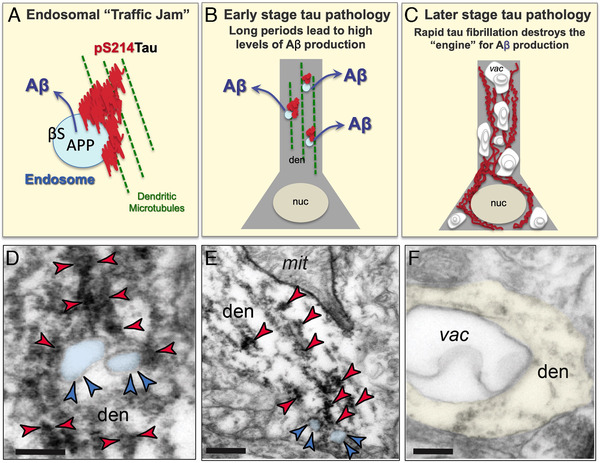
Endosomal “traffic jams” may drive amyloid beta (Aβ) pathology. (A) Schematic of an endosomal “traffic jam” similar to that described for genetic insults to retromer signaling, 45 showing an endosome containing amyloid precursor protein (APP) and β secretase trapped in aggregated pS214Tau on a microtubule. (B) A schematic diagram of early stage tau pathology in a dendrite (den), where aggregations of phosphorylated tau (red) on microtubules (green) trap endosomes (blue) and exacerbate the production of Aβ, which is then released into the extracellular space, e.g. after axonal transport. Real examples of aggregated pS214Tau on microtubules trapping endosomes are shown in panels D and E. nuc = nucleus. (C) A schematic diagram of later stage tau pathology, with fibrillated tau in the dendrite and soma now accompanied by extensive autophagic vacuoles (vac), where normal neuronal organelles are now lost, and the cellular engine for Aβ production has deteriorated. A real example of an autophagic vacuole in a dendrite is shown in panel F. (D, E) ImmunoEM of the aged monkey layer III dorsolateral prefrontal cortex (dlPFC) showing early‐stage, soluble pS214Tau (indicated by red arrowheads) aggregating on microtubules in dendrites (den) where it traps enlarged endosomes (pseudocolored in blue, and indicated by blue arrowheads). Aggregations of pS214Tau are often seen near dysmorphic mitochondria (mit), consistent with local calcium dysregulation. Note that the dlPFC is a site of extensive Aβ pathology in sporadic Altzheimer's disease. Interestingly, rhesus monkeys are apoE4 genotype, 52 which may increase the numbers of endosomes in this species. 53 Scale bar in B: 200 nm; scale bar in C: 200 nm. (F) With advanced age, pTau‐afflicted dendrites fill with autophagic vacuoles (vac), such as the one seen here in dlPFC, and lose normal organelles, including those needed to generate Aβ. Scale bar in D: 200 nm. Scale bars in D‐F: 200 nm
FIGURE 6.
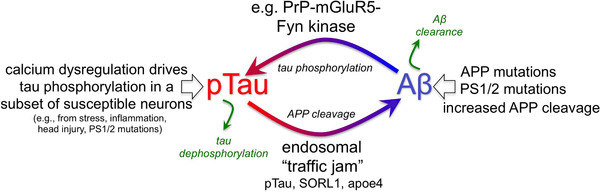
Potentially interacting vicious cycles in sporadic Altzheimer's disease (sAD). Cartoon showing pTau driving Aβ production and Aβ driving tau phosphorylation, setting up a vicious cycle. In sAD, cell biological changes in the aging association cortex (eg, calcium dysregulation) can increase early stage tau pathology that subsequently generates Aβ cleavage via trapping of endosomes in aggregated pTau. Genetic alterations in retromer signaling (eg, SORL1) may exacerbate the risk of sAD by further exacerbating these “endosomal traffic jams” which increase amyloid precursor protein (APP) cleavage to Aβ in endosomes. In contrast, in familial Altzheimer's, primary genetic perturbations in amyloid signaling (eg, duplications in APP) can initiate the degenerative process, which subsequently increases tau hyperphosphorylation. In both cases, vicious cycles would be set in motion that ultimately lead to amyloid plaques and neurofibrillary tangles. Thus, AD pathology could have multiple initiating factors that can lead to a similar phenotype
These data also caution that pTau can have destructive properties even before it can be detected by current PET imaging technologies, which only capture fibrillated pathology. Early stage tau pathology, for example, in aging dlPFC (Figure 5A‐B, D‐E), may drive Aβ production before there is any evidence of fibrillated tau. Thus, one would see an amyloid PIB PET signal prior to a fibrillated tau‐PET signal, although underlying, early‐stage pTau may be an “invisible” driving force for Aβps production. Future EM‐based, PET imaging, and seeding studies should aim to capture earlier forms of tau pathology in the human brain.
3.2. Future experiments and validation studies
Validation of the tau hypothesis requires non‐genetically‐altered animal models that recreate the molecular events occurring in the human brain. As mice do not develop AD pathology in the absence of genetic mutations, non‐human primates will be critical for this research. For instance, one could examine whether infusions of pTau into the rhesus monkey association cortex can initiate tau pathology and trafficking, and whether it is sufficient to induce Aβ accumulation in terminal fields. A key aspect will be to mimic the protracted time course of sAD, as rapid tau phosphorylation and degeneration likely lead to a different phenotype than the slow process that accrues in sAD. It would be less time‐intensive to perform in vitro experiments as well, for example, to test the roles of calcium dysregulation on tau phosphorylation and Aβ accumulation, but these would need to be performed in cell cultures with the same characteristics as those in the aging association cortex, a current challenge for the field.
4. MAJOR CHALLENGES FOR THE HYPOTHESIS
The causes of sAD have been difficult to assess, as research to date has relied heavily on rodent models utilizing autosomal dominant mutations to induce pathology. Monkey models are more suitable to study the natural course of sAD‐related pathology, but these studies are slow and expensive, and would greatly benefit from complementary in vitro experiments. However, cell cultures are normally made from perinatal cortical or hippocampal rodent neurons, as aged neurons do not survive in culture. Therefore, they may miss the very features supporting the development of sAD‐like pathology. Human stem cells may have promise but currently cannot be differentiated into association cortical pyramidal cells with the unique molecular aspects that drive tau pathology, and may also need to be “aged” to be more relevant to sAD. A key caveat for in vitro research is the need to perform subtle manipulations mimicking the protracted time course of aging human brain that differs from changes caused by the aggressive manipulations often used in cell culture experiments. For example, large increases in intracellular calcium induce apoptosis, which is not a characteristic of sAD, while more subtle calcium dysregulation may better mimic the changes that foster tau pathology.
5. LINKAGE TO OTHER MAJOR THEORIES
The amyloid hypothesis has dominated the AD field, positing that autosomal dominant AD is caused by genetic mutations that increase Aβ production, while sAD may be spurred by insults that reduce Aβ clearance. Our hypothesis does not refute a role for Aβ, but rather, states that multiple starting points can drive interacting vicious cycles and produce the final phenotype of sAD.
The importance of tau has often been dismissed because tau gene mutations produce tau pathology but do not cause AD. We propose that the conditions that induce rapidly advancing tau pathology destroy the cellular “engine” for Aβ production so quickly that neurons are unable to generate high levels of Aβ (Figure 5A‐C). This can be seen within the temporal allocortex in sAD, where tau pathology is especially aggressive, and in non‐AD tauopathies, such as chronic traumatic encephalopathy (CTE) or frontotemporal dementia (FTD), caused by environmental and/or genetic insults. Close examination of the human and monkey brains reveals surprisingly few Aβps in the ER and hippocampus compared to the high levels of Aβps in the association neocortex, and this may be owing to the swift advance of tau pathology, where neurons quickly fill with tau fibrils and degenerate, thereby destroying the organelles needed for Aβ production (Figures. 2B, 5C). Human and monkey data indicate that distal dendrites are an initial target of tau pathology and degeneration (eg, see vacuoles in a dendrite, Figure 5C, F), 16 , 17 , 20 and dendrites are also a major site for Aβ production in endosomes. 16 ImmunoEM in monkeys shows that autophagic degeneration quickly arises within dendrites of entorhinal layer pre‐α neurons, destroying a frequent site for Aβ production, 16 which may explain the low incidence of Aβps in this layer. If the rapid rise in tau pathology in disorders like CTE and FTD also destroys the machinery to manufacture Aβ, it may explain why these disorders do not develop the pattern of Aβps characteristic of sAD. In contrast, neurons that slowly accumulate pTau on microtubules would trap many endosomes and produce extensive Aβ (Figure 5B), for example, as occurs in the lateral dlPFC. These cellular dynamics deserve greater consideration, with factors such as intracellular microenvironment and the time course of pathological events (eg, slow vs rapid tau hyperphosphorylation) informing our hypotheses.
In summary, it is necessary to expand beyond simple, linear models of pathology and recognize the many interacting events that drive sAD pathology, including the important early rise in pTau that generates the scaffold for subsequent sAD pathology in vulnerable neuronal circuits.
CONFLICT OF INTEREST
Amy F.T. Arnsten and Yale receive royalties from the USA sales of Intuniv (extended release guanfacine). They do not receive royalties from non‐USA or generic sales of Intuniv. The other authors have no actual or potential conflicts of interest to declare.
ACKNOWLEDGMENTS
We dedicate this manuscript to Dr. Constantinos Paspalas, whose elegant work illuminates this field. We thank Mr. David Ewert (University of Ulm) for assistance with the layout for Figure 2, and Dr. Christopher van Dyck for helpful comments on the manuscript.
Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer's disease. Alzheimer's Dement. 2021;17:115–124. 10.1002/alz.12192
Funding Information
This work was supported by NIH grants Pioneer Award DP1AG047744‐01 and R01AG061190‐02 (AFTA); Alzheimer's Association Research Fellowship AARF‐17‐533294 (DD); American Federation on Aging Research/Diamond Postdoctoral Fellowship (DD); and the Hans & Ilse Breuer Foundation, Frankfurt am Main, Germany (HB).
REFERENCES
- 1. Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353‐356. [DOI] [PubMed] [Google Scholar]
- 2. Morris GP, Clark IA, Vissel B. Questions concerning the role of amyloid‐β in the definition, aetiology and diagnosis of Alzheimer's disease. Acta Neuropathol. 2018;136:663‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Braak H, Del Tredici K. Neuroanatomy and pathology of sporadic Alzheimer's disease. Adv Anat Embryol Cell Biol. 2015;215:1‐162. [PubMed] [Google Scholar]
- 4. Huang LK, Chao SP, Hu CJ. Clinical trials of new drugs for Alzheimer disease. J Biomed Sci. 2020;27:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Arboleda‐Velasquez JF, Lopera F, O'Hare M, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [11C]PIB‐PET Centiloid values and postmortem measures of Alzheimer's disease neuropathology. Alzheimers Dement. 2019;15:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giannakopoulos P, Herrmann FR, Bussière T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495‐1500. [DOI] [PubMed] [Google Scholar]
- 8. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 9. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960‐969. [DOI] [PubMed] [Google Scholar]
- 10. Bussière T, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR. Progressive degeneration of nonphosphorylated neurofilament protein‐enriched pyramidal neurons predicts cognitive impairment in Alzheimer's disease: stereologic analysis of prefrontal cortex area 9. J Comp Neurol. 2003;463:281‐302. [DOI] [PubMed] [Google Scholar]
- 11. Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015;138:2814‐2833. [DOI] [PubMed] [Google Scholar]
- 12. Buckmaster CA, Eichenbaum H, Amaral DG, Suzuki WA, Rapp PR. Entorhinal cortex lesions disrupt the relational organization of memory in monkeys. J Neurosci. 2004;24:9811‐9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braak H, Zetterberg H, Del Tredici K, Blennow K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid‐β changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. 2013;126:631‐641. [DOI] [PubMed] [Google Scholar]
- 14. Barthélemy NR, Mallipeddi N, Moiseyev P, Sato C, Bateman RJ. Tau phosphorylation rates measured by mass spectrometry differ in the intracellular brain vs. extracellular cerebrospinal fluid compartments and are differentially affected by Alzheimer's disease. Front Aging Neurosci. 2019;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arnsten AFT, Datta D, Leslie S, Yang ST, Wang M, Nairn AC. Alzheimer's‐like pathology in aging rhesus macaques: unique opportunity to study the etiology and treatment of Alzheimer's disease. Proc Natl Acad Sci U S A. 2019;116:26230‐26238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Paspalas CD, Carlyle BC, Leslie S, et al. The aged rhesus macaque manifests Braak‐stage III/IV Alzheimer's‐like pathology. Alzheimers Dement. 2018;14:680‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Carlyle BC, Nairn AC, Wang M, et al. cAMP‐PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A. 2014;111:5036‐5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kadavath H, Hofele RV, Biernat J, et al. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A. 2015;112:7501‐7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xia D, Gutmann JM, Götz J. Mobility and subcellular localization of endogenous, gene‐edited Tau differs from that of over‐expressed human wild‐type and P301L mutant Tau. Sci Rep. 2016;6:29074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Braak H, Del Tredici K. Spreading of tau pathology in sporadic Alzheimer's disease along cortico‐cortical top‐down connections. Cereb Cortex. 2018;28:3372‐3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Merino‐Serrais P, Benavides‐Piccione R, Blazquez‐Llorca L, et al. The influence of phospho‐tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer's disease. Brain. 2013;136:1913‐1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tai HC, Serrano‐Pozo A, Hashimoto T, Frosch MP, Spires‐Jones TL, Hyman BT. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin‐proteasome system. Am J Pathol. 2012;181:1426‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jicha GA, Weaver C, Lane E, et al. cAMP‐dependent protein kinase phosphorylations on tau in Alzheimer's disease. J Neurosci. 1999;19:7486‐7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Falcon B, Zhang W, Schweighauser M, et al. Tau filaments from multiple cases of sporadic and inherited Alzheimer's disease adopt a common fold. Acta Neuropathol. 2018;136:699‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edler MK, Sherwood CC, Meindl RS, et al. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer's disease. Neurobiol Aging. 2017;59:107‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu H, Hardy J, Duff KE. Selective vulnerability in neurodegenerative diseases. Nat Neurosci. 2018;21:1350‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khachaturian ZS. Overview of basic research on Alzheimer disease: implications for cognition. Alzheimer Dis Assoc Disord. 1991;5(S1):S1‐6. [DOI] [PubMed] [Google Scholar]
- 28. Mattson MP. Calcium and neurodegeneration. Aging Cell. 2007;6:337‐350. [DOI] [PubMed] [Google Scholar]
- 29. Lacampagne A, Liu X, Reiken S, et al. Post‐translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease‐like pathologies and cognitive deficits. Acta Neuropathol. 2017;134:749‐767. [DOI] [PubMed] [Google Scholar]
- 30. Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium‐activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628‐2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kurbatskaya K, Phillips EC, Croft CL, et al. Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer's disease brain. Acta Neuropathol Commun. 2016;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Arnsten AF. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. 2015;18:1376‐1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol. 1991;111:293‐301. [DOI] [PubMed] [Google Scholar]
- 34. Kondo H, Tanaka K, Hashikawa T, Jones EG. Neurochemical gradients along monkey sensory cortical pathways: calbindin‐immunoreactive pyramidal neurons in layers II and III. Eur J Neurosci. 1999;11:4197‐4203. [DOI] [PubMed] [Google Scholar]
- 35. Yang ST, Wang M, Paspalas CD, et al. Core differences in synaptic signaling between primary visual and dorsolateral prefrontal cortex. Cereb Cortex. 2018;28:1458‐1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kwon MS, Seo YJ, Shim EJ, Choi SS, Lee JY, Suh HW. The effect of single or repeated restraint stress on several signal molecules in paraventricular nucleus, arcuate nucleus and locus coeruleus. Neuroscience. 2006;142:1281‐1292. [DOI] [PubMed] [Google Scholar]
- 37. Glovaci I, Chapman CA. Dopamine induces release of calcium from internal stores in layer II lateral entorhinal cortex fan cells. Cell Calcium. 2019;80:103‐111. [DOI] [PubMed] [Google Scholar]
- 38. Liu F, Liang Z, Shi J, et al. PKA modulates GSK‐3β‐ and cdk5‐catalyzed phosphorylation of tau in site‐ and kinase‐specific manners. FEBS Lett. 2006;580:6269‐6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kobori N, Moore AN, Dash PK. Altered regulation of protein kinase a activity in the medial prefrontal cortex of normal and brain‐injured animals actively engaged in a working memory task. J Neurotrauma. 2015;32:139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johansson L, Guo X, Hällström T, et al. Common psychosocial stressors in middle‐aged women related to longstanding distress and increased risk of Alzheimer's disease: a 38‐year longitudinal population study. BMJ Open. 2013;3:e003142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bello‐Chavolla OY, Antonio‐Villa NE, Vargas‐Vázquez A, Ávila‐Funes JA, Aguilar‐Salinas CA. Pathophysiological mechanisms linking type 2 diabetes and dementia: review of evidence from clinical, translational and epidemiological research. Curr Diabetes Rev. 2019;15:456‐470. [DOI] [PubMed] [Google Scholar]
- 42. Liu L, Drouet V, Wu JW, et al. Trans‐synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kaufman SK, Del Tredici K, Thomas TL, Braak H, Diamond MI. Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho‐tau pathology in Alzheimer's disease and PART. Acta Neuropathol. 2018;136:57‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Braak H, Del Tredici K. Amyloid‐β may be released from non‐junctional varicosities of axons generated from abnormal tau‐containing brainstem nuclei in sporadic Alzheimer's disease: a hypothesis. Acta Neuropathol. 2013;126:303‐306. [DOI] [PubMed] [Google Scholar]
- 45. Small SA, Simoes‐Spassov S, Mayeux R, Petsko GA. Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer's disease. Trends Neurosci. 2017;40:592‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sadleir KR, Kandalepas PC, Buggia‐Prévot V, Nicholson DA, Thinakaran G, Vassar R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer's disease. Acta Neuropathol. 2016;132:235‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dai CL, Chen X, Kazim SF, et al. Passive immunization targeting the N‐terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm (Vienna). 2015;122:607‐617. [DOI] [PubMed] [Google Scholar]
- 48. Goñi‐Oliver P, Lucas JJ, Avila J, Hernández F. N‐terminal cleavage of GSK‐3 by Calpain—a new form of GSK‐3 regulation. J Biol Chem. 2007;282:22406‐22413. [DOI] [PubMed] [Google Scholar]
- 49. Jia L, Qiu Q, Zhang H, et al. Concordance between the assessment of Aβ42, T‐tau, and P‐T181‐tau in peripheral blood neuronal‐derived exosomes and cerebrospinal fluid. Alzheimers Dement. 2019;15:1071‐1080. [DOI] [PubMed] [Google Scholar]
- 50. Janelidze S, Stomrud E, Smith R, et al. Cerebrospinal fluid p‐tau217 performs better than p‐tau181 as a biomarker of Alzheimerr's disease. Nat Commun. 2020;11:1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu H, Suk HY, Yu RY, et al. Evolutionarily conserved role of calcineurin in phosphodegron‐dependent degradation of phosphodiesterase 4D. Mol Cell Biol. 2010;30:4379‐4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Poduri A, Gearing M, Rebeck GW, Mirra SS, Tigges J, Hyman BT. Apolipoprotein E4 and beta amyloid in senile plaques and cerebral blood vessels of aged rhesus monkeys. Am J Pathol. 1994;144:1183‐1187. [PMC free article] [PubMed] [Google Scholar]
- 53. Nuriel T, Peng KY, Ashok A, et al. The endosomal‐lysosomal pathway is dysregulated by APOE4 expression in vivo. Front Neurosci. 2017;11:702. [DOI] [PMC free article] [PubMed] [Google Scholar]