

This viewpoint provides an overview of applying genetic code expansion and click chemistry for labeling proteins for cellular imaging applications. This approach, which enables labeling of proteins in cells at a specific site with fluorescent dyes, has several advantages for cellular imaging. However, it also introduces limitations and challenges for cellular imaging. The current state of the field is discussed as well as the advantages and limitations of this unique approach.
Keywords: bioorthogonal reactions, click chemistry, fluorescent dyes, genetic code expansion, light microscopy, noncanonical amino acids, protein labeling
Abstract
Twenty‐five years ago, GFP revolutionized the field of cell biology by enabling scientists to visualize, for the first time, proteins in living cells. However, when it comes to current, state‐of‐the‐art imaging technologies, fluorescent proteins (such as GFP) have several limitations that result from their size and photophysics. Over the past decade, an elegant, alternative approach, which is based on the direct labeling of proteins with fluorescent dyes and is compatible with live‐cell and super‐resolution imaging applications, has been introduced. In this approach, an unnatural amino acid that can covalently bind a fluorescent dye is incorporated into the coding sequence of a protein. The protein of interest is thereby site‐specifically fluorescently labeled inside the cell, eliminating the need for protein‐ or peptide‐labeling tags. Whether this labeling approach will change cell biology research is currently unclear, but it clearly has the potential to do so. In this short review, a general overview of this approach is provided, focusing on the imaging of site‐specifically labeled proteins in mammalian tissue culture cells, and highlighting its advantages and limitations for cellular imaging.
Abbreviations
- AA
amino acid
- Fl‐dye
fluorescent dye
- GCE
genetic code expansion
- PM
plasma membrane
- POI
protein of interest
- SMLM
single‐molecule localization microscopy
- SPT
single‐particle tracking
- SR
super‐resolution
- STED
stimulated emission depletion
- STORM
stochastic optical reconstruction microscopy
- Tet‐Fl‐dye
tetrazine‐conjugated Fl‐dye
- unAAs
unnatural amino acids
How does it work?
The selective labeling of proteins via unnatural amino acids (unAAs) relies on the incorporation of an unAA into the amino acid (AA) sequence of the protein of interest (POI). This incorporation takes place in cells that were engineered to enable the addition of an unAA in response to a unique codon during ribosomal translation (Fig. 1A). Labeling then occurs by the formation of a covalent bond between the unAA and a chemically modified fluorescent dye (Fl‐dye) via highly specific chemical reactions, termed bioorthogonal or ‘click’ reactions (Fig. 1B). As a result, the POI is directly and site‐specifically labeled in live cells. The major steps in this labeling technique are briefly discussed below; for a more detailed description, please refer to Ref. [1, 2, 3].
Fig. 1.
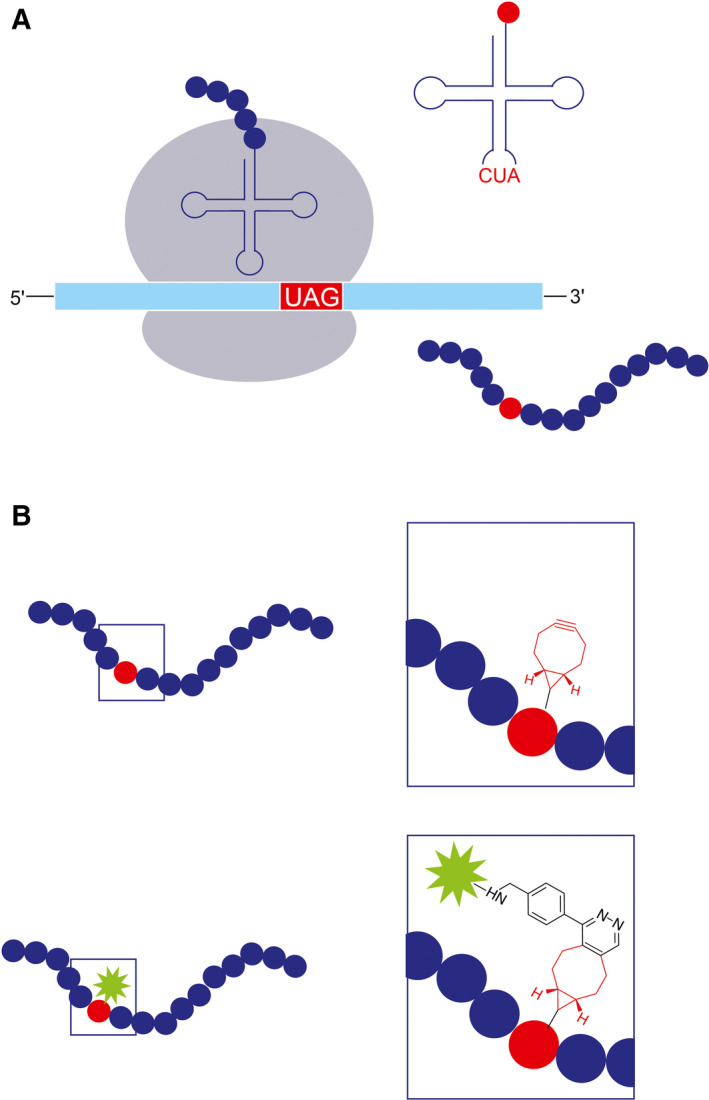
Labeling cellular proteins through genetic code expansion (GCE) and click chemistry. (A) Incorporating an unAA during ribosomal translation. A UAG codon is inserted in‐frame into the amino acid sequence of a POI. A unique tRNA, which carries the complementary codon to the UAG and was charged with the unAA by a unique tRNA synthetase, recognizes the UAG codon and incorporates the unAA into the newly formed polypeptide. Translation then continues until the full‐length polypeptide is released from the ribosome, giving rise to the synthesis of a full‐length protein that carries the unAA at a specific site. (B) Bioorthogonal labeling via a click reaction. The unAA carries a chemical modification; in this case, a BCN moiety (red) that specifically and rapidly reacts with the tetrazine moiety (black) that is attached to a fluorescent dye (green). Consequently, the protein is directly labeled with a fluorescent dye at a specific site.
Encoding an unAA in cellular proteins
Incorporating an unAA into a POI in the cell requires the unAA being recognized by the cellular translation machinery. One approach to accomplish this task is to use a modified AA that resembles a natural one, thus enabling its recognition by the endogenous tRNA and tRNA synthetase. This approach has been demonstrated using methionine analogs [4, 5, 6, 7, 8] and was successfully used to visualize newly synthesized proteins in mammalian cells [5, 6, 7]. However, this approach is not protein‐specific, as all endogenous methionine residues can potentially be substituted by the analogous unAA and, therefore, its applicability for the fluorescence imaging of specific proteins is limited.
Another approach, which allows the selective labeling of a POI within the cell, is to incorporate the unAA into the coding sequence of the POI by using the genetic code expansion (GCE) technique. To this end, a sequence of the POI with an in‐frame TAG codon (the amber stop codon) is added to the cell, together with the selected unAA. In addition, the cell is supplemented with a unique pair of tRNA/tRNA synthetase, which was evolved to facilitate the incorporation of the desired unAA in response to the amber stop codon (Fig. 1A). Notably, by allocating the amber stop codon to encode an additional, unnatural AA, the genetic code is expanded from 20 to 21 AAs.
Specifically attaching a fluorescent dye to the unAA in the cellular milieu
The most direct GCE approach for fluorescently labeling proteins in cells is to incorporate unAAs that bear fluorescent properties into the AA sequence of the POI [9]. This approach was used successfully to visualize intracellular proteins in bacteria and in mammalian cells, without affecting their function [10, 11, 12]. However, the currently available fluorescent unAAs are bulky, their incorporation efficiency is low, and they suffer from poor fluorescent properties, altogether hindering their utilization in cellular imaging [1, 2].
A more efficient and commonly used approach is to incorporate an unAA that carries a chemical moiety capable of covalently binding a Fl‐dye inside the cell via the fast and highly specific click reactions (Fig. 1B) (reviewed in Refs [1, 2, 13]). Notably, click reactions are not restricted to protein labeling and provide an attractive, superior tool for visualizing nucleotides, lipids, and sugars in mammalian cells, though this is beyond the scope of this review [1, 14, 15, 16, 17]. Currently, the preferred click reaction for protein labeling in live mammalian cells is the inverse electron‐demand Diels–Alder reaction (IEDDA) between strained alkynes or alkenes and tetrazines, which demonstrates fast kinetics at room temperature and a low cellular toxicity [2, 13, 18]. The unAAs that are most frequently used in this approach are trans‐cyclooctene (TCO)‐lysine and bicyclo[6.1.0]nonyne (BCN)‐lysine, and designated pairs of tRNA/tRNA synthetase have been evolved for their incorporation into proteins in mammalian cells [19, 20, 21]. Each of these two unAAs has its own advantages and limitations upon binding the Fl‐dye; BCN‐lysine exhibits a relatively high labeling efficiency (especially under low expression levels), while TCO‐lysine reacts faster and is more robust, but tends to produce higher background levels due to its hydrophobicity [22, 23].
Once the cells express a POI that carries the chosen unAA, a tetrazine‐conjugated Fl‐dye (Tet‐Fl‐dye) is added to the cells and specifically binds the unAA via the click reaction, resulting in a direct, highly specific labeling of the POI at a selected site (Fig. 1B). The click reaction is fluorogenic; the fluorescence of the Fl‐dye is quenched by the tetrazine moiety in the unbound form, and is increased, by several folds, upon binding to the unAA [13, 20, 24]. Essentially any Fl‐dye that is conjugated to tetrazine can be used to label the POI, and numerous Tet‐Fl‐dyes are available commercially. Of special interest are the silicon rhodamine (SiR)‐based dyes (SiR, HMSiR, and JF646), which are cell‐permeable and exhibit low background labeling in mammalian cells [15, 25, 26, 27, 28]. The fluorogenicity of the click reaction reduces background fluorescence that stems from the unspecific binding of the Fl‐dye—a known limitation of other labeling approaches based on Fl‐dyes, such as SNAP‐tag [29]. In general, when bound to methyl‐substituted tetrazines (Me‐Tet), Fl‐dyes are substantially more quenched than Fl‐dyes bound to H‐Tet [22, 24]. However, the fluorogenicity of the reaction also depends on the properties of the Fl‐dye itself and therefore should be examined on a case to case basis. The detailed analysis of the fluorescent properties of different Tet‐Fl‐dyes, recently completed by Beliu et al. can be used as a guideline [24].
What is it good for?
Genetic code expansion‐based labeling, namely, the labeling of cellular proteins through GCE and click chemistry, has several key advantages over other cellular imaging techniques. First, the Fl‐dye is attached directly to a specific residue, such that only the chemical handle and the Fl‐dye are added to the POI. This minimal compound is an order of magnitude smaller than a fluorescent protein (~ 0.5 nm, as compared with ~ 5 nm)—an advantage that is especially critical for preserving the functions of small POIs [22, 30]. Second, Fl‐dyes are generally brighter and more photostable than fluorescent proteins, enabling live‐cell imaging at lower laser powers and at higher temporal resolutions. Third, Fl‐dyes are available for the entire light spectrum, including the near‐infrared region (e.g., SiR and JF646), allowing imaging at longer wavelengths that are less phototoxic to the cells [26, 28, 31]. This is in contrast to fluorescent proteins, which exhibit poor photophysics at longer wavelengths. Fourth, the click reaction is stoichiometric (the Tet‐Fl‐dye binds the unAA at a 1 : 1 ratio), which enables protein quantification based on fluorescent intensity in heterologous expression systems [32]. These advantages are even more pronounced in single‐molecule applications and super‐resolution (SR) microscopy techniques, where cells are exposed to high laser powers for relatively long periods of time. The small size of the label and its 1 : 1 binding stoichiometry are especially attractive for single‐molecule SR techniques, as they set the ground for increasing localization accuracy and for molecule counting.
Taken together, GCE‐based labeling is advantageous for all fluorescence imaging applications and, in particular, for high‐spatiotemporal‐resolution quantitative analyses of proteins in live cells: The minimal label size helps preserve the nature of the POI; the 1 : 1 stoichiometry ratio allows quantification; and the ability to image at low laser powers and long wavelengths increases cell viability.
What has been done with it?
In 2012, the Chin laboratory demonstrated the efficient, site‐specific labeling of proteins in live mammalian cells by using the genetic encoding of BCN‐Lys or TCO‐Lys and click labeling with Tet‐Fl‐dyes [20]. As explained above, this GCE‐based labeling approach is fundamentally different from conventional protein‐based labeling approaches, such as fluorescent protein tags and protein self‐labeling tags (e.g., SNAP‐tags). Therefore, efforts have been made over the past years to validate this approach and evaluate its performance in different imaging modalities. In tissue culture cells, GCE‐based labeling was found to be compatible with live‐cell imaging and SR microscopy of both intra‐ and extracellular proteins. Moreover, whenever tested, the localization and function of the POIs were not affected by the presence of the unAA, further validating this approach [22, 30, 33]. An overview of the imaging applications tested so far with GCE‐based labeling is provided below.
Live‐cell imaging
Genetic code expansion‐based labeling has proven successful for the imaging of several receptors and channels in live cells [4, 20, 24, 32, 34] (Fig. 2). In addition, it has been used to label the HIV‐1 envelop glycoprotein Env, enabling the first‐ever recording of its dynamics on the plasma membrane (PM) [30]. This work is of special interest because prior attempts to visualize Env in live cells failed due to the inability to generate a functionally active labeled Env by using conventional protein tags. In all the above‐mentioned examples, the unAA was incorporated at the extracellular regions of the protein and labeling was performed using cell‐impermeable Tet‐Fl‐dyes, thus avoiding the relatively high background levels associated with the technique (see subsection The click reaction, below). After further optimization, the GCE‐based labeling technique was successfully applied for intracellular labeling, and various intracellular components—including intermediate filaments, microtubules, intracellular vesicles, the PM, lysosomes, and the endoplasmic reticulum—were labeled and imaged in live mammalian cells [22, 24, 35, 36, 37, 38]. Here, too, small proteins that could not be labeled using protein tags were successfully labeled and imaged. Peng and Hang, for example, recorded, for the first time, the dynamics of the IFITM3 protein, which comprises only 137 AAs [22].
Fig. 2.
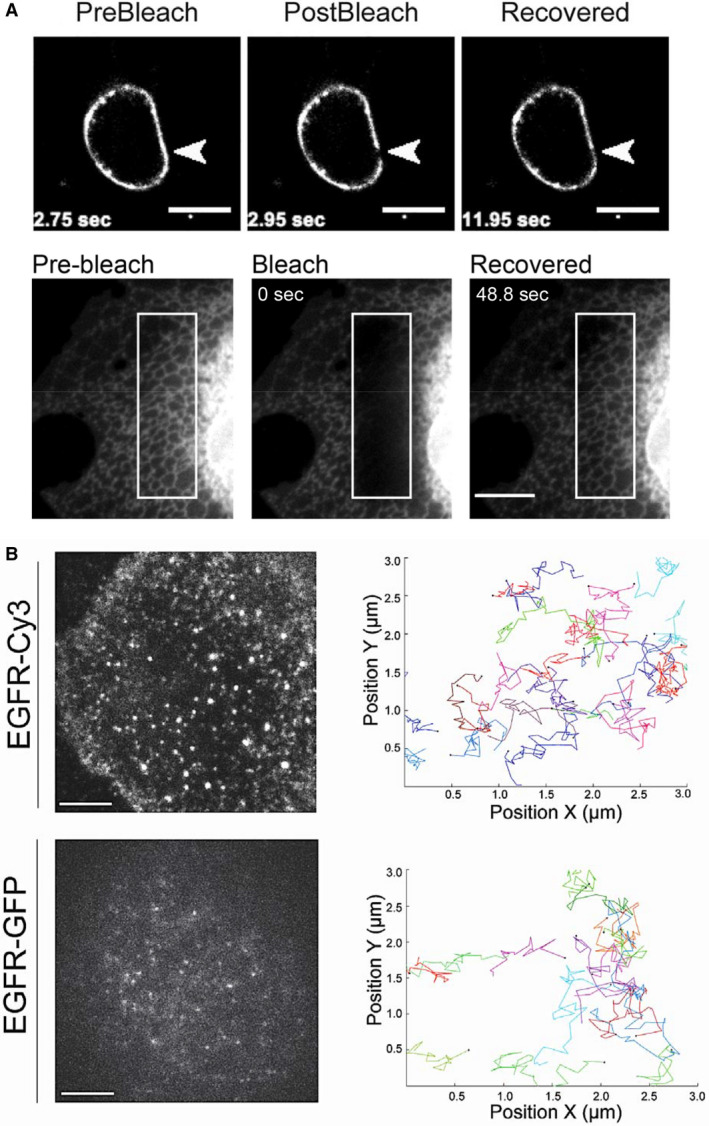
Live‐cell imaging of proteins labeled via GCE and click chemistry. (A) Photobleaching experiments employing cellular proteins labeled with unAAs. Top panel: HEK293 cells expressing the HIV protein Env, which carries a BCN‐lysine at position 407. The cells were labeled with the cell‐impermeable dye Tet‐Cy5 and imaged using a spinning disk confocal microscope. Reproduced with permission from Ref. [30]. Bottom panel: COS7 cells expressing the endoplasmic reticulum marker ERcb5TM, which was conjugated to a 14‐AA tag that carries BCN‐lysine. The cells were labeled with the cell‐permeable dye Tet‐TAMRA and imaged using a spinning disk confocal microscope. Reproduced from Ref. [36]. Scale bars, 10 μm. (B) SPT of EGFR in COS7 cells, imaged in TIRF mode over time. Top panel: The cells express EGFR, which carries BCN‐lysine at position 128, and are labeled with the cell‐impermeable dye Tet‐Cy3. Bottom panel: The cells express an EGFR‐GFP. In each case, individual particles obtained from the videos were segmented and tracked through time (right panels). Note that particles obtained for EGFR‐Cy3 were brighter than those obtained for EGFR‐GFP and that significantly more tracks were generated using this labeling approach. Reproduced from Ref. [34]. Scale bars, 10 μm.
Genetic code expansion‐based intra‐ and extracellular labeling was also used in photomanipulation techniques, such as FRET and FRAP [4, 30, 33, 34, 36, 38] (Fig. 2A). Single‐molecule FRET approaches were recently developed and applied for resolving the conformational changes of hemagglutinins on virus surfaces and of shaker Kv channels upon activation in Xenopus oocytes [4, 39]. Such high‐resolution experiments have only been made possible due to the site‐selective GCE‐based labeling.
Super‐resolution microscopy
Several SR techniques, including structured illumination microscopy (SIM), single‐molecule localization microscopy (SMLM; STORM and GSDIM), and stimulated emission depletion (STED), were performed in fixed cells on proteins labeled via GCE and click chemistry [3, 23, 30, 33, 34, 35, 37, 40, 41, 42] (Fig. 3). Rizzoli and colleagues systematically labeled 26 different proteins by using unAAs and visualized their multimolecular arrangements with both SMLM and STED [40]. A dSTORM imaging of click‐labeled NMDA receptors produced significantly higher labeling densities than those produced by antibody‐based NMDA labeling, suggesting that GCE‐based labeling is advantageous for SMLM [33] (Fig. 3A,B). In SIM images of GCE‐based labeled microtubules, adjacent fibers < 100 nm apart could be resolved, indicating that resolution in SIM may be improved by implementing GCE‐based labeling, potentially due to the small size of the label [35] (Fig. 3D). The technique was also successfully combined with DNA‐PAINT technology for the SMLM of the intermediate filament protein, vimentin, and the nuclear pore complex protein, Nup153 [37].
Fig. 3.
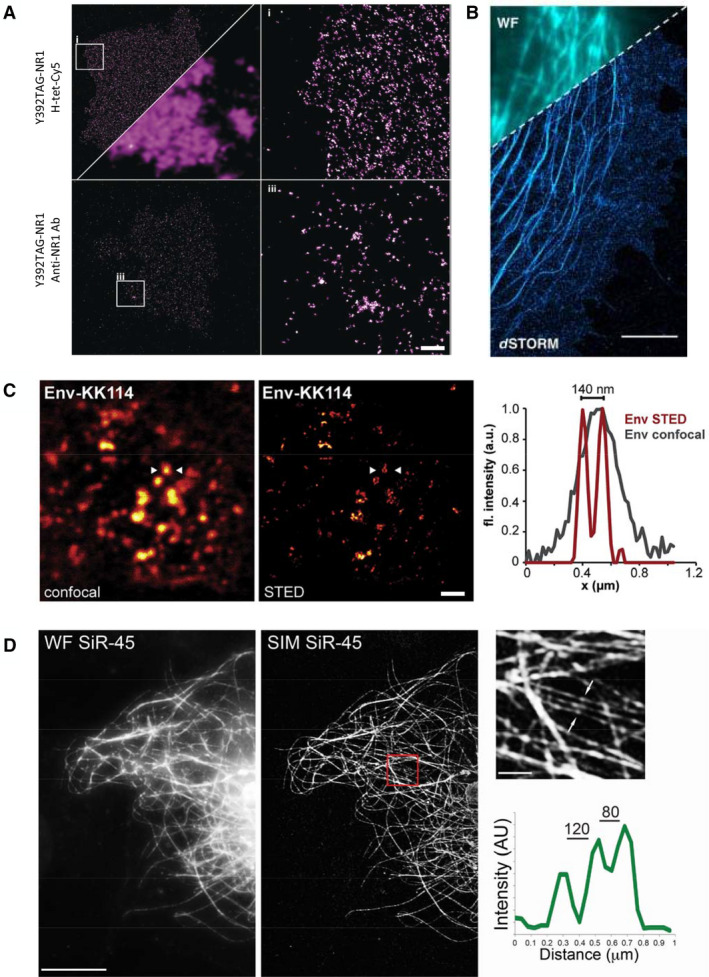
Super‐resolution microscopy imaging of proteins labeled via GCE and click chemistry. (A, B) SMLM performed on cellular proteins labeled with GCE and click chemistry. (A) Top panel: dSTORM imaging of NMDA receptors carrying a TCO‐lysine in position 392 and click‐labeled with Tet‐Cy5. Bottom panel: dSTORM imaging of NMDA receptors labeled with specific primary antibodies and Alexa 647 secondary antibodies. The right panels are zoomed‐in images of the areas marked with squares on the left panels. A wide‐field image is shown, for comparison, on the bottom right corner of the top left panel. Note that considerably more localizations were obtained using the click‐labeled NMDA receptor. Reproduced with permission from Ref. [33]. Scale bar, 2.5 μm. (B) dSTORM imaging of microtubules labeled with the microtubule‐binding protein EMTB carrying a TCO‐lysine at position 87 and click‐labeled with the cell‐permeable dye Tet‐HM‐SiR in COS7 cells. A wide‐field image is shown on the top left corner, for comparison. Reproduced from Ref. [24]. Scale bar, 1 μm. (C) Confocal (left) and STED (middle) images of the HIV protein Env, which carries a BCN‐lysine at position 407 and was labeled with Tet‐KK114 in HEK293 cells. Right panel: line intensity profiles obtained for an individual Env cluster (arrowheads in the left and middle panels), imaged via confocal microscopy or STED. The higher resolution obtained with STED can be clearly seen. Reproduced with permission from Ref. [30]. Scale bar, 1 μm. (D) SIM imaging of GCE‐labeled microtubules in COS7 cells. Microtubules, labeled in COS7 cells with tubulin that carry a BCN‐lysine at position 45 and Tet‐SiR, were imaged using wide‐field microscopy or SIM (left and middle panels, respectively). A zoomed‐in image of the red rectangle in the middle panel is shown in the top right panel. An intensity line profile across the region depicted by arrows in the top right panel is shown in the bottom right panel, demonstrating the ability to resolve fibers that are only 80 nm apart. Reproduced from Ref. [35]. Scale bars, 10 μm, zoomed‐in, 1 μm.
Single‐molecule techniques
More recently, GCE‐based labeling was applied to live‐cell single‐molecule applications, such as single‐particle tracking (SPT) and live STORM [32, 34] (Fig. 2B). By using image correlation spectroscopy, the fluorescence intensities obtained for GPCRs via click labeling were found to be linearly correlated with those obtained using GFP, indicating that the approach is quantitative. Measurements of the fluorescent intensity in peroxisomes colabeled with GCE‐based Tet‐SiR and GFP confirmed that the approach is quantitative also in an intracellular context [36]. SPT was used to measure the diffusion rates of click‐labeled GPCRs, EGFR, and the Shaker Kv channel [32, 34] (Fig. 2B). Notably, while the physiological response of the receptors appeared to be similar in click‐labeled and GFP‐labeled receptors, mild differences were measured in the confinement and rate of diffusion [32, 34]. On average, more tracks and longer trajectories were obtained for proteins labeled via unAAs, as compared with those tagged with GFP [34]. Therefore, GCE‐based labeling appears to be more reliable for tracking and, accordingly, superior over GFP labeling for measuring the diffusion of proteins in the cell. However, before switching from one labeling approach to the other, the cause for the differences in the diffusion of GFP‐ versus GCE‐based labeled proteins may need to be elucidated.
Substituting fluorescent proteins for Fl‐dyes using self‐labeling tags was shown to improve the temporal resolution of SMLM, enabling live‐SMLM imaging [43, 44, 45]. Similarly, live SMLM of GCE‐based labeled PM proteins was recently performed at a 30‐nm spatial resolution and a 2.5‐s temporal resolution [34]. Although the temporal resolution should be further improved, combining site‐specific labeling with STORM in live‐cell applications will surely advance our understanding of the dynamics and function of proteins in cells.
Multicolor labeling
Dual‐color labeling of different proteins in the same cell using GCE is currently a challenging task, as it requires exploiting two codons from the codon table and introducing two mutually orthogonal tRNA/tRNA synthetase pairs to the cells, which is extremely difficult in mammalian cells [46]. Although unAA‐based labeling can be readily combined with conventional protein tags for a dual‐color labeling [35, 47], several laboratories proposed strategies to overcome the challenge of GCE‐based dual‐color labeling [3, 46].
Rizzoli and colleagues incorporated different unAAs into different POIs in two separate cells and then fused the cells to one another by using inactivated virus particles [42]. This approach enabled dual‐color STED imaging, but its applicability to cell biology is limited because imaging is restricted to fixed cells and to protein pairs that can be studied in fused cells. By sequentially introducing two different unAAs and fine‐tuning the specificity of the click reactions, Lemke and colleagues were able to use two different unAAs to label the same POI at the same site with two different Fl‐dyes in live cells [41]. While this dual‐labeling approach is beneficial for some imaging applications (such as pulse‐chase analyses), it is limited to the labeling of a single POI. Another approach for labeling the same POI with dyes of two colors is to employ competitive labeling of the same unAA with two Tet‐conjugated Fl‐dyes bearing different fluorescent properties. This labeling strategy has been employed to study protein oligomerization, perform single‐molecule FRET, and simultaneously acquire data from a single cell by using different imaging modalities [32, 34, 39].
What should I consider before starting an experiment?
The factors that should be considered regarding GCE‐based labeling for cellular imaging can be generally divided according to the two main steps in the process: the incorporation of the unAA into the AA sequence of the POI, and the click reaction between the unAA and the Tet‐conjugated Fl‐dye. Critical considerations for each of these steps are discussed below.
Incorporation of the unAA
Conventionally, the incorporation of the unAA into the AA sequence of the POI is enabled by mutating one of the codons to TAG, such that one of the AAs in the POI is replaced by a noncanonical AA (in most cases, BCN‐Lys or TCO‐Lys). Therefore, when choosing the incorporation site, care should be taken to avoid replacing an AA that is crucial for either the structure or the function of the POI. Importantly, once a suitable location is found, the cellular localization and function of the protein do not seem to change by the presence of the unAA, suggesting that, if carefully considered, this is not a limitation of the approach [30, 33].
Another factor that should be considered is the incorporation of the unAA into endogenous TAG termination sites. Such cases may result in the unspecific labeling of endogenous proteins, which will increase background levels. The ratio between the specific labeling of the POI and the unspecific labeling of other proteins in the cell can be quantified by using in‐gel fluorescence [22, 23, 30, 36, 47]. In addition, the overall background level in cells can be evaluated by measuring the signal intensity after adding all the GCE and click reaction components to the cells—except the TAG‐modified POI [22, 23, 30, 35, 36, 38, 47]. Despite these potential background‐related limitations, GCE‐based labeling demonstrates relatively high signal‐to‐noise ratios (SNRs), which enable quantitative imaging in either fixed or live cells [24, 35, 36].
Adding an in‐frame TAG stop codon can also lead to a premature termination of translation. In fact, the newly introduced tRNA and the cellular translation termination factors (e.g., eRF1) constantly compete with each other over binding to the UAG stop codon in the mRNA. An interaction between eRF1 and the newly introduced stop codon will terminate translation and prevent the incorporation of the unAA. As a result, truncated versions of the POI will be expressed and the efficiency of unAA incorporation will be reduced. Expressing a dominant‐negative version of eRF1 can minimize this effect [48], but an eRF1 mutant is prone to perturb the termination of endogenous protein translation and, therefore, its expression is often avoided. The balance between the incorporation of the unAA and a premature termination appears to depend on the incorporation site, albeit the factors that govern incorporation efficiency remain elusive. Therefore, a traditional GCE‐based labeling experiment should begin by screening for efficient incorporation sites in which the full‐length POI is translated at relatively high levels and truncations are kept to a minimum. Choosing positions near the N terminus of the protein can potentially help to minimize the accumulation of protein truncations, due to the short length of the resulting polypeptide. However, in some cases, introducing a premature stop codon near the 5′ end of the mRNA promotes the re‐initiation of translation from a downstream AUG start codon [49]. To avoid such scenarios, the size of the POI should be verified by western blot analysis during the screening process.
The click reaction
An efficient click reaction requires that the unAA will be incorporated into the POI such that it is accessible to the freely diffusing Tet‐Fl‐dye [19, 35]. This requirement should be thoroughly considered, especially in the case of multicomplex proteins, and care should be taken to ensure that the unAA is accessible in both the monomeric and the multicomplex form, so as to avoid a biased visualization of only a subset of the protein population. Moreover, the addition of a Tet‐Fl‐dye can potentially affect the function of the protein and, therefore, functionality should also be verified in the labeled form.
Once an efficient, suitable labeling site is selected, background fluorescence should be minimized. In that respect, it should be recognized that any unAA present in the cell (not only the one that is incorporated into to the POI) will undergo the click reaction and contribute to background fluorescence. Sources for unAAs that are not bound to the POI include excess of soluble unAAs; unAAs that were incorporated into cellular proteins in response to endogenous TAG (as discussed above); and unAAs that are bound to tRNAs. Indeed, reducing the copy number of tRNA from four to one was shown to improve the SNR in mammalian cells without affecting the efficiency of unAA incorporation, and this strategy is, therefore, recommended for labeling purposes [38]. The unspecific labeling of charged tRNAs also appears to be the source for unspecific nuclear labeling observed in cells labeled via GCE [37, 38]. When labeling proteins outside the nucleus, sequestering unspecific labeling inside the nucleus can actually be an advantage, as it may reduce cytosolic background levels. When nuclear proteins need to be specifically labeled, the nuclear accumulation of tRNA can be avoided by introducing a nuclear export signal to the coding sequence of the orthogonal tRNA synthetase [37]. Extensive washes will minimize background labeling resulting from an excess of unAA [19, 35].
Labeling proteins through click reactions provides the flexibility to use a variety of Fl‐dyes that exhibit different chemical and fluorescence properties. Tailoring the characteristics of the Fl‐dye to the desired imaging application is a major advantage of GCE‐based labeling and can greatly improve the performance of advanced imaging techniques. While some dyes are cell‐permeable, others are not; some dyes may be more suited for labeling certain cellular components; and while some dyes are bright and photostable, others exhibit blinking properties, which are required in SMLM experiments [19, 36]. The cellular permeability and fluorogenicity of various Tet‐Fl‐dyes were recently reported and can be used as a guideline for a rational selection of the Fl‐dye [13, 24].
Notably, the various considerations regarding the incorporation of the unAA and the click reaction make this labeling approach somewhat more challenging than traditional labeling approaches. To overcome these challenges, we recently designed a short (14 AAs long) N‐terminal tag that encodes for the incorporation of an unAA, and demonstrated its applicability for GCE‐based labeling of proteins and organelles [36]. Although using a tag is not as elegant as incorporating the label directly into the POI, this approach provides a calibration‐free, easy‐to‐implement tool that retains most of the advantages associated with GCE‐based labeling.
What are the limitations?
Genetic code expansion‐based labeling relies on the manipulation of the cellular translation machinery, and the physiological consequences of such a manipulation have yet to be carefully considered. First, the cellular response to an exogenous tRNA/tRNA synthetase pair, and the effects on ribosomal activity, has not been characterized. Second, the cellular effects of protein read‐through resulting from the incorporation of an unAA at endogenous TAG codons have not been comprehensively analyzed. Although TAG is the least abundant stop codon in mammalian cells, there are over 25 000 open reading frames in the human genome with an annotated TAG stop codon. Among these are genes encoding for cytoskeleton, cell cycle, and cell adhesion proteins, and it remains largely unknown whether and how the read‐through of these proteins affects their cellular function. Moreover, Arbely and colleagues recently reported that the translation of sequences past the annotated stop codon promotes the aggregation and lysosomal targeting of the C‐terminally extended proteins [50]. It is still to be determined whether this is the fate of cellular proteins that carry a TAG codon in GCE‐modified cells, but this study stresses the need to begin investigating these processes in GCE‐modified cells. Potential cellular phenotypes associated with GCE can also explain why the efficiency of unAA incorporation varies across cell types. This observed variability may arise from different regulatory pathways and checkpoints associated with the origin of the tissue culture cells.
In addition to these global effects, introducing a premature stop codon into the natural AA sequence of the POI may affect cell physiology. Protein truncations—resulting either from an inefficient incorporation of the unAA into the genetic code of the POI or from a re‐initiation of translation at a downstream ATG codon—can affect the function and regulation of the POI. Such protein truncations should be avoided (or at least minimized), and care should be taken to verify that they do not affect the function of the POI or the overall cell physiology.
Despite these potential effects on cell physiology, GCE‐based labeling through click reactions has been successfully applied in several model organisms, including fish, flies, and mice, suggesting that the technique does not severely influence the overall viability of cells (reviewed in Ref. [51]). However, until these potential effects are carefully and systematically characterized, caution should be used when employing GCE‐based labeling to answer questions pertaining to cellular behavior and regulation.
Conclusions and perspectives
Genetic code expansion, together with click chemistry, offers an elegant tool for the labeling of proteins in both fixed‐ and live‐cell imaging applications. Previous studies indicate that this approach is suitable for the live‐cell imaging of both intra‐ and extracellular proteins and that it can be used with—and considerably contribute to—various advanced quantitative techniques, including photomanipulation, single‐molecule imaging, and super‐resolution techniques. However, care should be taken to follow optimized protocols, to validate the function of the POI, and to evaluate labeling specificity and efficiency by using relevant controls. Improving the performance and robustness of directly incorporated fluorescent unAAs for the visualization of proteins in live cells can overcome some of the background fluorescence issues currently associated with the approach. In the meantime, considerable time and effort can be saved by using previously published unAA incorporation positions in specific POIs, by adding the calibrated short peptide tag, and by adopting established protocols. Multicolor GCE‐based labeling is currently limited due to the challenge of exploiting two different codons for the incorporation of the unAAs. New concepts for genetic code engineering using four‐nucleotide codons instead of the traditional three‐nucleotide codons have been recently demonstrated in bacteria [52]. Although applying this approach to mammalian cell labeling is not trivial, such strategies can pave the way toward a multicolor, site‐specific labeling of cellular mammalian proteins. Lastly, the GCE‐based labeling technique requires the introduction of exogenous factors to the cellular translation machinery, and the effect that these factors may have on cellular physiology needs to be evaluated. Meanwhile, biophysical studies can be conducted to investigate the spatiotemporal organization of proteins in cells using an essentially ‘tag‐free’ labeling approach.
Conflict of interest
The author declare no conflict of interest.
Acknowledgements
I thank Dr Eyal Arbely and the members of his laboratory, as well as current and past members of the Elia laboratory, for their contribution to establishing the GCE‐based labeling system in our laboratory. I also thank Eyal Arbely, Dikla Nachmias, Andres Konig, and Shai Adar for their critical reading of the manuscript. The project has received funding from the European Research Council (ERC) Under The European Union's Horizon 2020 Research (starting grant, Natalie Elia).
References
- 1. Lee KJ, Kang D & Park HS (2019) Site‐specific labeling of proteins using unnatural amino acids. Mol Cells 42, 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lang K & Chin JW (2014) Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- 3. Nikic I & Lemke EA (2015) Genetic code expansion enabled site‐specific dual‐color protein labeling: superresolution microscopy and beyond. Curr Opin Chem Biol 28, 164–173. [DOI] [PubMed] [Google Scholar]
- 4. Gupta K, Toombes GE & Swartz KJ (2019) Exploring structural dynamics of a membrane protein by combining bioorthogonal chemistry and cysteine mutagenesis. Elife 8, e50776. 10.7554/eLife.50776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beatty KE, Liu JC, Xie F, Dieterich DC, Schuman EM, Wang Q & Tirrell DA (2006) Fluorescence visualization of newly synthesized proteins in mammalian cells. Angew Chem Int Ed Engl 45, 7364–7367. [DOI] [PubMed] [Google Scholar]
- 6. Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA & Schuman EM (2010) In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci 13, 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dieterich DC, Link AJ, Graumann J, Tirrell DA & Schuman EM (2006) Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc Natl Acad Sci USA 103, 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hinz FI, Dieterich DC, Tirrell DA & Schuman EM (2012) Non‐canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci 3, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nodling AR, Spear LA, Williams TL, Luk LYP & Tsai YH (2019) Using genetically incorporated unnatural amino acids to control protein functions in mammalian cells. Essays Biochem 63, 237–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chatterjee A, Guo J, Lee HS & Schultz PG (2013) A genetically encoded fluorescent probe in mammalian cells. J Am Chem Soc 135, 12540–12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Charbon G, Brustad E, Scott KA, Wang J, Lobner‐Olesen A, Schultz PG, Jacobs‐Wagner C & Chapman E (2011) Subcellular protein localization by using a genetically encoded fluorescent amino acid. ChemBioChem 12, 1818–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Charbon G, Wang J, Brustad E, Schultz PG, Horwich AL, Jacobs‐Wagner C & Chapman E (2011) Localization of GroEL determined by in vivo incorporation of a fluorescent amino acid. Bioorg Med Chem Lett 21, 6067–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oliveira BL, Guo Z & Bernardes GJL (2017) Inverse electron demand Diels‐Alder reactions in chemical biology. Chem Soc Rev 46, 4895–4950. [DOI] [PubMed] [Google Scholar]
- 14. Alamudi SH, Satapathy R, Kim J, Su D, Ren H, Das R, Hu L, Alvarado‐Martinez E, Lee JY, Hoppmann C et al. (2016) Development of background‐free tame fluorescent probes for intracellular live cell imaging. Nat Commun 7, 11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takakura H, Zhang Y, Erdmann RS, Thompson AD, Lin Y, McNellis B, Rivera‐Molina F, Uno SN, Kamiya M, Urano Y et al. (2017) Long time‐lapse nanoscopy with spontaneously blinking membrane probes. Nat Biotechnol 35, 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang H, English BP, Hazan RB, Wu P & Ovryn B (2015) Tracking surface glycans on live cancer cells with single‐molecule sensitivity. Angew Chem Int Ed Engl 54, 1765–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mateos‐Gil P, Letschert S, Doose S & Sauer M (2016) Super‐resolution imaging of plasma membrane proteins with click chemistry. Front Cell Dev Biol 4, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA & Bertozzi CR (2007) Copper‐free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci USA 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikic I, Kang JH, Girona GE, Aramburu IV & Lemke EA (2015) Labeling proteins on live mammalian cells using click chemistry. Nat Protoc 10, 780–791. [DOI] [PubMed] [Google Scholar]
- 20. Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM & Chin JW (2012) Genetic encoding of bicyclononynes and trans‐cyclooctenes for site‐specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels‐Alder reactions. J Am Chem Soc 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kozma E, Nikic I, Varga BR, Aramburu IV, Kang JH, Fackler OT, Lemke EA & Kele P (2016) Hydrophilic trans‐cyclooctenylated noncanonical amino acids for fast intracellular protein labeling. ChemBioChem 17, 1518–1524. [DOI] [PubMed] [Google Scholar]
- 22. Peng T & Hang HC (2016) Site‐specific bioorthogonal labeling for fluorescence imaging of intracellular proteins in living cells. J Am Chem Soc 138, 14423–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uttamapinant C, Howe JD, Lang K, Beranek V, Davis L, Mahesh M, Barry NP & Chin JW (2015) Genetic code expansion enables live‐cell and super‐resolution imaging of site‐specifically labeled cellular proteins. J Am Chem Soc 137, 4602–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Beliu G, Kurz AJ, Kuhlemann AC, Behringer‐Pliess L, Meub M, Wolf N, Seibel J, Shi ZD, Schnermann M, Grimm JB et al. (2019) Bioorthogonal labeling with tetrazine‐dyes for super‐resolution microscopy. Commun Biol 2, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lukinavicius G, Reymond L, Umezawa K, Sallin O, D'Este E, Gottfert F, Ta H, Hell SW, Urano Y & Johnsson K (2016) Fluorogenic probes for multicolor imaging in living cells. J Am Chem Soc 138, 9365–9368. [DOI] [PubMed] [Google Scholar]
- 26. Lukinavicius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Correa IR Jr, Luo ZG et al. (2013) A near‐infrared fluorophore for live‐cell super‐resolution microscopy of cellular proteins. Nat Chem 5, 132–139. [DOI] [PubMed] [Google Scholar]
- 27. Uno SN, Kamiya M, Yoshihara T, Sugawara K, Okabe K, Tarhan MC, Fujita H, Funatsu T, Okada Y, Tobita S et al. (2014) A spontaneously blinking fluorophore based on intramolecular spirocyclization for live‐cell super‐resolution imaging. Nat Chem 6, 681–689. [DOI] [PubMed] [Google Scholar]
- 28. Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott‐Schwartz J, Liu Z, Lionnet T et al. (2016) Bright photoactivatable fluorophores for single‐molecule imaging. Nat Methods 13, 985–988. [DOI] [PubMed] [Google Scholar]
- 29. van de Linde S, Heilemann M & Sauer M (2012) Live‐cell super‐resolution imaging with synthetic fluorophores. Annu Rev Phys Chem 63, 519–540. [DOI] [PubMed] [Google Scholar]
- 30. Sakin V, Hanne J, Dunder J, Anders‐Osswein M, Laketa V, Nikic I, Krausslich HG, Lemke EA & Muller B (2017) A versatile tool for live‐cell imaging and super‐resolution nanoscopy studies of HIV‐1 Env distribution and mobility. Cell Chem Biol 24, 635–645, e5. [DOI] [PubMed] [Google Scholar]
- 31. Lang K & Chin JW (2013) Fluorescent imaging: shining a light into live cells. Nat Chem 5, 81–82. [DOI] [PubMed] [Google Scholar]
- 32. Serfling R, Seidel L, Bock A, Lohse MJ, Annibale P & Coin I (2019) Quantitative single‐residue bioorthogonal labeling of G protein‐coupled receptors in live cells. ACS Chem Biol 14, 1141–1149. [DOI] [PubMed] [Google Scholar]
- 33. Neubert F, Beliu G, Terpitz U, Werner C, Geis C, Sauer M & Doose S (2018) Bioorthogonal click chemistry enables site‐specific fluorescence labeling of functional NMDA receptors for super‐resolution imaging. Angew Chem Int Ed Engl 57, 16364–16369. [DOI] [PubMed] [Google Scholar]
- 34. Konig AI, Sorkin R, Alon A, Nachmias D, Dhara K, Brand G, Yifrach O, Arbely E, Roichman Y & Elia N (2020) Live cell single molecule tracking and localization microscopy of bioorthogonally labeled plasma membrane proteins. Nanoscale 12, 3236–3248. [DOI] [PubMed] [Google Scholar]
- 35. Schvartz T, Aloush N, Goliand I, Segal I, Nachmias D, Arbely E & Elia N (2017) Direct fluorescent‐dye labeling of alpha‐tubulin in mammalian cells for live cell and superresolution imaging. Mol Biol Cell 28, 2747–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Segal I, Nachmias D, Konig A, Alon A, Arbely E & Elia N (2020) A straightforward approach for bioorthogonal labeling of proteins and organelles in live mammalian cells, using a short peptide tag. BMC Biol 18, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nikic I, Estrada Girona G, Kang JH, Paci G, Mikhaleva S, Koehler C, Shymanska NV, Ventura Santos C, Spitz D & Lemke EA (2016) Debugging eukaryotic genetic code expansion for site‐specific click‐PAINT super‐resolution microscopy. Angew Chem Int Ed Engl 55, 16172–16176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aloush N, Schvartz T, Konig AI, Cohen S, Brozgol E, Tam B, Nachmias D, Ben‐David O, Garini Y, Elia N et al. (2018) Live cell imaging of bioorthogonally labelled proteins generated with a single pyrrolysine tRNA gene. Sci Rep 8, 14527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das DK, Govindan R, Nikic‐Spiegel I, Krammer F, Lemke EA & Munro JB (2018) Direct visualization of the conformational dynamics of single influenza hemagglutinin trimers. Cell 174, 926–937, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vreja IC, Nikic I, Gottfert F, Bates M, Krohnert K, Outeiro TF, Hell SW, Lemke EA & Rizzoli SO (2015) Super‐resolution microscopy of clickable amino acids reveals the effects of fluorescent protein tagging on protein assemblies. ACS Nano 9, 11034–11041. [DOI] [PubMed] [Google Scholar]
- 41. Nikic I, Plass T, Schraidt O, Szymanski J, Briggs JA, Schultz C & Lemke EA (2014) Minimal tags for rapid dual‐color live‐cell labeling and super‐resolution microscopy. Angew Chem Int Ed Engl 53, 2245–2249. [DOI] [PubMed] [Google Scholar]
- 42. Saal KA, Richter F, Rehling P & Rizzoli SO (2018) Combined use of unnatural amino acids enables dual‐color super‐resolution imaging of proteins via click chemistry. ACS Nano 12, 12247–12254. [DOI] [PubMed] [Google Scholar]
- 43. Klein T, Loschberger A, Proppert S, Wolter S, van de Linde S & Sauer M (2011) Live‐cell dSTORM with SNAP‐tag fusion proteins. Nat Methods 8, 7–9. [DOI] [PubMed] [Google Scholar]
- 44. Wombacher R, Heidbreder M, van de Linde S, Sheetz MP, Heilemann M, Cornish VW & Sauer M (2010) Live‐cell super‐resolution imaging with trimethoprim conjugates. Nat Methods 7, 717–719. [DOI] [PubMed] [Google Scholar]
- 45. Jones SA, Shim SH, He J & Zhuang X (2011) Fast, three‐dimensional super‐resolution imaging of live cells. Nat Methods 8, 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng Y, Addy PS, Mukherjee R & Chatterjee A (2017) Defining the current scope and limitations of dual noncanonical amino acid mutagenesis in mammalian cells. Chem Sci 8, 7211–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Husen LS, Schedin‐Weiss S, Trung MN, Kazmi MA, Winblad B, Sakmar TP, Elsasser SJ & Tjernberg LO (2019) Dual bioorthogonal labeling of the amyloid‐beta protein precursor facilitates simultaneous visualization of the protein and its cleavage products. J Alzheimers Dis 72, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmied WH, Elsasser SJ, Uttamapinant C & Chin JW (2014) Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. J Am Chem Soc 136, 15577–15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cohen S, Kramarski L, Levi S, Deshe N, Ben David O & Arbely E (2019) Nonsense mutation‐dependent reinitiation of translation in mammalian cells. Nucleic Acids Res 47, 6330–6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kramarski L & Arbely E (2020) Translational read‐through promotes aggregation and shapes stop codon identity. Nucleic Acids Res 48, 3747–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brown W, Liu J & Deiters A (2018) Genetic code expansion in animals. ACS Chem Biol 13, 2375–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fredens J, Wang K, de la Torre D, Funke LFH, Robertson WE, Christova Y, Chia T, Schmied WH, Dunkelmann DL, Beranek V et al. (2019) Total synthesis of Escherichia coli with a recoded genome. Nature 569, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]