

Abstract
Fluoropyrimidines are widely used in the treatment of several types of solid tumors. Although most often well tolerated, severe toxicity is encountered in ~ 20–30% of the patients. Individualized dosing for these patients can reduce the incidence of severe fluoropyrimidine‐related toxicity. However, no consensus has been achieved on which dosing strategy is preferred. The most established strategy for individualized dosing of fluoropyrimidines is upfront genotyping of the DPYD gene. Prospective research has shown that DPYD‐guided dose‐individualization significantly reduces the incidence of severe toxicity and can be easily applied in routine daily practice. Furthermore, the measurement of the dihydropyrimidine dehydrogenase (DPD) enzyme activity has shown to accurately detect patients with a DPD deficiency. Yet, because this assay is time‐consuming and expensive, it is not widely implemented in routine clinical care. Other methods include the measurement of pretreatment endogenous serum uracil concentrations, the uracil/dihydrouracil‐ratio, and the 5‐fluorouracil (5‐FU) degradation rate. These methods have shown mixed results. Next to these methods to detect DPD deficiency, pharmacokinetically guided follow‐up of 5‐FU could potentially be used as an addition to dosing strategies to further improve the safety of fluoropyrimidines. Furthermore, baseline characteristics, such as sex, age, body composition, and renal function have shown to have a relationship with the development of severe toxicity. Therefore, these baseline characteristics should be considered as a dose‐individualization strategy. We present an overview of the current dose‐individualization strategies and provide perspectives for a future multiparametric approach.
5‐Fluorouracil (5‐FU), and its oral prodrug capecitabine, belong to the group of fluoropyrimidines and are the backbone of several treatment regimens in a wide range of cancer types, including colorectal cancer (CRC), breast cancer, and head and neck cancer. 1 Although fluoropyrimidines are reasonably well tolerated by patients, ~ 20–30% experience severe (Common Terminology Criteria for Adverse Events (CTC‐AE) grade 3–5) toxicity. The most common toxicities attributed to fluoropyrimidine‐based chemotherapy are diarrhea, nausea, vomiting, mucositis, neutropenia, and hand‐foot syndrome; the latter especially with capecitabine. 2 , 3 Severe fluoropyrimidine‐related toxicity can be fatal in up to 1% of patients. 4 Given the considerable number of patients (~ 2 million) treated with fluoropyrimidines worldwide every year, severe fluoropyrimidine‐related toxicity is a well‐recognized and significant clinical problem. Therefore, accurate biomarkers or methods that can predict and prevent severe fluoropyrimidine‐related toxicity are of high interest. Over the years, several approaches for prediction of toxicity and guidance of dose‐individualization of fluoropyrimidines have been studied. The probably most studied biomarker is the activity of the main catabolic enzyme dihydropyrimidine dehydrogenase (DPD), which is strongly correlated to the pharmacokinetics of 5‐FU. 5 Despite extensive research identifying biomarkers, predicting severe toxicity is challenging, and a consensus in approach for individualizing fluoropyrimidine dosing is lacking. In this review, we present an overview of the various possible strategies for dose‐individualization of fluoropyrimidine‐based chemotherapy (see Table 1 ). This review distinguishes itself from other reviews and guidelines by not only including strategies, such as DPYD‐genotyping and DPD‐phenotyping, but also discuss less‐known strategies, such as patient characteristics and multiparametric approaches in detail. Additionally, we will evaluate the level of evidence, discuss the feasibility, and provide recommendations regarding these dose‐individualization strategies. This review only focusses on 5‐FU and capecitabine, as the vast majority of studies have been conducted in patients receiving 5‐FU and capecitabine, excluding other fluoropyrimidines, such as tegafur.
Table 1.
Overview of dose‐individualization strategies, including their principles, advantages, and limitations
| Strategy | Principle | Advantages | Limitations |
|---|---|---|---|
| DPYD‐guided dose‐individualization | |||
| Dose‐modifications based on single nucleotide polymorphisms in the DPYD gene |
|
|
|
| DPD‐phenotype guided dose‐individualization | |||
| Endogenous uracil and dihydrouracil | Measurement of plasma/serum uracil and dihydrouracil concentration as a surrogate marker for a DPD deficiency |
|
|
| Administration of uracil | Measurement of pharmacokinetic parameters or metabolites |
|
|
| DPD enzyme activity | Measurement of the DPD enzyme activity in PBMCs |
|
|
| 5‐FU degradation rate | Measurement of the degradation of 5‐FU in PBMCs |
|
|
| Pharmacokinetically‐guided dosing | |||
| Dosing strategy based on the concentration and pharmacokinetic characteristics of 5‐FU |
|
|
|
| 5‐FU test dose | |||
| Administration of very low dose 5‐FU, after which blood samples are taken to determine the exposure |
|
|
|
| Patient characteristics at baseline | |||
| Sex | Dose adjustment of baseline characteristics that are associated with an increased risk of developing severe toxicity |
|
|
| Age | |||
| Body composition | |||
| Renal function | |||
5‐FU, 5‐Fluorouracil; DPD, dihydropyrimidine dehydrogenase; PBMCs, peripheral blood mononuclear cells.
METABOLISM OF FLUOROPYRIMIDINES
Capecitabine is metabolized into the active agent 5‐FU through three steps (see Figure 1 ). 6 First, capecitabine is converted to 5′‐deoxy‐5‐fluorocytidine by carboxylesterase, which is an enzyme located mainly in the liver. Second, 5′‐deoxy‐5‐fluorocytidine is converted to 5′‐deoxy‐5‐fluorouridine (5′‐dFUR) by cytidine deaminase, which is mainly located in the liver and tumor tissue. Third, 5′‐dFUR is converted to 5‐FU by thymidine phosphorylase. This last conversion primarily takes place in tumor tissue, due to higher concentrations of thymidine phosphorylase compared with normal, healthy tissue. 6 Thereupon, 5‐FU enters the cell through a facilitated transmembrane carrier. Subsequently, 5‐FU is enzymatically converted to the active intracellular cytotoxic metabolites 5‐fluoro‐2′‐deoxyuridine 5′‐monophosphate, 5‐fluorouridine 5′‐triphosphate, and 5‐fluoro‐2′‐deoxyuridine 5′ triphosphate. 7 Approximately 80–90% of 5‐FU is catabolized by DPD into metabolite 5‐dihydrofluorouracil (5‐FUH2), which is neither cytotoxic to the tumor cells nor toxic to normal cells. This conversion undergoes a circadian rhythm as DPD enzyme activity changes over time during the day. 8 Afterward, α‐fluoro‐β‐ureidopropionic and α‐fluoro‐β‐alanine (FBAL) are formed, which are excreted through the urine with the remaining ~ 10% of 5‐FU. 1 , 7
Figure 1.
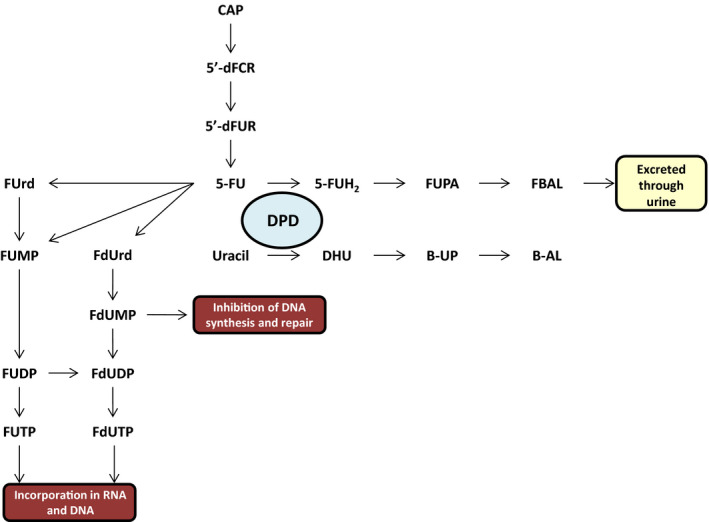
Metabolism of fluoropyrimidines. 5′‐dFCR, 5′‐deoxy‐5‐fluorocytidine; 5′‐dFUR, 5′‐deoxy‐5‐fluorouridine; 5‐FU, 5‐fluorouracil; 5‐FUH2, 5,6‐dihydro‐5‐fluorouracil; B‐AL, β‐ alanine; B‐UP, β‐ureidopropionate; DHU, Dihydrouracil; FBAL, α‐fluoro‐β‐alanine; FdUDP, 5‐fluoro‐2′‐deoxyuridine 5′‐diphosphate; FdUMP, 5‐fluoro‐2′‐deoxyuridine 5′‐monophosphate; FdUrd, 5‐fluoro‐2'‐deoxyuridine; FdUTP, 5‐fluoro‐2′‐deoxyuridine 5‐’triphosphate; FUDP, 5‐fluorouridine 5′‐diphosphate; FUMP, 5‐fluorouridine 5′‐monophosphate; FUPA, α‐fluoro‐β‐ureidopropionic acid; FUrd, 5‐fluorouridine; FUTP, 5‐fluorouridine 5′‐triphosphate. [Colour figure can be viewed at wileyonlinelibrary.com]
DOSING
Historically, most chemotherapeutic drugs are dosed based upon the patient’s body surface area (BSA). The same accounts for 5‐FU and capecitabine. BSA‐guided dosing intents to minimize interpatient variability in exposure due to differences in body size, resulting in less toxicity. 9 However, no correlation was found between BSA and 5‐FU plasma clearance by Gamelin et al. 10 Furthermore, Ratain addressed a few problems with dosing capecitabine, such as a large interpatient variability (> 85%) in 5‐FU concentration and area under the curve (AUC), the limited tablet strengths available (150 and 500 mg), and the lack of evidence for suggested dose modifications. 11 The interpatient variability in 5‐FU concentration and AUC are most likely caused by various enzymes involved in the conversion of capecitabine to 5‐FU. 12 In the summary of product characteristics (SmPCs), it is mentioned that a dose‐reduction of 25% is recommended for patients with grade ≥ 3 toxicity treated with capecitabine, although limited prospective research has been performed regarding dose modification in patients with grade ≥ 3 toxicity. It is questioned why a dose‐reduction of 25% is recommended whereas the calculation for the starting dose is very precise. Yet, alternative dosing strategies have been studied scarcely. 13 Recently, de Man et al. have shown that the tolerance and effectiveness of fixed‐dose capecitabine are comparable to BSA‐guided dosing and therefore fixed‐dosing could be an alternative for BSA‐guided dosing. However, fixed‐dosing of capecitabine did not lead to a decrease of severe toxicity. 14 Therefore, alternative strategies should be explored to optimize and individualize the treatment with fluoropyrimidines.
DPYD‐GUIDED DOSING
Dihydropyrimidine dehydrogenase
One of the main factors influencing drug exposure in fluoropyrimidine‐based chemotherapy is DPD enzyme activity. The DYPD gene encodes for the DPD enzyme. The availability of 5‐FU for conversion into cytotoxic metabolites is primarily determined by the activity of the DPD enzyme. 12 Reduced activity of DPD is one of the main causes of fluoropyrimidine‐related toxicity, due to the lower capacity to degrade 5‐FU into the inactive metabolites, resulting in higher exposure of 5‐FU and cytotoxic metabolites. 1 , 7 Most often, a DPD deficiency is the result of a deleterious single nucleotide polymorphism (SNP) in DPYD, altering the DPD enzyme activity. 15 A DPD deficiency is classified as partial if there is remaining DPD activity (e.g., 25–50% of normal) and as complete if no or almost no DPD enzyme activity (e.g., < 5%) is detectable. In the white population, ~ 3–7% have a DPD deficiency and 0.01–0.1% have a complete deficiency. 16 However, the frequency of DPD deficiencies can differ between ethnicities. For example, Mattison et al. found that ~ 8% of the African American population have a partial DPD deficiency. 17
DPYD variants
The first functionally relevant DPYD variant reported was the DPYD*2A (c.1905 + 1G>A; IVS14+1G>A; rs3918290) variant. 18 The DPD enzyme activity in heterozygous carriers of DPYD*2A is ~ 50% compared with wild types (WT). In addition to DPYD*2A, several other SNPs in DPYD have been reported that are associated with a reduced DPD enzyme activity, including c.1236G>A (rs56038477; Haplotype B3), c.2846A>T (D949V, rs67376798), and c.1679T>G (DPYD*13, I560S; rs55886062). 15 However, the decrease in DPD enzyme activity between these variants differ ranging from ~ 25% for c.1236G>A and c.2846a>T and 50% for c.1679T>G. 15 , 19
Furthermore, it is also possible that patients carry multiple DPYD variants simultaneously. Homozygous patients carry two identical DPYD variants, which results in reduced or inactive alleles and therefore a reduced or absent DPD enzyme activity. Compound heterozygous patients carry two or more DPYD variants either on one allele (in cis) or on different alleles (in trans) leading to differences in DPD enzyme activity. When two or more DPYD variants are present on different alleles, both alleles are impacted and DPD enzyme activity is impacted more severely. For example, patients that are compound heterozygous carriers of a c.1236G>A and DPYD*2A variants have ~ 75% reduced DPD enzyme activity, theoretically. If these DPYD variants were present on the same allele, the DPD enzyme activity would have been reduced by only ~ 50%. 15 , 20 This can make compound heterozygous genotypes difficult to interpret.
The relation between these DPYD variants and severe fluoropyrimidine‐related toxicity is widely accepted. Multiple meta‐analyses have shown that these variants are associated with severe fluoropyrimidine‐related toxicity. 2 , 21 , 22 Consequently, upfront genotyping for these variants and adjusting the dose according to the reduction in DPD enzyme activity was the next step.
DPYD‐GUIDED DOSING
Deenen et al. were the first to prospectively evaluate the safety of DPYD*2A‐guided dose‐individualization of fluoropyrimidines. 23 Before treatment with fluoropyrimidine‐based chemotherapy, patients (N = 2039) were prospectively screened for DPYD*2A and received a dose‐reduction of 50% if carrying DPYD*2A, followed by dose‐titration if tolerated. Toxicity was compared with a historical cohort of patients carrying a DPYD*2A variant treated with a standard dose and WTs treated with a standard dose in this study. The risk of developing severe fluoropyrimidine‐related toxicity was significantly reduced from 73% (95% confidence interval (CI) 58–85%) in the historical cohort (N = 48) to 28% (95% CI 10–53%) by DPYD‐guided dosing (P < 0.001). This was similar compared with WTs receiving the standard dose (23%; P = 0.64). Pharmacokinetic (PK) analysis showed that patients carrying DPYD*2A treated with a 50% dose‐reduction achieved similar 5‐FU exposure as WT patients treated with a standard dose, suggesting that dose‐reduction by 50% in DPYD*2A carriers does not lead to undertreatment. 23
Subsequently, a similar prospective study was conducted in which c.1236G>A, c.2846A>T and c.1679T>G were added to the screening panel. Patients carrying a DPYD variant received a dose‐reduction of either 50% (DPYD*2A and c.1679T>G carriers) or 25% (c.1236G>A or c.2846A>T carriers), after which the dose could be escalated when treatment was well‐tolerated. The incidence of toxicity was compared with a historical cohort similarly as described by Deenen et al. 16 , 23 A total of 1,103 patients were included and deemed evaluable of which ~ 8% (N = 85) were heterozygous carriers of 1 of the 4 DPYD variants. It was shown that the relative risk (RR) of developing severe fluoropyrimidine‐related toxicity was reduced in DPYD*2A (1.31 (0.63–2.72) vs. 2.87 (2.14–3.86)) and c.2846A>T (2.00 (1.19–3.34) vs. 3.11 (2.25–4.28)) carriers compared with a historical cohort. Furthermore, the 25% dose‐reduction for the c.1236G>A variant proved to be insufficient to reduce the RR (1.69 (1.18–2.42) vs. 1.72 (1.22–2.42)). Only one patient was included carrying the c.1679T>G variant and was treated safely with a dose‐reduction of 50%. PK analysis showed that the mean exposure to 5‐FU was similar between the group DPYD carriers treated with a reduced dose and WTs treated with a full dose. 16 Additionally, both Deenen et al. and Henricks et al. showed that upfront genotyping of DPYD and subsequent dose‐individualization is cost saving. 23 , 24
Although drug exposure is similar, uncertainty exists about the effectiveness of treatment with a reduced dose for variant carriers, as the often‐mentioned fear is that this dose‐reduction could result in underdosing. This was studied by Henricks et al. who compared DPYD*2A carriers treated with a 50% dose‐reduction with matched controls of WTs treated with a full dose (37 DPYD*2A carriers and 37 controls). The applied dose‐reduction did not negatively influence overall survival (OS; median 27 vs. 24 months, P = 0.47) nor progression‐free survival (median 14 vs. 10 months, P = 0.54). This suggests that a 50% dose‐reduction in DPYD*2A does not negatively impact effectiveness, while improving the patient safety. 25 However, this study only focused on DPYD*2A and had a relatively small sample size. The impact of dose‐reductions on the effectiveness of treatment remains to be studied for c.1236G>A, c.2846A>T, and c.1679T>G carriers.
These studies, among other published studies, have led to the update of the Clinical Pharmacogenetics Implementation Consortium (CPIC) and Dutch Pharmacogenetics Working Group (DPWG) guidelines for fluoropyrimidines and DPYD. These are evidence‐based guidelines focusing on the drug‐gene interaction of DPYD and fluoropyrimidines. The purpose of the CPIC guideline is to provide information for clinical interpretation of DPYD‐genotype test results to guide the dosing of fluoropyrimidines. 26 The DPWG aims to expedite pharmacogenetic implementation by developing evidence‐based guidelines to optimize pharmacotherapy. 27 Similar guidelines have been developed by the French Network of Pharmacogenetics (RNPGx) and the Italian Associazione Italiana di Oncologia Medica (AIOM) but are not available in English. A dose‐reduction of 50% (instead of 25%) for c.1236G>A or c.2846A>T carriers is now recommended in both the CPIC and the DPWG guideline. 27 , 28 Furthermore, information about DPYD‐genotyping has been added to the SmPC of capecitabine and the European Medicines Agency (EMA) has recently recommended that patients treated with fluoropyrimidines should be tested for the lack of DPD before the start of treatment. 13 , 29 Similarly, the US Food and Drug Administration (FDA) added statements to the label of 5‐FU and capecitabine warning for the increased risk of severe toxicity in patients with a DPD deficiency. 30
DPYD‐guided genotyping has shown to be an effective and cost‐saving strategy for individualized dosing of fluoropyrimidine‐based chemotherapy. Other advantages of DPYD‐guided dosing are that genotyping of the DPYD gene is relatively simple and gives unequivocal results. In addition, dosing‐guidelines based on DPYD‐genotype are readily available and have been implemented in routine clinical care. 31 However, there are also a few drawbacks. The first and main drawback is that only a part of severe fluoropyrimidine‐related toxicity can be traced back to genetic variants of the DPYD gene. 32 Meulendijks et al. reported that ~ 17% of the patients experiencing severe fluoropyrimidine‐related toxicity are identified by genotyping for the 4 DPYD variants. 21 Furthermore, these DPYD variants are most likely only predictive of severe toxicity in the Western population. It has been shown by Elraiyah et al. that these variants were not present in patients of the East African descent. However, 12 nonsynonymous DPYD variants were identified in this study, of which 7 variants showed a significantly decreased DPD enzyme activity in vitro. 33 In addition, Offer et al. also showed that patients of African American descent carry unique variants, such as DPYD‐Y186C, which was not present in patients of European American descent. 34 Furthermore, Hariprakash et al. studied DPYD variants associated with toxicity in south‐Asian populations and showed that certain variants (e.g., rs1801160 and rs12022243) are observed in higher frequency in south‐Asia compared with other populations. 35 This problem has been acknowledged and further research regarding DPYD variants in patients of non‐Western descent is being conducted (NCT04300361). Last, another disadvantage of DPYD‐guided dosing is the lack of options for patients with a homozygous or compound heterozygous DPYD‐genotype. These patients are generally not treated with fluoropyrimidines.
PHENOTYPE‐GUIDED DOSING
Endogenous uracil and dihydrouracil
The variability in DPD enzyme activity can only partly be traced back to SNPs in the DPYD gene. Therefore, DPD‐phenotyping could be useful to identify more patients with a DPD deficiency. Several DPD‐phenotyping methods have been described over the years and are mostly based on the conversion of the endogenous substrate of DPD, uracil (U), to dihydrouracil (DHU; see Figure 1 ). It is thought that a DPD‐deficiency decreases the conversion rate of U to DHU, resulting in higher U concentrations in DPD‐deficient patients. Pretreatment serum U concentrations have been measured in 550 patients and the predictive value of U for early severe fluoropyrimidine‐related toxicity were compared. It was shown that a high pretreatment serum U concentration (> 16 ng/mL) was strongly associated with global severe toxicity (odds ratio (OR) 5.3, P = 0.009). 36 In addition to this, Etienne‐Grimaldi et al. have shown that patients with a U concentration above 16 ng/mL were significantly prone to develop grade 4 toxicity compared with patients with a lower U concentration (RR 20.6, P = 0.021). 37 Moreover, a significant correlation was found by Boisdron‐Celle et al. between U plasma concentrations and 5‐FU toxicity with a threshold value of 15 ng/mL for toxicity. 38 Furthermore, an abstract of a prospective pilot study showed an association between U and DHU concentration and the development of severe fluoropyrimidine‐related toxicity (median concentration 12.7 ng/mL (U) and 110 ng/mL (DHU) vs. 10.2 ng/mL (U) and 93 ng/mL (DHU) in patients with and without toxicity, P = 0.014 (U) and P = 0.011 (DHU)). Receiver operating characteristic analysis showed that these differences were too small to use as predictors for toxicity. 39
The endogenous U concentration is an interesting biomarker for the prediction of severe fluoropyrimidine‐related toxicity, yet most phenotyping studies conducted have been aimed towards DHU/U ratio rather than U concentration alone. Several studies have shown that there is an association between DHU/U ratio and 5‐FU plasma concentration and severe fluoropyrimidine‐related toxicity. 10 , 40 , 41 , 42 , 43 On the contrary, no correlation was found between DHU/U ratio and 5‐FU clearance by Boisdron‐Celle et al., whereas a significant correlation was found with severe toxicity (P < 0.001) with a threshold of 6. 38 In addition, Etienne‐Grimaldi et al. could not establish correlation between DHU/U ratio and toxicity (median 9.1 vs. 9.6 in patients with and without toxicity, P = 0.80). 39 The earlier mentioned retrospective study by Meulendijks et al. also showed that the DHU/U ratio was a less accurate in predicting severe toxicity compared with the pretreatment U concentration. 36
It has been shown that there is most likely an association between these phenotypes and severe toxicity. However, the major concern with the use of these phenotyping methods is the lack of prospective validation confirming that dose adjustments based upon U or DHU/U ratio lead to a decreased incidence of severe toxicity. Despite the lack of prospective validation, the French National Authority for Health and French National Cancer Institute recently recommended testing for DPD deficiency by determination of U concentration for patients treated with fluoropyrimidines in France. 44 Recently, a study in the Netherlands has started (NCT04194957, The Alpe2U‐study) in which patients are prospectively screened for pretreatment serum U concentration and a dose‐reduction of 50% is applied to patients with a pretreatment serum U concentration above 16 ng/mL. Another important issue is the limited information concerning the sensitivity and specificity of U as a biomarker. It has been mentioned that the sensitivity of U is better compared with DPYD‐genotyping by Captain et al. 45 However, this analysis was performed on selected patients with severe toxicity. This influences the results, as no information is available on patients with no severe toxicity and high U concentrations (> 16 ng/mL) and vice versa, which would reduce the sensitivity of U as a biomarker. Furthermore, U is measured in low concentrations, which requires specific equipment. This equipment is not readily available at all hospitals, which complicates the implementation in the clinic. In addition, the limited stability of U and DHU has to be taken into account. It has been shown that the concentration of U and DHU increases over time at room temperature after samples have been taken. 46 , 47 This could significantly influence the possible dose‐individualization based on these methods and indicates that samples need to be processed as soon as possible to minimize the increase of U and DHU concentration. This could be challenging in clinical practice where samples most often are not processed immediately. Last, the conditions under which blood samples are taken for determination of U and DHU should be chosen carefully as U is influenced by circadian rhythm and food. 8 , 48 It has been shown that U levels were higher in fasted state compared with fed state. It is recommended that sampling should be performed preferably between 8:00 and 9:00 am after overnight fasting to avoid bias introduced by circadian rhythm and food effects. 48
Administration of uracil
Other phenotypic methods based on the conversion of U to DHU are the U loading dose and the U breath test. The U loading dose consists of oral administration of U and blood sampling at specific time points. After sampling, the concentrations of U and DHU are measured. Staveren et al. have shown that PK‐parameters, such as the AUC and the maximum concentration (Cmax) of U and DHU, significantly differ between subjects with a DPD deficiency and without. 49 Additional research was performed to assess the sensitivity and specificity of this test to identify patients with a DPD deficiency. A sensitivity and specificity of 80% and 98%, respectively, was obtained for the DHU/U ratio a t = 120 minutes to discriminate between subjects with a normal DPD activity and DPD‐deficient subjects. This shows that DPD‐deficient patients can be accurately identified using this method. 50 An advantage of this strategy is that the DPD enzyme temporarily is saturated and therefore U is eliminated following zero‐order kinetics. This is a better representation of the DPD enzyme activity than measuring endogenous U concentrations as under normal conditions the elimination of U follows first‐order kinetics. This suggests that the rate of U elimination is more dependent on the amount of U and not primarily on the amount of DPD enzyme activity. 51 However, the administration of U followed by a blood draw after 2 hours is relatively patient‐unfriendly and demanding on the clinical staff and resources. More research is needed to further establish the correlation between the U loading dose and the prediction of severe toxicity. Furthermore, a prospective study in which dose‐adaptions are applied based on this method needs to be conducted to see if the incidence of severe toxicity can be reduced.
Another phenotypic method in which U is administered orally is the U breath test. This method is based on the production 13CO2 from 2‐13C‐uracil by enzymes in the metabolism of U. First, baseline samples of patients are taken by collecting breath samples in bags. Second, 2‐13C‐uracil is ingested orally in an aqueous solution after which breath samples are taken. Third, concentrations of 13CO2 and 12CO2 are measured by infrared spectrometry and expressed as a delta‐over‐baseline (DOB) ratio. This ratio represents a change in the ratio of 13CO2/ 12CO2 of the samples collected before and after administration of 2‐13C‐uracil. 52 , 53 Mattison et al. have shown that the concentration of exhaled 13CO2 is reduced in patients with a DPD deficiency. 52 This was based on a single time point determination at 50 minutes after administration. 52 In addition to this, it has also been shown that the U breath test correlates with DPD enzyme activity in peripheral blood mononuclear cells (PBMCs; R = 0.78) and plasma [2‐13C]‐uracil AUC (R = −0.73). 54 In addition to this, Cunha‐Junior et al. studied the ability of the U breath test to identify patients at risk of severe toxicity. Mean DOB50min significantly differed between patients with grade 0–1 and grade 3–4 toxicity. A DOB50min cutoff of ≤ 161.4 was found, which could fairly accurately discriminate individuals who experienced severe toxicity from those who did not (sensitivity = 61%; specificity = 85%). 55 However, DPD is not the only enzyme involved in the conversion of [2‐13C]‐uracil to 13CO2. Several other enzymes are involved in the complete conversion and therefore could influence the outcome. Furthermore, due to the complex and laborious logistics, clinical implementation of the breath test could be hampered.
DPD enzyme activity in PBMCs
A more direct way of determining a DPD deficiency is by measuring the DPD enzyme activity in PBMCs. DPD enzyme activity can be detected in multiple human tissues, with the highest activity found in the liver and lymphocytes. 56 A prospective study was conducted with 27 patients in which a significant linear correlation was found between DPD enzyme activity in the liver and in PBMCs (R = 0.59, P = 0.002). This indicates that DPD enzyme activity measured in PBMCs reflects DPD enzyme activity expressed in the liver. 57 Therefore, PBMCs are often used to measure the DPD enzyme activity and identify patients with a DPD deficiency. Kuilenburg et al. demonstrated that in ~ 60% of the cases with severe toxicity a decreased DPD enzyme activity could be detected in PBMCs. In addition, 55% of patients with decreased DPD enzyme activity developed severe grade 4 neutropenia vs. 13% in patients with a normal DPD enzyme activity (P = 0.01). Moreover, the onset of toxicity was significantly faster in patients with a decreased DPD enzyme activity compared with patients with a normal DPD enzyme activity (10.0 ± 7.6 vs. 19.1 ± 15.3 days, P < 0.05). 58
Over the years, several assays have been developed for the determination of the DPD enzyme activity in PBMCs and this has led to different thresholds for DPD deficiency. By our knowledge, no consensus has been reached about a uniform threshold to determine DPD deficiency based on DPD enzyme activity, making it hard to properly interpret and compare results. A pragmatic approach for determination of the threshold is described by Milano et al. who define a significant DPD deficiency as the DPD enzyme activity in PBMCs < 70% of the mean population value. 59 In addition, as earlier mentioned for U, DPD enzyme activity is influenced by a circadian rhythm, which could influence the measured activity and therefore the subsequent dose‐adaption. 8 Furthermore, the clinical implementation of the measurement of DPD enzyme activity in PBMCs is hampered by its complex and laborious sample processing, which makes it also time‐consuming and expensive. In addition, not all laboratories (especially in smaller hospitals) have the specific equipment to perform this assay, which also does not add to a widespread implementation. However, in the rare case of a homozygous or compound heterozygous DPYD‐genotype, the DPD enzyme activity test in PBMCs could still be extremely useful. Patients with these genotypes most likely have very low DPD enzyme activity (or a complete DPD deficiency) and in general will not be treated with fluoropyrimidine‐based chemotherapy, as these genotypes are difficult to interpret and the risk of severe toxicity is too high. For these rare cases, the DPD enzyme activity could be determined and treatment could be tailored based on the remaining DPD enzyme activity compared with a normal DPD enzyme activity, as described by Henricks et al. 60
5‐FU degradation rate
Another method to predict the risk of severe toxicity based on PBMCs is the determination of 5‐FU degradation rate (5‐FUDR). This assay measures the rate of 5‐FU degradation in intact PBMCs. The 5‐FUDR distinguishes itself from DPD enzyme activity measured in PBMCs by incorporating the complete metabolism involved in drug catabolism instead of focusing on a specific enzyme. 61 This phenotypic method was tested and three metabolic classes were identified: poor metabolizers (5‐FUDR ≤ 0.85 ng/mL/106 cells/min), normal metabolizers (0.85 ng/mL/106 cells/min < 5‐FUDR ≤ 2.20 ng/mL/106 cells/min), and ultra‐rapid metabolizers (5‐FUDR > 2.20 ng/mL/106 cells/min). As expected, poor metabolizers showed an increased risk of developing severe toxicity compared to normal metabolizers. However, it was also seen that ultra‐rapid metabolizers were at increased risk of developing severe toxicity. It was hypothesized that this could be caused by an increased activity of the enzymes producing the active and cytotoxic metabolites. 62 , 63 Two retrospective studies also showed a similar association between low and high (OR 11.14, 95% CI 1.09–113.77 (low) and OR 9.63, 95% CI 1.70–54.55 (high), P = 0.002) 5‐FUDR and severe toxicity. 64 , 65 Furthermore, due to low costs (mentioned to be only €10 per sample), noninvasive sampling and quick test results (within 1 working day) 5‐FUDR seems suitable for clinical implementation. 65 Although promising, 5‐FUDR has similar disadvantages, as measurement of DPD enzyme activity in PBMCs requires specific equipment. 61 Furthermore, 5‐FUDR lacks prospective validation, which makes it difficult to assess clinical utility. More research is needed to assess the ability to predict severe toxicity and how fluoropyrimidine treatment should be individualized based on 5‐FUDR.
PHARMACOKINETICALLY GUIDED DOSING
In addition to DPYD‐genotyping and DPD‐phenotyping, PK‐guided dosing of fluoropyrimidines has been studied extensively as a measure to individualize dosing. Use of a PK‐based dosing approach could assist in dose‐individualization of fluoropyrimidines and optimal systemic exposure, which would be ultimately more effective and less toxic for the patient. PK‐guided dosing is better known as therapeutic drug monitoring (TDM). As mentioned earlier, no correlation has been found between BSA and the 5‐FU clearance. 10 Therefore, an alternative could be to adjust the dose based on direct monitoring of the blood levels of 5‐FU, as it has been shown that there is a relationship among 5‐FU plasma concentration and biological effect, toxicity, and efficacy. 66 , 67 , 68
It should be mentioned that limited data are available for TDM of capecitabine and therefore only 5‐FU will be discussed in this subsection. Although capecitabine shares the same metabolic pathway, it is hypothesized that TDM is most likely not applicable for capecitabine in a clinical setting due to the complex PKs.
Over the years, several studies have been performed in which PK‐guided dosing was applied. 67 , 69 , 70 , 71 , 72 Fety et al. conducted a randomized clinical trial in which 122 patients with head and neck cancer were treated with a continuous infusion of 5‐FU (96 hours). 72 Patients received a standard dose (4 g/m2), after which the dose was modified based on either toxicity (St‐arm) or PK parameters (PK‐arm). In the PK‐arm (N = 49), the AUC and 5‐FU doses were significantly reduced during cycles 2 and 3 compared with the St‐arm (P < 0.001), whereas maintaining a comparable response rate. In addition, grades 3 and 4 neutropenia and thrombopenia were significantly more frequent in the St‐arm compared with the PK‐arm (17.5% vs. 7.6%, P = 0.013). 72 In another study by Gamelin et al., a PK‐guided dosing approach in 280 patients with metastatic CRC was studied. 67 Patients were randomly assigned to either arm A (BSA‐guided dosing of 5‐FU) or arm B (PK‐guided dosing of 5‐FU). The initial dose was 1500 mg/m2 5‐FU plus 200 mg/m2 folinic acid during a continuous 8‐hour infusion. In arm B, 5‐FU doses were adjusted weekly based on single point measurements of 5‐FU plasma concentrations at steady‐state until the therapeutic range of 2.5–3.0 mg/L (AUC range of 20–24 mg*h/L) was reached. 67 This range was established by Gamelin et al. in previous studies. 73 , 74 It was shown that patients in arm A received a mean 5‐FU dose of 1,500 mg/m2 throughout treatment compared with 1,790 mg/m2 in arm B, whereas significantly more patients experienced severe toxicity in arm A (P = 0.003). Furthermore, a trend toward a better median OS was seen in arm B compared with arm A (22 months vs. 16 months, P = 0.08). This showed that arm B was treated with a higher dose‐intensity without experiencing more toxicity and most likely improved OS. 67
Dosing based on the proposed range by Gamelin et al. of 2.5–3.0 mg/L has shown to reduce toxicity without the loss of efficacy. 67 However, this range is rather small, especially knowing that there is a large intrapatient variability in PK of 5‐FU. This could lead to unnecessary or incorrect dose adjustments. Therefore, Kaldate et al. proposed a wider AUC0‐≥18h range of 20–30 mg*h/L. 70 Furthermore, a dosing algorithm was proposed for AUC0‐≥18h values of 8 mg*h/L to values higher than 40 mg*h/L, with corresponding dose adjustments. 70 This algorithm was prospectively validated by Wilhelm et al. in 75 patients with metastatic CRC. 71 After the fourth cycle, 54% of patients had an AUC within the target range and the incidence of severe fluoropyrimidine‐related toxicity was significantly reduced compared with historical data, despite 55% of patients receiving an increased dose. 71 In addition, Goldstein et al. have shown that PK‐guided dose‐individualization is a cost‐effective strategy compared with conventional BSA‐guided dosing. 75
These studies show that PK‐guided dosing of 5‐FU is a viable strategy to individualize dosing of 5‐FU, which can reduce toxicity while maintaining adequate exposure to 5‐FU and efficacy. However, patients are still initially treated with a full dose. Severe fluoropyrimidine‐related toxicity can occur rapidly (especially in DPD‐deficient patients) and PK‐guided dose‐individualization does not prevent that. Furthermore, additional blood samples need to be taken, which is relatively patient unfriendly and could require an additional visit to the hospital, depending on the 5‐FU scheme. In addition, PK‐guided dosing only applies to treatment with 5‐FU, which limits the application of this method. Nevertheless, PK‐guided follow‐up of patients in combination with another dosing strategy could improve the safety and efficacy. An initial dose‐reduction could be applied based on, for example, the DPYD‐genotype, after which the AUC could be evaluated every cycle and dose adjustments can be made to achieve maximal safe exposure.
5‐FU test dose
A more direct way to identify patients at risk of toxicity is by administrating a very low dose of 5‐FU or capecitabine followed by blood sampling to assess the exposure to treatment with the fluoropyrimidine drug. This was first tested by Bocci et al. in 20 patients with CRC who were given 2 dose‐levels of 5‐FU, 250 and 370 mg/m2 administered by i.v. bolus. Afterward, 5‐FU and 5‐FUH2 were determined in plasma samples obtained at baseline and several time points between 5 minutes and 4 hours after i.v. bolus. Significant differences in the plasma PK‐parameters (AUC, Cmax, and total body clearance) of 5‐FU and 5‐FUH2 were found between the test‐dose and the treatment dose. This is expected as these parameters are influenced by the administered dose. In contrary, no correlations were found between 5‐FU or 5‐FUH2 at the 2 dose‐levels and the DPD enzyme activity in PBMCs. 76 This was further studied by Bocci et al. in 188 patients with gastrointestinal cancer who were treated with 5‐FU. Patients were given a 5‐FU test‐dose of 250 mg/m2 2 weeks before starting initial treatment with 370 mg/m2. The 5‐FU test dose was well‐tolerated in all patients. In 3 of 188 patients, marked reduced drug clearance was seen in the presence of a normal DPD enzyme activity. Therefore, these patients were treated with irinotecan instead of 5‐FU, which was well‐tolerated. An association was found between 5‐FUH2tmax values higher than 30 minutes and the risk of moderate to severe neutropenia and diarrhea (P = 0.0323 and P = 0.0138). This suggests that a 5‐FU test dose might be useful for the identification of patients at risk of severe fluoropyrimidine‐related toxicity. 77 However, very limited data are available and more research is needed. In addition, to our knowledge, no studies have been conducted in which a test dose of capecitabine has been studied. This could limit the use of a test dose as in certain countries capecitabine is used more frequently than 5‐FU. Furthermore, it is not certain that both 5‐FU or capecitabine will behave similarly when given at such low dose levels compared with normal dose levels. Last, administration of a test dose of 5‐FU to patients with a complete DPD deficiency could lead to possibly life‐threatening toxicity. Therefore, the 5‐FU test dose should be combined with at least one other method that can detect a DPD deficiency upfront before administration.
PATIENT CHARACTERISTICS AT BASELINE
Sex
Although numerous studies have explored the use of the above‐mentioned methods to predict severe fluoropyrimidine‐related toxicity, few have studied the use of patient characteristics at baseline. Sex‐dependent differences in response rates and the probability of toxicity in patients treated with chemotherapy have been seen. It has been suggested that these differences are explained by variation in expression levels of metabolic enzymes and differences in body composition leading to different PKs. It has often been seen that the half‐life of drug therapy for oncologic diseases are longer in women compared with men, which is associated with improved survival, however, also with increased toxicity. 78
In the SmPC of capecitabine, it has been stated the AUC and Cmax of FBAL are ~ 10% and 20%, respectively, higher in women compared with men. 13 This suggests that capecitabine is catabolized slower in women compared with men. Yet, sex did not have any clinical significant effect on the PKs of the main metabolites of capecitabine (5′‐dFUR, 5‐FU, and FBAL). 13 The PKs of fluoropyrimidines have been studied by several researchers and showed different results. Milano et al. determined the 5‐FU clearance for 380 patients (301 men and 79 women) treated for head and neck cancer with a 5‐day continuous intravenous infusion. 79 The 5‐FU clearance levels showed a large variation in both men and women, but was significantly lower in women (median 155 L/h/m2 vs. 179 L/h/m2, P = 0.0005). When adjusted for age and dose, the influence of sex remained significant (P = 0.013). 79 This indicates that women have less capacity to clear 5‐FU compared with men, and are more likely to develop severe fluoropyrimidine‐related toxicity. 79 These differences in 5‐FU clearance were later also shown by Mueller et al. 80 PK‐sampling was performed at baseline for 32 patients receiving a 46‐hour continuous infusion of 5‐FU and showed that men had a higher elimination of both 5‐FU and 5‐FUH2 (26% and 18% higher, respectively). In addition, a significant lower AUC was found in men (18 vs. 22 mg*h/L, P = 0.04), independent of weight or BSA, indicating that exposure to fluoropyrimidines is higher in women compared with men. 80 Another study by Stein et al., in which the toxicity of 331 patients was analyzed, showed that sex is an independent risk predictor, which strengthen the findings of Milano et al. 79 , 81 In addition, two meta‐analyses of North Central Cancer Treatment Group trials have been undertaken. 82 , 83 The first meta‐analysis included data from 731 patients (402 men and 329 women) and focused on the incidence of 5‐FU‐induced stomatitis. Stomatitis was more frequently reported for women and with greater severity compared with men. The incidence of severe or very severe stomatitis for women and men was 22% and 16% (P = 0.0006), respectively. Additionally, women were also more likely to experience grade ≥ 3 leukopenia (18% vs. 11%, P = 0.004). 82 The second meta‐analysis included data from 2,348 patients (1,093 men and 1,093 women) and focused on the incidence of stomatitis, leukopenia, alopecia, diarrhea, nausea, and vomiting. Significant differences were found between incidence of severe toxicity between women and men (51% vs. 38%, P < 0.0001) across cycles 1 to 3 adjusted for study, dose body mass index, and age. 83 Several other studies have also reported the association between sex and severe fluoropyrimidine‐related toxicity. 80 , 84 , 85 , 86
These studies indicate that women have a decreased 5‐FU clearance leading to an increased exposure to fluoropyrimidines and an increased risk of developing severe fluoropyrimidine‐related toxicity. Therefore, sex‐based dose‐individualization should be considered. To our knowledge, this has not been studied yet. In future studies, women could be treated with an initially reduced dose, after which, according to toxicity or PK, the dose could be increased. A major advantage of this is that no additional tests or blood sampling are initially required. However, as not all studies have adjusted the results for body size, it cannot be stated that the increased risk of developing severe fluoropyrimidine‐related toxicity is caused by a decreased 5‐FU clearance. Furthermore, prospective studies are needed to confirm the clinical significance of sex‐based dosing.
Age
Age has also been studied as a risk factor of developing severe fluoropyrimidine‐related toxicity. The decision to treat elderly patients with a reduced dose due to being more fragile and therefore more prone to develop severe fluoropyrimidine‐related toxicity has been frequently discussed. Milano et al. and Stein et al. both studied the influence of age on severe toxicity. 79 , 81 Interestingly, mixed results were found. Milano et al. did not find an association between age and risk of developing severe toxicity, whereas Stein et al. found that age was a significant risk factor for severe toxicity (P < 0.0001). 79 , 81 Furthermore, Meulendijks et al. retrospectively studied the relationship between age and the risk of developing severe toxicity in 1,463 patients of which 231 (16%) experienced early severe toxicity and 132 (9%) were hospitalized. 87 They found that age was a predictor of early severe toxicity, yet not statistically significant (OR 1.14 per 10 years, P = 0.0891). However, age was significantly associated with fatal treatment‐related toxicity (OR 5.75, P = 0.0008). 87 Recently, a large retrospective study was published in which the impact of age on toxicity and efficacy of 5‐FU‐based combination chemotherapy was studied. 85 A total of 3,223 patients were included of which 2,488 patients were < 70 years and 735 were ≥ 75 years. Older age was associated with a higher probability of serious adverse events (AEs; OR 0.649; 95% CI 0.545–0.772; P < 0.001) and separate toxicities, such as all‐grade diarrhea, high‐grade diarrhea, high‐grade stomatitis, high‐grade thrombocytopenia, all‐grade neutropenia, and high‐grade neutropenia. 88 Another study showed that older age was associated with a higher risk of hospitalization. A total of 2,533 patients were included of which 1,010 experienced at least one serious AE. In total, 945 (39.9%) patients were hospitalized one or more times and 148 (5.8%) patients had fatal events. It was shown that older age was predictive of hospitalization (P < 0.001). Older age might be associated with a higher risk of developing severe fluoropyrimidine‐related toxicity, however, limited information is available. More research is necessary to properly establish the relationship between age and severe toxicity.
Body composition
Another patient characteristic that has been associated with an increased risk of severe toxicity is body composition. Gusella et al. have studied the relationship between body composition parameters, including body cell mass, total body water, and lean body mass (LBM), and 5‐FU PKs. 89 This relationship was studied in 34 patients with CRC (13 women and 21 men) treated with intravenous 5‐FU. This study showed that the clearance of 5‐FU better correlated with the LBM than the standard measures, such as body weight and BSA. 89 This was further studied by Prado et al. who used data from a prospective study to determine if the highest doses of 5‐FU per kilogram LBM would be associated with dose‐limiting toxicity in patients with colon cancer treated with 5‐FU and leucovorin. 90 A cutoff point of 20 mg 5‐FU/kg LBM was found as the threshold for developing severe toxicity (P = 0.005). This was only found in women (OR 16.73, P = 0.021), which had a relatively low proportion LBM compared with their body weight. 90 This could explain the difference in the relationship between men and women and severe toxicity found in other studies. Other body composition parameters, such as (skeletal) muscle mass, have also been studied as predictors of severe toxicity. Williams et al. examined the association of low skeletal muscle (sarcopenia) on PK‐parameters of 5‐FU. 91 No significant differences in AUC were found between patients with sarcopenia and those without sarcopenia. However, LBM was also studied and a significant association was found between 5‐FU per kg LBM and hematological toxicities (110 vs. 94 mg/kg, P = 0.002). Yet, no correlation between the dose/LBM and 5‐FU AUC was found. 91 Another study examined the association of sarcopenia and dose‐limiting toxicity during treatment with capecitabine combination therapy in patients with metastatic CRC. In contrary to Williams et al., sarcopenia and/or muscle loss was associated with increased risk of dose‐limiting toxicities. 92 Furthermore, Jung et al. reviewed the data of 229 patients with colon cancer treated with 5‐FU, oxaliplatin, and leucovorin, and studied the association of muscle mass and toxicity. 93 It was shown that a decreased muscle mass was associated with an increased risk of grade 3–4 toxicity and poor prognosis. 93 These studies suggest that body composition parameters, such as LBM and muscle mass, could be an interesting marker to predict severe toxicity. However, more research is needed to confirm these associations and to determine the corresponding dose modifications.
Renal function
The 5‐FU is predominantly metabolized in the liver and tumor tissues. 6 Therefore, at first, it is not expected that renal impairment would influence the exposure to 5‐FU. However, pooled data from phase I studies showed that creatinine clearance has a significant influence on the AUC of 5‐FU. On the contrary, a population PK analysis of phase III trials did not reveal a significant effect of the creatinine clearance on the PKs of 5‐FU and 5‐FUH2. A significant effect was observed for FBAL, and a positive relationship was seen between AUC of FBAL and treatment‐related grade 3–4 diarrhea and Cmax of FBAL and treatment‐related grade 3–4 AEs. However, this does not necessarily mean that FBAL causes these AEs. FBAL might be a marker of the amount of 5‐FU that is formed in tissues. Meaning that patients with high FBAL concentrations might be patients with a high exposure to 5‐FU. Renal impairment leads to a major increase in the systemic exposure to FBAL, but did not significantly impact the PKs of capecitabine and 5‐FUH2. 6 Another study by Cassidy et al. showed that creatinine clearance is inversely correlated to risk of toxicity and recommended a dose reduction of 25% for patients with moderate renal impairment (calculated creatinine clearance 30–50 mL/min) and contraindicate capecitabine for patients with a severe renal impairment (< 30 mL/min). 94 This recommendation was followed up and taken up in the SmPC in 2005. 13 Furthermore, Meulendijks et al. also found that renal function is a clinically relevant predictor of severe fluoropyrimidine‐related toxicity in a dataset of 1,463 patients treated with capecitabine or 5‐FU. 21 However, the precise mechanism by which renal impairment increases risk of severe fluoropyrimidine‐related toxicity is unclear.
MULTIPARAMETRIC APPROACHES
Information about patient characteristics, such as sex, age, and renal function are easily obtained or measured and have shown to most likely have a relationship with the development of severe fluoropyrimidine‐related toxicity. Therefore, the logical next step would be to combine these patient characteristics with the more established strategies, such as DPYD‐genotyping and DPD‐phenotyping to develop a dosing algorithm. In 2007, a decision‐tree was described by Boisdron‐Celle et al. in which DPYD‐genotyping was combined with the measurement of endogenous U concentration, DHU/U‐ratio, and individual PK follow‐up. 38 This algorithm was further developed and a multicenter prospective cohort study was performed to assess the clinical benefit of this new multiparametric approach. In this study, two parallel cohorts were treated with 5‐FU‐based chemotherapy. In arm A, patients were screened upfront for DPD deficiency with the multiparametric approach, whereas no screening for DPD deficiency was performed in arm B. In total 1,142 patients were included, of which 718 were in arm A and 398 were in arm B. The percentage of patients experiencing grade 4–5 toxicity in arm A was 1.2% vs. 3.0% in arm B (P = 0.0406) and 10.9% vs. 17.6% (P = 0.497) for grade 3–5 toxicity, respectively. It was concluded that this multiparametric approach significantly reduced the risk of developing severe fluoropyrimidine‐related toxicity. 95 Although promising, some serious questions are raised regarding the methodology of this study, as mentioned by Etienne‐Grimaldi et al. in a letter to the editor. 96 It was noted that the prevalence of DPD deficiency based on the multiparametric approach and DHU/U ratio in arm A was 2.5‐fold (P = 0.00017) and 4‐fold fewer (P = 0.00007) compared with arm B, respectively. This means that the two arms were incomparable at baseline resulting in less toxicity in arm A. 96 The most important factor that makes it difficult to properly interpret these results is the fact that this multiparametric approach is protected by a patent, therefore, it is unknown what this approach consists out of and could be seen as a so called “blackbox.” It is mentioned that DPYD‐genotyping is combined with DPD‐phenotyping (DHU/U ratio) and that demographic parameters are used, but how this is converted into a dose‐recommendation is not described.
Similarly, Botticelli et al. aimed to develop a nomogram that could accurately predict toxicity. 97 This nomogram consisted of metabolic parameters and clinical patient characteristics. Fluoropyrimidine‐related toxicity was correlated with patient‐specific and treatment‐related factors. Univariate logistic regression analyses were performed to identify predictive variables. Variables with a P value < 0.10 in the univariate model were entered into a multivariate model. Multivariate logistic regression showed that age, DPYD status, the number of drugs administered, and 5‐FUDR value were associated with severe fluoropyrimidine‐related toxicity (P values below 0.05). Based on these findings, a nomogram was structured to assess a score to predict the probability of developing severe fluoropyrimidine‐related toxicity before starting treatment. However, no corresponding dose‐modification is mentioned. Therefore, it is unclear how much the dose should be reduced if a patient has a certain probability of developing severe fluoropyrimidine‐related toxicity. Furthermore, it is unclear why the chosen variables were selected to include in the univariate analysis. In addition, this nomogram has not been validated either internally or externally, therefore, it is difficult to assess how accurate this nomogram can predict the probability of developing severe fluoropyrimidine‐related toxicity. 97
Recently, Etienne‐Grimaldi et al. presented the results of the FUSAFE meta‐analysis in which the performance of DPYD‐genotyping to predict fluoropyrimidine‐related toxicity was studied. 98 A clinical model was developed to assess the prognostic value of consensual deleterious DPYD variants on grade 4–5 toxicity. This model was based on data of 6,403 white patients from 7 studies and included age, sex, body mass index, fluoropyrimidine administration mode, and associated anticancer drugs as predictors of grade 4–5 toxicity. The presence of DPYD*2A, c.2846A>T, and c.1679T>G improved the model and showed to be relevant in predicting grade 4–5 toxicity. Despite its association with toxicity, c.1236G>A did not improve the ability of the model to identify patients at risk of grade 4–5 toxicity. 98
CONCLUSION AND FUTURE PERSPECTIVES
Numerous strategies for dose‐individualization have been discussed in this review. However, the level of evidence and feasibility differs a lot between these strategies. Currently, the most established and evidence‐based strategy for dose‐individualization of fluoropyrimidine‐based chemotherapy is DPYD‐guided dosing. It has been shown that this strategy significantly reduces the incidence of severe fluoropyrimidine‐related toxicity, does not negatively impact efficacy, and is cost‐effective. 16 , 23 , 24 , 25 Therefore, we think that DPYD‐guided dosing should be the cornerstone in dose‐individualization of fluoropyrimidines and recommend that this strategy is implemented in routine clinical care. However, only a limited number of patients experiencing severe toxicity can be identified with the four current variants and these variants are most likely only predictive for severe toxicity in patients of western descent. Therefore, additional screening methods are needed and more research should be conducted in ethnicities that are under‐represented in genetic studies. The major issue with these additional screening methods is the lack of prospective validation. Multiple screening methods (e.g., DPD‐phenotyping) have shown to be promising, but due to the lack of prospective studies are scarcely being implemented. Measuring the DPD enzyme activity in PBMCs would probably be the choice for which most evidence is available, yet due to the complicated and laborious method is not recommended for application in clinical routine care. Measurement of U or the DHU/U ratio could be a good alternative. Previous studies have shown that U could be an accurate predictor of severe fluoropyrimidine‐related toxicity. Therefore, results of the recently started prospective clinical trial, which combines DPYD‐genotyping and U measurements (NCT04194957) are awaited. In addition to these methods, PK‐guided follow‐up of patients could further improve the safety of treatment with fluoropyrimidine‐based chemotherapy, especially for 5‐FU‐treated patients. Set dose adjustments based on DPYD‐genotype or DPD‐phenotype can reduce the incidence of severe toxicity but are not suited for all patients in a similar manner. With PK‐guided follow‐up, patients could be monitored and treatment could be altered if concentrations are outside of the therapeutic range. However, this is only possible for patients treated with 5‐FU due to the complex metabolism of capecitabine. An interesting addition to these dosing strategies could be the use of patient characteristics at baseline. Patient characteristics, such as age, sex, and renal function are easily obtained or measured and have shown to most likely have a relationship with the development of severe fluoropyrimidine‐based toxicity. However, only limited information is available. Studies in which the dose of fluoropyrimidines are individualized based on these characteristics are needed.
All the strategies described in this review have shown to have potential, however, the limitations of these strategies need to be overcome by conducting additional research before combining of strategies is possible. In an ideal world, all the proposed strategies could be combined into an algorithm or model that could accurately predict the probability of developing severe fluoropyrimidine‐related toxicity and translate this probability into a dose recommendation (Figure 2 ). By combining all these strategies all known factors that have been associated with severe fluoropyrimidine‐related toxicity are covered, which could significantly improve the safety of fluoropyrimidine‐based chemotherapy.
Figure 2.
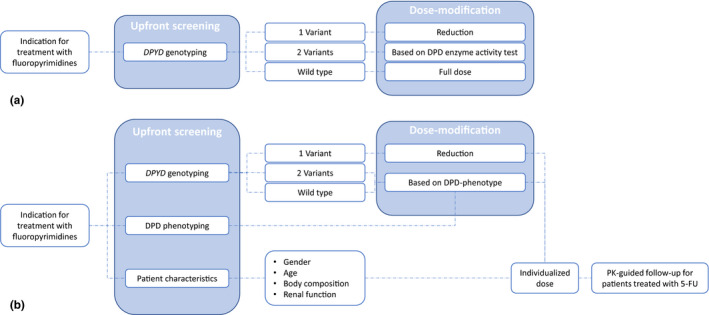
Overview of the current dosing strategy and a suggestion for a potential future dosing strategy. (a) Current dosing strategy. (b) Potential future dosing strategy in which upfront screening is performed which includesDPYD‐genotyping, DPD‐phenotyping and screening of baseline characteristics and PK‐guided follow‐up. 5‐FU, 5‐fluorouracil; DPD, dihydropyrimidine dehydrogenase; PK, pharmacokinetic. [Colour figure can be viewed at wileyonlinelibrary.com]
Funding
No funding was received for this work.
Conflict of Interest
The authors declared no competing interests for this work.
References
- 1. Longley, D.B. , Harkin, D.P. & Johnston, P.G. 5‐Fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 (2003). [DOI] [PubMed] [Google Scholar]
- 2. Rosmarin, D. et al. Genetic markers of toxicity from capecitabine and other fluorouracil‐based regimens: Investigation in the QUASAR2 study, systematic review, and meta‐analysis. J. Clin. Oncol. 32, 1031–1039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Twelves, C. et al. Capecitabine as adjuvant treatment for stage III colon cancer. N. Engl. J. Med. 352, 2696–2704 (2005). [DOI] [PubMed] [Google Scholar]
- 4. Hoff, P.M. et al. Comparison of oral capecitabine versus intravenous fluorouracil plus leucovorin as first‐line treatment in 605 patients with metastatic colorectal cancer: results of a randomized phase III study. J. Clin. Oncol. 19, 2282–2292 (2017). [DOI] [PubMed] [Google Scholar]
- 5. Van Kuilenburg, A.B.P. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5‐fluorouracil. Eur. J. Cancer 40, 939–950 (2004). [DOI] [PubMed] [Google Scholar]
- 6. European Medicines Agency (EMA) . Capecitabine Scientific Discussion <https://www.ema.europa.eu/en/documents/scientific‐discussion‐variation/avastin‐h‐c‐582‐ii‐0008‐epar‐scientific‐discussion‐variation_en.pdf> 2005. [Google Scholar]
- 7. Diasio, R.B. & Harris, B.E. Clinical pharmacology of 5‐fluorouracil. Clin. Pharmacokinet. 16, 215–237 (1989). [DOI] [PubMed] [Google Scholar]
- 8. Jacobs, B.A.W. et al. Pronounced between‐subject and circadian variability in thymidylate synthase and dihydropyrimidine dehydrogenase enzyme activity in human volunteers. Br. J. Clin. Pharmacol. 82, 706–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reilly, J.J. & Workman, P. Normalisation of anti‐cancer drug dosage using body weight and surface area: is it worthwhile? Cancer Chemother. Pharmacol. 32, 411–418 (1993). [DOI] [PubMed] [Google Scholar]
- 10. Gamelin, B.E. et al. Correlation between uracil and dihydrouracil plasma ratio, fluorouracil (5‐FU) pharmacokinetic parameters, and tolerance in patients with advanced colorectal cancer: a potential interest for predicting 5‐FU toxicity and determining optimal 5‐FU dosage. J. Clin. Oncol. 17, 1105–1110 (1999). [DOI] [PubMed] [Google Scholar]
- 11. Ratain, M.J. Dear doctor: we really are not sure what dose of capecitabine you should prescribe for your patient. J. Clin. Oncol. 20, 1434–1435 (2002). [DOI] [PubMed] [Google Scholar]
- 12. Reigner, B. , Blesch, K. & Weidekamm, E. Clinical pharmacokinetics of capecitabine. Clin. Pharmacokinet. 40, 85–104 (2001). [DOI] [PubMed] [Google Scholar]
- 13. Xeloda [SmPC] . Hoffmann‐La Roche Inc [Internet] <https://www.ema.europa.eu/documents/product‐information/xeloda‐epar‐product‐information_en.pdf> (2017).
- 14. de Man, F.M. et al. Comparison of toxicity and effectiveness between fixed‐dose and body surface area‐based dose capecitabine. Ther. Adv. Vaccines 11, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henricks, L.M. et al. Translating DPYD genotype into DPD phenotype: using the DPYD gene activity score. Pharmacogenomics 16, 1277–1286 (2015). [DOI] [PubMed] [Google Scholar]
- 16. Henricks, L.M. et al. DPYD genotype‐guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 19, 1459–1467 (2018). [DOI] [PubMed] [Google Scholar]
- 17. Mattison, L.K. , Fourie, J. , Desmond, R.A. , Modak, A. , Saif, M.W. & Diasio, R.B. Increased prevalence of dihydropyrimidine dehydrogenase deficiency in African‐Americans compared with Caucasians. Clin. Cancer Res. 12, 5491–5495 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Wei, X. , McLeod, H.L. , McMurrough, J. , Gonzalez, F.J. & Fernandez‐Salguero, P. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5‐fluorouracil toxicity. J. Clin. Invest. 98, 610–615 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meulendijks, D. et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine‐associated toxicity: a systematic review and meta‐analysis of individual patient data. Lancet Oncol. 16, 1639–1650 (2015). [DOI] [PubMed] [Google Scholar]
- 20. Lunenburg, C.A.T.C. et al. Diagnostic and therapeutic strategies for fluoropyrimidine treatment of patients carrying multiple DPYD variants. Genes (Basel) 9, 1–13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meulendijks, D. , Cats, A. , Beijnen, J.H. & Schellens, J.H.M. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity – ready for clinical practice? Cancer Treat. Rev. 50, 23–34 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Terrazino, S. , Cargnin, S. , Del Re, M. , Danesi, R. , Canonico, P.L. & Genazzani, A.A. DPYD IVS14 + 1G > A and 2846A > T genotyping for the prediction of severe fluoropyrimidine‐related toxicity: a meta‐analysis. Pharmacogenomics 14, 1255–1272 (2013). [DOI] [PubMed] [Google Scholar]
- 23. Deenen, M.J. et al. Upfront genotyping of DPYD∗2A to individualize fluoropyrimidine therapy: a safety and cost analysis. J. Clin. Oncol. 34, 227–234 (2016). [DOI] [PubMed] [Google Scholar]
- 24. Henricks, L.M. et al. A cost analysis of upfront DPYD genotype–guided dose individualisation in fluoropyrimidine‐based anticancer therapy. Eur. J. Cancer 107, 60–67 (2019). [DOI] [PubMed] [Google Scholar]
- 25. Henricks, L.M. et al. Effectiveness and safety of reduced‐dose fluoropyrimidine therapy in patients carrying the DPYD*2A variant: a matched pair analysis. Int. J. Cancer 144, 2347–2354 (2019). [DOI] [PubMed] [Google Scholar]
- 26. Amstutz, U. et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 103, 210–216 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lunenburg, C.A.T.C. et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene–drug interaction of DPYD and fluoropyrimidines. Eur. J. Hum. Genet. 28, 508–517 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Fluoropyrimidines and DPYD [Internet] <https://cpicpgx.org/guidelines/guideline‐for‐fluoropyrimidines‐and‐dpyd/> (2018).
- 29. European Medicines Agency . EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine <https://www.ema.europa.eu/en/news/ema‐recommendations‐dpd‐testing‐prior‐treatment‐fluorouracil‐capecitabine‐tegafur‐flucytosine> (2020).
- 30.US Food and Drug Administration (FDA) . Fluorouracil Highlights of Prescribing Information [Internet] <https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/012209s040lbl.pdf> (2016).
- 31. Lunenburg, C.A.T.C. , Van Staveren, M.C. , Gelderblom, H. & Swen, J.J. Pharmacogenomics of prospective DPYD genotyping in. Pharmacogenomics 17, 721–729 (2016). [DOI] [PubMed] [Google Scholar]
- 32. Falvella, F.S. et al. DPD and UGT1A1 deficiency in colorectal cancer patients receiving triplet chemotherapy with fluoropyrimidines, oxaliplatin and irinotecan. Br. J. Clin. Pharmacol. 80, 581–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elraiyah, T. et al. Novel deleterious dihydropyrimidine dehydrogenase variants may contribute to 5‐fluorouracil sensitivity in an East African population. Clin. Pharmacol. Ther. 101, 382–390 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Offer, S.M. , Lee, A.M. , Mattison, L.K. , Fossum, C. , Wegner, N.J. & Diasio, R.B. A DPYD variant (Y186C) in individuals of African ancestry associated with reduced DPD enzyme activity. Clin. Pharmacol. Ther. 94, 158–166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hariprakash, J.M. et al. Pharmacogenetic landscape of DPYD variants in south Asian populations by integration of genome‐scale data. Pharmacogenomics 19, 227–241 (2018). [DOI] [PubMed] [Google Scholar]
- 36. Meulendijks, D. et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine‐associated toxicity. Br. J. Cancer 116, 1415–1424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Etienne‐Grimaldi, M.C. et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS One 12, 1–19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boisdron‐Celle, M. et al. 5‐Fluorouracil‐related severe toxicity: a comparison of different methods for the pretherapeutic detection of dihydropyrimidine dehydrogenase deficiency. Cancer Lett. 249, 271–282 (2007). [DOI] [PubMed] [Google Scholar]
- 39. Etienne‐Grimaldi, M.C. et al. A French prospective pilot study for identifying dihydropyrimidine dehydrogenase (DPD) deficiency in breast cancer patients receiving capecitabine. J. Clin. Oncol. 31, e13519 (2013). [Google Scholar]
- 40. Kristensen, M.H. , Pedersen, P. & Mejer, J. The value of dihydrouracil/uracil plasma ratios in predicting 5‐fluorouracilrelated toxicity in colorectal cancer patients. J. Int. Med. Res. 38, 1313–1323 (2010). [DOI] [PubMed] [Google Scholar]
- 41. Jiang, H. , Lu, J. , Jiang, J. & Hu, P. Important role of the dihydrouracil/uracil ratio in marked interpatient variations of fluoropyrimidine pharmacokinetics and pharmacodynamics. J. Clin. Pharmacol. 44, 1260–1272 (2004). [DOI] [PubMed] [Google Scholar]
- 42. Zhou, Z.W. et al. The dihydrouracil/uracil ratios in plasma and toxicities of 5‐fluorouracil‐based adjuvant chemotherapy in colorectal cancer patients. Chemotherapy 53, 127–131 (2007). [DOI] [PubMed] [Google Scholar]
- 43. Ben Fredj, R. , Gross, E. , Ben Ahmed, S. , Hassine, H. & Saguem, S. The dihydrouracil/uracil ratio in plasma, clinical and genetic analysis for screening of dihydropyrimidine dehydrogenase deficiency in colorectal cancer patients treated with 5‐fluorouracil. Pathol. Biol. 57, 470–476 (2009). [DOI] [PubMed] [Google Scholar]
- 44. French National Authority for Health . Screening for dihydropyrimidine dehydrogenase deficiency to decrease the risk of severe toxicities related to fluoropyrimidines (5‐fluorouracil or capecitabine. Ina Br <https://www.has‐sante.fr/jcms/c_2891090/fr/methodes‐de‐recherche‐d‐un‐deficit‐en‐dihydropyrimidine‐deshydrogenase‐visant‐a‐prevenir‐certaines‐toxicites‐severes‐associees‐aux‐traitements‐incluant‐une‐fluoropyrimidine‐5‐fluorouracile‐ou‐capecitabine> (2018). [Google Scholar]
- 45. Capitain, O. et al. Screening patients for fluoropyrimidine‐related toxicity risk: the most effective method to save lives <https://www.odpm.fr/uploads/rte/File/Poster%20ASCO%202019_FINAL‐1.pdf> (2019).
- 46. Coudoré, F. et al. Validation of an ultra‐high performance liquid chromatography tandem mass spectrometric method for quantifying uracil and 5,6‐dihydrouracil in human plasma. J. Chromatogr. Sci. 50, 877–884 (2012). [DOI] [PubMed] [Google Scholar]
- 47. Jacobs, B.A.W. et al. Journal of pharmaceutical and biomedical analysis development and validation of a rapid and sensitive UPLC – MS/MS method for determination of uracil and dihydrouracil in human plasma. J. Pharm. Biomed. Anal. 126, 75–82 (2016). [DOI] [PubMed] [Google Scholar]
- 48. Henricks, L.M. et al. Food‐effect study on uracil and dihydrouracil plasma levels as marker for dihydropyrimidine dehydrogenase activity in human volunteers. Br. J. Clin. Pharmacol. 84, 2761–2769 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Van Staveren, M.C. , Theeuwes‐Oonk, B. , Guchelaar, H.J. , Van Kuilenburg, A.B.P. & Maring, J.G. Pharmacokinetics of orally administered uracil in healthy volunteers and in DPD‐deficient patients, a possible tool for screening of DPD deficiency. Cancer Chemother. Pharmacol. 68, 1611–1617 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van Staveren, M.C. et al. Evaluation of an oral uracil loading test to identify DPD‐deficient patients using a limited sampling strategy. Br. J. Clin. Pharmacol. 81, 553–561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Kuilenburg, A.B.P. et al. Phenotypic and clinical implications of variants in the dihydropyrimidine dehydrogenase gene. Biochim. Biophys. Acta 1862, 754–762 (2016). [DOI] [PubMed] [Google Scholar]
- 52. Mattison, L.K. , Ezzeldin, H. , Carpenter, M. , Modak, A. , Johnson, M.R. & Diasio, R.B. Rapid identification of dihydropyrimidine dehydrogenase deficiency by using a novel 2–13C‐uracil breath test. Clin. Cancer Res. 10, 2652–2658 (2004). [DOI] [PubMed] [Google Scholar]
- 53. Opdam, F.L. , Modak, A.S. , Gelderblom, H. & Guchelaar, H.J. Breath tests to phenotype drug disposition in oncology. Clin. Pharmacokinet. 52, 919–926 (2013). [DOI] [PubMed] [Google Scholar]
- 54. Mattison, L.K. et al. The uracil breath test in the assessment of dihydropyrimidine dehydrogenase activity: Pharmacokinetic relationship between expired13CO2and plasma [2‐13C] dihydrouracil. Clin. Cancer Res. 12, 549–555 (2006). [DOI] [PubMed] [Google Scholar]
- 55. Cunha‐Junior, G.F. et al. 13C‐uracil breath test to predict 5‐fluorouracil toxicity in gastrointestinal cancer patients. Cancer Chemother. Pharmacol. 72, 1273–1282 (2013). [DOI] [PubMed] [Google Scholar]
- 56. Van Kuilenburg, A.B.P. , Van Lenthe, H. , Blom, M.J. , Mul, E.P.J. & Van Gennip, A.H. The activity of dihydropyrimidine dehydrogenase in human blood cells. Adv. Exp. Med. Biol. 431, 823–826 (1998). [DOI] [PubMed] [Google Scholar]
- 57. Chazal, M. , Etienne, M.C. , Renée, N. , Bourgeon, A. , Richelme, H. & Milano, G. Link between dihydropyrimidine dehydrogenase activity in peripheral blood mononuclear cells and liver. Clin. Cancer Res. 2, 507–510 (1996). [PubMed] [Google Scholar]
- 58. Van Kuilenburg, A.B.P. , De Abreu, R.A. & Van Gennip, A.H. Pharmacogenetic and clinical aspects of dihydropyrimidine dehydrogenase deficiency. Ann. Clin. Biochem. 40, 41–45 (2003). [DOI] [PubMed] [Google Scholar]
- 59. Milano, G. , Etienne, M.C. , Pierrefite, V. , Barberi‐Heyob, M. , Deporte‐Fety, R. & Renée, N. Dihydropyrimidine dehydrogenase deficiency and fluorouracil‐related toxicity. Br. J. Cancer 79, 627–630 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Henricks, L.M. et al. Treatment algorithm for homozygous or compound heterozygous DPYD variant allele carriers with low‐dose capecitabine. JCO Precis. Oncol. 1, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 61. Lostia, A.M. et al. A liquid chromatography‐tandem mass spectrometry method for the determination of 5‐fluorouracil degradation rate by intact peripheral blood mononuclear cells. Ther. Drug Monit. 31, 482–488 (2009). [DOI] [PubMed] [Google Scholar]
- 62. Mazzuca, F. et al. Pre‐treatment evaluation of 5‐fluorouracil degradation rate: association of poor and ultra‐rapid metabolism with severe toxicity in a colorectal cancer patients cohort. Oncotarget 7, 20612–20620 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Onesti, C.E. et al. 5‐Fluorouracil degradation rate could predict toxicity in stages II‐III colorectal cancer patients undergoing adjuvant FOLFOX. Anticancer Drugs 28, 322–326 (2016). [DOI] [PubMed] [Google Scholar]
- 64. Roberto, M. et al. Evaluation of 5‐fluorouracil degradation rate and pharmacogenetic profiling to predict toxicity following adjuvant capecitabine. Eur. J. Clin. Pharmacol. 73, 157–164 (2017). [DOI] [PubMed] [Google Scholar]
- 65. Borro, M. et al. Pre‐treatment assay of 5‐fluorouracil degradation rate (5‐ FUDR) to improve prediction of 5‐fluorouracil toxicity in gastroesophageal cancer. Oncotarget 8, 14050–14057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gamelin, E.C. et al. Intensity and therapeutic response in patients with advanced colorectal cancer receiving infusional therapy containing 5‐FU. Cancer 77, 441–451 (1996). [DOI] [PubMed] [Google Scholar]
- 67. Gamelin, E. et al. Individual fluorouracil dose adjustment based on pharmacokinetic follow‐up compared with conventional dosage: results of a multicenter randomized trial of patients with metastatic colorectal cancer. J. Clin. Oncol. 26, 2099–2105 (2008). [DOI] [PubMed] [Google Scholar]
- 68. Milano, G. et al. Relationship between fluorouracil systemic exposure and tumor response and patient survival. J. Clin. Oncol. 12, 1291–1295 (1994). [DOI] [PubMed] [Google Scholar]
- 69. Patel, J.N. et al. A community‐based multicenter trial of pharmacokinetically guided 5‐fluorouracil dosing for personalized colorectal cancer therapy. Oncologist 19, 959–965 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kaldate, R.R. , Haregewoin, A. , Grier, C.E. , Hamilton, S.A. & McLeod, H.L. Modeling the 5‐fluorouracil area under the curve versus dose relationship to develop a pharmacokinetic dosing algorithm for colorectal cancer patients receiving FOLFOX6. Oncologist 17, 296–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wilhelm, M. et al. Prospective, multicenter study of 5‐fluorouracil therapeutic drug monitoring in metastatic colorectal cancer treated in routine clinical practice. Clin. Colorectal Cancer 15, 381–388 (2016). [DOI] [PubMed] [Google Scholar]
- 72. Fety, R. et al. Clinical patients impact with of pharmacokinetically‐guided results locally from advanced a multicentric head dose randomized neck trial in of carcinomas. Clin. Cancer Res. 4, 2039–2045 (1998). [PubMed] [Google Scholar]
- 73. Gamelin, E.C. et al. Relationship between 5‐fluorouracil (5‐FU) dose intensity and therapeutic response in patients with advanced colorectal cancer receiving infusional therapy containing 5‐FU. Cancer 77, 441–451 (1996). [DOI] [PubMed] [Google Scholar]
- 74. Gamelin, E. et al. Long‐term weekly treatment of colorectal metastatic cancer with fluorouracil and leucovorin: Results of a multicentric prospective trial of fluorouracil dosage optimization by pharmacokinetic monitoring in 152 patients. J. Clin. Oncol. 16, 1470–1478 (1998). [DOI] [PubMed] [Google Scholar]
- 75. Goldstein, D.A. et al. Cost effectiveness analysis of pharmacokinetically‐guided 5‐fluorouracil in FOLFOX chemotherapy for metastatic colorectal cancer. Clin. Colorectal Cancer 13, 219–225 (2014). [DOI] [PubMed] [Google Scholar]
- 76. Bocci, G. et al. Comparative pharmacokinetic analysis of 5‐fluorouracil and its major metabolite 5‐fluoro‐5,6‐dihydrouracil after conventional and reduced test dose in cancer patients. Clin. Cancer Res. 6, 3032–3037 (2000). [PubMed] [Google Scholar]
- 77. Bocci, G. et al. A pharmacokinetic‐based test to prevent severe 5‐fluorouracil toxicity. Clin. Pharmacol. Ther. 80, 384–395 (2006). [DOI] [PubMed] [Google Scholar]
- 78. Arnold, A.P. , Chen, X. & Itoh, Y. Sex differences in drug therapy for oncologic diseases [Internet]. Sex Gender Diff. Pharmacol 214, 67–88 (2012). [Google Scholar]
- 79. Milano, G. et al. Influence of sex and age on fluorouracil clearance. J. Clin. Oncol. 10, 1171–1175 (1992). [DOI] [PubMed] [Google Scholar]
- 80. Mueller, F. et al. Gender‐specific elimination of continuous‐infusional 5‐fluorouracil in patients with gastrointestinal malignancies: results from a prospective population pharmacokinetic study. Cancer Chemother. Pharmacol. 71, 361–370 (2013). [DOI] [PubMed] [Google Scholar]
- 81. Stein, B.N. , Petrelli, N.J. , Douglass, H.O. , Driscoll, D.L. , Arcangeli, G. & Meropol, N.J. Age and sex are independent predictors of 5‐fluorouracil toxicity. Analysis of a large scale phase III trial. Cancer 75, 11–17 (1995). [DOI] [PubMed] [Google Scholar]
- 82. Sloan, B.J.A. , Loprinzi, C.L. , Novotny, P.J. , Okuno, S. , Nair, S. & Barton, D.L. Sex differences in fluorouracil‐induced stomatitis. J. Clin. Oncol. 18, 412–420 (2000). [DOI] [PubMed] [Google Scholar]
- 83. Sloan, J.A. et al. Women experience greater toxicity with fluorouracil‐based chemotherapy for colorectal cancer. J. Clin. Oncol. 20, 1491–1498 (2002). [DOI] [PubMed] [Google Scholar]
- 84. Wettergren, Y. , Carlsson, G. , Odin, E. & Gustavsson, B. Pretherapeutic uracil and dihydrouracil levels of colorectal cancer patients are associated with sex and toxic side effects during adjuvant 5‐fluorouracil–based chemotherapy. Cancer 118, 2953–3043 (2012). [DOI] [PubMed] [Google Scholar]
- 85. Abdel‐Rahman, O. Impact of sex on chemotherapy toxicity and efficacy among patients with metastatic colorectal cancer: pooled analysis of 5 randomized trials. Clin. Colorectal Cancer 18, 110–115.e2 (2019). [DOI] [PubMed] [Google Scholar]
- 86. Lim, H. et al. Sex‐dependent adverse drug reactions to 5‐fluorouracil in colorectal cancer. Biol. Pharm. Bull. 42, 594–600 (2019). [DOI] [PubMed] [Google Scholar]
- 87. Meulendijks, D. et al. Renal function, body surface area, and age are associated with risk of early‐onset fluoropyrimidine‐associated toxicity in patients treated with capecitabine‐based anticancer regimens in daily clinical care. Eur. J. Cancer 54, 120–130 (2016). [DOI] [PubMed] [Google Scholar]
- 88. Abdel‐Rahman, O. & Karachiwala, H. Impact of age on toxicity and efficacy of 5‐FU‐based combination chemotherapy among patients with metastatic colorectal cancer; a pooled analysis of five randomized trials. Int. J. Colorectal Dis. 34, 1741–1747 (2019). [DOI] [PubMed] [Google Scholar]
- 89. Gusella, M. , Toso, S. , Ferrazzi, E. , Ferrari, M. & Padrini, R. Relationships between body composition parameters and fluorouracil pharmacokinetics. Br. J. Clin. Pharmacol. 54, 131–139 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Prado, C.M.M. et al. Body composition as an independent determinant of 5‐fluorouracil‐based chemotherapy toxicity. Clin. Cancer Res. 13, 3264–3268 (2007). [DOI] [PubMed] [Google Scholar]
- 91. Williams, G.R. et al. The impact of skeletal muscle on the pharmacokinetics and toxicity of 5‐fluorouracil in colorectal cancer. Cancer Chemother. Pharmacol. 81, 413–417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kurk, S. et al. Skeletal muscle mass loss and dose‐limiting toxicities in metastatic colorectal cancer patients. J. Cachexia Sarcopenia Muscle 10, 803–813 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jung, H.W. et al. Effect of muscle mass on toxicity and survival in patients with colon cancer undergoing adjuvant chemotherapy. Support Care Cancer 23, 687–694 (2014). [DOI] [PubMed] [Google Scholar]
- 94. Cassidy, J. et al. First‐line oral capecitabine therapy in metastatic colorectal cancer: a favorable safety profile compared with intravenous 5‐fluorouracil/leucovorin. Ann. Oncol. 13, 566–575 (2002). [DOI] [PubMed] [Google Scholar]
- 95. Boisdron‐Celle, M. et al. Prevention of 5‐fluorouracil‐induced early severe toxicity by pre‐therapeutic dihydropyrimidine dehydrogenase deficiency screening: assessment of a multiparametric approach. Semin. Oncol. 44, 13–23 (2017). [DOI] [PubMed] [Google Scholar]
- 96. Etienne‐Grimaldi, M.C. et al. Prevention of 5‐fluorouracil‐induced early severe toxicity by pre‐therapeutic dihydropyrimidine dehydrogenase (DPD) deficiency screening: The multiparametric approach is not convincing. Semin. Oncol. 44, 13–23 (2017). [DOI] [PubMed] [Google Scholar]
- 97. Botticelli, A. et al. A nomogram to predict 5‐fluorouracil toxicity: when pharmacogenomics meets the patient. Anticancer Drugs 28, 551–556 (2017). [DOI] [PubMed] [Google Scholar]
- 98. Etienne‐Grimaldi, M.C. et al. FUSAFE individual patient data meta‐analysis (MA) to assess the performance of dihydropyrimidine dehydrogenase (DPD) gene polymorphisms for predicting grade 4–5 fluoropyrimidine (FP) toxicity (Abstract). Ann. Oncol. 30, 4–5 (2019). [Google Scholar]