

Abstract
Composite lymphoma is the rare simultaneous manifestation of two distinct lymphomas. Chronic lymphocytic leukemia (CLL) has a propensity for occurring in composite lymphomas, a phenomenon that remains to be elucidated. We applied cytogenetics, droplet digital polymerase chain reaction, and massively parallel sequencing to analyze longitudinally a patient with CLL, who 3 years later showed transformation to a hairy cell leukemia‐variant (HCL‐V). Outgrowth of the IGHV4‐34‐positive HCL‐V clone at the expense of the initially dominant CLL clone with trisomy 12 and MED12 mutation started before CLL‐guided treatment and was accompanied by a TP53 mutation, which was already detectable at diagnosis of CLL. Furthermore, deep sequencing of IGH showed a composite lymphoma with presence of both disease components at all analyzed timepoints (down to a minor clone: major clone ratio of ~1:1000). Overall, our analyses showed a disease course that resembled clonal dynamics reported for malignancies with intratumoral heterogeneity and illustrate the utility of deep sequencing of IGH to detect distinct clonal populations at diagnosis, monitor clonal response to therapy, and possibly improve clinical outcomes.
Keywords: CLL, clonal dynamics, composite lymphoma, HCL‐V, IGHV
1. INTRODUCTION
Chronic lymphocytic leukemia (CLL) is characterized by clonal expansion of mature B‐cells and is the most common leukemia in Western countries. The clinical course is often indolent and in a large proportion of patients, surveillance may suffice for many years before treatment is indicated. 1 Nonetheless, CLL is accompanied by perturbations of the immune system that potentially contribute to the development of secondary malignancies (eg, skin cancer or hematological malignancy), which pose a significant mortality risk in CLL. 2 , 3 CLL and a second lymphoma occurring concurrently are referred to as either composite lymphoma or Richter syndrome. 4 In the majority of composite lymphomas, composed of two or more B‐cell non‐Hodgkin's lymphomas (NHLs), the components harbor different immunoglobulin heavy chain variable (IGHV) genes (defined as clonally unrelated lymphomas), whereas Richter syndrome consists of a transformation of CLL typically into a diffuse large B‐cell lymphoma with the same rearranged IGHV gene (ie, clones are related). 3 , 4 Massively parallel sequencing of the IG receptor has allowed new insights into the clonal composition and subclonal architecture of CLL. 5 As an example, multiple productive clonally unrelated IGHV rearrangements have been detected by deep sequencing of IGH in ~25% of CLL cases. 6 The clinical implication of this finding has to be addressed in more detail. 5 Some of these cases may represent oligoclonal CLL; others may have a composite lymphoma as underlying cause. We here report on a patient who was first diagnosed with CLL harboring a trisomy 12, who developed a rare hairy cell leukemia‐variant (HCL‐V) with a complex karyotype 3 years later, suggesting consecutive disease outbreak of two B‐cell NHLs. However, retrospective deep sequencing of IGH showed two productive clonally unrelated IGHV rearrangements in the peripheral blood (PB) already at CLL diagnosis. Longitudinal studies over 4.6 years demonstrated expansion of the initially minor IGHV4‐34‐positive HCL‐V clone at the expense of the incumbent CLL clone with IGHV1‐3 gene usage.
2. MATERIAL AND METHODS
2.1. Cytogenetic analysis
Cytogenetic analysis was performed using standard procedures. Interphase fluorescence in situ hybridization FISH (iFISH) included the probes for the chromosomal regions 7cen (D7Z1), 7q31 (D7S522), t(8;21)(q22;q22) (RUNX1/RUNX1T1), 8q24 (MYC rearrangement), 11q22.3 (ATM), 13q14.3 (D13S319), 13q34 (LAMP1), 17p13.1 (TP53), 17cen (D17Z1) (all Abbott Molecular, Downers Grove, IL), and 12cen (D12Z3) (Abbott Molecular or MetaSystems, Altlussheim, Germany). At least 100 nuclei were counted per test for each probe. Multicolor FISH (mFISH) was performed with the 24XCyte human multicolor FISH probe kit (MetaSystems).
2.2. IGH deep sequencing
IGHV mutational status and IGH VDJ rearrangements were assessed by means of targeted deep sequencing using the LymphoTrack Dx IGHV Leader Somatic Hypermutation Assay Kit A (Invivoscribe, San Diego, CA) according to the manufacturer's instructions. Libraries of amplified genomic DNA were sequenced (2 × 301 bp) on a MiSeq platform (Illumina, San Diego, CA). Sequencing data were analyzed using LymphoTrack DX bioinformatics software (Invivoscribe). IGHV mutational status (mutated vs unmutated) was defined using a cut‐off of 98% of nucleotide identity to germline. To infer the percentage of clone, reads with the same VJ rearrangement were merged.
2.3. Whole exome sequencing
Libraries were captured using the Agilent SureSelect Human All Exon kit V6 (Agilent, Santa Clara, CA) and sequenced (2 × 151 bp) on an HiSeq 4000 instrument (Illumina). Using DRAGEN somatic pipeline on BaseSpace (Illumina), paired‐end reads were mapped to the human reference genome (hg38). Variant Interpreter (Illumina) was used to annotate passed variants with coding consequences. Mutations recurrently reported as somatic in the catalog of somatic mutations in cancer (COSMIC) and nonsense, frameshift, and splice site (± 2 intronic bp) mutations in genes implicated as putative tumor suppressors in lymphoid malignancies were considered as oncogenic variants. DRAGEN CNV Baseline Builder and DRAGEN Enrichment were used to call copy number variants. Mean coverage of CLL and HCL‐V component was 146x and 123x, respectively (Supporting Information Tables 1 and 2). Circular genomic (“circos”) plots and fish plot were generated using circlize R package and fishplot R package, respectively. Computational analyses were performed using R version 4.0.2 (www.r-project.org/).
2.4. Droplet digital polymerase chain reaction
A custom droplet digital polymerase chain reaction (PCR) assay was obtained from Bio‐Rad Laboratories (Bio‐Rad Laboratories, Hercules, CA) for the sensitive detection of TP53 p.Ser94Ter. PCR reactions in duplicate were partitioned using a QX200 droplet generator (Bio‐Rad) according to the manufacturer's instructions. After PCR amplification, plates with droplets were read on a QX200 droplet reader (Bio‐Rad) and quantified with QuantaSoft software (Bio‐Rad).
3. RESULTS
3.1. Clinical features and cytogenetics
A 74 year old male patient presented with unexplained mild anemia and otherwise normal blood counts (Figure 1A). Bone marrow (BM) examinations showed 39% lymphocytes and revealed a monoclonal population of B‐cells expressing CD19, CD5, and CD23. For a further 2 years, the patient remained asymptomatic. However, an increase in lymphocytes was then observed in the PB (14.9 G/L), compatible with the diagnosis of CLL clinical stage Binet A/Rai 0. At this time, cytogenetics showed a main clone with an isolated trisomy 12 and a small subclone with concomitant trisomies 12 and 21 (Figure 1B). Nine‐months later, the patient presented with disease‐related symptoms and rapidly increasing lymphocytosis (84.9 G/L), progressive lymphadenopathy, and prominent hepatosplenomegaly. Cytogenetics of the PB again revealed two clones, the clone with isolated trisomy 12 and a newly detected clone showing a complex karyotype (defined as ≥3 chromosomal aberrations; Figure 1B,C), suggestive of clonal evolution of the CLL clone during “watch and wait” and fitting well with the observed clinical progression. Subsequently, the patient received first‐line treatment with bendamustine and rituximab, which resulted in partial remission. Approximately 2 years later, the patient presented again with B symptoms, painful splenomegaly, and lymphocytosis (7.7 G/L). Surprisingly, the PB smear showed infiltration with hairy cells. Flow cytometry confirmed disease transformation by showing a population (CD11c+, CD103+, and CD25‐) consistent with a HCL‐V diagnosis. 7 , 8 As expected, molecular analysis showed no BRAF p.Val600Glu, which is absent in HCL‐V, but detectable in almost all cases of classical HCL. Cytogenetics exclusively showed the complex karyotype; iFISH was negative for del(17p). Results of cytogenetics and iFISH are summarized for all five different timepoints in Supporting Information Table 3. About two and a half years after the HCL‐V diagnosis, the patient was lost to follow‐up.
FIGURE 1.
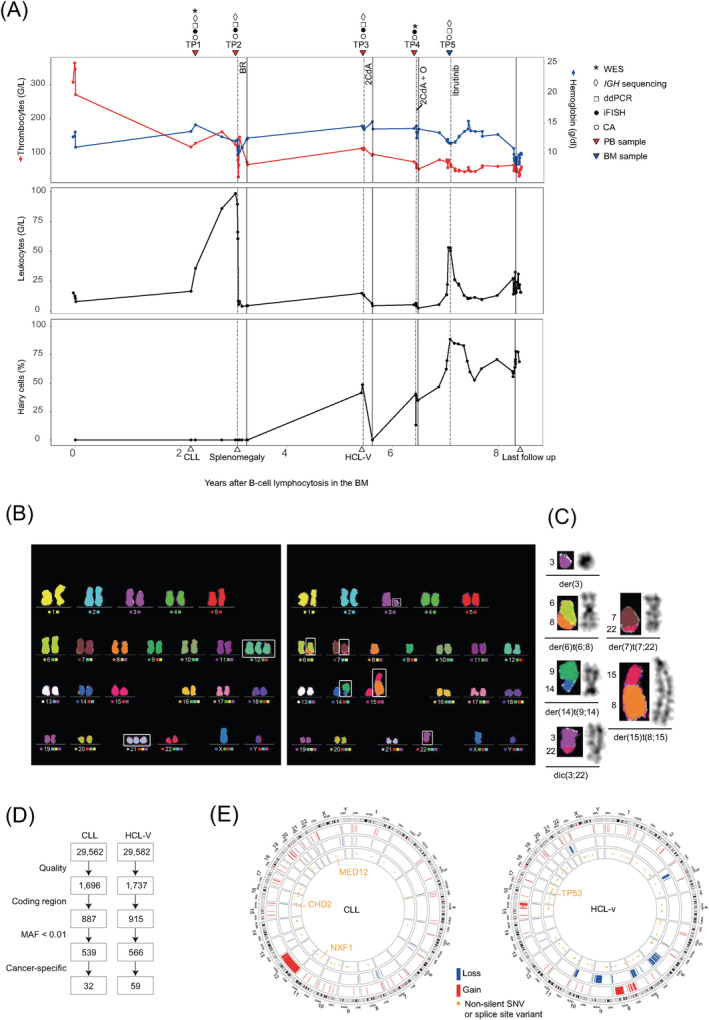
A, Peripheral blood (PB) counts during the clinical course. Dashed and solid vertical lines denote commencement and discontinuation of therapy, respectively. Therapy was discontinued because of toxicity (BR), unknown reasons (2CdA), inadequate response and toxicity (2CdA + O), or disease progression (Ibrutinib). Down‐pointing triangles indicate sequential samples; up‐pointing triangles indicate CLL diagnosis, first presentation with splenomegaly, HCL‐V diagnosis, and last follow‐up (left to right). B, Twenty‐four color multicolor FISH (mFISH) karyograms of peripheral PB showing the CLL subclone with concomitant trisomy 12 and trisomy 21 (TP1; left) and the complex karyotype of the HCL‐V clone (TP3; right). C, Derivative chromosomes of the HCL‐V clone is visualized with mFISH on the left and with Giemsa banding on the right. D, Numbers on the flow chart refer to single‐nucleotide variants and indels detected by whole exome sequencing. Quality: duplicate and orientation bias reads were removed; variants were excluded if they occurred in the “panel of normals.” Variants affecting the coding region, having a minor allele frequency (MAF) of <0.01 in the Genome Aggregation Database and being cancer‐specific (present in the CLL and absent in the HCL‐V component, and vice versa) were considered. E, Circos plots summarizing the genetic lesions of the patient's untreated CLL sample (TP1) and relapsed HCL‐V sample (TP4). The outer ring shows the chromosome ideogram oriented clockwise. The next three rings indicate gains or amplifications shown in red, deletions in blue, and cancer‐specific nonsilent single‐nucleotide variants or splice site variants in orange (genes with oncogenic variants are depicted). BM, bone marrow; BR, bendamustine plus rituximab; CA, chromosomal analysis; ddPCR, droplet digital PCR; iFISH, interphase fluorescence in situ hybridization; O, ofatumumab; SNV, single‐nucleotide variant; WES, whole exome sequencing; 2CdA, 2‐chloro‐2′‐deoxyadenosine (cladribine)
3.2. Whole exome sequencing
To explore the mutational landscape of both malignancies, we performed whole exome sequencing (WES) of the untreated CLL sample and the relapsed HCL‐V sample (Figure 1A,D). To identify tumor‐specific mutations despite the lack of matched normal DNA, we subtracted the variants (n = 507) that were common in both samples. Subsequently, we identified 30 nonsynonymous single‐nucleotide variants (SNVs), one splice site variant, and one frameshift variant in the CLL sample (Figure 1D,E; Supporting Information Table 4). Nonsense variants in CHD2 (p.Arg114Ter; variant allele frequency [VAF] 8%) and NXF1 (p.Gln160Ter; VAF 22%) and a recurrent missense mutation in MED12 (p.Gly44Asp; VAF 63%; COSM131596) were detected. Copy number gains over the whole chromosome 12 were detected (Supporting Information Table 5), consistent with trisomy 12 detected in cytogenetic analysis. Fifty‐three nonsilent SNVs, three splice site variants and three frameshift variants were detected in the HCL‐V sample (Figure 1D,E; Supporting Information Table 6). The sample showed a nonsense mutation in the TP53 gene (p.Ser94Ter) reported in the IARC TP53 and COSMIC (COSM45653) databases. The high VAF of 93% indicated a bi‐allelic inactivation of the TP53 gene. In addition, a missense variant KDM6A (p.Asn1169Lys; VAF 79%) was found. This variant of unknown significance, found in a gene reported as being repeatedly mutated in HCL, 8 , 9 was not listed in COSMIC, but was considered to be damaging by both polymorphism phenotyping (PolyPhen) and sorting intolerant from tolerant (SIFT) in silico prediction tools. A MAP2K1 mutation was not detected. The HCL‐V sample was characterized by several losses in chromosome arms 3p, 6q, 7q, 9q, 10q, and 22q and gains in chromosome arms 8p, 8q (eg, MYC), and 15q (Supporting Information Table 7). The telomeric end of 8q (MAPK15) was deleted as previously described in classical HCL and HCL‐V samples. 9 To test whether we could detect a somatic or germline mutation, which may have contributed to the development of both cancers, we compared the 507 rare variants present in both diseases with a list of 152 genes recurrently mutated in CLL, HCL‐V, or other lymphoid malignancies and 276 genes associated with major DNA damage‐response mechanisms (Supporting Information Table 8). In total, we identified six variants, none of which could be classified as oncogenic. Mutational signature 1, a signature associated with aging processes and CLL, 10 made a prominent contribution to the mutational profile of the CLL sample as extracted by the R package deconstructSig (Supporting Information Figure 1). The mutational signature of the HCL‐V was more flattened: a signature 3 contribution was extracted, which is attributable to DNA repair deficiency and has been detected in other B‐cell malignancies such as multiple myeloma. 11 Both samples also showed a contribution of signature 6 that is characterized by C > T at NpCpG mutations and associated with microsatellite instability. 10
3.3. Clonal dynamics investigated by means of deep sequencing of IGH, droplet digital PCR, and iFISH
Longitudinal deep sequencing of IGH demonstrated replacement of the initially dominant IGH rearrangement of the CLL clone (IGHV1‐3*01/IGHD2‐15*01/IGHJ6*02) that carried an unmutated IGHV, namely with the IGH rearrangement of the HCL‐V clone (IGHV4‐34*01/IGHD3‐3*01/IGHJ6*02), which also had an unmutated IGHV (Figure 2A; Supporting Information Table 9). The transformation occurred within 37 months. During this period the patient received his first‐line therapy. At diagnosis of CLL, the major CLL clone and the minor HCL‐V clone had a frequency of 62.9% and 0.15% of all reads, respectively. Nine‐months later at the time of progression and directly before treatment start, both the CLL and the HCL‐V clone (61.3% and 3.2%, respectively) were simultaneously detectable in the PB above the proposed cutoff of 2.5%. 6 Thus, the HCL‐V clone had already expanded before treatment. After 28 months and therapy with bendamustine and rituximab, the clonal dominance was completely changed: the previously minor HCL‐V clone had a frequency of 62.0%, whereas the CLL clone was almost eradicated, which resulted in the HCL‐V diagnosis. However, the CLL clone persisted at a low cellular fraction in PB at HCL‐V diagnosis (0.09%) and in the BM ~18 months after HCL‐V diagnosis (0.04% vs 44.8% [HCL‐V clone]). Droplet digital PCR demonstrated the presence of the TP53 p.Ser94Ter mutation of the HCL‐V clone at all investigated timepoints including already at CLL diagnosis (Figure 2B and Supporting Information Figure 2). Outgrowth of the HCL‐V clone correlated with the abundance of the TP53 mutation (Supporting Information Figure 3). Clonal shift and simultaneous presence of CLL and HCL‐V clone in the PB were confirmed at the cellular level by iFISH targeting regions on chromosomes 7, 8, 12, and 21 (Figure 2C and Supporting Information Figure 4).
FIGURE 2.
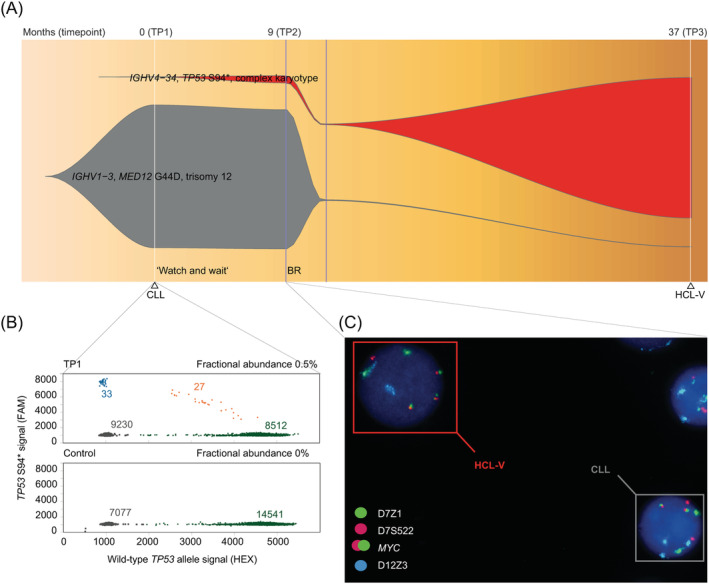
A, Fish plot of the CLL and the HCL‐V clone was inferred by deep sequencing of IGH and morphological examination (at partial remission). Timepoints of the IGH sequencing are shown at the top, history of diagnoses and treatment below. Start and discontinuation of treatment are indicated by vertical lines in blue. Clones are annotated by the IGHV genes, driver mutations harboring a high variant allele frequency, and cytogenetic characteristics. B, Droplet digital PCR showing the number of TP53 p.Ser94Ter (S94*) positive (blue), TP53 S94*/wild‐type TP53 allele positive (orange), wild‐type TP53 allele positive (green) signals, and empty droplets (black) of the patient sample at TP1 and of a control sample. Fractional abundance of TP53 S94* droplets are shown. C, Interphase FISH at TP2 shows the simultaneous presence of HCL‐V and CLL in the peripheral blood. The nucleus of the HCL‐V clone shows three MYC fusion signals indicating a gain of 8q24 and one signal of D7S522 indicating a deletion of 7q31. The nucleus of the CLL clone shows three 12cen signals indicating trisomy 12. BR, bendamustine plus rituximab
4. DISCUSSION
Over 20 cases of composite CLL and HCL, two of them with apparent HCL‐V involvement, have been reported in the literature. 12 , 13 In the composite lymphoma described by Jain et al 12 the clonal dynamics and genetic characteristics (eg, trisomy 12 and a TP53 mutation) were similar to those in the present case. In both cases, the CLL clone persisted after outgrowth of the HCL‐V component at a very low level. Because in our patient the two diseases manifested themselves at separate times, we were able to unambiguously assign the genetic lesions. The CLL component showed a trisomy 12 with a MED12 hotspot mutation (p.Gly44Asp); MED12 is mutated in approximately 1% to 9% of the CLL cases and plays a role in NOTCH signaling. 14 In contrast to CLL, the mutational landscape of HCL‐V is less well known due to its low incidence. We here report a HCL‐V component with biallelic TP53 inactivation and a complex karyotype predominantly with chromosomal losses.
Longitudinal analyses conducted with sensitive sequencing approaches showed the importance of subclonal heterogeneity in treatment‐naive CLL for the course of the disease. 15 , 16 , 17 , 18 This heterogeneity may include mutations in unrelated minor clones in the context of oligoclonal CLL or of composite lymphoma 16 , 19 ; trisomy 12 CLL might predispose to the latter condition. 20 In patients with only CLL 16 , 18 , 21 and with composite CLL and HCL(‐V) 12 , 22 therapy initiation could lead to a change in the clonal architecture. Clones with preceding del(17p) and/or TP53 mutation may harbor greater fitness, particularly under chemotherapy 19 , 21 , 23 , 24 and may eventually contribute to the expansion of a subclone in a treated CLL or, as in our case, to the expansion of a minor clone in a treated composite lymphoma. From a cytogenetic perspective, emergence of the complex karyotype could be misinterpreted as CLL evolution, if results of other laboratory methods are not known or are ambiguous. However, detection of two cytogenetically unrelated clones 25 together with the absence of an early driver lesion such as trisomy 12 in the new clone might heighten awareness for the relatively rare possibility that there is also a secondary cancer. Morphologic examinations may also have limited sensitivity: in the present case at timepoint 2, PB showed a few prolymphocytes, most probably HCL‐V cells, that were noted but could not be attributed to HCL‐V due to the lack of further evidence. With deep sequencing of IGH an additional tool has become available that allows sensitive detection of unrelated clones and could provide further evidence for a secondary malignancy.
Our case shows that TP53 mutations may not only play a relevant role in the selection of a subclone over a main clone in the case of intratumoral heterogeneity, but also in the expansion of an unrelated minor clone in a composite lymphoma. Deep sequencing of IGH may be a useful tool for confirming and monitoring unrelated clones and helping guide therapy, particularly in cases where the minor clone carries high‐risk features (eg, TP53 mutations) associated with inferior responses to standard therapy. Although treatment of composite lymphomas may not be standardized, early detection of two distinct diseases and close follow‐up for disease progression may ultimately improve patient outcome.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
We would like to thank the staff at the Institute of Human Genetics of the Medical University of Innsbruck. Library preparation and sequencing (WES) were performed at the Helmholtz Zentrum Munich, Neuherberg, Germany.
Locher M, Jukic E, Bohn J‐P, et al. Clonal dynamics in a composite chronic lymphocytic leukemia and hairy cell leukemia‐variant. Genes Chromosomes Cancer. 2021;60:287–292. 10.1002/gcc.22925
Contributor Information
Johannes Zschocke, Email: johannes.zschocke@i-med.ac.at.
Normann Steiner, Email: normann.steiner@i-med.ac.at.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Strati P, Jain N, O'Brien S. Chronic lymphocytic leukemia: diagnosis and treatment. Mayo Clin Proc. 2018;93(5):651‐664. [DOI] [PubMed] [Google Scholar]
- 2. Strati P, Parikh SA, Chaffee KG, et al. Relationship between co‐morbidities at diagnosis, survival and ultimate cause of death in patients with chronic lymphocytic leukaemia (CLL): a prospective cohort study. Br J Haematol. 2017;178(3):394‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zheng G, Chattopadhyay S, Sud A, et al. Second primary cancers in patients with acute lymphoblastic, chronic lymphocytic and hairy cell leukaemia. Br J Haematol. 2019;185(2):232‐239. [DOI] [PubMed] [Google Scholar]
- 4. Küppers R, Dührsen U, Hansmann M‐L. Pathogenesis, diagnosis, and treatment of composite lymphomas. Lancet Oncol. 2014;15(10):e435‐e446. [DOI] [PubMed] [Google Scholar]
- 5. Davi F, Langerak AW, de Septenville AL, et al. Immunoglobulin gene analysis in chronic lymphocytic leukemia in the era of next generation sequencing. Leukemia. 2020;34(10):2545‐2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stamatopoulos B, Timbs A, Bruce D, et al. Targeted deep sequencing reveals clinically relevant subclonal IgHV rearrangements in chronic lymphocytic leukemia. Leukemia. 2017;31(4):837‐845. [DOI] [PubMed] [Google Scholar]
- 7. Matutes E, Martínez‐Trillos A, Campo E. Hairy cell leukaemia‐variant: disease features and treatment. Best Pract Res Clin Haematol. 2015;28(4):253‐263. [DOI] [PubMed] [Google Scholar]
- 8. Maitre E, Cornet E, Troussard X. Hairy cell leukemia: 2020 update on diagnosis, risk stratification, and treatment. Am J Hematol. 2019;94(12):1413‐1422. [DOI] [PubMed] [Google Scholar]
- 9. Maitre E, Bertrand P, Maingonnat C, et al. New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget. 2018;9(48):28866‐28876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexandrov LB, Nik‐Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoang PH, Cornish AJ, Dobbins SE, Kaiser M, Houlston RS. Mutational processes contributing to the development of multiple myeloma. Blood Cancer Journal. 2019;9(8):60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jain P, Kanagal‐Shamanna R, Konoplev S, Zuo Z, Estrov Z. Biclonal IGHV‐4‐34 hairy cell leukemia variant and CLL‐successful treatment with ibrutinib and venetoclax. Am J Hematol. 2018;93(12):1568‐1569. [DOI] [PubMed] [Google Scholar]
- 13. Obiorah IE, Francischetti IMB, Wang H‐W, et al. Concurrent chronic lymphocytic leukemia/small lymphocytic lymphoma and hairy cell leukemia: clinical, pathologic and molecular features. Leuk Lymphoma. 2020;61(13):3177‐3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wu B, Słabicki M, Sellner L, et al. MED12 mutations and NOTCH signalling in chronic lymphocytic leukaemia. Br J Haematol. 2017;179(3):421‐429. [DOI] [PubMed] [Google Scholar]
- 15. Schuh A, Becq J, Humphray S, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120(20):4191‐4196. [DOI] [PubMed] [Google Scholar]
- 16. Rose‐Zerilli MJJ, Gibson J, Wang J, et al. Longitudinal copy number, whole exome and targeted deep sequencing of “good risk” IGHV‐mutated CLL patients with progressive disease. Leukemia. 2016;30(6):1301‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Leeksma AC, Taylor J, Wu B, et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia. 2019;33(2):390‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Plevova K, Francova HS, Burckova K, et al. Multiple productive immunoglobulin heavy chain gene rearrangements in chronic lymphocytic leukemia are mostly derived from independent clones. Haematologica. 2014;99(2):329‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Strati P, Abruzzo LV, Wierda WG, O'Brien S, Ferrajoli A, Keating MJ. Second cancers and Richter transformation are the leading causes of death in patients with trisomy 12 chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2015;15(7):420‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Malcikova J, Stano‐Kozubik K, Tichy B, et al. Detailed analysis of therapy‐driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia. 2015;29(4):877‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cavalli M, Ilari C, Del Giudice I, et al. A case of concomitant chronic lymphocytic leukaemia and hairy cell leukaemia evaluated for IGHV‐D‐J rearrangements and BRAF‐V600E mutation: lack of evidence for a common origin. Br J Haematol. 2016;174(2):329‐331. [DOI] [PubMed] [Google Scholar]
- 23. Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia‐update on methodological approaches and results interpretation. Leukemia. 2018;32(5):1070‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lazarian G, Guièze R, Wu CJ. Clinical implications of novel genomic discoveries in chronic lymphocytic leukemia. J Clin Oncol. 2017;35(9):984‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGowan‐Jordan J, Simons A, Schmid M. An International System for Human Cytogenetic Nomenclature (2016). Vol 2016. Karger Ag S.; 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.