

Abstract
Background and Aim
The incidence of non‐alcoholic steatohepatitis (NASH)‐related hepatocellular carcinoma (HCC) is progressively increasing. However, the pathophysiology and etiology of NASH progression to HCC are unknown. We hypothesized that steatosis was the key factor in NASH‐related hepatocarcinogenesis and aimed to evaluate the effects of long‐term liver X receptor (LXR) agonist stimulation on hepatic steatosis induced by a high‐fat diet and oxidative stress.
Methods
We used an LXR agonist (T0901317) and CCl4 to induce hepatic steatosis and oxidative stress, respectively. C57BL/6 mice fed with a high‐fat diet were treated with either T0901317 + CCl4 (T09 + CCl4 group) or CCl4 alone (CCl4 group). T0901317 (2.5 mg/kg) and CCl4 (0.1 mL/kg) were intraperitoneally administered twice weekly for 24 weeks.
Results
The liver‐to‐body weight ratio was significantly higher in the T09 + CCl4 group than in the CCl4 group. Mice in the T09 + CCl4 group exhibited abnormal lipid metabolism and NASH‐like histopathological features. Additionally, all mice in the T09 + CCl4 group developed liver tumors diagnosed as well‐differentiated HCC. The genes identified via microarray analysis were related to NASH and HCC development.
Conclusions
By combining long‐term LXR agonist stimulation with oxidative stress and a high‐fat diet, we successfully reproduced liver conditions in mice similar to those in humans with NASH and progression to HCC. Our results provide new insight into NASH‐related HCC progression and therapy.
Keywords: Liver X receptor, NASH‐related HCC, Non‐alcoholic steatohepatitis
Introduction
Because of the growing epidemic of obesity and diabetes, the number of patients suffering from non‐alcoholic steatohepatitis (NASH) is increasing. Correspondingly, the percentage of hepatocellular carcinoma (HCC) cases related to NASH is increasing exponentially, and NASH may become a leading etiology of HCC. 1 However, the pathophysiology of NASH in the progression to HCC is unknown. 2 We previously reported the establishment of a novel NASH model by the administration of T0901317 in a high‐fat diet‐induced and CCl4‐induced mouse model. 3 T0901317 is a liver X receptor (LXR) agonist for treating hyperlipidemia; however, severe hepatic steatosis is a side effect. 4 The pathological features of the novel NASH model resembled those of human NASH, and the severity of steatosis was remarkable. We thus hypothesized that lipid deposition plays an important role in NASH‐related hepatocarcinogenesis. Lipid deposition is the result of abnormal lipid metabolism in the liver. LXR is a nuclear receptor and plays an important role in lipid metabolism. LXR is involved in the control of major metabolic pathways for cholesterol homeostasis and lipogenesis. Indeed, in a human NASH patient study, LXR expression was correlated with the degree of hepatic fat deposition, hepatic inflammation, and fibrosis. 5 As is known from the literature, both hepatic steatosis and fibrosis represent independent risk factors for HCC development. 6 , 7 Additionally, insulin resistance results in increased levels of toxic metabolites and inflammatory cytokines in the tissue environment. Hepatocytes with increased inflammatory cytokine levels may contribute to HCC by activating the AKT signaling pathway and could have prognostic value in HCC. 8 , 9 To evaluate the long‐term effect of steatosis, we combined a low dose of T0901317 with a high‐fat diet and a low dose of CCl4 for 24 weeks. The aim of this study was to investigate the effects of long‐term stimulation by T0901317, thereby revealing the influence of steatosis on the progression of NASH to HCC.
Methods
Animals
Six‐week‐old male C57BL/6 mice were purchased from Charles River Laboratories (Yokohama, Japan) and were acclimated for 1 week before the start of the experiments. All mice were housed in a temperature‐controlled, humidity‐controlled, and ventilation‐controlled vivarium and maintained on a 12‐h light–dark cycle under specific pathogen‐free conditions. All mice were allowed ad libitum access to water and a high‐fat diet (5.24 kcal/g, 60 kcal% fat and 20 kcal% carbohydrate; D12492, Research Diets, New Brunswick, NJ, USA). Mice were divided into two groups (12 animals each): (i) Mice in the T09 + CCl4 group were intraperitoneally injected with T0901317 (Cayman Chemical Co., Ann Arbor, MI, USA) and CCl4 (Wako Pure Chemical Industries, Ltd., Osaka, Japan) solubilized in corn oil, and (ii) mice in the CCl4 group were intraperitoneally injected with CCl4 alone. All mice in both groups were administered with intraperitoneal (i.p.) injections twice weekly. The CCl4 dose was 0.1 mL/kg (CCl4 : corn oil = 1:39). The T0901317 dose was 2.5 mg/kg (T0901317 : Dimethyl sulfoxide, phosphate‐buffered saline = 1:99). Mice were weighed twice weekly and periodically assessed for signs of disease or morbidity. Mice were sacrificed by exsanguination under isoflurane anesthesia. Blood samples were collected from the inferior vena cava and centrifuged at 1200 g for 10 min to isolate serum. Each sample was stored at −80 °C until analysis. The liver was quickly removed and immediately fixed in 10% neutral buffered formalin for further histological examination. Animal experiments were carried out humanely after approval was obtained from the Institutional University Experiment Committee of the University of Tsukuba and in accordance with the Regulations for Animal Experiments of the university and the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology.
Histological analysis
Fixed liver tissues were processed and embedded in paraffin using standard methods. Liver tissues were assessed in 2‐μm paraffin sections stained with hematoxylin and eosin (HE) to assess the extent of steatosis, inflammation, hepatocellular ballooning, and nuclear atypia. Fibrosis was evaluated by Masson's trichrome (MT) staining. Lipid droplets in hepatocytes were evaluated by Oil Red O staining. Immunohistochemical staining for F4/80 (ab100790; Abcam, Cambridge, MA, USA), 4‐HNE (ab46545), CD34 (ab81289), AFP (ab46799), and glypican 3 (ab66596) was performed. Liver specimens were evaluated by an experienced pathologist to assess the severity of hepatocellular steatosis, inflammation, and ballooning using the following non‐alcoholic fatty liver disease (NAFLD) activity scores. To assess hepatocellular steatosis, specimens were classified as grade 0–3 according to the percentage of the hepatic parenchyma occupied by steatosis (grade 0: < 5%; grade 1: 6–33%; grade 2: 34–66%; and grade 3: more than 66%). To assess inflammatory cell infiltration, specimens were classified as grade 0–3 according to the number of foci per 200× field (grade 0: 0; grade 1: 1–2; grade 2: 3–4; and grade 3: more than 4). To assess hepatocellular ballooning, specimens were classified as grade 0–2 (grade 0: no balloon cells; grade 1: few balloon cells; and grade 2: many cells/prominent ballooning). The area positive of Oil Red O staining was measured using Image J software (U.S. National Institutes of Health, Bethesda, Maryland, USA). 10
Insulin resistance
To assess insulin resistance, we performed an insulin tolerance test (ITT) and calculated the homeostasis model assessment of insulin resistance (HOMA‐R) score. 11
Insulin tolerance test
Mice were fasted for 6 h before the injection of insulin (Humulin R, 0.75 U/kg, i.p.) (Eli Lilly Co., Kobe, Japan). The blood glucose level in tail vein blood was measured 0, 30, 60, 90, and 120 min after insulin injection.
Homeostasis model assessment of insulin resistance
As another insulin resistance indicator, the HOMA‐R score was calculated as follows: HOMA‐R score = (fasting plasma glucose value × fasting serum insulin value)/405.
The serum level of insulin was measured with an ultrasensitive mouse insulin ELISA kit (Morinaga Institute of Biological Science, Kanagawa, Japan).
Western blot analysis
Liver tissue was collected and immediately frozen at −30 °C until use. Total protein was extracted from liver tissue with a Minute Total Protein Extraction Kit for Animal Cultured Cells and Tissues (#SD‐001/SN‐002; Invent Biotechnologies, Inc., Eden Prairie, MN, USA). Protein concentrations were estimated using densitometry. Samples were boiled at 95 °C for 3 min, and 15‐μL aliquots of each sample were loaded on 10% and 12% sodium dodecylsulfate–polyacrylamide gel electrophoresis gels and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). Anti‐FASN (#3180), anti‐pGSK‐3b (#9336), anti‐ERK (#9102), anti‐cyclin D1 (#2922), anti‐p38 MAPK (#9211), and anti‐GAPDH (#2118) (all from Cell Signaling Technology, Beverly, MA, USA) primary antibodies were used. A horseradish peroxidase‐conjugated goat anti‐rabbit secondary antibody was purchased from Zymed Laboratories (San Francisco, CA, USA).
Gene expression analysis
Liver tissue samples were freshly collected and immediately frozen at 30 °C until use. The frozen liver samples were homogenized, and total RNA was isolated from whole cells using a NucleoSpin RNA kit (Takara Bio, Inc., Otsu, Japan). The RNA concentrations were determined by measuring the absorbance at 260/280 nm with a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Inc., Wilmington, DE, USA). The synthesis of complementary DNA was performed using PrimeScript RT Master Mix (Takara, Shiga, Japan). The primers for real‐time polymerase chain reaction (PCR) were designed using primer express software for Real‐Time PCR ver. 3.0 (Applied Biosystems, Inc., Foster City, CA, USA) based on the sequences available in GenBank. The primers were purchased from Takara Bio, Inc. The Lpl primer sequences were 5′‐AGAGGCTATAGCTGGGAGCAGAAAC‐3′ and 5′‐GCAAGGGCTAACATTCCAGCA‐3′. The Lepr primer sequences were 5′‐TCATGTGCCGGTACCCAGAG‐3′ and 5′‐ACCTAAGGGTGGATCGGGTTTC‐3′, and the Src primer sequences were 5′‐CAGCGGCGGTTTCTACATCA‐3′ and 5′‐GCTTGGATGTGGGACATACGG‐3′. The glyceraldehyde 3‐phosphate dehydrogenase (Gapdh) primer sequences were 5′‐TGTGTCCGTCGTGGATCTGA‐3′ and 5′‐TTGCTGTTGAAGTCGCAGGAG‐3′. Gapdh was used as an endogenous control. Quantitative real‐time PCR was conducted with 400‐nM primers, 25 ng of cDNA template, and TB Green Premix Ex Tax II (Takara, RR820A) in an Applied Biosystems 7300 Real‐Time PCR system.
Microarray analysis
Frozen liver samples were homogenized, and total RNA was isolated from whole cells using a NucleoSpin RNA kit (Takara Bio, Inc.). The RNA concentration was determined by the absorbance at 260/280 nm with a NanoDrop Spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA was synthesized using AMV Reverse Transcriptase (Promega Corp., Madison, WI, USA) and random primers (Takara Bio, Inc.). Briefly, a mixture of 1‐mM dNTPs (Fermentas Life Sciences, Inc., Burlington, ON, Canada), 0.025 μg/mL of random primers, 0.25 U/mL of reverse transcriptase, and 500 ng of total RNA was incubated at 30 °C for 10 min, 37 °C for 60 min, 95 °C for 5 min, and 4 °C prior to storage at −80 °C. DNA microarray analysis was conducted using RNA samples isolated from both groups. Labeled cDNA was synthesized from 100 ng of total RNA using a GeneChip 3′ IVT Plus Reagent Kit (Affymetrix, Inc., Santa Clara, CA, USA) according to the manufacturer's recommended protocol. Fragmented and labeled cDNA (600 ng) was hybridized to a SurePrint G3 Mouse Gene Expression ver. 2.0 8×60K Microarray Kit (Agilent, Inc., Santa Clara, CA, USA) for 17 h at 65 °C. The strips were washed and stained using a Low Input Quick Amp Labeling Kit (Agilent, Inc.) and scanned using a DNA microarray scanner (Agilent, Inc.). Probe‐level analysis, including background subtraction and quantile normalization, was conducted using the percentile shift algorithm with GeneSpring GX 14.5 (Agilent, Inc.). The gene expression profile of the T09 + CCl4 group was compared with that of the CCl4 group. Genes with a change in expression of greater than twofold were classified as upregulated genes, and those with a change in expression of less than 0.5‐fold were classified as downregulated genes.
Statistical analysis
All data are expressed as the mean ± standard deviation. Statistical analysis was conducted using Graph Pad Prism 8 (Graphpad Software, Inc., San Diego, CA). The Mann–Whitney U‐test was used for comparisons between two groups. P‐values less than 0.05 were considered significant. Intergroup differences were compared by one‐way analysis of variance with Tukey's post hoc test.
Results
Body and liver weight
Figure 1a indicates the liver‐to‐body weight ratio and body weight of the mice. The liver weight and liver‐to‐body weight ratio were significantly higher in the T09 + CCl4 group than in the CCl4 group (Fig. S1: liver weight at 24 weeks). The final body weight was increased in both groups relative to the initial body weight, although mice in the T09 + CCl4 group exhibited a gradual reduction in body weight gain after 20 weeks.
Figure 1.
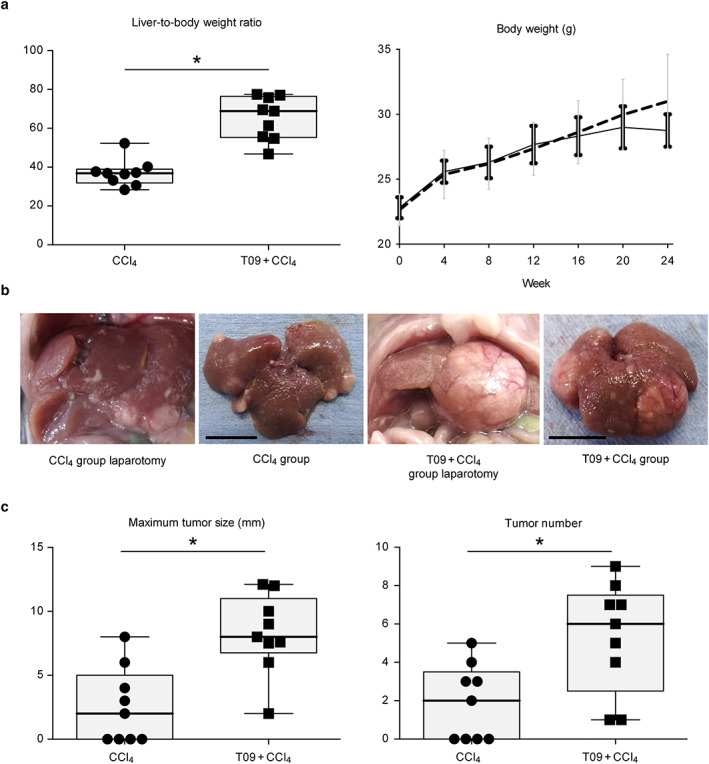
Liver‐to‐body weight ratio and gross appearance of livers and tumors. (a) Body weight and liver‐to‐body weight ratio (n = 9). Change in body weight over 24 weeks (n = 6).  , CCl4;
, CCl4;  , T09 + CCl4. (b) Photographs of livers and tumors. Left panel, photographs taken at laparotomy; right panel, photographs taken immediately after removal (bar length = 1 cm). (c) Maximum tumor size and tumor number (n = 9). The results are expressed as the mean ± SD and were compared with the Mann–Whitney U‐test. [Color figure can be viewed at wileyonlinelibrary.com]
, T09 + CCl4. (b) Photographs of livers and tumors. Left panel, photographs taken at laparotomy; right panel, photographs taken immediately after removal (bar length = 1 cm). (c) Maximum tumor size and tumor number (n = 9). The results are expressed as the mean ± SD and were compared with the Mann–Whitney U‐test. [Color figure can be viewed at wileyonlinelibrary.com]
High efficiency of tumorigenesis
Figure 1b shows the gross appearance of tumors from both groups. Mice in both groups developed tumors; however, mice in the CCl4 group had fewer and smaller tumors. In addition, all mice in the T09 + CCl4 group developed tumors. Figure 1c shows the increased efficiency of tumorigenesis in the T09 + CCl4 group relative to that in the CCl4 group. In addition, the maximum size and number of tumors were significantly different between the groups.
T09 + CCl4 group mice developed fatty liver, steatohepatitis, and fibrosis in the background liver tissue
Figure 2 shows the liver histological findings in both groups. The normal liver structure was disrupted in the T09 + CCl4 group. Based on the Oil Red O‐stained and MT‐stained liver tissue slides, a greater extent of steatosis and more severe fibrosis were observed in the T09 + CCl4 group than in the CCl4 group (Fig. 2a). Mice in the T09 + CCl4 group developed macrovesicular hepatic steatosis, ballooned hepatocytes with Mallory–Denk bodies, lobular inflammation, and fibrosis (Fig. 2b). In addition, on the Oil Red O‐stained liver slides, the Oil Red O‐positive area indicating steatosis, as calculated by imagej software, was significantly higher in the T09 + CCl4 group than in the CCl4 group (Fig. 2c). Oxidative stress was measured by immunostaining for 4‐HNE, which is a marker of lipid peroxidation. Oxidative stress in the CCl4 group was observed in zone 1. In contrast, oxidative stress in the T09 + CCl4 group was observed in the central vein area (zone 3) (Fig. 2d). We also assessed the severity of fibrosis by calculating the blue‐stained area on the MT‐stained liver slides and found that the area of fibrosis was significantly higher in the T09 + CCl4 group than in the CCl4 group. Clinically, the severity of NASH is assessed by the NAFLD activity score. In this study, the average NAFLD activity score was significantly higher in the T09 + CCl4 group than in the CCl4 group (Fig. 2c).
Figure 2.
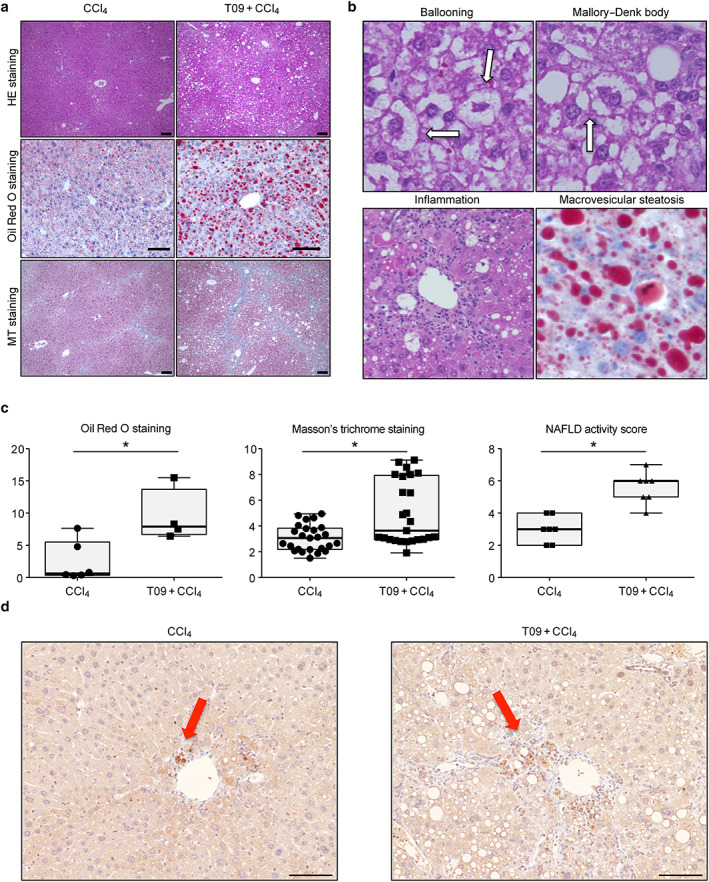
Histopathological features and typical scores of non‐alcoholic steatohepatitis. (a) Hematoxylin and eosin (HE), Oil Red O, and Masson's trichrome (MT) staining of tissues from representative mice in the CCl4 and T09 + CCl4 groups. Scale bar = 100 μm. (b) Representative HE staining of liver tissue from the T09 + CCl4 group depicting the individual components of steatohepatitis—ballooning, Mallory–Denk bodies, inflammation, and macrovesicular steatosis. (c) Quantification of histological scores for steatosis, fibrosis, and non‐alcoholic fatty liver disease (NAFLD) activity. The results are expressed as the mean ± SD and were compared with the Mann–Whitney U‐test. (a) Oil Red O staining (n = 5). (b) MT staining (n = 5). (c) NAFLD activity score (n = 7). (d) Representative 4‐HNE immunostaining of liver tissue from the T09 + CCl4 and CCl4 groups. [Color figure can be viewed at wileyonlinelibrary.com] [Color figure can be viewed at wileyonlinelibrary.com]
Abnormal lipid metabolism and insulin resistance
To assess the severity of lipid metabolism abnormalities in the T09 + CCl4 group, we performed histological F4/80 immunostaining and evaluated FASN expression by Western blotting. Figure 3a shows the crown‐like structures of inflammatory cells in abdominal adipose tissue and liver tissue. Western blot analysis of FASN, a regulator of lipid metabolism, showed significantly upregulated Fasn expression in the T09 + CCl4 group compared with FASN expression in the CCl4 group (Fig. 3b). Insulin resistance was assessed by an ITT and the HOMA‐R score and was compared among the three groups (the T09 + CCl4, CCl4 and normal chow groups). The glucose level at 30, 60, 90, and 120 min after insulin injection was significantly higher in the T09 + CCl4 group than in the normal chow group (Fig. 3c). Additionally, the area under the curve in the T09 + CCl4 group was significantly higher than that in the normal chow group. We also assessed insulin resistance by the HOMA‐R score. The HOMA‐R score in the T09 + CCl4 group was significantly higher than that in the normal chow group. Moreover, analysis of the pGSK‐3 protein level showed significant differences among the groups (Fig. 3d).
Figure 3.
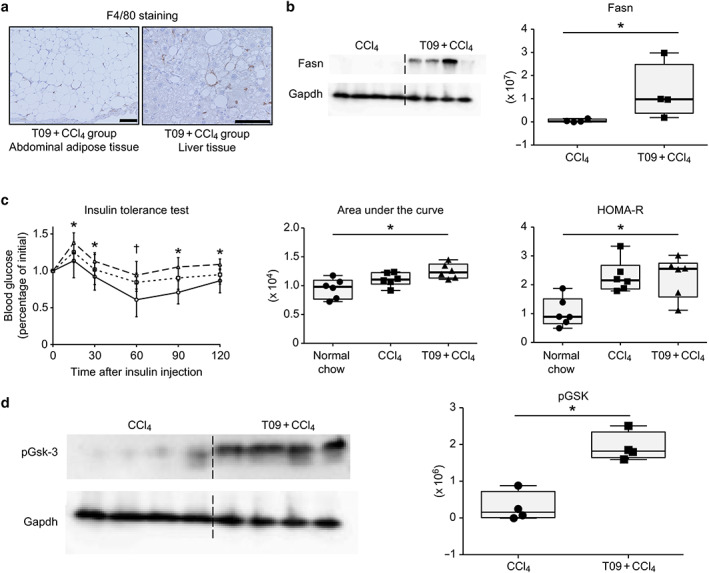
Abnormal lipid metabolism and insulin resistance. (a) Representative F4/80 immunostaining of adipose tissue and liver tissue from the T09 + CCl4 group. (b) Fatty acid synthetase (Fasn) protein expression in the CCl4 and T09 + CCl4 groups. (c) Insulin tolerance test, area under the curve, and homeostasis model assessment of insulin resistance (HOMA‐R) score.  , T09 + CCl4;
, T09 + CCl4;  , CCl4;
, CCl4;  , normal chow. (d) Phospho‐glycogen synthetase kinase 3 (pGsk‐3) protein expression in the CCl4 and T09 + CCl4 groups. The results are expressed as the mean ± SD and were compared with the Mann–Whitney U‐test. Scale bars: 100 μm. [Color figure can be viewed at wileyonlinelibrary.com]
, normal chow. (d) Phospho‐glycogen synthetase kinase 3 (pGsk‐3) protein expression in the CCl4 and T09 + CCl4 groups. The results are expressed as the mean ± SD and were compared with the Mann–Whitney U‐test. Scale bars: 100 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Pathological findings in tumors from the T09 + CCl4 group
Figure 4a shows the pathological findings from HE, CD34, AFP, and glypican 3 staining of tumors from the T09 + CCl4 group. HE staining of tumors revealed steatohepatitic features, considerable cellular and nuclear pleomorphism, and atypical mitoses. Sinusoidal capillarization was ascertained in tumors stained for CD34. Additionally, tumors from the T09 + CCl4 group exhibited positive staining for AFP and glypican 3.
Figure 4.
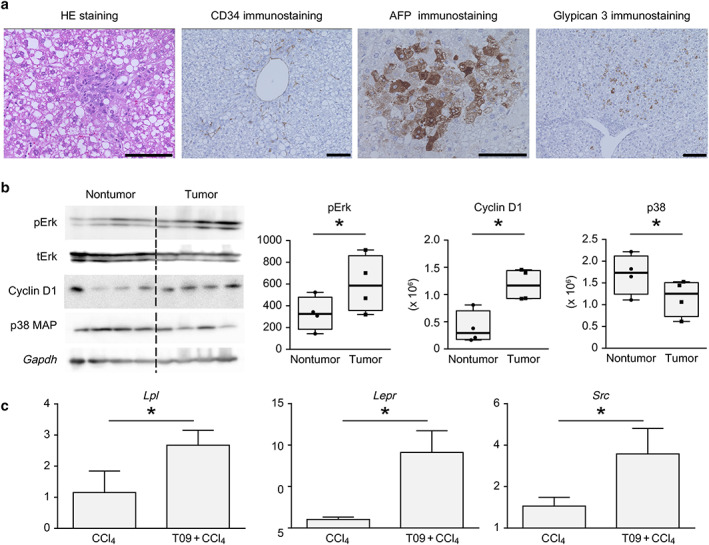
Pathological findings and analysis of protein levels in tumors. (a) Representative hematoxylin and eosin (HE) staining and CD34, AFP, and glypican 3 immunostaining in tumors from the T09 + CCl4 group. (b) pErk, cyclin D1, and p38 MAP protein levels in nontumor and tumor tissues from the T09 + CCl4 group. The results are expressed as the mean ± SD and were compared with the Mann–Whitney U‐test. P < 0.05 for the comparison of nontumor tissue versus tumor tissue. Scale bars: 100 μm. (c) Upregulated expression of genes involved in non‐alcoholic steatohepatitis‐related carcinogenesis, shown by microarray analysis. (a) mRNA expression of Lpl in the liver. (b) mRNA expression of Lepr in the liver. (c) mRNA expression of Src in the tumor. The data are expressed as the mean ± SD (n = 5). P < 0.05 versus the CCl4 group. [Color figure can be viewed at wileyonlinelibrary.com]
Measurement of tumor formation‐related protein levels
Figure 4b shows the results of the analysis of the pERK, tERK, cyclin D1, and p38 MAPK levels in nontumor tissue and tumor tissue from the T09 + CCl4 group. Figure 4b shows the increased levels of proliferation‐related and cell cycle‐related proteins. Furthermore, the expression of a stress response‐related protein was suppressed in tumor tissue.
Microarray analysis comparing the CCl4 group with the T09 + CCl4 group
Tables 1, 2, 3, 4 show the results of the microarray analysis and the Ingenuity Pathway Analysis. The results in the disease and disorders category for the background liver tissue between the two groups indicated inflammatory and connective tissue disorders related to fibrosis in the liver (Table 1). In addition, the hepatoxicity results in the top toxicity function analysis suggested that all index items listed were related to pathological conditions associated with NASH (Table 1). Table 2 shows the results of the gene expression analysis of background liver tissue between the two groups. Table 3 indicates the results of the gene expression analysis of tumor tissue and adjacent non tumor tissue in T09 + CCl4 group. The expression of Cd5l, which has been reported to be a promoter gene in human HCC, was upregulated in the tumor tissue. Lpl, a mediator of free fatty acid metabolism, and Ucp2, a regulator of energy metabolism, are considered to be related to human NASH; indeed, the expression of these genes was upregulated in the T09 + CCl4 group. In addition, the expression of apoptosis suppressor genes, such as Src and Fgr, was upregulated. Table 4 indicates the results of the gene expression analysis of tumor tissue in both group. The expression of Src and Fam83e, which are involved in Raf1/Ampk signaling, was upregulated. In addition, the expression of NASH‐related genes, such as Lpl and Ucp2, was upregulated.
TABLE 1.
Microarray analysis of genes related to NASH and NASH‐associated HCC: The disease and disorders category and top toxicity functions of hepatotoxicity were detected by IPA
| P‐value range | No. of molecules | |
|---|---|---|
| Disease and disorders | ||
| Inflammatory response | 3.02 × 10−6 to 3.93 × 10−27 | 278 |
| Immunological disease | 1.95 × 10−6 to 1.94 × 10−22 | 270 |
| Organismal injury and abnormalities | 2.37 × 10−6 to 4.18 × 10−20 | 517 |
| Connective tissue disorders | 9.50 × 10−7 to 3.38 × 10−16 | 126 |
| Inflammatory disease | 1.11 × 10−6 to 3.38 × 10−16 | 199 |
| Top toxicity functions of hepatotoxicity | ||
| Liver inflammation/hepatitis | 1.00 × 100 to 5.92 × 10−8 | 39 |
| Liver damage | 1.00 × 100 to 3.67 × 10−6 | 33 |
| Liver steatosis | 6.18 × 10−1 to 7.62 × 10−5 | 28 |
| Liver necrosis/cell death | 5.51 × 10−1 to 2.23 × 10−4 | 25 |
| Glutathione depletion in liver | 4.52 × 10−1 to 9.06 × 10−4 | 8 |
Liver samples from two to four mice in the T09 + CCl4 and CCl4 groups were used for the microarray analysis.
HCC, hepatocellular carcinoma; IPA, Ingenuity Pathway Analysis; NASH, non‐alcoholic steatohepatitis.
TABLE 2.
Microarray analysis of genes related to NASH and NASH‐associated HCC: Eleven genes related to NASH were differentially expressed between the T09 + CCl4 and CCl4 groups
| Relation | Gene symbol | Gene name (description) | Fold change | P‐value | Regulation |
|---|---|---|---|---|---|
| Oxidative stress | Cd14 | CD14 antigen | 2.525 | 0.042 | Upregulation |
| Dnmt3b | DNA methyltransferase 3B | 2.043 | 0.014 | Upregulation | |
| Gstm2 | Glutathione S‐transferase Mu 2 | 2.101 | 0.044 | Upregulation | |
| Pil3cd | Phosphatidylinositol 3‐kinase catalytic delta polypeptide | 2.088 | 0.011 | Upregulation | |
| Hepatic steatosis | Acot1 | Acyl‐CoA thioesterase 1 | 2.124 | 0.014 | Upregulation |
| Lpl | Lipoprotein lipase | 6.301 | 0.005 | Upregulation | |
| Trib3 | Tribbles homolog 3 (Drosophila) | 0.395 | 0.044 | Downregulation | |
| Hepatic fibrosis | Lepr | Leptin receptor | 6.455 | 0.038 | Upregulation |
| Lgals3 | Lectin, galactose binding, soluble 3 | 2.049 | 0.038 | Upregulation | |
| Inflammation | Tfrc | Transferrin receptor | 2.725 | 0.005 | Upregulation |
| Fcgr4 | Fc receptor, IgG, low affinity IV | 2.379 | 0.004 | Upregulation |
Liver samples from two to four mice in the T09 + CCl4 and CCl4 groups were used for the microarray analysis.
HCC, hepatocellular carcinoma; NASH, non‐alcoholic steatohepatitis.
TABLE 3.
Microarray analysis of genes related to NASH and NASH‐associated HCC: Eight genes related to HCC and NASH were differentially expressed between tumor tissue and adjacent nontumor tissue in the T09 + CCl4 group
| Relation | Gene symbol | Gene name (description) | Fold change | P‐value | Regulation |
|---|---|---|---|---|---|
| Hepatocellular carcinoma | Cfp | Complement factor properdin | 2.688 | 0.009 | Upregulation |
| Cd5l | CD5 antigen‐like | 2.371 | 0.003 | Upregulation | |
| Vsig4 | V‐set and immunoglobulin domain containing 4 | 2.346 | 0.014 | Upregulation | |
| Sirt3 | Sirtuin 3 | 0.487 | 0.036 | Downregulation | |
| NAFLD | Lpl | Lipoprotein lipase | 5.12 | 0.009 | Upregulation |
| Ucp2 | Uncoupling protein 2 | 2.121 | 0.042 | Upregulation | |
| Apoptosis ↓ | Src | Rous sarcoma oncogene | 3.755 | 0.025 | Upregulation |
| Fgr | Gardner–Rasheed feline sarcoma viral (Fgr) oncogene homolog | 3.142 | 0.004 | Upregulation |
Liver samples from two to four mice in the T09 + CCl4 and CCl4 groups were used for the microarray analysis.
HCC, hepatocellular carcinoma; NASH, non‐alcoholic steatohepatitis.
TABLE 4.
Microarray analysis of genes related to NASH and NASH‐associated HCC: Four genes related to HCC and NASH were differentially expressed in tumor tissue between the T09 + CCl4 and CCl4 groups
| Relation | Gene symbol | Gene name (description) | Fold change | P‐value | Regulation |
|---|---|---|---|---|---|
| Hepatocellular carcinoma | Fam83e | Family with sequence similarity 83, member E | 9.419 | 0.004 | Upregulation |
| Src | Rous sarcoma oncogene | 3.755 | 0.003 | Upregulation | |
| NAFLD | Lpl | Lipoprotein lipase | 3.790 | 0.048 | Upregulation |
| Hepatic fibrosis | Lepr | Leptin receptor | 2.455 | 0.033 | Upregulation |
Liver samples from two to four mice in the T09 + CCl4 and CCl4 groups were used for the microarray analysis.
HCC, hepatocellular carcinoma; NASH, non‐alcoholic steatohepatitis.
Real‐time polymerase chain reaction analysis of genes associated with non‐alcoholic steatohepatitis‐related hepatocarcinogenesis
Figure 4c shows the results of the real‐time PCR analysis. The expression of three genes (Lpl, Lepr, and Src), which were detected by microarray analysis, was upregulated in the T09 + CCl4 group. The mRNA expression of these genes was significantly upregulated in the T09 + CCl4 group compared with the CCl4 group.
Discussion
Non‐alcoholic steatohepatitis is an emerging major cause of nonviral HCC and may account for a large percentage of HCC cases worldwide. 12 NASH is histologically characterized by the presence of steatosis, inflammation, hepatocyte injury (ballooning), and/or fibrosis. In particular, hepatic steatosis plays a crucial role in the progression of NASH. 13 Hepatic steatosis is caused by the activity of several nuclear receptors, such as FXR, PPAR, and LXR. 14 Ahn et al. 5 and Zhou et al. 15 reported that LXR plays a key role in hepatic steatosis via the induction of lipogenesis in NASH patients. Xiong reported that T0901317 inhibited the development of HCC in HCC cell line‐derived xenografts. However, the amount of T0901317 used was 25 mg/kg/day, which was 10 times greater than that used in our study. 16 In the present study, T0901317 was a very important reagent in reproducing the metabolic disorder observed in human NASH to cause NASH‐related hepatocarcinogenesis. Various studies have shown the importance of insulin resistance and oxidative stress in NASH‐related hepatocarcinogenesis. However, the role of LXR activation in the development of NASH‐related HCC has been less thoroughly investigated. We investigated the contribution of LXR agonists to the etiology of NASH and NASH‐related hepatocarcinogenesis.
We previously conducted similar experiments to establish a novel mouse model of NASH using an LXR agonist, a high‐fat diet, and a low dose of CCl4. 3 Liver specimens in past models exhibited pathological features resembling those of human NASH; however, carcinogenesis did not occur. Moreover, the tissues of mice subjected to the previous protocol exhibited severe damage after 4 weeks. Thus, we reduced the volume of T0901317 and CCl4 and decreased the frequency of T0901317 administration to make a completely different protocol for modeling hepatocarcinogenesis in NASH. In previous reports, models consisting of an LXR agonist and a high‐fat diet did not exhibit fat accumulation. Thus, we compared the CCl4–high‐fat diet model and the CCl4–high‐fat diet–LXR agonist model.
Various mouse models of NASH‐related HCC have been reported. However, few models have recapitulated all of the metabolic and histological features. For example, mice treated with a methionine/choline‐deficient diet have traditionally been used in NASH studies. However, these mice exhibit body weight loss and do not develop insulin resistance. 17 Additionally, combined chronic treatment with CCl4 and a choline/l‐amino acid‐deficient diet for up to 9 months can induce NASH and HCC. 18 However, these reports lacked a blinded quantitative assessment of the three features of the NAFLD activity score, which is used routinely in human studies. Furthermore, only 30% of these mice developed tumors in the liver at 24 weeks. In this study, mice in the T09 + CCl4 group did not exhibit weight loss and had a high liver‐to‐body weight ratio. Furthermore, all mice in the T09 + CCl4 group developed multiple tumors in the liver that were diagnosed as HCC by an experienced pathologist.
In clinical settings, NASH is diagnosed only by the histological findings of liver biopsy specimens. We compared the NAFLD activity score and the degree of fibrosis between the two groups to assess the influence of LXR. High NAFLD activity scores and specific histological findings of human NASH, such as ballooning and Mallory–Denk bodies, were observed only in the T09 + CCl4 group. Ahn et al. 5 reported that the intensity and extent of LXR expression were positively correlated with lobular inflammation, hepatocellular ballooning, the NAFLD activity score, and the degree of intrahepatic fibrosis. Additionally, the results of 4‐HNE immunostaining suggested that the oxidative stress observed in zone3 in the T09 + CCl4 group may be associated with the pathological features of NASH in humans, in which the fibrotic area is likely to be observed in zone 3. 19 Our results show that the hepatic phenotype of mice with NASH could be similar to that of humans with NASH with LXR activity. Therefore, LXR activity is presumed to be a key factor in the etiology and development of NASH.
Non‐alcoholic fatty liver disease, including NASH, is considered to be a hepatic component of metabolic syndrome because it is closely associated with metabolism. 20 , 21 Abnormal lipid metabolism causes excessive lipid flux, resulting in direct lipotoxicity and the generation of reactive oxygen species. 22 We evaluated local macrophage recruitment reflecting abnormal lipid metabolism in the liver by F4/80 staining. The characteristic finding of crown‐like structures was observed in the liver under conditions of abnormal lipid metabolism. 23 Under these conditions, macrophages are found around dead adipose tissue. Indeed, mice in the T09 + CCl4 group showed crown‐like structures in the liver. Additionally, the accumulation of Fasn was revealed by Western blotting. Such metabolic dysregulation can promote chronic inflammation in adipose tissue and cause insulin resistance, 24 , 25 which also leads to an imbalance in the gain and loss of fat and disrupted cholesterol and lipid homeostasis in the liver. 26 , 27 Additionally, insulin resistance influences glucose homeostasis. GSK‐3, a key enzyme in glycogen synthesis, regulates transcription factors to modulate insulin signaling and glucose homeostasis. 28 In this study, after administration of the LXR agonist, increased levels of p‐GSK3 and FASN were observed, and the ITT showed an increase in the severity of insulin resistance. These results suggest that LXR agonists may be the key factor in disrupting glucose and lipid homeostasis to recapitulate NASH etiology.
Compared with patients with simple steatosis, patients with NASH have a greatly increased risk of progression to HCC. 29 Clinically, NASH‐related HCC is often diagnosed as moderately or well differentiated. The diagnosis of well‐differentiated HCC is frequently difficult, even for expert pathologists. 2 To assess tumors accurately, we used immunostaining for CD34, AFP, and glypican 3. Because HCC is a hypervascular tumor, the vascular endothelium typically stains positive for CD34. 30 Additionally, the combination of glypican 3 and AFP was helpful for accurately distinguishing HCC from adenoma. 30 We verified the tumors from the T09 + CCl4 group as well‐differentiated HCC by HE staining and immunostaining for CD34, AFP, and glypican 3. However, the tumors from the CCl4 group did not exhibit staining (data not shown). Thus, mice in the T09 + CCl4 group could have developed well‐differentiated HCC tumors. Clinical reports on NASH‐related HCC have noted upregulated ERK signaling in carcinoma tissue. 31 The ERK signaling pathway normally transduces extracellular signals to regulate fundamental cellular processes, including proliferation, differentiation, and cell survival. 32 In contrast, dysregulated ERK signaling may induce hepatocarcinogenesis through the overexpression of oncogenes, such as cyclin D1. 33 In this study, we found the upregulated expression of both ERK and cyclin D1 by Western blotting. Moreover, p38 MAPK expression was elevated in the tumor tissue. p38 MAPK usually plays an important role in the coordination of cellular stress responses in normal cells. 34 In contrast, the stress response mediated by p38 MAPK could be altered in tumor cells, favoring tumor progression. 35 Thus, these changes were presumed to lead to the contribution of the inappropriate stress response to hepatocarcinogenesis in mice in the T09 + CCl4 group.
Moreover, our microarray analysis of noncancerous background liver tissue compared with cancerous tissue showed significantly upregulated expression of genes related to NASH. Oxidative stress has been verified to play an important role in mediating liver injury through at least two mechanisms: direct cell injury and indirect changes in cell signaling pathways. 36 , 37 These findings suggest that the transcriptomic features of NASH were represented by the administration of LXR. In the microarray analysis of tumor tissue from the T09 + CCl4 and CCl4 groups, the expression of Src and Fam83e, which are involved in Raf1/Ampk signaling, was upregulated (Table 4). These changes have also been reported in human HCC. 38 Additionally, in tumor tissue, the expression of NASH‐related genes, such as Lpl and Ucp2, was upregulated; thus, the tumor tissue exhibited features of NASH. Furthermore, we confirmed that the expression of three genes (Lpl, Lepr, and Src) associated with NASH‐related hepatocarcinogenesis was significantly upregulated in the T09 + CCl4 group (Fig. 4c). Lpl is related to lipid metabolism, and its overexpression causes liver‐specific insulin resistance and exacerbates the accumulation of TG in the mouse liver. 39 Leptin modulates appetite and body weight, and Lepr (leptin receptor) has been shown to be positively correlated with the stage of hepatic fibrosis and insulin resistance in NASH. 40 Numerous reports have shown that Src is a key messenger in many cellular pathways related to RAS/RAF/MEK/ERK signaling, which plays important roles in regulating proliferation, differentiation, survival, invasion, metastasis, and angiogenesis. The expression and activity of Src are correlated with advanced malignancy and poor prognosis in a variety of human cancers. 41 Thus, the expression of several genes related to the development of NASH and hepatocarcinogenesis was found to be upregulated in the T09 + CCl4 group. Therefore, these data support the conclusion that mice in the T09 + CCl4 group exhibited transcriptomic features of NASH‐related HCC.
There are possible limitations to this study. First, we could not assess when carcinogenesis began. We assessed the liver at 12 and 22 weeks to determine when carcinogenesis began. Compared with mice at 22 weeks, mice at 12 weeks showed relatively normal liver cells and sinusoidal structures, and more severe fibrosis was observed in the liver at 22 weeks than at 12 weeks. Additionally, two‐thirds of the mice had a tumor in the liver (Fig. S2: histopathological findings in mouse liver 12 and 22 weeks). However, these tumors did not show immunostaining for CD34 (data not shown). Thus, these tumors were considered benign tumors. Therefore, the liver of mice at 22 weeks exhibited features of NASH; however, it is unknown when hepatocarcinogenesis began. Second, the trigger of hepatocarcinogenesis remains unclear. We assessed the differences in the tumors between the CCl4 group and the T09 + CCl4 group and detected upregulated Src expression in the T09 + CCl4 group. However, there were many differences that were determined not to be the main trigger of carcinogenesis. When and how carcinogenesis begins are subjects for future research.
Supporting information
Figure S1. Liver weight. Liver weight in the CCl4 and T09‐CCl4 groups (n = 9). The results are expressed as the mean ± SD and were compared with the Mann–Whitney U test. P < 0.05 vs the CCl4 group.
Figure S2. Histological findings of livers from the T09 + CCl4 group at 12 weeks and 22 weeks. Representative MT staining of liver tissue from the T09 + CCl4 group at 12 weeks and 22 weeks. The open triangle indicates steatosis, and the closed triangle indicates fibrosis. Scale bars: 100 μm
Shimizu, Y. , Tamura, T. , Kemmochi, A. , Owada, Y. , Ozawa, Y. , Hisakura, K. , Matsuzaka, T. , Shimano, H. , Nakano, N. , Sakashita, S. , Oda, T. , and Ohkohchi, N. (2021) Oxidative stress and Liver X Receptor agonist induce hepatocellular carcinoma in Non‐alcoholic steatohepatitis model. Journal of Gastroenterology and Hepatology, 36: 800–810. 10.1111/jgh.15239.
Declaration of conflict of interest: None.
Author contribution: Y. Shimizu, T. Tamura, K. Hisakura, and N. Ohkohchi designed the study and wrote the initial draft of the manuscript. Y. Shimizu, A. Kemmochi, Y. Owada, and Y. Ozawa performed the animal experiments, biochemical analysis, and gene expression analysis. Y. Shimizu performed the insulin tolerance test. Histological analysis results were evaluated by N. Nakano and S. Sakashita. Y. Shimizu, T. Tamura, T. Matsuzaka, and H. Shimano contributed to the analysis and interpretation of the data. All other authors have critically reviewed the manuscript. The final version of the manuscript was approved by all the authors.
Financial support: This study is supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan, KAKENHI, Nos. 26861059 and 16K10489.
Data availability statement
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.
References
- 1. Cholankeril G, Patel R, Khurana S, Satapathy SK. Hepatocellular carcinoma in non‐alcoholic steatohepatitis: current knowledge and implications for management. World J. Hepatol. 2017; 9: 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non‐alcoholic fatty liver disease: an emerging menace. J. Hepatol. 2012; 56: 1384–1391. [DOI] [PubMed] [Google Scholar]
- 3. Owada Y, Tamura T, Tanoi T et al. Novel non‐alcoholic steatohepatitis model with histopathological and insulin‐resistant features. Pathol. Int. 2018; 68: 12–22. [DOI] [PubMed] [Google Scholar]
- 4. Kirchgessner TG, Sleph P, Ostrowski J et al. Beneficial and adverse effects of an LXR agonist on human lipid and lipoprotein metabolism and circulating neutrophils. Cell Metab. 2016; 24: 223–233. [DOI] [PubMed] [Google Scholar]
- 5. Ahn SB, Jang K, Jun DW, Lee BH, Shin KJ. Expression of liver X receptor correlates with intrahepatic inflammation and fibrosis in patients with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2014; 59: 2975–2982. [DOI] [PubMed] [Google Scholar]
- 6. Pekow JR, Bhan AK, Zheng H, Chung RT. Hepatic steatosis is associated with increased frequency of hepatocellular carcinoma in patients with hepatitis C‐related cirrhosis. Cancer 2007; 109: 2490–2496. [DOI] [PubMed] [Google Scholar]
- 7. Baud V, Karin M. Is NF‐κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009; 8: 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang SN, Yang SF, Tsai HH, Lee KT, Yeh YT. Increased adiponectin associated with poor survival in hepatocellular carcinoma. J. Gastroenterol. 2014; 49: 1342–1351. [DOI] [PubMed] [Google Scholar]
- 9. Chen MJ, Yeh YT, Lee KT, Tsai CJ, Lee HH, Wang SN. The promoting effect of adiponectin in hepatocellular carcinoma. J. Surg. Oncol. 2012; 106: 181–187. [DOI] [PubMed] [Google Scholar]
- 10. Rasband WS. ImageJ. Bethesda, Maryland: National Institutes of Health, 1997. –2015. [Google Scholar]
- 11. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412–419. [DOI] [PubMed] [Google Scholar]
- 12. Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 2010; 51: 1820–1832. [DOI] [PubMed] [Google Scholar]
- 13. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin. Gastroenterol. Hepatol. 2015; 13: 643–654.e641–649 quiz e639–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cave MC, Clair HB, Hardesty JE et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016; 1859: 1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou J, Febbraio M, Wada T et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARγ in promoting steatosis. Gastroenterology 2008; 134: 556–567. [DOI] [PubMed] [Google Scholar]
- 16. Xiong T, Li Z, Huang X et al. TO901317 inhibits the development of hepatocellular carcinoma by LXRα/Glut1 decreasing glycometabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2019; 316: G598–G607. [DOI] [PubMed] [Google Scholar]
- 17. Machado MV, Michelotti GA, Xie G et al. Mouse models of diet‐induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015; 10: e0127991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Minicis S, Agostinelli L, Rychlicki C et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014; 9: e97136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takahashi Y, Fukusato T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014; 20: 15539–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell. Mol. Life Sci. 2016; 73: 1969–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marchesini G, Bugianesi E, Forlani G et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003; 37: 917–923. [DOI] [PubMed] [Google Scholar]
- 22. McClain CJ, Barve S, Deaciuc I. Good fat/bad fat. Hepatology 2007; 45: 1343–1346. [DOI] [PubMed] [Google Scholar]
- 23. Itoh M, Suganami T, Nakagawa N et al. Melanocortin 4 receptor‐deficient mice as a novel mouse model of nonalcoholic steatohepatitis. Am. J. Pathol. 2011; 179: 2454–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Invest. 2006; 116: 1784–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanyal AJ, Campbell‐Sargent C, Mirshahi F et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120: 1183–1192. [DOI] [PubMed] [Google Scholar]
- 26. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell. Mol. Life Sci. 2019; 76: 99–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muscat GE, Wagner BL, Hou J et al. Regulation of cholesterol homeostasis and lipid metabolism in skeletal muscle by liver X receptors. J. Biol. Chem. 2002; 277: 40722–40728. [DOI] [PubMed] [Google Scholar]
- 28. Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res. Clin. Pract. 2007; 77: S49–S57. [DOI] [PubMed] [Google Scholar]
- 29. Sun B, Karin M. Obesity, inflammation, and liver cancer. J. Hepatol. 2012; 56: 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang F, Jing X, Wang T et al. Differential diagnostic value of GPC3‐CD34 combined staining in small liver nodules with diameter less than 3 cm. Am. J. Clin. Pathol. 2012; 137: 937–945. [DOI] [PubMed] [Google Scholar]
- 31. Tsuboi Y, Ichida T, Sugitani S et al. Overexpression of extracellular signal‐regulated protein kinase and its correlation with proliferation in human hepatocellular carcinoma. Liver Int. 2004; 24: 432–436. [DOI] [PubMed] [Google Scholar]
- 32. Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 2000; 351: 289–305. [PMC free article] [PubMed] [Google Scholar]
- 33. Patil MA, Lee SA, Macias E et al. Role of cyclin D1 as a mediator of c‐Met‐ and β‐catenin‐induced hepatocarcinogenesis. Cancer Res. 2009; 69: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gutierrez‐Uzquiza A, Arechederra M, Bragado P, Aguirre‐Ghiso JA, Porras A. p38α mediates cell survival in response to oxidative stress via induction of antioxidant genes: effect on the p70S6K pathway. J. Biol. Chem. 2012; 287: 2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Igea A, Nebreda AR. The stress kinase p38α as a target for cancer therapy. Cancer Res. 2015; 75: 3997–4002. [DOI] [PubMed] [Google Scholar]
- 36. Anstee QM, Goldin RD. Mouse models in non‐alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006; 87: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Magee N, Zou A, Zhang Y. Pathogenesis of nonalcoholic steatohepatitis: interactions between liver parenchymal and nonparenchymal cells. Biomed. Res. Int. 2016; 2016: 5170402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cipriano R, Miskimen KL, Bryson BL, Foy CR, Bartel CA, Jackson MW. Conserved oncogenic behavior of the FAM83 family regulates MAPK signaling in human cancer. Mol. Cancer Res. 2014; 12: 1156–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim JK, Fillmore JJ, Chen Y et al. Tissue‐specific overexpression of lipoprotein lipase causes tissue‐specific insulin resistance. Proc. Natl. Acad. Sci. U. S. A. 2001; 98: 7522–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Medici V, Ali MR, Seo S et al. Increased soluble leptin receptor levels in morbidly obese patients with insulin resistance and nonalcoholic fatty liver disease. Obesity (Silver Spring) 2010; 18: 2268–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wheeler DL, Iida M, Dunn EF. The role of Src in solid tumors. Oncologist 2009; 14: 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Liver weight. Liver weight in the CCl4 and T09‐CCl4 groups (n = 9). The results are expressed as the mean ± SD and were compared with the Mann–Whitney U test. P < 0.05 vs the CCl4 group.
Figure S2. Histological findings of livers from the T09 + CCl4 group at 12 weeks and 22 weeks. Representative MT staining of liver tissue from the T09 + CCl4 group at 12 weeks and 22 weeks. The open triangle indicates steatosis, and the closed triangle indicates fibrosis. Scale bars: 100 μm
Data Availability Statement
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request.