

Abstract
Cyclobutanones hold a privileged role in enantioselective desymmetrization because their inherent ring strain allows for a variety of unusual reactions to occur. Current strategies include α‐functionalization, rearrangement, and C−C bond activation to directly convert cyclobutanones into a wide range of enantiomerically enriched compounds, including many biologically significant scaffolds. This Minireview provides an overview of state‐of‐the‐art methods that generate complexity from prochiral cyclobutanones in a single operation.
Keywords: C−C bond activation, cyclobutanone, desymmetrization, quaternary stereocenters
Take the strain: The ring strain of cyclobutanones makes them ideal for enantioselective desymmetrization. α‐Functionalization, rearrangement, and C−C bond activation can be used to directly convert cyclobutanones into enantiomerically enriched compounds, including many biologically significant scaffolds. This Minireview provides an overview of state‐of‐the‐art methods that generate complexity from prochiral cyclobutanones in a single operation.
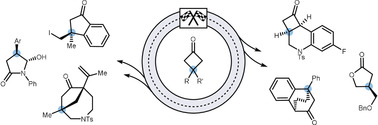
1. Introduction
Chirality not only adds a new dimension to the complexity of molecular frameworks, but it also provides a handle for interaction with large structural networks, which are found in all living organisms. [1] The asymmetric introduction of new stereocenters is considered one of the most stimulating challenges in organic chemistry, and has a great impact on related fields such as medicine and biology. [2] Point chirality, the most prominent element of chirality, is traditionally introduced by energetically delineating an addition to two prochiral faces of a planar entity (Scheme 1).[ 3 , 4 ] In an alternative approach, a symmetric three‐dimensional structure can be desymmetrized to establish a new stereocenter. [5] In terms of synthetic applicability, desymmetrization offers several advantages, as the type of reaction and the nature of the stereocenter are not interconnected and sterically congested stereocenters can be introduced at a distal, sterically accessible position of the molecule.
Scheme 1.
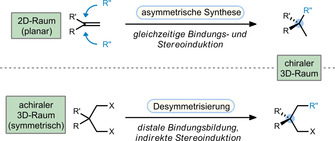
Establishing point chirality through asymmetric synthesis and desymmetrization of a prochiral molecule.
With these specific advantages in mind, desymmetrization of cyclobutanones offers a range of attractive features for method development. [6] For example, the ring strain [7] enables unusual and otherwise difficult reaction trajectories, including polymerizations. [8] Ring expansions are energetically downhill processes, and the corresponding five‐ and six‐membered rings are among the most prominent ring sizes found in nature. In addition, an exergonic energy profile provides several possibilities for the design of cascade reactions and the generation of multiple stereocenters in a single transformation. [9]
This vast build‐up of molecular complexity makes cyclobutanone desymmetrization an ideal tool for the synthesis of natural products.[ 5b , 5e ] Moreover, highly congested positions are addressable, including quaternary stereocenters. [5c] This Minireview is intended to highlight recent developments and stimulate synthetic applications of this powerful reaction ensemble.
2. α‐Functionalization
A viable method for cyclobutanone desymmetrization is α‐functionalization with an appropriate electrophile via formation of an initial enolate (Scheme 2, left). Such an endeavor leaves the core cyclobutanone motif intact and sets two consecutive stereocenters within the ring. Critical for reaction design is the high reactivity of the ketone function, [10] which poses an unusual challenge when compared to simple ketone functionalizations.
Scheme 2.
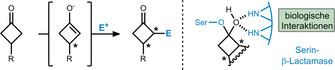
Left: Cyclobutanone desymmetrization by α‐functionalization. E=electrophile. Right: Mode of action of a cyclobutanone as a serine β‐lactamase inhibitor.
As a consequence of the inherent strain and the precise arrangement of substituents, cyclobutanones tend to form hemiketals, which are key for their activity as β‐lactamase inhibitors, as studied by Dmitrienko and co‐workers in 2008 (Scheme 2, right).[ 11 , 12 ] These predictable interactions with known biological residues provide further incentive for method development for the enantioselective synthesis of cyclobutanones in the future.
2.1. Aldol Reaction
Honda et al. conducted pioneering work related to enantioselective cyclobutanone desymmetrization through aldol reactions in the early 1990s (Scheme 3, top). [13] Their strategy was based on an enantioselective deprotonation of 3‐phenylcyclobutanone (1) using chiral lithium amide 2 at low temperatures. The chiral enolate was trapped with triethylsilyl (TES) chloride to give silyl enol ether 3 in an enantiomeric ratio (er) of 96:4. The addition of valeraldehyde and tetrabutylammonium fluoride (TBAF) led to the formation of aldol 4 as a mixture of diastereoisomers.
Scheme 3.
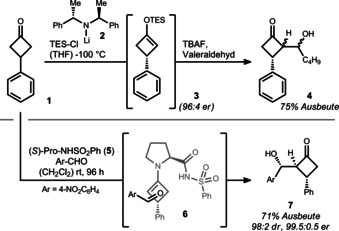
Top: The enantioselective deprotonation strategy of Honda. Bottom: The organocatalyzed approach of Aitken and Frongia.
In 2012, the groups of Aitken and Frongia developed an organocatalyzed desymmetrization of the same cyclobutanone 1 using proline‐based catalyst 5 to tackle previous issues regarding diastereoselectivity and cryogenic temperatures (Scheme 3, bottom). [14] Catalyst 5 has a dual role: it activates cyclobutanone 1 by formation of a chiral enamine, and orchestrates the approach of the aldehyde through hydrogen bonding (6 shows the preferred approach). Thus, good control of the diastereo‐ and enantioselectivity was achieved for activated aromatic aldehydes such as 4‐nitrobenzaldehyde. Product 7, containing three contiguous stereocenters, was isolated in 71 % yield, thus highlighting the fast assembly of complexity with this method. Later, the same groups were able to extend the scope to nitrostyrenes by switching to a different type of organocatalyst (not shown). [15]
2.2. α‐Arylation
Based on seminal work by the groups of Jia [16] and Britton, [17] Lu and co‐workers initiated a study towards the desymmetrization of cyclobutanones through intramolecular α‐arylation. [18] This strategy relies on synergistic palladium/enamine activation, which conveniently provides two handles for control of the enantioselectivity.
Cyclobutanone 8 with a pendent ortho‐bromoaryl group was treated with chiral phosphine 9 and chiral amine 10 under palladium catalysis (Scheme 4, top). The necessity of both chiral sources provides credibility for an intermediate such as enamine 11. For the final C−C bond formation to afford tricyclic cyclobutanone 12, the authors propose a Heck‐type mechanism.
Scheme 4.
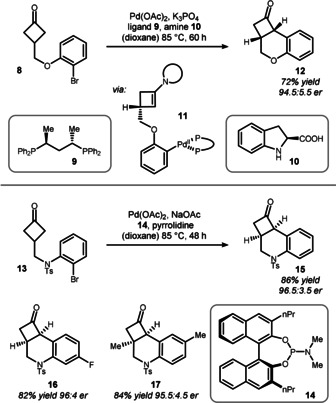
Synergistic palladium/enamine catalysis used by Lu. Ts=p‐toluenesulfonyl.
Interestingly, for starting materials bearing a nitrogen‐based linker such as 13, chiral information on the palladium center was sufficient to achieve good enantioselectivity (Scheme 4, bottom). The best ligand was found to be phosphoramidite 14, which allowed differently substituted arylcyclobutanones 15–17 to be accessed.
In 2019, Zhang, Dong, and co‐workers showed with a single example that a similar dual activation strategy was also viable for reductive Heck reactions to access formally α‐alkylated cyclobutanones. [19]
3. Rearrangement
Whereas ring contractions of cyclobutanones are energetically uphill processes, the opposite is true for ring expansions. The release of the inherent ring strain makes such an endeavor particularly favorable, and cyclobutanones are privileged structures for a range of ring extensions.
For example, Furstoss and co‐workers relied on enzymes from the fungus E. echinulata to conduct an asymmetric Baeyer–Villiger [20] reaction of cyclobutanone 18 (Scheme 5).[ 21 , 22 , 23 , 24 ] The authors then employed the corresponding lactone 19 in an enantiodivergent synthesis of (S)‐ and (R)‐proline.
Scheme 5.
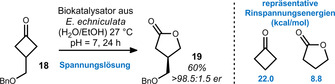
Biocatalytic desymmetrization of cyclobutanone 18 according to Furstoss. Bn=benzyl.
Interestingly, rearrangements of this type are not limited to enzymes, and the upcoming section will focus on non‐enzymatic methods. Since rearrangements are generally initiated upon orbital alignment, precise control of a substrate's conformation is key for achieving selectivity, and many solutions have been inspired by nature. The following section gives an overview of non‐enzymatic transformations.
3.1. Baeyer–Villiger Oxidation
Pioneering studies related to the asymmetric Baeyer–Villiger oxidation of cyclobutanones were conducted by the groups of Lopp, Bolm, and Kotsuki in the late 1990s and early 2000s. [25] As a result of their energetic bias, cyclobutanone desymmetrizations recently became a platform to test new strategies for chiral induction in Baeyer–Villiger‐type reactions. Scheme 6 summarizes recent methods that utilize different types of activation for the reaction of 3‐phenylcyclobutanone (1) to lactone 20.
Scheme 6.
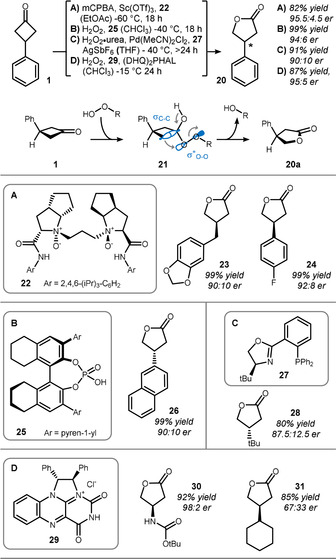
Stereoelectronic rationale behind the Baeyer–Villiger oxidation and recent examples of enantioinduction: A) Lewis acid catalyzed approach of Feng; B) organocatalyzed method of Ding using chiral phosphoric acids; C) transition‐metal‐catalyzed system of Stoltz; D) method of Yamamoto using a self‐assembled flavinium‐(DHQ)2PHAL ion pair. mCPBA=meta‐chloroperoxybenzoic acid, (DHQ)2PHAL=hydroquinine 1,4‐phthalazinediyl diether.
Mechanistically, a peroxide species adds to the electrophilic carbonyl group to form Criegee intermediate 21. Based on studies by the groups of Chandrasekhar, Kishi, and Calhoun, [26] rearrangement only happens when the σ‐C−C bond and the σ‐O−O bond are aligned in an antiperiplanar fashion, thereby providing numerous possibilities for catalyst design. Feng and co‐workers developed a Lewis acid based approach, in which chiral ligand 22 induces the respective enantioselectivity.[ 27 , 28 ] Structurally diverse lactones such as 23 and 24 were accessible by this sequence in ≥90:10 er (Scheme 6 A). Ding and co‐workers used BINOL‐derived phosphoric acid 25 as a bulky organocatalyst for their enantioinduction (Scheme 6 B). [29] Interestingly, hydrogen peroxide was reactive enough to furnish products such as naphtholactone 26 in good yield and selectivity. Cationic palladium bound to chiral PHOX ligand 27 is also viable, thereby allowing the assembly of alkyl lactones such as 28 (Scheme 6 C, Peterson and Stoltz).[ 30 , 31 ]
Yamamoto and co‐workers discovered that flavinium 29 self‐assembles with (DHQ)2PHAL to allow the enantioselective formation of carbamate 30 and aliphatic lactone 31 (Scheme 6 D).[ 32 , 33 ] The mechanism is most likely related to enzymatic processes, involving an initial addition of hydrogen peroxide to flavinium 29 from where it is then transferred to cyclobutanone 1. [34]
Small peptides with an embedded phosphothreonine unit are also capable catalysts for asymmetric Baeyer–Villiger oxidations. Their mode of action is closely related to BINOL‐derived phosphoric acid 25, but offers multiple sides for contact with substrates through hydrogen bonding, thus mimicking enzymatic processes. In 2019, Miller and co‐workers introduced oligopeptide 32 as a competent catalyst for such an endeavor (Scheme 7). [35] Depending on the position of the carbamate within the phenyl ring, substrates 33 and 34 underwent Baeyer–Villiger oxidation with reversal of the absolute configuration. Thus, R‐configured lactam 35 and S‐configured lactam 36 were accessible in excellent yield and good enantioselectivity with the same catalyst 32. On the basis of structure–selectivity studies, the authors propose hydrogen bonding between the acyl‐protected amine and the polar carbamate group to be crucial for this reversal. Thus, inherent structural biases were overridden and opportunities provided for the functionalization of more complex molecules in the future.
Scheme 7.
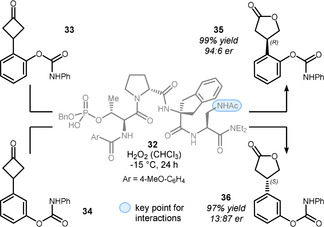
Miller's multifunctional peptide catalyst for the asymmetric Baeyer–Villiger oxidation.
3.2. Schmidt and Beckmann‐Type Reactions
The nitrogen analogues of the Baeyer–Villiger oxidation have been less studied in the context of cyclobutanone desymmetrization. Aubé and co‐workers developed an elegant approach for an asymmetric Schmidt reaction through the use of an “in situ tethering” strategy (Scheme 8, top). [36] Boron trifluoride initiates the formation of an oxocarbenium ion 37 from 1 and chiral azidopropanol 38. This highly electrophilic species is then intramolecularly trapped by the azide to give spirocycle 39. The chiral information at the six‐membered ring preferentially aligns one of the cyclobutanone's C−C bonds in an antiperiplanar relation to the leaving group. Thus, rearrangement to 40 and subsequent hydrolysis yields lactam 41. This study primarily focused on cyclohexanone desymmetrization, and a strong correlation between linker length and diastereoselectivity was found during the optimization. Only modest diastereoselectivity was observed for cyclobutanone 1 under these unoptimized conditions. Nevertheless, this investigation represents the first example of the formation of an asymmetric lactam from a cyclobutanone using a cleverly installed linker to initiate the attack of the poorly nucleophilic azide function.
Scheme 8.
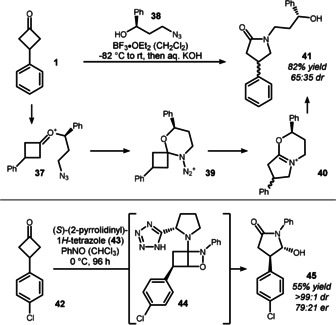
Top: The asymmetric Schmidt reaction developed by Aubé. Bottom: Approach towards enantioenriched lactams used by Piras and Frongia.
An interesting rearrangement to hydroxylactams was observed by Frongia and co‐workers when they treated 3‐chlorophenylcyclobutanone (42) with tetrazole catalyst 43 in the presence of nitrosobenzene (Scheme 8, bottom). [37] Initially, the authors aimed for α‐hydroxylation at the four‐membered ring, as seen in related organocatalytic reactions with unstrained ketones and aldehydes. [38] However, the cyclobutanone iminium collapses prior to hydrolysis and forms the corresponding bicycle 44, which ultimately reassembles to lactam 45. The authors were able to prove the structure and absolute configuration of 45 by a formal synthesis of the amino acid baclofen.
4. C−C Bond Cleavage
In general, C−C bonds are relatively inert to chemical transformations because of their high bond energy and steric inaccessibility. Nevertheless, C−C bonds have the potential to engage in reactions with transition metals under certain conditions and when appropriately activated. [39] The distortion of orbitals through molecular strain and the directing effects of adjacent functional groups enable transition metals to overcome the kinetic barrier and successfully initiate C−C bond cleavage (Figure 1). In addition to the kinetic barrier, thermodynamically favored processes that cleave C−C bonds are rare. One possibility to overcome this issue is through substrate design, as evident by a large number of “spring‐loaded” starting materials and/or intramolecular reactions.
Figure 1.
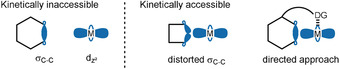
Kinetic barriers for interactions between σ‐C−C bonds and transition metals (M).
Cyclobutanones fulfill many of the aforementioned criteria for successful C−C bond cleavage. In addition to the incorporated molecular strain, the α‐C−C bond is further activated by the neighboring carbonyl group. In 1994, Ito and co‐workers were the first to report successful α‐C−C bond cleavage by treating alkyl cyclobutanones with Rh at elevated temperatures (Scheme 9). [40]
Scheme 9.
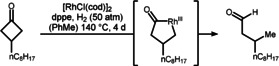
First report of transition‐metal‐catalyzed C−C bond cleavage of cyclobutanones. cod=1,5‐cycloctadiene, dppe=1,2‐bis(diphenylphosphino)ethane.
The prochiral nature of the two α‐C−C bonds in 3‐substituted cyclobutanones provides opportunities for enantioselective desymmetrization. In the past decade, various downstream reactions have been discovered that give rise to a diverse range of complex structures.
4.1. Rhodium Catalysis
Historically, rhodium was not only the first, but is also the most important transition metal for the cleavage of cyclobutanone C−C bonds. [41] In 2006, Murakami and co‐workers reported an enantioselective Rh‐catalyzed cyclobutanone desymmetrization in which SEGPHOS was utilized as the source of the enantioinduction (Scheme 10). [42] Initially, the transmetalation of boryl cyclobutanone 46 to rhodium delivers phenyl‐rhodium species 47, thereby placing the metal in proximity to the C−C bond of interest. The authors propose C−C bond cleavage to occur through a two‐step process involving the addition of the arylrhodium compound to the carbonyl group (47→48) and site‐selective elimination of a β‐carbon atom. Thus, chiral rhodacyclopentanone 49 is formed, which releases the product 50 after protonolysis. The authors were able to use this method to access cyclopentanone 51, which served as a starting point in an enantioselective synthesis of (−)‐herbertenol with a highly congested quaternary stereocenter.
Scheme 10.
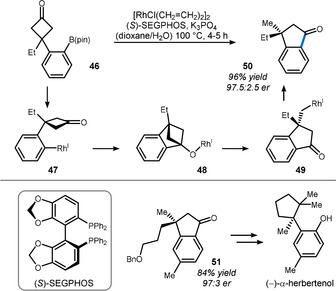
RhI‐catalyzed desymmetrization of arylcyclobutanone boronic esters according to Murakami.
Later, the same group expanded this reaction sequence to hydroxyphenylcyclobutanones such as 52, thereby enabling the synthesis of dihydrocoumarin 53 and analogues (Scheme 11). [43] The slightly modified BINAP derivative (S)‐Tol‐BINAP was found to be a superior candidate for enantioinduction in this case.
Scheme 11.
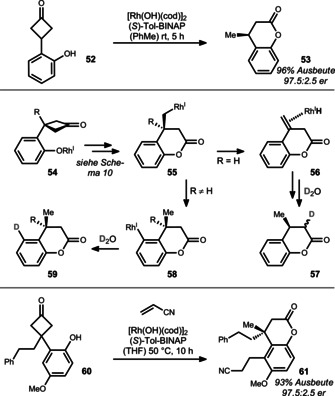
Method used by Murakami to access chiral dihydrocoumarins.
Detailed mechanistic studies through deuterium labeling uncovered the following mechanism: Phenoxyrhodium 54 undergoes an addition/β‐carbon elimination sequence that is closely related to that elucidated in Scheme 10 to give rhodadehydrocoumarin 55.
In the case of R=H, a sequence of β‐hydride elimination/re‐insertion outcompetes protoderhodation and thus enables the rhodium to migrate—notably with high face fidelity—to the thermodynamically favored enolate (sequence via intermediate 56). A final deuterium abstraction releases product 57. On the other hand, when R≠H, the rhodium migrates to the aryl group (55→58) instead, as indicated by the final position of the deuterium in product 59. Based on this mechanistic detail, the authors were able to replace the protoderhodation by another migratory insertion through addition of a Michael acceptor. In this remarkable cascade, cyclobutanone 60 was directly converted in 93 % yield and excellent enantioselectivity into polysubstituted coumarin 61 bearing a quaternary stereocenter.
In 2014, Cramer and co‐workers reported a formal C−C bond addition across an alkene (Scheme 12).[ 44 , 45 ] Mechanistically, precoordination of rhodium to styrenyl cyclobutanone 62 initiates oxidative addition to one of the enantiotopic C−C bonds of complex 63. The authors propose that the corresponding rhodacyclopentanone 64 undergoes migratory insertion to give bicyclic rhodacycle 65, followed by a final reductive elimination.
Scheme 12.
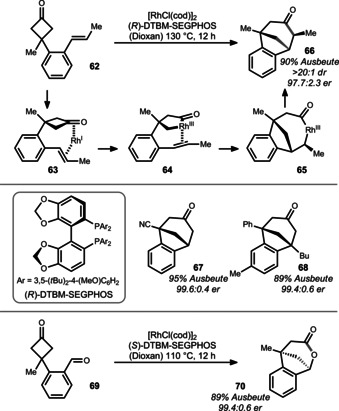
Approach used by Cramer to access benzocycloketones and lactones.
High stereospecificity together with low epimerization rates allow the synthesis of thermodynamically unfavored 66 in high diastereoselectivity. Consecutive treatment with base allowed epimerization to epi‐66. The overall high enantioselectivity was secured by DTBM‐SEGPHOS, thereby making optically active nitrile 67 and ketone 68 also readily available.
Interestingly, when aldehyde 69 was subjected to the reaction conditions at 110 °C, a formal alkylacylation was observed and the corresponding lactone 70 isolated. [46] The mechanism likely follows a similar course as the alkenyl case before. Overall, this method is very powerful as it establishes two stereocenters, both (if desired) quaternary, in a single step. In addition, the bicyclic ketone or lactone structures provide plenty of opportunities for further functionalizations.
In contrast to alkenes, allenes undergo intramolecular insertion into cyclobutanones in a [4+1] fashion (Scheme 13). [47] In their detailed study, Zhou and Dong propose an initial oxidative addition of cyclobutanone 71 to rhodium. Intermediate 72 then forms allylic rhodacycle 73, which is reluctant to undergo reductive elimination. Instead, β‐hydride elimination gives rhodium hydride 74, whose existence was corroborated by deuterium labeling of the methyl group. Finally, re‐insertion of either Rh−H or Rh−C followed by reductive elimination releases the [4.2.1] bicyclic structure 75. The authors also excluded an initial allene isomerization through control experiments. The excellent level of enantioselectivity (>99:1 er) can be ascribed to the addition of phosphoramidite 76 as a ligand. Structurally divergent products such as benzyl ether 77 and cyclopentene 78 were also well‐tolerated in this sequence.
Scheme 13.
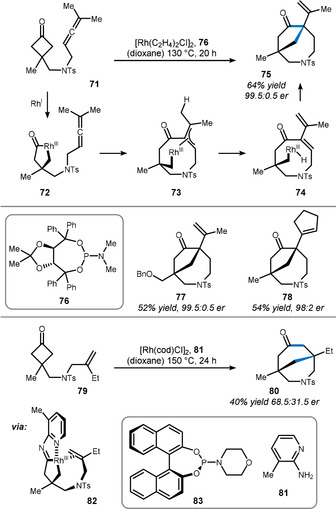
Asymmetric method used by Dong to access bridged ring systems.
In contrast to allenes, the insertion of unactivated alkenes such as 79 occurs in a [4+2] fashion to provide [3.3.1] bicycle 80, in close analogy to Cramer's study with styrenyl cyclobutanones. In this study, [48] Ko and Dong relied on the transient directing group 2‐aminopyridine 81 to assist with the oxidative addition. [49] In contrast to the previous examples, inferior precoordination and a relatively flexible alkene linker kinetically hinder the smooth oxidative addition. Thus, intermediate 82 is a likely intermediate en route to azepane 80. Even though ligand 83 only allowed for moderate enantioselectivity, this reaction represents a rare example of the successful utilization of an unactivated alkene in such a complex setting. In general, this type of [4+2] cyclobutanone–alkene fusion resembles a complementary approach to the intramolecular Diels–Alder‐type disconnection, further highlighting its importance.
4.2. Nickel Catalysis
Nickel often exhibits complemental reactivity to rhodium and has become important for the enantioselective desymmetrization of cyclobutanones. For example, styrenyl cyclobutanone 84 undergoes a different type of skeletal rearrangement (Scheme 14), as seen previously for rhodium in Scheme 12. The mechanistic rationale behind this divergence is based on metal‐specific preferences for oxidative addition versus oxidative cyclization.[ 50 , 51 ] Thus, a plausible mechanism comprises precoordination of nickel(0) to the C=C and C=O π‐bonds (intermediate 85) triggering oxidative cyclization to oxa‐nickelacycle 86. [52] It is important to note that this step establishes benzylic stereocenters, which only render selective β‐carbon elimination to 87 possible. In this study by Murakami and co‐workers, phosphoramidite 88 enables selective cyclization by energetically discriminating one of the prochiral faces of the alkene to bind to nickel.
Scheme 14.
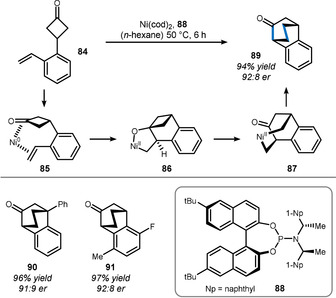
Method used by Murakami to access chiral benzobicyclo[2.2.2]octenones.
Thus, excellent enantioselectivity was achieved and bicyclo[2.2.2]octenone 89 was isolated in 94 % yield. Quaternary stereocenters (phenyloctenone 90) as well as substitution at the ring (fluoroarene 91) were well‐tolerated, thus making this procedure valuable—particularly because of the importance of bicyclo[2.2.2]octanols as calcium channel blockers. [53]
A similar metal‐dependent regiodivergence was observed by Zhou and Dong when allenyl cyclobutanone 71 was subjected to nickel(0) instead of rhodium (Scheme 15). [54] Here, a sequence of oxidative cyclization (71→92) and β‐carbon elimination (92→93) explains the formal [4+2] cyclization to azepane 94 (versus the [4+1] cyclization in Scheme 13). In this study, ligand 76 gave the best enantioselectivity, as illustrated through the differently decorated products 95, 96, and 97. Overall, this example highlights how achiral starting material 71 serves as a precursor for the fast assembly of constitutional isomers 75 and 94 with multiple stereocenters.
Scheme 15.
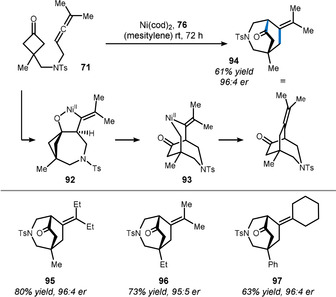
Nickel‐catalyzed desymmetrization using allenes according to Dong.
4.3. Palladium Catalysis
Given the plethora of data on palladium‐catalyzed C−C bond formation, [55] it is surprising how little is known about C−C bond cleavage, specifically in the context of cyclobutanones. [56] Recently, the Xu group developed an enantioselective σ‐bond reshuffling for cyclobutanones (Scheme 16; e.g. using cyclobutanone 98). [57] Enantioinduction was provided in this reaction by TADDOL‐derived phosphoramidite 99 a. Mechanistically, the authors originally proposed a reaction sequence closely related to the rhodium example in Scheme 10. [56a] DFT calculations suggest an alternative mechanism in which all the common oxidation states of palladium are energetically favored. [56b] Herein, aryl iodide 98 oxidatively adds to Pd0 to form arylpalladium iodide 100.
Scheme 16.
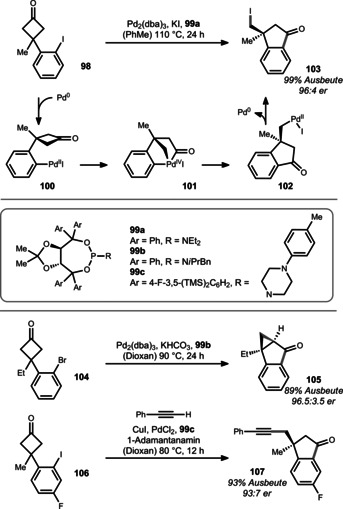
Cascade used by Xu towards benzocyclopentanones bearing a quaternary stereocenter. dba=dibenzylidene, TMS=trimethylsilyl.
Oxidative addition to the α‐C−C bond of the cyclobutanone subunit leads to palladium(IV) species 101, which subsequently undergoes reductive elimination to cyclopentanone 102. The neopentylic palladium species is prone to a second reductive elimination to form the C−I bond of product 103. The authors found that the same product 103 could also be accessed from an aryl bromide substrate by addition of potassium iodide through an in situ bromine–iodine exchange. When no iodide was present (e.g. with bromide 104 and ligand 99 b), reductive elimination occurred from an intramolecular carbon‐bound enolate to furnish cyclopropane 105 in excellent yield. Interestingly, intermediate 102 could also undergo Suzuki‐ and Sonogashira‐type coupling when the appropriate nucleophile was added and a slightly modified ligand used. An illustrative reaction is the conversion of fluoroaryl cyclobutanone 106 into cyclopentanone 107 using ligand 99 c, which establishes a quaternary stereocenter with good efficiency. The flexibility of palladium to allow the formation of different products is remarkable and highlights its ability for divergent reaction development.
5. Conclusions
Cyclobutanones are unique building blocks that allow a variety of unusual reactions and the generation of vast molecular complexity. The combination of the overall downhill energy profile and enantioselective desymmetrization enables multiple stereocenters to be accessed in a single operation. The asymmetric functionalization of cyclobutanones provides rigid 3D structures that are interesting as potential drug candidates in medicinal chemistry. Whereas the Baeyer–Villiger rearrangement has been studied extensively, asymmetric versions of the nitrogen and carbon analogues are mostly unknown. The recent introduction of alternative transition metals for C−C bond cleavage opens new avenues for divergent syntheses, and many metals have never been studied in this context. The same holds true for β‐C−C bond breaking and ring‐opening reactions, which could also be powerful approaches. Inspiration for synthetic endeavors, especially in terms of total synthesis, might be worth considering, since breaking the symmetry enables the sophisticated and atom‐economic construction of molecular frameworks.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Jan Sietmann obtained his B.Sc. in chemistry from the Westfälische Wilhelms‐Universität Münster (2016). During his Master studies in 2017 he worked at Armacell GmbH, where he focused on the storage stability of adhesives. He completed his M.Sc. on the synthesis of a ganglioside in the group of Prof. Ryan Gilmour in Münster in 2018. He is currently a graduate student in the group of Dr. Johannes Wahl, where he is working on the enantioselective ring expansion of cyclobutanones.

Biographical Information
Johannes M. Wahl obtained his M.Sc. in 2012 from the University of Basel, Switzerland. After research with Prof. Donna G. Blackmond at The Scripps Research Institute, he moved to Technische Universität München to pursue his Ph.D. with Prof. Thorsten Bach. After obtaining his Ph.D. in 2016 and a subsequent postdoctoral appointment with Prof. M. Kevin Brown at Indiana University, he started his independent career as a junior research group leader at the Westfälische Wilhelms‐Universität Münster in 2019. His research focuses on strain‐mediated reaction development and application in natural product synthesis.

Acknowledgements
We acknowledge generous financial support from the Westfälische Wilhelms‐Universität Münster and the Fonds der chemischen Industrie (Liebig fellowship to J.M.W.). Open access funding enabled and organized by Projekt DEAL.
J. Sietmann, J. M. Wahl, Angew. Chem. Int. Ed. 2020, 59, 6964.
References
- 1.For a stimulating review on chirality, see Cintas P., Angew. Chem. Int. Ed. 2007, 46, 4016–4024; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 4090–4099. [Google Scholar]
- 2.
- 2a. Crossley R., Tetrahedron 1992, 48, 8155–8178; [Google Scholar]
- 2b. Brooks W. H., Guida W. C., Daniel K. G., Curr. Top. Med. Chem. 2011, 11, 760–770; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Cintas P., Biochirality: Origins, Evolution and Molecular Recognition, Vol. 1, Springer, Berlin, Heidelberg, 2013. [Google Scholar]
- 3.
- 3a. Seebach D., Prelog V., Angew. Chem. Int. Ed. Engl. 1982, 21, 654–660; [Google Scholar]; Angew. Chem. 1982, 94, 696–702; [Google Scholar]
- 3b. Christmann M., Bräse S., Asymmetric Synthesis: The Essentials, Vol. 2, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 4.For terminology, see
- 4a. Prelog V., Helmchen G., Angew. Chem. Int. Ed. Engl. 1982, 21, 567–583; [Google Scholar]; Angew. Chem. 1982, 94, 614–631; [Google Scholar]
- 4b. Mislow K., Siegel J., J. Am. Chem. Soc. 1984, 106, 3319–3328; [Google Scholar]
- 4c. Moss G. P., Pure Appl. Chem. 1996, 68, 2193–2222. [Google Scholar]
- 5.For desymmetrization as a tool in synthesis, see
- 5a. Willis M. C., J. Chem. Soc. Perkin Trans. 1 1999, 1765–1784; [Google Scholar]
- 5b. Wang M., Feng M., Tang B., Jiang X., Tetrahedron Lett. 2014, 55, 7147–7155; [Google Scholar]
- 5c. Zeng X.-P., Cao Z.-Y., Wang Y.-H., Zhou F., Zhou J., Chem. Rev. 2016, 116, 7330–7396; [DOI] [PubMed] [Google Scholar]
- 5d. Borissov A., Davies T. Q., Ellis S. R., Fleming T. A., Richardson M. S. W., Dixon D. J., Chem. Soc. Rev. 2016, 45, 5474–5540; [DOI] [PubMed] [Google Scholar]
- 5e. Merad J., Candy M., Pons J.-M., Bressy C., Synthesis 2017, 49, 1938–1954. [Google Scholar]
- 6.For the synthetic utility of cyclobutane derivatives, see
- 6a. Belluš D., Ernst B., Angew. Chem. Int. Ed. Engl. 1988, 27, 797–827; [Google Scholar]; Angew. Chem. 1988, 100, 820–850; [Google Scholar]
- 6b. Namyslo J. C., Kaufmann D. E., Chem. Rev. 2003, 103, 1485–1538; [DOI] [PubMed] [Google Scholar]
- 6c. Salaün J., in Science of Synthesis, Vol. 26 (Ed.: Cossy J.), Thieme, Stuttgart, 2004, pp. 557–606; [Google Scholar]
- 6d. Seiser T., Saget T., Tran D. N., Cramer N., Angew. Chem. Int. Ed. 2011, 50, 7740–7752; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7884–7896; [Google Scholar]
- 6e. Secci F., Frongia A., Piras P. P., Molecules 2013, 18, 15541–15572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.For a brief overview of molecular strain, see
- 7a. Liebman J. F., Greenberg A., Chem. Rev. 1976, 76, 311–365; [Google Scholar]
- 7b. Wiberg K. B., Angew. Chem. Int. Ed. Engl. 1986, 25, 312–322; [Google Scholar]; Angew. Chem. 1986, 98, 312–322. [Google Scholar]
- 8. Duda A., Kowalski A., in Handbook of Ring-Opening Polymerization, Vol. 1 (Eds: Dubois P., Coulembier O., Raquez J.-M.), Wiley-VCH, Weinheim, 2009, pp. 1–45. [Google Scholar]
- 9.For a review on cascade reactions, see Tietze L. F., Beifuss U., Angew. Chem. Int. Ed. Engl. 1993, 32, 131–163; [Google Scholar]; Angew. Chem. 1993, 105, 137–170. [Google Scholar]
- 10. Li Z., Jangra H., Chen Q., Mayer P., Ofial A. R., Zipse H., Mayr H., J. Am. Chem. Soc. 2018, 140, 5500–5515. [DOI] [PubMed] [Google Scholar]
- 11. Johnson J. W., Evanoff D. P., Savard M. E., Lange G., Ramadhar T. R., Assoud A., Taylor N. J., Dmitrienko G. I., J. Org. Chem. 2008, 73, 6970–6982. [DOI] [PubMed] [Google Scholar]
- 12.For a review on the topic, see Devi P., Rutledge P. J., ChemBioChem 2017, 18, 338–351. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Honda T., Kimura N., Tsubuki M., Tetrahedron: Asymmetry 1993, 4, 1475–1478; [Google Scholar]
- 13b. Honda T., Kimura N., J. Chem. Soc. Chem. Commun. 1994, 77–78; [Google Scholar]
- 13c. Honda T., Kimura N., Sato S., Kato D., Tominaga H., J. Chem. Soc. Perkin Trans. 1 1994, 1043–1046. [Google Scholar]
- 14. Aitken D. J., Bernard A. M., Capitta F., Frongia A., Guillot R., Ollivier J., Piras P. P., Secci F., Spiga M., Org. Biomol. Chem. 2012, 10, 5045–5048. [DOI] [PubMed] [Google Scholar]
- 15. Capitta F., Frongia A., Ollivier J., Aitken D. J., Secci F., Pirasa P. P., Guillot R., Synlett 2015, 26, 123–126. [Google Scholar]
- 16. Liu R.-R., Li B.-L., Lu J., Gao J.-R., Jia Y.-X., J. Am. Chem. Soc. 2016, 138, 5198–5201. [DOI] [PubMed] [Google Scholar]
- 17. Chang S., Holmes M., Mowat J., Meanwell M., Britton R., Angew. Chem. Int. Ed. 2017, 56, 748–752; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 766–770. [Google Scholar]
- 18. Wang M., Chen J., Chen Z., Zhong C., Lu P., Angew. Chem. Int. Ed. 2018, 57, 2707–2711; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 2737–2741. [Google Scholar]
- 19. Shen H.-C., Zhang L., Chen S.-S., Feng J., Zhang B.-W., Zhang Y., Zhang X., Wu Y.-D., Gong L.-Z., ACS Catal. 2019, 9, 791–797. [Google Scholar]
- 20.For the historic context, see
- 20a. Baeyer A., Villiger V., Ber. Dtsch. Chem. Ges. 1899, 32, 3625–3633; [Google Scholar]
- 20b. Friess S. L., Frankenburg P. E., J. Am. Chem. Soc. 1952, 74, 2679–2680. [Google Scholar]
- 21.
- 21a. Gagnon R., Grogan G., Groussain E., Pedragosa-Moreau S., Richardson P. F., Roberts S. M., Willetts A. J., Alphand V., Lebreton J., Furstoss R., J. Chem. Soc. Perkin Trans. 1 1995, 2527–2528; [Google Scholar]
- 21b. Mazzini C., Lebreton J., Alphand V., Furstoss R., J. Org. Chem. 1997, 62, 5215–5218. [Google Scholar]
- 22.For related biocatalytic examples, see
- 22a. Mihovilovic M. D., Rudroff F., Grötzl B., Kapitan P., Snajdrova R., Rydz J., Mach R., Angew. Chem. Int. Ed. 2005, 44, 3609–3613; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 3675–3679; [Google Scholar]; Angew. Chem. 2005, 117, 3675–3679; [Google Scholar]
- 22b. Mihovilovic M. D., Rudroff F., Winninger A., Schneider T., Schulz F., Reetz M. T., Org. Lett. 2006, 8, 1221–1224; [DOI] [PubMed] [Google Scholar]
- 22c. Torres Pazmiño D., Snajdrova R., Baas B., Ghobrial M., Mihovilovic M., Fraaije M., Angew. Chem. Int. Ed. 2008, 47, 2275–2278; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2307–2310; [Google Scholar]
- 22d. Rudroff F., Rydz J., Ogink F. H., Fink M., Mihovilovic M. D., Adv. Synth. Catal. 2007, 349, 1436–1444; [Google Scholar]
- 22e. Rial D. V., Bianchi D. A., Kapitanova P., Lengar A., van Beilen J. B., Mihovilovic M. D., Eur. J. Org. Chem. 2008, 1203–1213; [Google Scholar]
- 22f. Rodríguez-Mata M., Lavandera I., Gotor-Fernández V., Gotor V., García-Cerrada S., Mendiola J., de Frutos Ó., Collado I., Tetrahedron 2016, 72, 7268–7275. [Google Scholar]
- 23.For representative strain energies, see
- 23a. Wolf G., Helv. Chim. Acta 1972, 55, 1446–1459; [Google Scholar]
- 23b. Benson S. W., Thermochemical Kinetics, Vol. 2, Wiley, New York, 1976; [Google Scholar]
- 23c. Wiberg K. B., Waldron R. F., J. Am. Chem. Soc. 1991, 113, 7697–7705. [Google Scholar]
- 24.The strain energy of cyclobutanone was determined using the increment theory developed by the Benson group with the respective heat of formation extracted from Wolf's report (Ref. [23a,b]).
- 25.For seminal examples, see
- 25a. Lopp M., Paju A., Pehk T., Tetrahedron Lett. 1996, 37, 7583–7586; [Google Scholar]
- 25b. Bolm C., Khanh Luong T. K., Schlingloff G., Synlett 1997, 10, 1151–1152; [Google Scholar]
- 25c. Shinohara T., Fujioka S., Kotsuki H., Heterocycles 2001, 55, 237–242. [Google Scholar]
- 26.
- 26a. Chandrasekhar S., Roy C. D., Tetrahedron Lett. 1987, 28, 6371–6372; [Google Scholar]
- 26b. Chandrasekhar S., Roy C. D., J. Chem. Soc. Perkin Trans. 2 1994, 2141–2143; [Google Scholar]
- 26c. Goodman R. M., Kishi Y., J. Am. Chem. Soc. 1998, 120, 9392–9393; [Google Scholar]
- 26d. Crudden C. M., Chen A. C., Calhoun L. A., Angew. Chem. Int. Ed. 2000, 39, 2851–2855; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2973–2977; [Google Scholar]
- 26e. Vil′ V. A., dos Passos Gomes G., Bityukov O. V., Lyssenko K. A., Nikishin G. I., Alabugin I. V., Terent′ev A. O., Angew. Chem. Int. Ed. 2018, 57, 3372–3376; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3430–3434. [Google Scholar]
- 27. Zhou L., Liu X., Ji J., Zhang Y., Hu X., Lin L., Feng X., J. Am. Chem. Soc. 2012, 134, 17023–17026. [DOI] [PubMed] [Google Scholar]
- 28.For a related study on Lewis acid activation, see Drożdż A., Foreiter M. B., Chrobok A., Synlett 2014, 25, 559–563. [Google Scholar]
- 29.
- 29a. Xu S., Wang Z., Zhang X., Zhang X., Ding K., Angew. Chem. Int. Ed. 2008, 47, 2840–2843; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2882–2885; [Google Scholar]
- 29b. Xu S., Wang Z., Zhang X., Ding K., Chin. J. Chem. 2010, 28, 1731–1735; [Google Scholar]
- 29c. Xu S., Wang Z., Li Y., Zhang X., Wang H., Ding K., Chem. Eur. J. 2010, 16, 3021–3035. [DOI] [PubMed] [Google Scholar]
- 30. Petersen K. S., Stoltz B. M., Tetrahedron 2011, 67, 4352–4357. [Google Scholar]
- 31.For related studies using transition metals, see
- 31a. Malkov A. V., Friscourt F., Bell M., Swarbrick M. E., Kočovský P., J. Org. Chem. 2008, 73, 3996–4003; [DOI] [PubMed] [Google Scholar]
- 31b. Cavarzan A., Bianchini G., Sgarbossa P., Lefort L., Gladiali S., Scarso A., Strukul G., Chem. Eur. J. 2009, 15, 7930–7939. [DOI] [PubMed] [Google Scholar]
- 32. Poudel P. P., Arimitsu K., Yamamoto K., Chem. Commun. 2016, 52, 4163–4166. [DOI] [PubMed] [Google Scholar]
- 33.For related flavin-based studies, see
- 33a. Murahashi S.-I., Ono S., Imada Y., Angew. Chem. Int. Ed. 2002, 41, 2366–2368; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2472–2474; [Google Scholar]
- 33b. Riebel A., Fink M. J., Mihovilovic M. D., Fraaije M. W., ChemCatChem 2014, 6, 1112–1117; [Google Scholar]
- 33c. Arakawa Y., Yamanomoto K., Kita H., Minagawa K., Tanaka M., Haraguchi N., Itsuno S., Imada Y., Chem. Sci. 2017, 8, 5468–5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.For mechanistic insights, see
- 34a. Walsh C., Acc. Chem. Res. 1980, 13, 148–155; [Google Scholar]
- 34b. Bruice T. C., Acc. Chem. Res. 1980, 13, 256–262; [Google Scholar]
- 34c. Murahashi S., Oda T., Masui Y., J. Am. Chem. Soc. 1989, 111, 5002–5003; [Google Scholar]
- 34d. Kamerbeek N. M., Janssen D. B., van Berkel W. J. H., Fraaije M. W., Adv. Synth. Catal. 2003, 345, 667–678. [Google Scholar]
- 35. Featherston A. L., Shugrue C. R., Mercado B. Q., Miller S. J., ACS Catal. 2019, 9, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Sahasrabudhe K., Gracias V., Furness K., Smith B. T., Katz C. E., Reddy D. S., Aubé J., J. Am. Chem. Soc. 2003, 125, 7914–7922; [DOI] [PubMed] [Google Scholar]
- 36b. Ribelin T. P., Aubé J., Nat. Protoc. 2008, 3, 137–143; [DOI] [PubMed] [Google Scholar]
- 36c. Aubé J., Wang Y., Ghosh S., Langhans K. L., Synth. Commun. 1991, 21, 693–701. [Google Scholar]
- 37. Capitta F., Frongia A., Ollivier J., Piras P. P., Secci F., Synlett 2011, 1, 89–93. [Google Scholar]
- 38.For selected examples, see
- 38a. Brown S. P., Brochu M. P., Sinz C. J., MacMillan D. W. C., J. Am. Chem. Soc. 2003, 125, 10808–10809; [DOI] [PubMed] [Google Scholar]
- 38b. Momiyama N., Torii H., Saito S., Yamamoto H., Proc. Natl. Acad. Sci. USA 2004, 101, 5374–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.For an overview, see
- 39a. Seiser T., Cramer N., Org. Biomol. Chem. 2009, 7, 2835–2840; [DOI] [PubMed] [Google Scholar]
- 39b. Souillart L., Cramer N., Chem. Rev. 2015, 115, 9410–9464; [DOI] [PubMed] [Google Scholar]
- 39c. Murakami M., Ishida N., J. Am. Chem. Soc. 2016, 138, 13759–13769; [DOI] [PubMed] [Google Scholar]
- 39d. Fumagalli G., Stanton S., Bower J. F., Chem. Rev. 2017, 117, 9404–9432; [DOI] [PubMed] [Google Scholar]
- 39e. Chen P.-H., Billett B. A., Tsukamoto T., Dong G., ACS Catal. 2017, 7, 1340–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murakami M., Amii H., Ito Y., Nature 1994, 370, 540–541. [Google Scholar]
- 41.For reviews on rhodium-catalyzed ring cleavage, see
- 41a. Winter C., Krause N., Angew. Chem. Int. Ed. 2009, 48, 2460–2462; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 2497–2499; [Google Scholar]
- 41b. Murakami M., Chem. Rec. 2010, 10, 326–331; [DOI] [PubMed] [Google Scholar]
- 41c. Souillart L., Cramer N., Chimia 2015, 69, 187–190; [DOI] [PubMed] [Google Scholar]
- 41d. Kondo T., Eur. J. Org. Chem. 2016, 1232–1242; [Google Scholar]
- 41e. Shaw M. H., Bower J. F., Chem. Commun. 2016, 52, 10817–10829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Matsuda T., Shigeno M., Makino M., Murakami M., Org. Lett. 2006, 8, 3379–3381; [DOI] [PubMed] [Google Scholar]
- 42b. Matsuda T., Shigeno M., Makino M., Murakami M., Org. Lett. 2004, 6, 1257–1259. [DOI] [PubMed] [Google Scholar]
- 43. Matsuda T., Shigeno M., Murakami M., J. Am. Chem. Soc. 2007, 129, 12086–12087. [DOI] [PubMed] [Google Scholar]
- 44. Souillart L., Parker E., Cramer N., Angew. Chem. Int. Ed. 2014, 53, 3001–3005; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3045–3049. [Google Scholar]
- 45.See also: Xu T., Min Ko H., Savage N. A., Dong G., J. Am. Chem. Soc. 2012, 134, 20005–20008. [DOI] [PubMed] [Google Scholar]
- 46. Souillart L., Cramer N., Angew. Chem. Int. Ed. 2014, 53, 9640–9644; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9794–9798. [Google Scholar]
- 47. Zhou X., Dong G., J. Am. Chem. Soc. 2015, 137, 13715–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ko H. M., Dong G., Nat. Chem. 2014, 6, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.For seminal examples using the transient directing group aminopicoline, see
- 49a. Suggs J. W., J. Am. Chem. Soc. 1979, 101, 489; [Google Scholar]
- 49b. Jun C.-H., Lee D.-Y., Lee H., Hong J.-B., Angew. Chem. Int. Ed. 2000, 39, 3070–3072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3214–3216. [Google Scholar]
- 50.For a review, see Montgomery J. M., Angew. Chem. Int. Ed. 2004, 43, 3890–3908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3980–3998. [Google Scholar]
- 51.For selected examples, see
- 51a. Hratchian H. P., Chowdhury S. K., Gutiérrez-García V. M., Amarasinghe K. K. D., Heeg M. J., Schlegel H. B., Montgomery J., Organometallics 2004, 23, 4636–4646; [Google Scholar]
- 51b. Murakami M., Ashida S., Matsuda T., J. Am. Chem. Soc. 2005, 127, 6932–6933; [DOI] [PubMed] [Google Scholar]
- 51c. Murakami M., Ashida S., Matsuda T., J. Am. Chem. Soc. 2006, 128, 2166–2167. [DOI] [PubMed] [Google Scholar]
- 52.
- 52a. Liu L., Ishida N., Murakami M., Angew. Chem. Int. Ed. 2012, 51, 2485–2488; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2535–2538; [Google Scholar]
- 52b. Murakami M., Ashida S., Chem. Commun. 2006, 4599–4601. [DOI] [PubMed] [Google Scholar]
- 53.For a patent on the importance of bicyclo[2.2.2]octanols, see Hilpert K., Hubler F., Renneberg D. (Actelion Pharmaceuticals Ltd), WO2010046855 A1, 2010.
- 54. Zhou X., Dong G., Angew. Chem. Int. Ed. 2016, 55, 15091–15095; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15315–15319. [Google Scholar]
- 55.For an overview, see Johansson Seechurn C. C. C., Kitching M. O., Colacot T. J., Snieckus V., Angew. Chem. Int. Ed. 2012, 51, 5062–5085; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5150–5174. [Google Scholar]
- 56.For selected examples, see
- 56a. Matsuda T., Shigeno M., Murakami M., Org. Lett. 2008, 10, 5219–5221; [DOI] [PubMed] [Google Scholar]
- 56b. Ishida N., Ikemoto W., Murakami M., Org. Lett. 2012, 14, 3230–3232; [DOI] [PubMed] [Google Scholar]
- 56c. Ishida N., Ikemoto W., Murakami M., J. Am. Chem. Soc. 2014, 136, 5912–5915; [DOI] [PubMed] [Google Scholar]
- 56d. Okumura S., Sun F., Ishida N., Murakami M., J. Am. Chem. Soc. 2017, 139, 12414–12417. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Cao J., Chen L., Sun F.-N., Sun Y.-L., Jiang K.-Z., Yang K.-F., Xu Z., Xu L.-W., Angew. Chem. Int. Ed. 2019, 58, 897–901; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 907–911; [Google Scholar]
- 57b. Sun Y.-L., Wang X.-B., Sun F.-N., Chen Q.-Q., Cao J., Xu Z., Xu L.-W., Angew. Chem. Int. Ed. 2019, 58, 6747–6751; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6819–6823; [Google Scholar]
- 57c. Sun F.-N., Yang W.-C., Chen X.-B., Sun Y.-L., Cao J., Xu Z., Xu L.-W., Chem. Sci. 2019, 10, 7579–7583. [DOI] [PMC free article] [PubMed] [Google Scholar]