

Abstract
Background:
Protein interactions between voltage-gated sodium (Nav) channels and accessory proteins play an essential role in neuronal firing and plasticity. However, a surprisingly limited number of kinases have been identified as regulators of these molecular complexes. We hypothesized that numerous as-of-yet unidentified kinases indirectly regulate the Nav channel via modulation of the intracellular fibroblast growth factor 14 (FGF14), an accessory protein with numerous unexplored phosphomotifs and required for channel function in neurons.
Methods:
Here we present results from an in-cell high-throughput screening (HTS) against the FGF14:Nav1.6 complex using >3,000 diverse compounds targeting an extensive range of signaling pathways. Regulation by top kinase targets was then explored using in vitro phosphorylation, biophysics, mass-spectrometry and patch-clamp electrophysiology.
Results:
Compounds targeting Janus kinase 2 (JAK2) were over-represented among HTS hits. Phosphomotif scans supported by mass spectrometry revealed FGF14Y158, a site previously shown to mediate both FGF14 homodimerization and interactions with Nav1.6, as a JAK2 phosphorylation site. Following inhibition of JAK2, FGF14 homodimerization increased in a manner directly inverse to FGF14:Nav1.6 complex formation, but not in the presence of the FGF14Y158A mutant. Patch-clamp electrophysiology revealed that through Y158, JAK2 controls FGF14-dependent modulation of Nav1.6 channels. In hippocampal CA1 pyramidal neurons, the JAK2 inhibitor Fedratinib reduced firing by a mechanism that is dependent upon expression of FGF14.
Conclusions:
These studies point toward a novel mechanism by which levels of JAK2 in neurons could directly influence firing and plasticity by controlling the FGF14 dimerization equilibrium, and thereby the availability of monomeric species for interaction with Nav1.6.
Keywords: FGF14, Sodium channels, tyrosine kinases, JAK2, signaling
Graphical Abstract:
1. Introduction
The voltage-gated Na+ (Nav) channel forms the basis of neuronal excitability[1]. As molecular determinant of the action potential, the Nav channel underlies the major electrical signaling in the brain mediating neuronal firing, synaptic transmission and plasticity[2]. Due to the critical role they play, Nav channels are understandably subject to intense regulation by accessory proteins including β-IV spectrin, ankyrin G, and intracellular fibroblast growth factors (iFGFs)[3]. In turn, signaling pathways downstream of transmembrane receptors modulate protein:protein interactions (PPI) between these accessory proteins and the Nav channel through phosphorylation, which can confer functional specificity to neuronal firing in response to extracellular stimuli. Not only do these regulatory mechanisms play fundamental roles in neuronal plasticity, but dysregulation of these processes has been associated with increased risk for neuropsychiatric and neurological disorders[3–8] spurring a great interest in searching for novel kinase signaling pathways that control the Nav channel complex.
It has been demonstrated that phosphorylation plays a critical role in regulating Nav channels, particularly for Ser/Thr kinases[9–14]. For example, protein kinase A (PKA) and protein kinase C (PKC) have been shown to phosphorylate multiple serine residues on the interdomain I-II and III-IV linkers of Nav1.2, significantly reducing current and increasing firing thresholds[13,15,16]. Glycogen synthase kinase 3β (GSK3β) phosphorylates the Nav1.2 C-terminal tail at T1966, suppressing Na+ currents and channel trafficking to the plasma membrane[17], an effect that was found to be opposite for the Nav1.6 channel isoform[18]. Casein-kinase II (CK2), a priming kinase of GSK3β, has been shown to phosphorylate Nav channels at the II-III intracellular loop with implications for ankyrin G-dependent trafficking at the axon initial segment (AIS)[19], the site of axon potential initiation where Nav channels are abundantly expressed.
Our laboratory has also demonstrated that FGF14 is a key accessory protein that binds to the intracellular Nav1.6 C-terminal tail, and that GSK3β can phosphorylate FGF14 both in vitro and in vivo at S226[20] in an experimental model of Alzheimer’s disease (AD). In addition, GSK3β was found to be the converging node of a signaling network that includes the PI3K/Akt pathway, the cell-cycle regulator Wee1 kinase, and PKC as modulators of the FGF14:Nav1.6 complex[21]. However, these signaling cascades are highly complex and the mechanisms for how phosphorylation specifically changes PPI between these complexes is not well understood.
Furthermore, despite the essential functions that phosphorylation plays in modulating Nav channel functions, a surprisingly limited number of kinases have been identified as regulators of PPI within the Nav channel complex. This is particularly true for Tyr kinases, although some cases are known. For example, studies have shown that the protein Tyr kinases Fyn and Src directly phosphorylate Nav channels intracellularly, contributing to synaptic plasticity[22–24]. However, the Nav1.6 channel and FGF14 sequences are abundant in predicted phosphorylation sites for both Tyr and Ser/Thr kinases, but evidence for or against phosphorylation of these sites is lacking. Thus, we hypothesized that numerous as-of-yet unidentified kinases regulate the FGF14:Nav1.6 channel complex through mechanisms that are relevant for neuronal plasticity. We sought to discover new regulators and explore potential phosphorylation networks regulating the FGF14:Nav1.6 complex by first conducting a high-throughput screening (HTS) campaign of diverse chemical libraries that was significantly expanded compared to previous studies[20,21]. We have recently developed and optimized an in-cell, luminescence-based assay for HTS against PPI at the FGF14:Nav1.6 complex for this purpose, and have validated its ability to detect potent inhibitors and enhancers of this interaction[25]. Using this robust form of the split-luciferase complementation assay (LCA), here we screened three libraries comprising a total of ~3,000 well-characterized kinase inhibitors, FDA-approved drugs, and natural products. As promiscuity of kinase inhibitors is a well-known phenomenon, observing multiple structurally diverse hits with a common kinase target provides stronger evidence for target relevance. Thus, we selected libraries to provide coverage for a comprehensive array of targets representative of the known “kinome”, while also including a high degree of target overlap to ensure confidence and reproducibility in preliminary screening results (i.e., validation studies not based on a single hit kinase inhibitor).
Following exclusion of toxic compounds identified through cell viability screening run in parallel, we discovered that inhibitors of Janus kinase 2 (JAK2), a Tyr kinase downstream of transmembrane receptors[26], were over-represented among hits. Phosphomotif scans, molecular modeling, and in-cell counter-screening suggested a regulatory mechanism dependent on changes affecting residues at the PPI interface that were common to both the FGF14:Nav1.6 and FGF14:FGF14 homodimer complexes[27,28]. Subsequent biophysical studies including mass-spectrometry (MS) and surface plasmon resonance (SPR) revealed that JAK2 phosphorylates FGF14 at Y158, a site critical in mediating high-affinity dimerization[28,29]. Functionally, JAK2 inhibition prevents FGF14-dependent regulation of Na+ currents, resulting in reduced firing in hippocampal CA1 pyramidal neurons, where FGF14 and Nav1.6 are abundantly expressed [20,30]. Based on these results, we concluded that by regulating the equilibrium between FGF14 homodimerization, activation of JAK2 might enable neurons to dynamically adjust firing in response to JAK2-mediated receptor signaling.
2. Materials and Methods
2.1. Chemicals
D-luciferin was purchased from Gold Biotechnologies (St. Louis, MO). Screened compounds are described below. Repurchased hits, including Momelotinib, TG101209, Fedratinib, Pacritinib, Danusertib, Saracatinib, Ibrutinib, and Bosutinib were obtained from Selleck (Houston, TX). For mass spectrometric experiments, LC–MS grade acetonitrile (ACN) and water were from J.T. Baker (Philipsburg, NJ). Formic acid was obtained from Pierce (Rockford, IL) and iodoacetamide (IAA) and dithiothreitol (DTT) were purchased from Sigma-Aldrich (St. Louis, MO). Sequencing grade trypsin was supplied by Promega (Madison, WI).
2.2. DNA constructs
The CLuc-FGF14WT, CLuc-FGF14Y158A, FGF14WT-NLuc, FGF14Y158A-NLuc, CD4-Nav1.6-NLuc, pQBI-FGF14-GFP and pQBI-GFP constructs were engineered and characterized as previously described [20,25,28,29,31–33]. The corresponding gene ID numbers are as follows: NM_175929.2 (human FGF14–1b), NT_009759.16 (human CD4), and NM_014191.3 (human Nav1.6). The plasmid pGL3 expressing full-length firefly (Photinus pyralis) luciferase was a gift from Dr. P. Sarkar (Department of Neurology, UTMB). For protein purification, the pET bacterial expression vectors (pET28a-FGF14; pET30a-Nav1.6) were used as previously described[18,28], and encode FGF14 (accession number NP_787125; aa 64–252) or the C-terminal tail of Nav1.6 (accession number #NP_001171455; aa 1767–1912).
2.3. Cell culture
HEK293 cells were incubated at 37 °C with 5% CO2 in medium composed of equal volumes of Dulbecco modified essential medium (DMEM) and F12 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin. For transfection, cells were seeded in 24-well CELLSTAR® tissue culture plates (Greiner Bio-One, Monroe, NC) at 4.5×105 cells per well and incubated overnight to give monolayers at 90%–100% confluency. The cells were then transiently transfected with LCA construct pairs (i.e., CLuc-FGF14 and CD4-Nav1.6-C-tail-NLuc or FGF14-NLuc) constructs or the full-length P. pyralis luciferase construct (pGL3) using Lipofectamine 3000 (Invitrogen), according to the manufacturer’s instructions. 1 μg of each plasmid was used per transfection per well. The double stable HEK293 cell line expressing CD4-Nav1.6C-tail-Nluc and Cluc-FGF14 was described in a previous study[25] and was maintained using selective antibiotics (0.5 mg/mL G418 and 5 μg/mL puromycin). HEK293 cells stably expressing the Nav1.6 channel (HEK-Nav1.6) were maintained under 80 μg/ml G418.
2.4. Split-luciferase Complementation Assay
96-well plate assay:
Cells were trypsinized (0.25%), triturated in medium, and seeded in white, clear-bottom CELLSTAR μClear® 96-well tissue culture plates (Greiner Bio-One) at ~0.9×105 cells per well in 200 μL of medium. For transiently transfected cells, the trypsinization occurred 48 h post-transfection. The cells were incubated for 24 h, and the growth medium was subsequently replaced with 100 μL of serum-free, phenol red–free DMEM/F12 medium (Invitrogen) containing inhibitors (0.25–50 μM). The final concentration of DMSO was maintained at 0.3% for all wells. Following 2 h incubation at 37 °C, the reporter reaction was initiated by injection of 100 μL substrate solution containing 1.5 mg/mL of D-luciferin dissolved in PBS (final concentration = 0.75 mg/mL) by the Synergy™ H4 Multi-Mode Microplate Reader (BioTek). Luminescence readings were performed at 2-min intervals for 20 min, integration time 0.5 s, and the cells were maintained at 37 °C throughout the measurements. Signal intensity for each well was calculated as a mean value of peak luminescence; the calculated values were expressed as percentage of mean signal intensity of the per plate control samples.
384-well plate assay:
Cells were trypsinized (0.25%), triturated in a medium, and seeded in white, clear-bottom CELLSTAR μClear® 384-well tissue culture plates (Greiner Bio-One) at 3×104 cells per well in 40 μL of serum-free, phenol red–free DMEM/F12 medium using the Multidrop Combi (Thermo Fisher). The LabCyte Echo 550 was used to acoustically deliver nanoliter volumes of compounds, TNF-α, and DMSO. The final concentration of DMSO was maintained at 0.3% for all wells excluding the positive control wells containing medium alone. Following 2 h incubation at 37 °C, the reporter reaction was initiated by injection of 40 μL substrate solution containing 1.5 mg/mL of D-luciferin (final concentration = 0.75 mg/mL) by the Multidrop Combi. After 1 h incubation, the Tecan Infinite M1000 was used to detect luminescence. Detailed methods for LCA can be found in previous studies[21,25,27,28,31,33]. Statistical parameters of assay performance were calculated as described previously[25,34] according to the following formulas:
| (1) |
| (2) |
| (3) |
| (4) |
where δp and δn are standard deviation of the positive control group p and the negative control group n, and μp and μn are the arithmetic means of the two groups, respectively; S:B, signal to background; S:N, signal-to-noise; and SW, signal window. Z-scores were calculated for each screened compound using the following formula:
| (5) |
where μi is the luminescent signal of the sample (i.e., any particular screened compound), and μDMSO and δDMSO are the mean and standard deviation, respectively, of the per plate 0.3% DMSO controls for that sample. Each compound replicate was on an independent 384-well plate, and a percent luminescence and Z-score was calculated separately using each replicate’s respective per plate controls. The three replicate percent luminescence values and Z-scores for each compound were subsequently averaged. For hit dose-response validation studies, compounds were tested between 0.25 – 50 μM using n = 8 per concentration over two 384-well plates per compound. Luminescence was normalized to per plate 0.3% DMSO controls, and dose-response curves were obtained using GraphPad Prism 8 by fitting the data with a non-linear regression:
| (6) |
where x is log10 of the compound concentration in M, x0 is the inflection point (EC50 or IC50), A is the bottom plateau effect, B is the top plateau effect, and H is the Hill slope. Kinase inhibitors that increased FGF14:Nav1.6 interaction with increasing doses were classified as agonists; inhibitors that decreased FGF14:Nav1.6 interaction were classified as antagonists.
Screened libraries:
Three libraries provided by the Gulf Coast Consortium (GCC) were screened using the LCA, including the Broad Collection, Selleck Bioactive Collection, and UT Austin Combined Kinase Collection (UTKinase), for a total of 3,121 compounds. All compounds were provided as 10 mM in DMSO and were screened at a final concentration of 30 μM. The Broad Collection has been previously described as the “Informer Set,”[35] which targets nearly 250 distinct proteins, encompassing a broad range of cell circuitry relevant to cancer cell line growth and survival. The collection screened here uses 406 FDA-approved agents, clinical candidates, and small-molecule probes from the Informer Set that were commercially available. The Selleck Bioactive Collection contains some compounds that have been approved by the FDA, have bioactivity and safety confirmed by preclinical research and clinical trials and includes most Selleck inhibitors, active pharmaceutical ingredients, natural products, and chemotherapeutic agents. The collection is structurally diverse, medicinally active, and cell permeable. The UTKinase collection is comprised of over 1,400 well-characterized, cell permeable, potent and reversible protein kinase inhibitors, the majority of which are ATP-competitive, less cytotoxic, stable in DMSO/H2O, and structurally diverse. Inhibitors target a broad spectrum of >100 kinases, including but not limited to, RSTK (Receptor Serine/Tyrosine kinase), TK (Tyrosine Kinase), TKL (Tyrosine Kinase like), AGC (PKA, PKG, and PKC family), CMGC (CDK, MAPK, GSK-3, and CLK family), RTP (Receptor Tyrosine Phosphatase), TP (Tyrosine Phosphatase), CAMK (Ca2+/Calmodulin Dependent Protein Kinases), STE (Yeast Sterile Protein Kinases), Atypical, MAPK signaling, PI3-kinase/Atk Signaling, and Transferase. The useful applications of this particular collection are target identification in drug discovery, biochemical pathway analysis, and screening new protein kinases.
2.5. Cell viability assay
The CellTiter-Blue (CTB) Cell Viability Assay (Promega) was used as a control to detect compounds causing cellular toxicity. Immediately following luminescence reading, 10 μL of 1X CTB reagent was dispensed into 384-well plates, incubated overnight (16 h) at 37 °C, and fluorescence was detected using the Tecan Infinite M1000 reader (excitation λ =560 nm, emission λ = 590 nm). Cell viability was expressed as percent mean fluorescent signal intensity in the control samples from the same experimental plate.
2.6. Phosphomotif Searches
To search for potential phosphorylation motifs (both S/T and Y) and tyrosine binding motifs, the FGF14–1b (aa 1–252) and Nav1.6 C-tail (aa 1763 – 1968) sequences were input to the Human Protein Reference Database (HPRD) PhosphoMotif Finder (http://hprd.org/PhosphoMotif_finder)[36], NetPhos 3.1[37], and NetPhorest 2.1[38]. The HPRD PhosphoMotif Finder contains known kinase/phosphatase substrate as well as binding motifs that are curated from the published literature, and this program reports the presence of any literature-derived motif without making any predictions as to whether it will truly exist. NetPhos predicts serine, threonine, and tyrosine phosphorylation sites using a neural network. Note that while NetPhos 3.1 does not currently have the capability to predict JAK2 phosphomotifs, the HPRD PhosphoMotif Finder was able to detect JAK2 consensus motifs. NetPhorest predicts kinase binding sites based on an atlas of consensus sequence motifs for kinases and phosphorylation-dependent binding domains
2.7. Protein Expression and Purification
The pET28a-FGF14 or pET30a-Nav1.6 plasmids for protein expression and purification of FGF14 or Nav1.6 C-tail, respectively, were transformed into E. coli BL21 (DE3) pLys (Invitrogen). Cells were grown until OD600 = 0.7, and the recombinant proteins were expressed after induction with 0.1 mM isopropyl thio-β-D-galacto-pyranoside (IPTG) for 24 h at 16 °C. Cells were harvested and lysed by sonication at 4°C in lysis/binding buffer containing following components (mM): 10 sodium phosphate (prepared from 0.5 M of Na2HPO4 and NaH2PO4), 25 HEPES, 150 NaCl, phenyl methyl sulphonyl fluoride (PMSF) 0.1, CHAPS 0.1% pH 7.0 (for FGF14), and with glycerol 10% (for Nav1.6 C-tail) pH 7.5. The respective proteins were centrifuged at 40,000 × g for 1 h at 4°C. For purification of FGF14, the supernatant was applied to pre-equilibrated heparin and the proteins were then eluted with NaCl 0.2–2.0 M (sodium phosphate 10 mM, NaCl 0.2–2.0 M, pH 7.0) buffer. For purification of Nav1.6 C-tail, the supernatant was first applied to a cobalt column (Thermo Fisher Scientific) and eluted with imidazole (200 mM). The Nav1.6 C-tail was further purified using HiTrap QFF-sepharose column (GE Healthcare) using a buffer containing Tris-HCl 50 mM and eluted with NaCl (10–500 mM) at pH 7.5. Finally, all concentrated proteins were purified on an AKTA FPLC using a Superdex 200 HiLoad 16 × 60 column and equilibrated in Tris-HCl 50 mM + NaCl 150 mM, pH 7.5 (GE Healthcare). Protein concentrations were determined using UV absorbance with a Thermo NanoDrop.
2.8. In Vitro Phosphorylation and Sample Preparation
In vitro phosphorylation of the FGF14 peptide [KFKESVFENYYVIYSSMLYR-NH2] (aa149–169) by baculovirus-produced recombinant human JAK2 or Src kinase protein (SignalChem) was performed in the presence of 50 μM FGF14 peptide, 200 nM kinase (JAK2 or Src), 10 mM Tris-HCl, 25 mM NaCl, 1.5 mM Glutathione, 0.5 mM EDTA, 0.25 mM DTT, 5 mM MOPS, 5 mM MgCl2, 1 mM ATP and 15 ng/μL BSA. Reactions with Src also included 2.5 mM MnCl2. Reactions were incubated at 30 °C for 30 min, followed by overnight incubation at 4 °C. Control studies were performed under identical conditions but lacking the addition of either the kinase or ATP to the reaction solution. Peptide samples for SPR were then buffer exchanged and concentrated into running buffer (HBS-P+ supplemented with 2% DMSO). For in vitro phosphorylation of purified recombinant proteins, FGF14 was purified as described above, and phosphorylation by JAK2 or Src was performed identically. To confirm phosphorylation status by mass spectrometry, samples were reduced with 10 mM DTT, alkylated with 5 mM IAA, and digested with modified sequencing grade trypsin 1:50 (w/w) overnight at 37 °C.
2.9. Mass spectrometry and data analysis
Digested peptide samples were desalted using C18 ZipTips (Millipore) and 1 μl of this solution was combined with 1 μl of a 3 mg/ml α-cyano-4-hydroxycinnamic acid (60% acetonitrile, 1 mM ammonium diphosphate) and spotted onto MALDI targets. All MALDI-MS experiments were performed using a 5800 MALDI-TOF/TOF (Applied Biosystems). The MS data were acquired using the reflectron detector in positive mode (700–4500 Da, 1900 Da focus mass) using 300 laser shots (50 shots per sub-spectrum). Collision induced dissociation tandem MS spectra were acquired on the ions found in the MS1 experiment, using 1 kV of collision energy. Identified phosphopeptide spectra were manually sequenced and annotated using the MS-Product tool on the Protein Prospector website (prospector.ucsf.edu) to generate and compare theoretical m/z values for all fragment ions against observed fragment ions. Phosphorylation sites were identified manually by locating all present site-identifying b and y ions in the sequence.
2.10. Molecular modeling
The FGF14:FGF14 homodimer model was built with the FGF13 dimer crystal structure (PDB ID: 3HBW)[29] as template, as described previously[27]. The FGF14:Nav1.6 homology model was generated using the FGF13:Nav1.5:CaM ternary complex crystal structure (PDB ID: 4DCK)[39] as template, as described previously[28].
2.11. Surface Plasmon Resonance Spectroscopy
SPR experiments were performed on a Biacore T100 instrument (GE Healthcare, Pittsburgh, PA). Proteins were immobilized on CM5 sensor chips using 10 mM sodium acetate buffer (pH 5.5) with the Amine Coupling Kit (GE Healthcare) as per the manufacturer’s instructions. For studies assessing the interaction of recombinant proteins with peptides, chips with FGF14 bound to final RU values of 16,045 and 17,895 were used. For studies assessing the interaction of recombinant proteins, chips with FGF14 bound to final values of 930 and 1,130 were used. No protein was coupled to the control flow channels of the chip (Lanes 1 and 3). The interaction of analytes against FGF14 and Nav1.6 proteins were studied at 25 °C using a flow rate of 50 μl/min. Recombinant protein or peptide samples were serially diluted (10 – 2000 nM or 31 – 6000 nM, respectively) in HBS-P+ (HBS supplemented with Tween-20 0.005%). Each sample was injected over the chip for 60–120 s followed by a dissociation period of 250 s and finally chip surface regeneration (1.5 M NaCl, 3% DMSO) for 120 s. Peptides were tested with concentrations of 31, 62.5, 125, 250, 500, 750, 1000, 1500, 2000, 3000, and 6000 nM. Recombinant proteins were tested with concentrations of 10, 50, 100, 200, 300, 400, 500, 750, 1000, 1500, and 2000 nM. Each sample group also included a minimum of two blanks (buffer prepared similarly to samples). For experiments using protein phosphorylated in vitro, recombinant FGF14 protein (50 μM) was incubated with ATP and either 100 nM BSA (control), active JAK2 kinase, or active Src kinase (100 nM) for 30 min at 30 °C, followed by buffer exchange into SPR running buffer (HBS-P+). The serial dilution and buffer exchange concentration was such that the maximal possible concentration of JAK2 or Src in the FGF14 samples for SPR was 4 nM for the highest concentration of FGF14 (2 μM), and thus should yield negligible signal. Additional controls included blanks for each sample group that were prepared identically as the in vitro phosphorylation reaction, but lacking FGF14 protein (i.e., 0 μM FGF14, 4 nM JAK2 or Src). These controls yielded similar or identical response signal (RU) as buffer alone, and thus we ruled out the effects of JAK2 or Src on SPR signal. For each compound injection, nonspecific responses (buffer alone) were subtracted from experimental sensorgrams/traces prior to data analysis. Kinetic data were analyzed using the Biacore T100 Analysis software. Following visual inspection of the binding curves, the equilibrium constant (KD) was calculated using two methods: (1) maximal responses were plotted against compound concentration, and the steady state KD was calculated from the fitted saturation binding curve; (2) a kinetic analysis of each ligand/analyte interaction was obtained by fitting the response data to the simplest Langmuir 1:1 interaction model (KD=koff/kon). The kinetic constants generated from the fitted binding curves were assessed for accuracy based on the distribution of the residuals (even and near zero to baseline). Graphs were plotted in GraphPad Prism 8 Software (La Jolla, CA).
2.12. Animals.
Fgf14−/− mice were maintained on an inbred C57/BL6J background (greater than ten generations of backcrossing to C57/BL6J). All genotypes described were confirmed by Charles River Laboratories International, Inc. (Houston, TX). Fgf14+/+ wild-type controls (C57/BL6J) were either Fgf14−/− littermates or were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed, n ≤ 5 per cage, with food and water ad libitum. Mice were closely monitored for health and overall well-being daily by veterinary staff and the investigators. Animal maintenance and experiments were performed in accordance with US National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee.
2.13. Electrophysiology
HEK-Nav1.6 cells transiently transfected with GFP or FGF14-GFP were plated at low density on glass cover slips for 3–4 hours and subsequently transferred to the recording chamber. Recordings were performed at room temperature (20–22°C) 24 h post-transfection using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA). The composition of recording solutions consisted of the following salts; extracellular (mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 10 glucose, pH 7.3; intracellular (mM): 130 CH3O3SCs, 1 EGTA, 10 NaCl, 10 HEPES, pH 7.3. Additionally, cells were treated with either compounds or vehicle alone (DMSO), with all conditions having a final concentration of 0.01% DMSO. Membrane capacitance and series resistance were estimated by the dial settings on the amplifier and compensated for electronically by 70–75%. Data were acquired at 20 kHz and filtered at 5 kHz prior to digitization and storage. All experimental parameters were controlled by Clampex 9.2 software (Molecular Devices) and interfaced to the electrophysiological equipment using a Digidata 1200 analog-digital interface (Molecular Devices). Voltage-dependent inward currents for HEK-Nav1.6 cells were evoked by depolarization to test potentials between −100 mV and +60 mV from a holding potential of −70 mV followed by a voltage pre-step pulse of −120 mV (Nav1.6). Steady-state (fast) inactivation of Nav channels was measured with a paired-pulse protocol. From the holding potential, cells were stepped to varying test potentials between −120 mV (Nav1.6) and +20 mV (pre-pulse) prior to a test pulse to −20 mV.
Current densities were obtained by dividing Na+ current (INa) amplitude by membrane capacitance. Current–voltage relationships were generated by plotting current density as a function of the holding potential. Conductance (GNa+) was calculated by the following equation:
| (7) |
where INa+ is the current amplitude at voltage Vm, and Erev is the Na+ reversal potential.
Activation curves were derived by plotting normalized GNa+ as a function of test potential and fitted using the Boltzmann equation:
where GNa,+Max is the maximum conductance, Va is the membrane potential of half-maximal activation, Em is the membrane voltage and k is the slope factor. For steady-state inactivation, normalized current amplitude (INa+/INa,+Max) at the test potential was plotted as a function of prepulse potential (Vm) and fitted using the Boltzmann equation:
where Vh is the potential of half-maximal inactivation, Em is the membrane voltage, and k is the slope factor.
To determine effects on long-term inactivation (LTI), a four-sweep protocol composed of four 20 ms-long 0 mV pulses separated by 40 ms interpulse recovery phases from a −90 mV holding potential was used. For direct comparison of cells of various size, current densities were calculated by dividing INa amplitude/membrane Capacitance (Cm). For LTI, the fraction of channels recovered after the nth depolarization cycle was defined as INa peak (n + 1) / INa-peak 1st pulse.
For ex vivo patch-clamp electrophysiology, coronal brain slices containing the hippocampus were prepared from Fgf14−/− mice and C57BL/6J mice aged 21–40 days. Mice were anesthetized with isoflurane (Baxter) and quickly decapitated before brains were dissected, and 300 μm coronal slices containing the hippocampus were prepared with a vibratome (Leica Biosystems) in a continuously oxygenated (mixture of 95%/5% O2/CO2) chilled tris-based artificial cerebrospinal fluid (aCSF), consisting of the following: 72 mM Tris-HCl, 18 mM Tris-Base, 1.2 mM NaH2PO4, 2.5 mM KCl, 20 mM HEPES, 20 mM sucrose, 25 mM NaHCO3, 25 mM glucose, 10 mM MgSO4, 3 mM Na-pyruvate, 5 mM Na-ascorbate and 0.5 mM CaCl2 (Sigma-Aldrich); 300–310 mOsm, pH 7.4. Slices were transferred to a 31°C recovery chamber with fresh tris-based aCSF for 15 minutes before being transferred to a 31°C chamber with continuously- oxygenated (mixture of 95%/5% O2/CO2) standard aCSF consisting of the following: 123.9 mM NaCl, 3.1 mM KCl, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2, 24 mM NaHCO3, and 1.16 mM NaH2PO4 (Sigma-Aldrich); 300–310 mOsm, pH 7.4. Slices were incubated in standard aCSF with 20 μM Fedratinib or 0.02% DMSO for 1 hr before recording. Hippocampal CA1 pyramidal neuron somatic recordings in standard aCSF were performed using recording electrodes filled with an internal solution containing 145 mM K-gluconate, 2 mM MgCl2, 0.1 mM EGTA, 2.5 mM Na2ATP, 0.25 mM Na2GTP, 5 mM phosphocreatine, and 10 mM HEPES (pH 7.2; 290 mOsm). After giga-seal formation and cell membrane rupture, pyramidal neurons were held in I=0 mode for approximately 1 minute to determine resting membrane potential before switching to current clamp mode to assess neuronal activity. Electrophysiological brain slice data analysis was performed as previously described[18]. Intrinsic neuronal excitability was assessed by measuring evoked action potentials with a range of current injections ranging from 10 pA to 200 pA with 800 msec 10 pA pulses and the maximum number of action potentials fired was determined. Statistical significance was determined with a t-test with Welch Correction when data met assumptions for normality. Data that did not meet assumptions for normality were tested with a Mann Whitney test.
3. Results
3.1. High-throughput screening of kinase inhibitors to discover new regulators
We have previously developed and reported an in-cell, high-throughput assay that can be used to identify targets that inhibit and/or enhance the FGF14:Nav1.6 complex assembly[25]. This adapted form of the luciferase complementation assay (LCA) is based on a double stable HEK293 cell line expressing CLuc-FGF14 and CD4-Nav1.6 C-tail-NLuc recombinant proteins that, upon binding, produce luminescence in the presence of the substrate luciferin[31,40]. Based on this assay, we sought to identify potential regulators of the FGF14:Nav1.6 complex by screening a large library of well-characterized and structurally diverse kinase inhibitors targeting an extensive range of cell signaling pathways. Compound screening was subsequently validated using previously established orthogonal screening methods (i.e., cell viability and full-length luciferase assays)[25] to identify artifacts, followed by target-based hit selection (Figure 1A). Top ranking targets were then counter-screened against the FGF14:FGF14 homodimer to identify pathways of biological relevance for FGF14 signaling.
Figure 1. HTS pipeline and results for discerning mechanisms of Nav channel complex regulation by kinases.
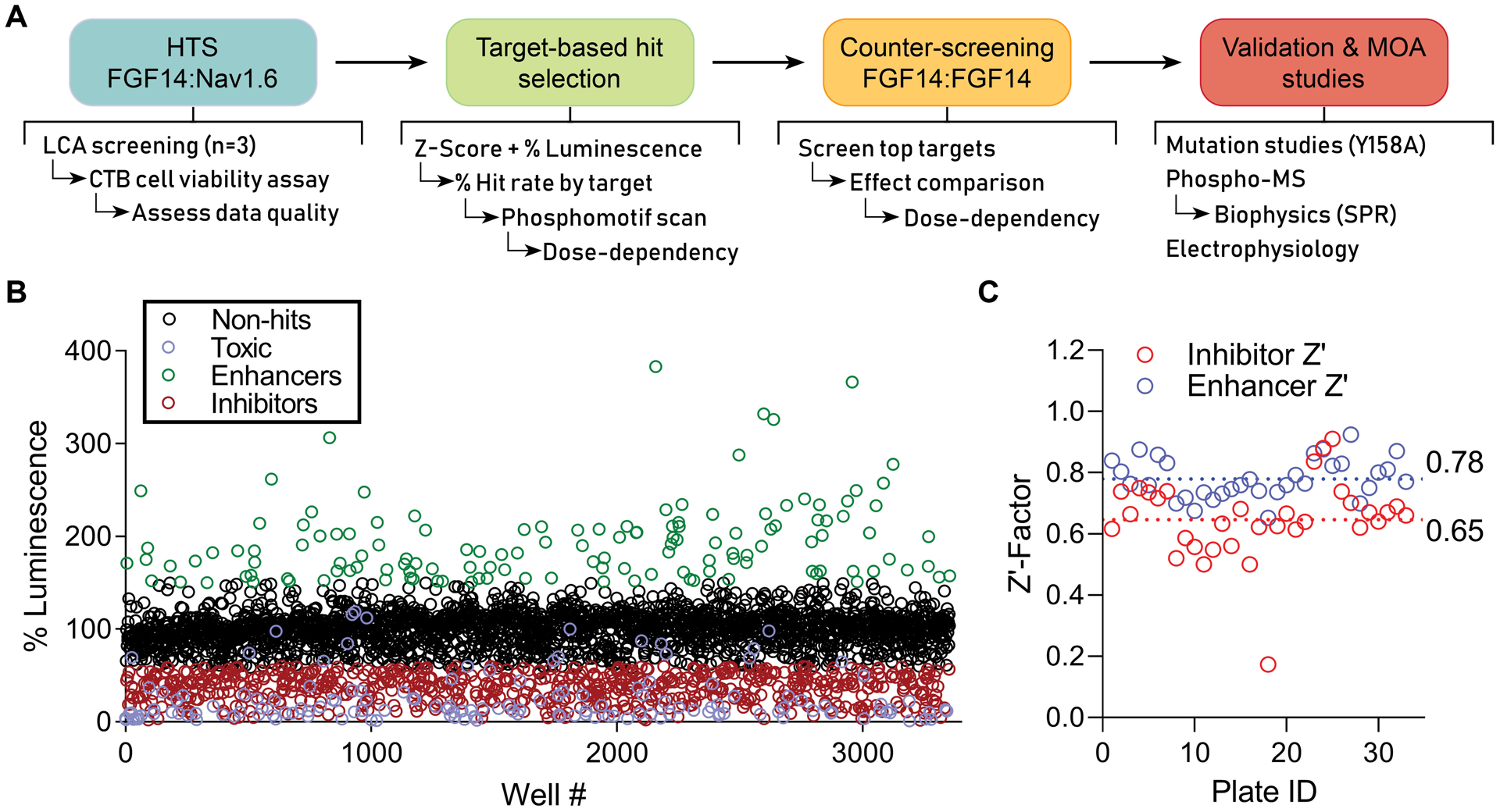
(A) Screening and validation pipeline. (B) Double stable HEK293 cells expressing CLuc-FGF14 and CD4-Nav1.6-NLuc were plated in 384-well plates and treated with kinase inhibitors (n = 1 compound/well) from the Broad, Selleck, and UTKinase collections, with each plate screened in triplicate. The mean percent luminescence (normalized to on-plate 0.3% DMSO controls) is shown for each compound. Following exclusion of toxic compounds (purple), hits were initially selected using unbiased criteria of change in FGF14:Nav1.6 complex assembly by at least 40% (i.e., % luminescence > 140% or < 60%) and Z-score ≥ 3 (enhancers, green) or Z-score ≤ −4 (inhibitors, red). (C) Z’-Factor (Z’) for each screened library plate, calculated using either the inhibitor (red) or enhancer (blue) positive controls as described previously[25]. A total of 33 plates were screened, including 6 from Broad, 12 from Selleck, and 15 from UTKinase, for a total of 3,120 compounds.
With this identification and validation pipeline, we screened three libraries: the Broad Institute Collection (406 compounds), the Selleck Bioactive Collection (1,280 compounds), and the UT Austin Combined Kinase Collection (1,434 compounds) (Figure 1B). This library included a high degree of overlap between kinase targets to ensure broad “kinome” coverage and to increase confidence in results (i.e., rule out promiscuity by observing multiple hits with a common target). Cells were seeded in 384-well plates containing compounds at a screening concentration of 30 μM (n = 1 compound per well; 320 compounds/plate), as well as negative controls (0.3% DMSO, n = 16; cells alone, n = 8 wells), and previously established[25] inhibitory positive controls (MNS, concentration range: 2.5 – 30 μM, n = 24), and enhancer positive controls (TNF-α, n = 16) that were used to calculate Z’-factor to assess assay robustness throughout the screening campaign. Each plate of compounds was screened in triplicate. Immediately following luminescence reading, the CellTiter-Blue® (CTB) cell viability assay was initiated by dispensing 10 μL of CTB reagent per well. Fluorescence was read after 16 hrs, and cut-offs were set at a Z-score of < −3 (relative to DMSO controls) to identify and exclude toxic compounds. The coefficient of variation (CV) and Z’ factor for our in-cell HTS assay were found to be within acceptable range (Broad (n = 6 plates): CV = 0.09 ± 0.005; inhibitor Z’ = 0.66 ± 0.02; enhancer Z’ = 0.78 ± 0.05; Selleck (n = 12 plates): CV = 0.08 ± 0.02; inhibitor Z’ = 0.64 ± 0.09; enhancer Z’ = 0.77 ± 0.06; UTKinase (n = 15 plates): CV = 0.07 ± 0.02; inhibitor Z’ = 0.65 ± 0.17; enhancer Z’ = 0.78 ± 0.07; data are mean ± SD) (Fig 1C of Z’ graph and Supplementary Table 1 of Z’/CV values) [25]. Following exclusion of toxic compounds (Supplementary Figure 1), hits were initially selected using unbiased criteria of change in FGF14:Nav1.6 complex assembly by at least 40% (i.e., % luminescence > 140% or < 60%) and Z-score ≥ 3 (enhancers, green) or Z-score ≤ −4 (inhibitors, red). Note that this combination of Z-scores and % luminescence was used to ensure that hits were not preferentially selected from plates with lower control standard deviation, which could artificially over-inflate a given compound’s rank despite lack of biological relevance[25].
We observed clusters of hits targeting kinases including Akt, GSK3, PKC, PI3K, MEK, p38 MAPK, and NF-kB, supporting findings of previous smaller scale studies[20,21]. However, this expanded screening campaign identified two previously unexplored targets that had the highest proportion of hits to compounds screened: the JAK2 (28 hits out of 47 compounds screened) and Src (20 hits out of 31 screened) tyrosine kinases (Table 1). Phosphomotif scans (Table 2) using the HPRD PhosphoFinder and NetPhos 3.1 revealed that the FGF14 sequence contains possible phosphorylation sites for several of these kinases, including GSK3, PI3K, PKC, p38 MAPK, JAK2, and Src. Additionally, NetPhorest 2.1 identified probable Src homology 2 (SH2) domains at residues 155–164 (corresponding to the FGF14 sequence VFENYYVIYSS) and residues 206–216 (LEVAMpYREPSL).
Table 1.
Total number of HTS hits compared with total number of compounds screened by kinase target.
| Target | # Screened Compounds | # Hits | Hit % |
|---|---|---|---|
| Src | 31 | 20 | 0.65 |
| JAK | 47 | 28 | 0.60 |
| PKC | 21 | 12 | 0.57 |
| VEGFR | 64 | 34 | 0.53 |
| GSK3 | 33 | 17 | 0.52 |
| NF-kB | 36 | 18 | 0.50 |
| Raf | 24 | 11 | 0.46 |
| FLT3 | 11 | 5 | 0.45 |
| STAT3 | 7 | 3 | 0.43 |
| Akt | 29 | 11 | 0.38 |
| p38 MAPK | 27 | 10 | 0.37 |
| MEK | 28 | 9 | 0.32 |
| Raf | 23 | 7 | 0.30 |
| Wee1 | 7 | 2 | 0.29 |
| Syk | 18 | 5 | 0.28 |
| PI3K | 64 | 16 | 0.25 |
| EGFR | 75 | 13 | 0.17 |
| mTOR | 37 | 6 | 0.16 |
Table 2. FGF14 phosphomotifs correspond to hit targets identified by HTS.
Subsequent investigation using the HPRD PhosphoMotif Finder, NetPhos 3.1, and NetPhorest 2.1 revealed potential JAK2 and Src phosphorylation sites at Y158 and Y162, respectively, as well as other sites for kinases identified in the HTS.
| FGF14 Position | FGF14 Sequence | Kinase or Phosphatase | FGF14 Position | FGF14 Sequence | Tyrosine Binding Motifs |
|---|---|---|---|---|---|
| 81 – 86 | YCRQGY | ALK | 81 – 84 | pYCRQ | STAT3 |
| 145 – 147 | TPE | p38 MAPK | 86 – 89 | pYYLQ | STAT3 |
| 158 – 161 | YYVI | JAK2 | 129 – 132 | pYIAM | PI3K p85 |
| 162–163 | YS | Src | 156 – 161 | SVFENpYYVIYS | SHC, SH2 |
| 162 – 167 | YSSMLY | ALK, INSR | 154 – 165 | VFENYpYVIYSS | SH2, PTP |
| 193 – 195 | KKT | PKC | 162 – 165 | pYSSM | PI3K p85 |
| 211 – 212 | YR | Src | 167 – 170 | pYRQQ | STAT3 |
| 226 – 230 | SKSTS | GSK3 | 206 – 216 | LEVAMpYREPSL | SH2, PTP |
Despite there being a disproportionately high number of PI3K inhibitors (64) in the screening set, only 16 compounds ranked as hits, and the primary target IP3K isoform was distributed between hits, reducing our interest in this target/suggesting this was due to off-target effects/more complex mechanism.
The GSK3 and CK2 sites have previously been thoroughly explored[20,41], the identified JAK2 and Src phosphorylation and binding sites, Y158 and Y162, are in line with the observed screening data. Previous studies have shown the importance of Y158 in mediating both FGF14 dimerization and binding to the Nav1.6 C-terminal tail[27,28], and Y158 was found at the PPI interface for both the FGF14 dimer and the FGF14:Nav1.6 complex[27,28] (Suppl. Fig 2). Conversely, despite its relative vicinity to Y158, Y162 is more buried in the complex at a location that may render kinase binding and phosphorylation at this site more challenging. Thus, Y162 is less likely to be involved in structurally relevant regulation of the PPI interface (> 10Å from the PPI surfaces).
Primary hits that fulfilled the following two criteria were promoted for further studies: (1) inhibition or stimulation of the FGF14:Nav1.6 complex by at least 40% (equivalent to 60% or 140% luminescence when normalized to DMSO controls, respectively) and Z-score ≥ 3 for enhancers or ≤ −5 for inhibitors, and (2) the primary kinase target was targeted by two or more compounds meeting the prior hit selection criteria (i.e., at least two inhibitors of JAK2 observed to modulate FGF14:Nav1.6 complex assembly by ≥ 40%). Compounds with known promiscuity were avoided where possible (alternative hit inhibitors of the same target).
3.2. Initial validation of hits against JAK2 and Src
Prior to further mechanistic studies, we conducted dose response validation of selected hits targeting JAK2 and Src against the double stable cell line using screening library compounds to determine which to proceed with for additional testing. These studies confirmed initial findings, revealing low micromolar potencies (1–15 μM), and validated JAK2 and Src inhibitors from structurally distinct families (Figure 2). JAK inhibitors with a preference for the JAK3 isoform had varied effects by LCA (Suppl. Table 2), and the most significant JAK3 inhibitors, such as 420121, have numerous additional targets. This combined with the observation that the JAK3 inhibitor 420126 failed to validate during initial dose dependency studies (Suppl. Fig. 3), suggested that JAK3 was unlikely to be a key regulator of the FGF14:Nav1.6 complex. Additionally, the enhancing effect of JAK inhibitors with a preference toward JAK1 (INCB424, XL019) may suggest a different role for JAK1-mediated regulation of the FGF14:Nav1.6 complex.
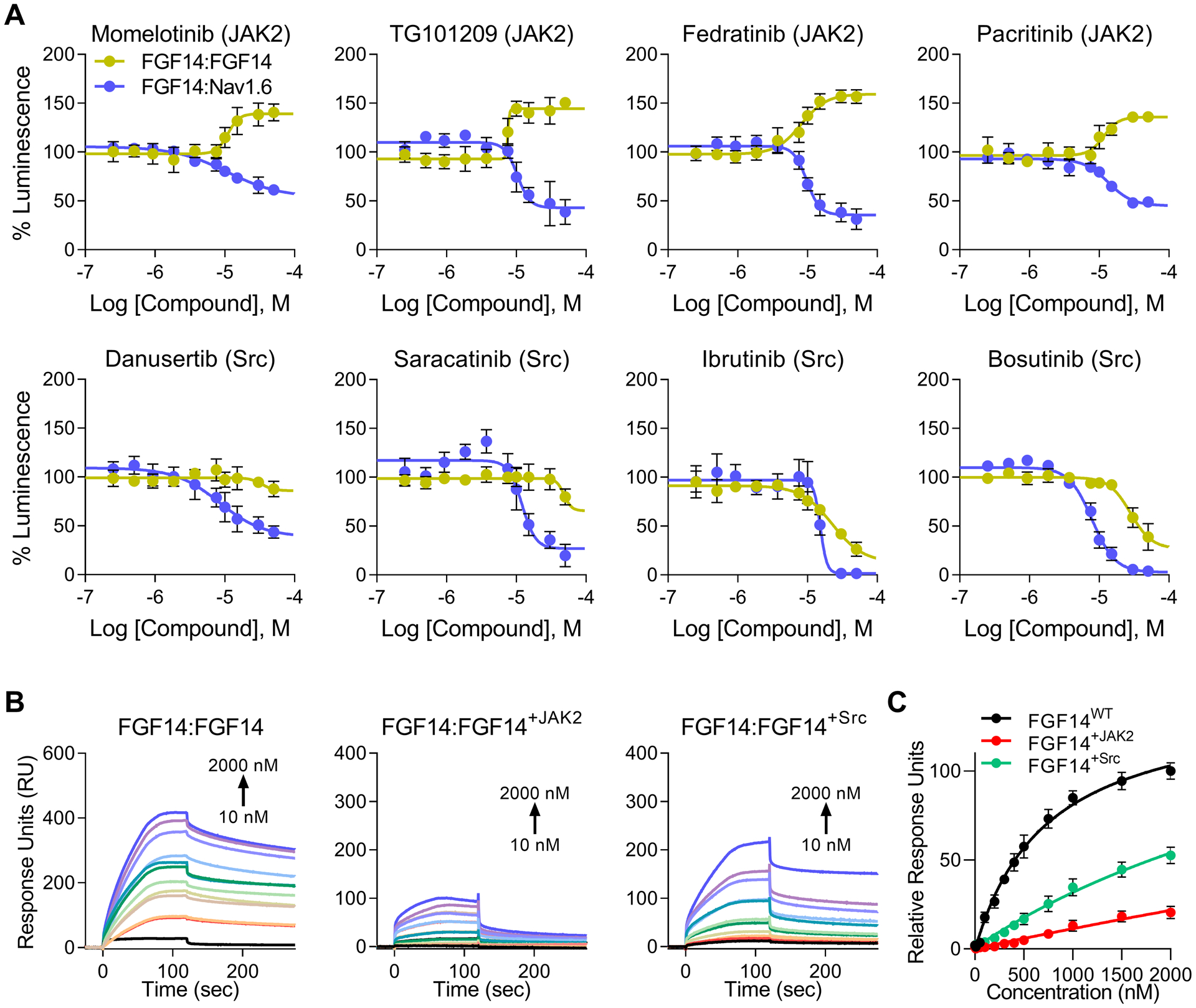
Figure 2. Identification of JAK and Src as regulators of the Nav1.6 complex by HTS.
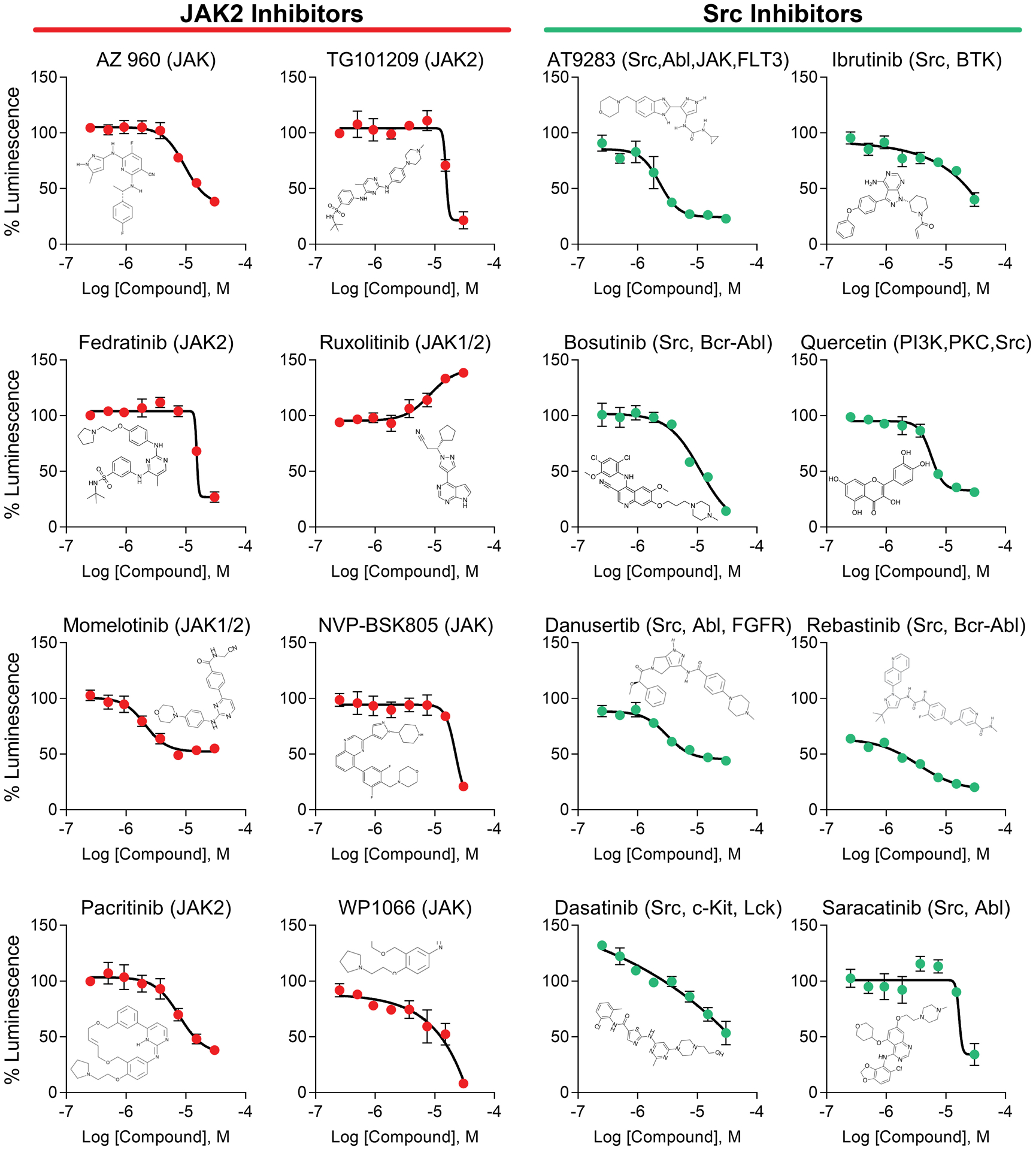
JAK and Src kinase inhibitors were consistently ranked among the highest scoring non-toxic compounds from an HTS of kinase inhibitors against the FGF14:Nav1.6 complex. Dose response plots are shown using screening library compound for the top eight inhibitors targeting JAK2 (orange) or Src (teal). Each compound’s structure is inlaid into its respective plot, demonstrating structural diversity among hits. Data are mean percent luminescence ± SD (n = 4 per treatment over two 384-well plates) with a non-linear regression curve fitting.
As to whether the effects of JAK inhibitors are driven in part by signal transducer and the activator of transcription 3 (STAT3) signaling, a pathway that often co-exists with JAK2 in cell regulatory mechanisms[26], we also identified several STAT3 inhibitors in the screening (Table 1), and STAT3 tyrosine binding motifs were identified in the FGF14 sequence (Table 2). From the Selleck library, two STAT3 inhibitors (S3I-201 and Ursolic Acid) had minimal impact (115.3% and 103.5% luminescence, respectively), while the inhibitor Stattic resulted in almost complete inhibition of the FGF14:Nav1.6 complex (6.0% luminescence), but was excluded due to cell toxicity (57.2% fluorescence from the CTB assay). In the Broad library, two top scoring STAT3 inhibitors Cucurbitacin I and Niclosamide (5.6% and 18.7% luminescence, respectively) were identified. Initial dose-dependency studies for the natural product Cucurbitacin I revealed highly potent but undesirable curve shape (linear decrease in luminescence) (Suppl. Fig. 3), possibly due to additional inhibition of the NF-kB pathway[42]. Although this odd behavior for the FGF14:Nav1.6 complex was less prevalent in follow-up studies with repurchased compound, a similar pattern of linear direction for enhancing the FGF14:FGF14 dimer was observed using Cucurcitabine I, but not S3I-201, which had minimal effect against FGF14:Nav1.6. Altogether, the results suggest that STAT3 may be involved, but further studies would be required to fully discern the mechanism. One possible reason for the less clear patterns observed may be due to fewer available inhibitors specific for STAT3, as well as that changes in STAT3 regulation of the FGF14:Nav1.6 complex may be a less potent form of regulation (i.e. indirect) than that of phosphorylation by JAK2.
Inhibitors of other Src-family kinases were also identified as hits. Five out of 18 inhibitors targeting Lck kinase were hit inhibitors, including PRT062607, Syk Inhibitor III, and ER 27319, with % luminescence ranging from 22.2 to 50.5%. Two out of three compounds targeting Lck, were also hits, including AMG-47a and 428205. Of these, PRT062607 and 428205 demonstrated promising concentration dependency (Suppl. Fig. 3), but ER 27319 was less ideal. Although these findings support the role of Src family kinases in regulation of the FGF14:Nav1.6 complex, Lck and Syk were not further pursued due to both proportionately low number of hits (relative to total # screened compounds targeting that kinase), as well as lack of observed phosphorylation motifs in FGF14.
3.3. Counter-screening and differential regulation of FGF14:FGF14 homodimer by JAK2, but not Src
Select kinases targeted by ≥4 hits from the HTS against the FGF14:Nav1.6 complex were counter-screened against the FGF14:FGF14 homodimer, using ≥2 selected compounds per target based on hypothesis-driven target analysis, as well as inhibitor selectivity, potency, and availability. GSK3, NF-kB, Akt, MEK, and PI3K inhibitors were tested based on our previous studies showing regulation of the FGF14:Nav1.6 complex by these pathways[20,21,41], and direct phosphorylation of FGF14 at S226 by GSK3β[43]. Conversely, Src, JAK2, and p38 MAPK inhibitors were tested based on phosphomotifs identified in the FGF14 sequence (Table 2). HEK293 cells transiently transfected with CLuc-FGF14 and FGF14-NLuc (Figure 3A) were treated with inhibitors (30 μM) in 384-well plates in triplicate, similarly to the primary screening against the FGF14:Nav1.6 complex. For simplicity, the results are shown as a heat map for both the individual compound screening results (Figure 3B, left), as well as the average effect from all inhibitors for a given kinase target (Figure 3B, right). More detailed compound screening results with statistical analysis are shown in Suppl. Figure 4. Counter-screening revealed that JAK2 was the only target that differentially regulated the two complexes (change in complex assembly in opposing directions) (mean from all four JAK2 inhibitors: FGF14:Nav1.6, 43.65%; FGF14:FGF14, 144.6% luminescence). We also proceeded with investigations of Src due to the high confidence of FGF14:Nav1.6 complex regulation, as indicated by the high proportion of hits versus screened compounds (65%; ranked #1 out of all kinase targets). Additionally, the three predicted phosphorylation sites in FGF14 (Y158, Y162, and Y211; Table 2), as well as the moderate inhibitory effect of Src inhibitors on FGF14:FGF14 dimerization (mean from all four Src inhibitors: FGF14:Nav1.6, 32.6%; FGF14:FGF14, 66.7% luminescence) was indicative of regulation by Src on both complexes.
Figure 3. Counter-screening of inhibitors for top kinase targets against the FGF14:FGF14 dimer.
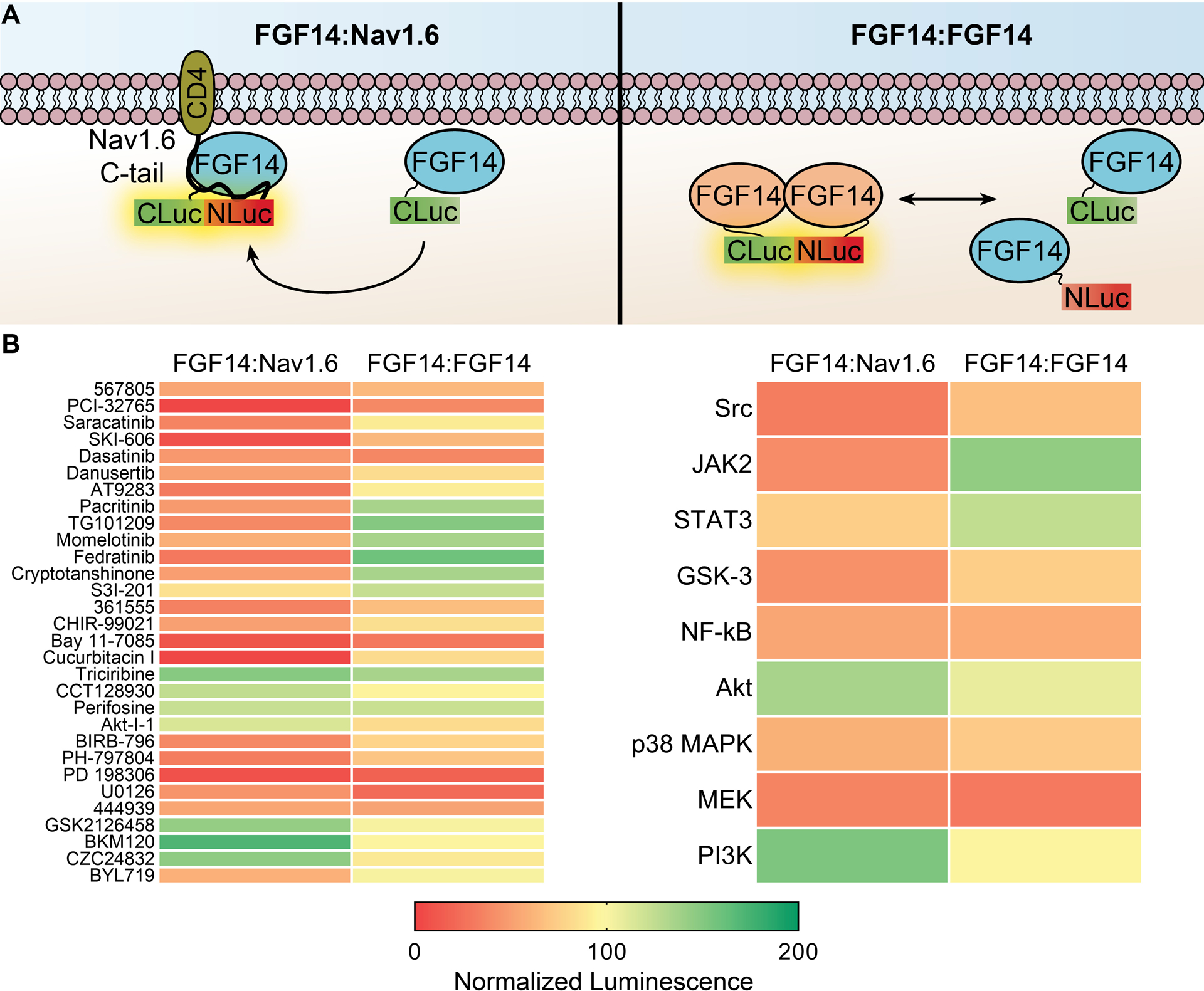
(A) Cartoon representation of hypothesized interactions occurring in LCA for the FGF14:Nav1.6 (left) vs. FGF14:FGF14 dimer (right). (B) Kinases targeted by ≥4 hits from the HTS against the FGF14:Nav1.6 complex that also revealed phosphorylation or binding motifs (or those of upstream pathways, such as Akt) in FGF14 were counter-screened against the FGF14:FGF14 dimer, using ≥2 selected compounds per target based on selectivity, potency, and availability. Left, heatmap of mean normalized luminescence for individual compounds tested against either the FGF14:Nav1.6 complex (represented in panel A, left) or FGF14:FGF14 dimer (represented in panel A, right). For counter-screening, transiently transfected HEK293 cells were seeded in 384-well plates and treated with 0.3% DMSO (n = 32) or kinase inhibitors (30 μM; n = 3 per compound). Right, the mean percent luminescence from all compounds for each kinase group is shown as a heat map for the two complexes. Note that only JAK2 inhibitors demonstrated a consistent and opposing response between the FGF14:Nav1.6 complex and FGF14:FGF14 dimer. Individual values and statistical analysis for these data are shown in Suppl. Fig. 4.
Based upon potency, efficacy, curve shape from the initial concentration dependency experiments, as well as inhibitor selectivity, we repurchased top JAK2 and Src inhibitors for counter-screening and validation studies. While virtually all kinase inhibitors have numerous off-target effects, we proceeded with compounds where no unspecific activity has been reported, and to not have identical off-target effects. Fedratinib, Pacritinib, and TG101209 preferentially inhibit JAK2 over other JAK isoforms, but Fedratinib also binds TYK2, and the latter two also target FLT3 at higher concentrations[44–46]. Momelotinib (also known as CYT387) inhibits both JAK1/2, but is not known for significant effects against either FLT3 or TYK2[45,47]. The lack of identical off-target effects among inhibitors enabled us to rule out the effect of these alternate targets.
For Src, we did not further pursue AT9283, Quercetin, or Dasatinib due to known promiscuity for many kinase targets[48]. Interestingly, the Src inhibitor KX2–391, present in all three screened libraries, was the only compound targeting Src that acted as an enhancer of the FGF14:Nav1.6 complex (Suppl. Table 2 and Suppl. Fig 3). KX2–391 is a non-ATP competitive peptide mimetic, differentiating it from the majority of available Src inhibitors, but this compound also promotes tubulin polymerization[49], and we have previously demonstrated that microtubule inhibitors act as potent enhancers of the LCA[25], which may not be biologically relevant. Therefore, we continued with Danusertib, Saracatinib, Ibrutinib, and Bosutinib for further studies targeting Src.
All eight of these compounds, including the JAK2 inhibitors Momelotinib, TG101209, Fedratinib, and Pacritinib, as well as the Src inhibitors Danusertib, Saracatinib, Ibrutinib, and Bosutinib, were additionally counter-screened against the full-length luciferase to rule out that observed LCA effects were due to modulation of luciferase alone (Suppl. Fig 5).
Following inhibition of JAK2, but not Src, FGF14 homodimerization increased in a manner directly inverse to FGF14:Nav1.6 complex formation with a comparable degree of both efficacy (minimum vs. maximum percent luminescence for FGF14:Nav1.6 compared to FGF14:FGF14 dimerization, respectively), as well as potency (inhibitor IC50 against the FGF14:Nav1.6 complex vs. EC50 against the FGF14:FGF14 dimer) (Figure 4A and Table 3). The most potent JAK2 inhibitor was Fedratinib (FGF14:Nav1.6, IC50 = 9.7 μM; FGF14:FGF14, EC50 = 8.2 μM), also exhibiting strong maximal but inverse effects for each complex (FGF14:Nav1.6, 35.7% luminescence; FGF14:FGF14, EC50 = 156.7% luminescence).
Figure 4. Differential regulation of the FGF14:FGF14 dimer and FGF14:Nav1.6 complex by JAK2, but not Src.
(A) Dose responses (10-point, n = 8 per concentration over two 384-well plates) were conducted against the FGF14:Nav1.6 complex (purple) for promising hits using repurchased compounds in order to validate HTS results. Positive hits were then counter-screened against the FGF14:FGF14 dimer (yellow), with the hypothesis that changes in FGF14 dimerization could be associated with inverse changes in FGF14:Nav1.6 binding. Inhibition of JAK2 but not Src, increases FGF14 dimerization in a manner directly inverse to FGF14:Nav1.6 complex formation. Estimated efficacy and potency are shown in Table 3. Luminescence for each well was normalized to per plate 0.3% DMSO controls (n = 32 per plate), and the mean normalized luminescence ± SD is shown. (B) Representative SPR sensorgrams from proteins flown across a chip with FGF14 bound (1,030 RU) using a flow rate of 50 μL/min. Purified FGF14 protein was phosphorylated in vitro by pre-incubation with either JAK2 or Src tyrosine kinases as indicated above each panel. The resulting equilibrium dissociation constants (KD), as well as kinetic association (kon) and dissociation (koff) rates are provided in Table 4. (C) Steady-state saturation plot for comparison of wild-type (WT) versus phosphorylated protein binding to FGF14 with response units (RU) relative to the maximal binding response of the WT protein. Data are mean normalized response units ± SD.
Table 3. Potency and efficacy against the FGF14:FGF14 dimer and FGF14:Nav1.6 complex by JAK2 and Src inhibitors.
Estimated potency (IC50 or EC50, μM) and efficacy (minimal (IMin) or maximal (EMax) percent luminescence at the bottom plateau for inhibition and top plateau for stimulation, respectively) and for JAK2 and Src inhibitors based on the LCA data in Figures 4 and 6. Luminescence for each well was normalized to per plate 0.3% DMSO controls (n = 32 per plate).
| Inhibitor | Target | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| IMin/EMax | IC/EC50 | IMin/EMax | IC/EC50 | IMin/EMax | IC/EC50 | IMin/EMax | IC/EC50 | ||
| Momelotinib | JAK2 | 53.5 | 10.9 | 138.6 | 10.8 | 101.8 | N/A | 90.9 | N/A |
| TG101209 | JAK2 | 43.0 | 10.3 | 150.6 | 7.6 | 78.0 | 98.0 | 73.4 | 69.4 |
| Fedratinib | JAK2 | 35.7 | 9.7 | 156.7 | 8.2 | 76.8 | 27.1 | 72.8 | 28.5 |
| Pacritinib | JAK2 | 45.0 | 13.5 | 136.0 | 11.6 | 110.6 | N/A | 79.3 | 5.5 |
| Danusertib | Src | 38.6 | 8.4 | 86.0 | 34.1 | ||||
| Saracatinib | Src | 27.0 | 12.1 | 65.7 | 48.3 | ||||
| Ibrutinib | Src | 1.6 | 15.3 | 12.8 | 22.6 | ||||
| Bosutinib | Src | 3.2 | 9.7 | 26.8 | 28.1 |
For Src inhibitors, FGF14:Nav1.6 complex formation was potently inhibited (IC50 range: 8.4 – 15 μM) to a high degree (range: 1.6 – 38.6% luminescence). Varying degrees of inhibition were also observed for the FGF14 dimer (range: 12.8 – 86.0% luminescence), but the potency for these compounds was greatly increased compared to the FGF14:Nav1.6 complex (IC50 range: 28 – 48 μM).
3.4. High affinity FGF14:FGF14 dimerization is efficiently abolished by JAK2 phosphorylation
Next, we used surface plasmon resonance (SPR) to determine the impact of phosphorylation on the FGF14:FGF14 dimer formation using unphosphorylated recombinant FGF14 protein bound to the chip surface (1,030 RU). This revealed that the high affinity dimerization of recombinant FGF14 protein (KD = 440 nM) was abolished upon pre-incubation with (and presumably phosphorylation by) JAK2 (KD = 2.6 μM) or Src (KD = 1.3 μM) kinases, with notable change in kinetics (Figure 4B and Table 4). FGF14 flowing over the chip appeared to remain tightly bound following injection stop (koff = 0.000934 s−1), and similar kinetics are observed for FGF14+Src, although a lesser degree of FGF14 remained bound (dissociated more quickly; koff = 0.00109 s−1); the association rate between the two appeared similar. Following incubation with JAK2, however, FGF14 dissociated rapidly (koff = 0.00459 s−1) from the FGF14 protein bound to the chip surface. In terms of overall binding, FGF14 had the highest overall binding (~400 RU), followed by ~200 RU for FGF14+Src and ~100 RU for FGF14+JAK2. We concluded that FGF14 phosphorylated by JAK2 had overall less affinity for FGF14WT and bound much more transiently. For FGF14 phosphorylated by Src, we concluded that the dimerization event became overall less favorable, as well as that the dimer stability was moderately reduced. Given the much stronger phenotype with JAK2, we pursued it for further mechanistic validation studies, beginning with identifying the phosphorylation site(s).
Table 4. High affinity FGF14:FGF14 dimerization is abolished by phosphorylation by JAK2, and by Src to a lesser extent.
Kinetic constants calculated from data represented in Figure 3B. The KD represents the average between the kinetic KD, calculated using the simplest Langmuir 1:1 interaction model (KD=koff/kon), and the steady-state saturation (affinity) KD. Data are mean ± SD.
| KD (nM) | kon (M−1 s−1) | koff (s−1) | |
|---|---|---|---|
| FGF14:FGF14 | 440 ± 57 | 1.36×104 ± 4.9×102 | 9.04×10−4 ± 9.2×10−5 |
| FGF14:FGF14+JAK2 | 2701 ± 97 | 4.02×103 ± 5.6×102 | 4.52×10−3 ± 3.1×10−4 |
| FGF14:FGF14+Src | 1312 ± 84 | 9.06×103 ± 3.5×102 | 1.33×10−3 ± 4.9×10−4 |
3.5. JAK2 phosphorylates FGF14Y158
Phosphomotif scans revealed Y158 as a potential JAK2 phosphorylation site (Table 2), and homology modeling here as well as in previous studies[27,28] has demonstrated that this site is at the PPI interface of both the FGF14:FGF14 dimer and the FGF14:Nav1.6 complex (Figure 5A and Suppl. Fig. 2). Thus, hypothesizing that this was the site of interest, we used a 20aa peptide derived from FGF14 (aa149–168 of FGF14–1b, corresponding to the sequence KFKESVFENYYVIYSSMLYR) containing the predicted JAK2 substrate motif to confirm this as the phosphorylation site. Peptides were incubated with 200 nM recombinant human JAK2 kinase at 30 °C for 30 min, followed by overnight incubation at 4 °C. Mass spectrometry (MALDI TOF-MS/MS) confirmed Y158 as the site of phosphorylation by JAK2 in vitro (Figure 5B), as identified by the presence of y10 (theoretical m/z of 1293.66, observed m/z of 1293.56) and y11 (theoretical m/z of 1536.69, observed m/z of 1536.63) ions (1536.63–1293.56 = 243.07, corresponding to the MW of Y(PO3).
Figure 5. MALDI TOF-MS/MS validation of JAK2 phosphorylation of Y158 on FGF14.
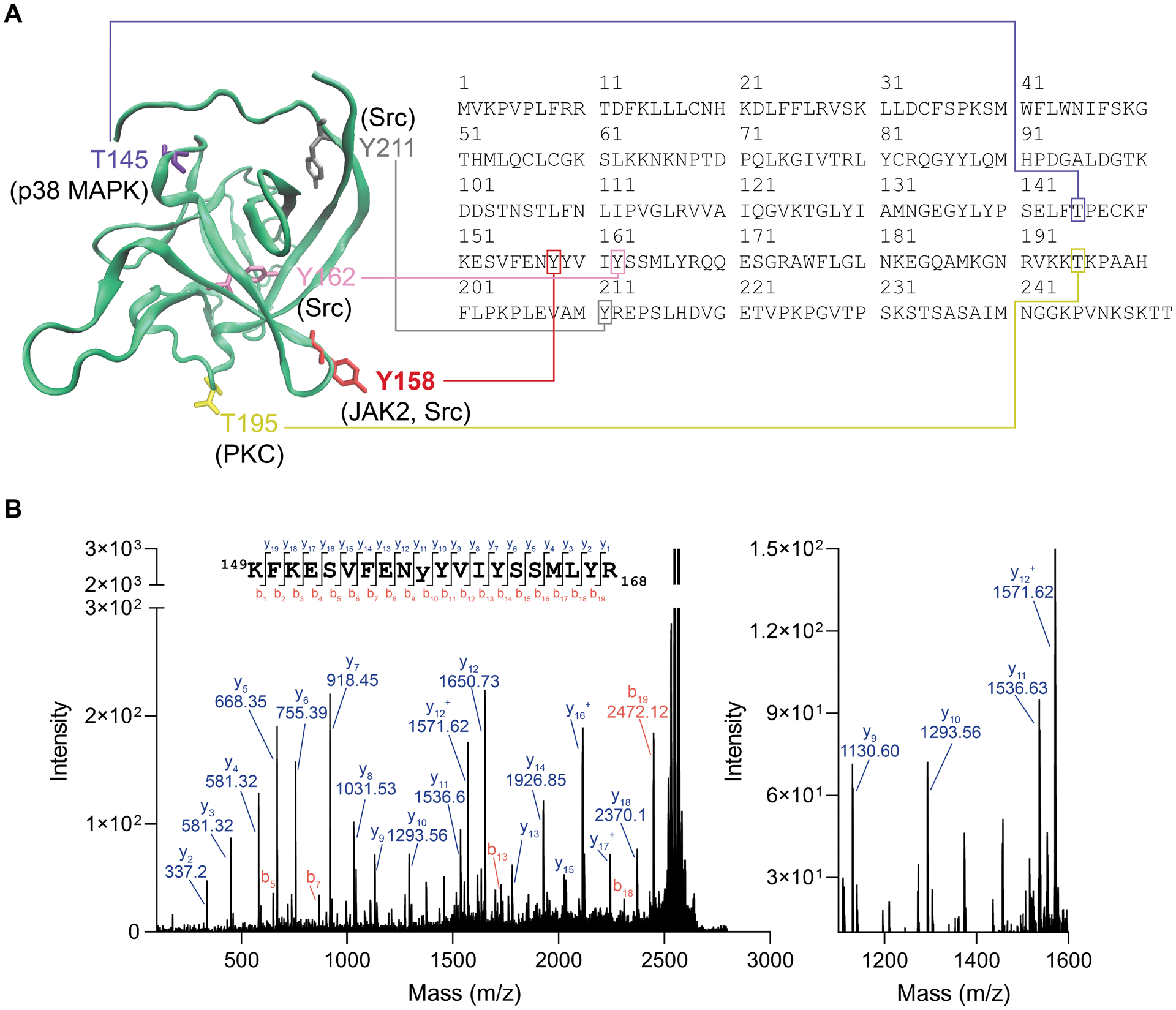
(A) Homology model of an FGF14 monomer showing potential phosphorylation sites and their corresponding motif in the FGF14–1b sequence (accession number NP_787125). Y158, red, while Y162 is shown as purple. Also showing other predicted phosphorylation sites that are not at the protein:protein interaction interface, including T145, T195, and Y211. (B) MALDI TOF-MS/MS fragmentation spectrum of the phosphopeptide KFKESVFENyYVIYSSMLYR (y = phosphotyrosine), encompassing residues 149–168 of FGF14–1b. The presence of y10 (theoretical m/z of 1293.66, observed m/z of 1293.56) and y11 (theoretical m/z of 1536.69, observed m/z of 1536.63) ions confirms Y158 as the site of phosphorylation (1536.63–1293.56 = 243.07, corresponding to the MW of Y(PO3).
3.6. Y158 mediates both JAK2 regulation of FGF14, as well as high affinity FGF14 dimerization
To validate Y158 as the site for JAK2-dependent regulation of FGF14 and Nav1.6, an alanine point mutation (Y158A) was introduced in the CLuc-FGF14 LCA construct. This construct was used to compare the effect of JAK2 inhibition on the mutant FGF14:FGF14Y158A heterodimer and FGF14Y158A:Nav1.6 complexes to the corresponding FGF14 wild-type complexes (Figure 4). As expected, the effects of JAK2 inhibitors on both the FGF14:Nav1.6 complex and the FGF14:FGF14 dimer were abolished or reversed in the presence of FGF14Y158A (Figure 6A), the site of JAK2 phosphorylation in vitro. As shown in Table 3, for the FGF14Y158A:Nav1.6 complex, the effects of Momelotinib and Pacritinib were abolished, while those of TG101209 and Fedratinib were greatly reduced (78.0% and 76.8% luminescence, respectively, with these remnant inhibitory effects being observed only at much higher concentrations compared to FGF14WT (i.e., Fedratinib IC50 shift from 9.7 μM to 27 μM).
Figure 6. Y158 mediates both JAK2 regulation of FGF14, as well as high affinity dimerization.
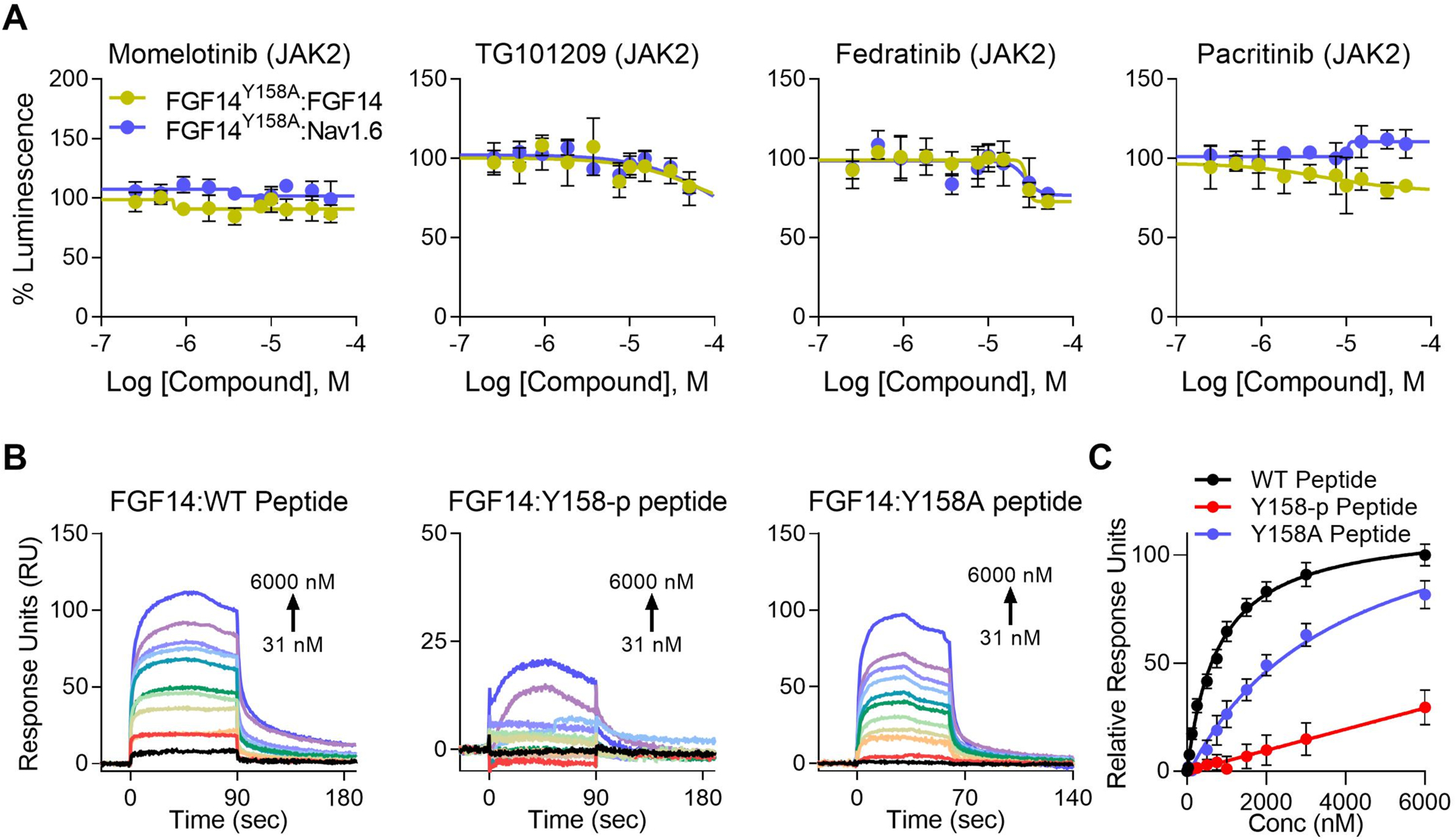
(A) Dose responses (10-point, n = 8 per concentration over two 384-well plates). Differential regulation of JAK2 inhibitors between the FGF14:Nav1.6 complex and FGF14:FGF14 dimer is almost completely abolished when tested against FGF14Y158A:Nav1.6 and FGF14Y158A:FGF14 mutant complexes using LCA. Furthermore, the effect of Fedratinib was reversed for the FGF14Y158A:FGF14 heterodimer, with high concentrations mildly decreasing dimerization. Estimated efficacy and potency are shown in Table 3. Data are mean normalized luminescence ± SD. (B) Representative SPR sensorgrams showing increasing concentrations of non-phosphorylated wild-type FGF14 peptide (WT peptide, left), FGF14 peptide phosphorylated at Y158 (Y158-p peptide, middle), or FGF14Y158A mutant peptide (Y158A peptide, right) flown over recombinant FGF14 protein bound to CM5 chips (16,045 RU) using a flow rate of 60 μL/min and concentrations ranging from 31 – 6000 nM. Phosphorylation (or lack thereof) at Y158 was verified by mass spectrometry (Figure 5). Kinetic analysis of each ligand:analyte interaction was obtained by fitting the response data to the simplest Langmuir 1:1 interaction model (KD=koff/kon). The resulting equilibrium dissociation constants (KD), as well as kinetic association (kon) and dissociation (koff) rates are provided in Table 5. The phosphorylated FGF14Y158-p peptide has reduced binding affinity for FGF14. (C) Steady-state saturation plot for comparison of wild-type (WT, black) versus phosphorylated (Y158-p, orange) and mutant (Y158A) peptide binding (black, WT; orange, phosphorylated (Y158-p); teal, Y158A mutant) to FGF14 with response units (RU) relative to the maximal binding response of the WT peptide. Data are mean normalized response units ± SD.
Similarly, the enhancing effect of JAK2 inhibitors on the FGF14:FGF14 dimer was reversed in the presence of FGF14Y158, with high concentrations inhibiting dimerization by non-significant (Momelotinib) to moderate degrees (TG101209, Fedratinib, and Pacritinib). This may signify off-target effects due to the higher concentrations and IC50 values observed.
We next sought to determine how phosphorylation at Y158 changes FGF14 self-interaction (homodimerization) using SPR. Phosphorylated and non-phosphorylated peptides were flown across a chip with FGF14 bound (16,045 RU). All peptides were incubated with JAK2 kinase, but only the phospho-peptide sample received ATP. Following the phosphorylation reaction, peptides were buffer exchanged into SPR running buffer (HBS-P+). Kinetic analysis revealed completely different binding kinetics and vastly reduced binding affinity of the phosphorylated FGF14 peptide (KD = 147 μM compared to 1 μM for the non-phosphorylated peptide) (Figure 6B and Table 5). Note that fitting kinetic data for the phosphorylated peptide was difficult due to kinetic constants approaching limits of instrument detection, and thus the estimated KD value should be interpreted only qualitatively. The reduced binding affinity of the Y158A peptide compared to the WT peptide demonstrates that Y158 is a key residue in mediating tight FGF14 homodimerization, as predicted by previous studies[27,28]. However, the binding kinetics are not fundamentally different compared to the WT peptide, confirming that other residues near Y158 are also important in mediating self-interaction.
Table 5. Equilibrium and kinetic constants for FGF14 self-interaction by SPR.
Kinetic constants were calculated based on data represented in Figure 5B. The KD represents the average between the kinetic KD, calculated using the simplest Langmuir 1:1 interaction model (KD=koff/kon), and the steady-state saturation (affinity) KD. Data are mean ± SD. Note that fitting kinetic data for the phosphorylated peptide was difficult due to kinetic constants approaching limits of instrument detection, and thus the estimated values should be interpreted only qualitatively.
| KD (μM) | kon (M−1 s−1) | koff (s−1) | |
|---|---|---|---|
| FGF14WT Peptide | 1.01 ± 0.07 | 1.69×104 ± 8.1×102 | 4.64×10−2 ± 7.2×10−3 |
| FGF14Y158-p Peptide | 181.1 ± 109 | 3.29×102 ± 1.2×102 | 8.97×10−2 ± 3.9×10−2 |
| FGF14Y158A Peptide | 7.02 ± 0.21 | 2.32×104 ± 1.6×103 | 1.58×10−1 ± 7.6×10−3 |
3.7. JAK2 inhibition abolishes FGF14-dependent functional modulation of Nav1.6 channels
To test the modulatory effects of JAK2 inhibition on Nav1.6-mediated Na+ currents, we used whole-cell patch-clamp electrophysiology on HEK293 cells stably expressing Nav1.6 (HEK-Nav1.6) that were transiently transfected with either GFP (HEK-Nav1.6 GFP) or FGF14-GFP (HEK-Nav1.6 FGF14); each group was treated with either Fedratinib (20 μM) or vehicle (0.01% DMSO) (Figure 7 and Suppl. Table 3). Fedratinib was selected based on the LCA dose response studies demonstrating it as having the greatest potency and efficacy compared to other JAK2 inhibitors (as represented in Figure 4A and Table 3). In agreement with previous studies[28,33], expression of FGF14-GFP suppressed Nav1.6-mediated peak transient Na+ current (INa) density (−24.87 ± 2.99 pA/pF, n = 16 vs. −59.43 ± 6.0 pA/pF, n = 15; p < 0.0001; Student t-test; Figure 7A–C). In the presence of Fedratinib, the FGF14-mediated suppression of Nav1.6 current was abolished relative to controls (Fedratinib: −81.27 ± 11.3 pA/pF, n = 13; DMSO: −24.87 ± 2.99 pA/pF, n = 16, p < 0.0001; Student t test). Crucially, Fedratinib had no effect on the channel alone (HEK-Nav1.6 GFP), as the peak current density was not significantly different (−59.43 ± 6.0 pA/pF, n = 15) compared DMSO (−51.5 ± 3.69 pA/pF, n = 13, p < 0.2848).
Figure 7. Inhibition of JAK2 abolishes FGF14-dependent modulation of Nav1.6 currents.
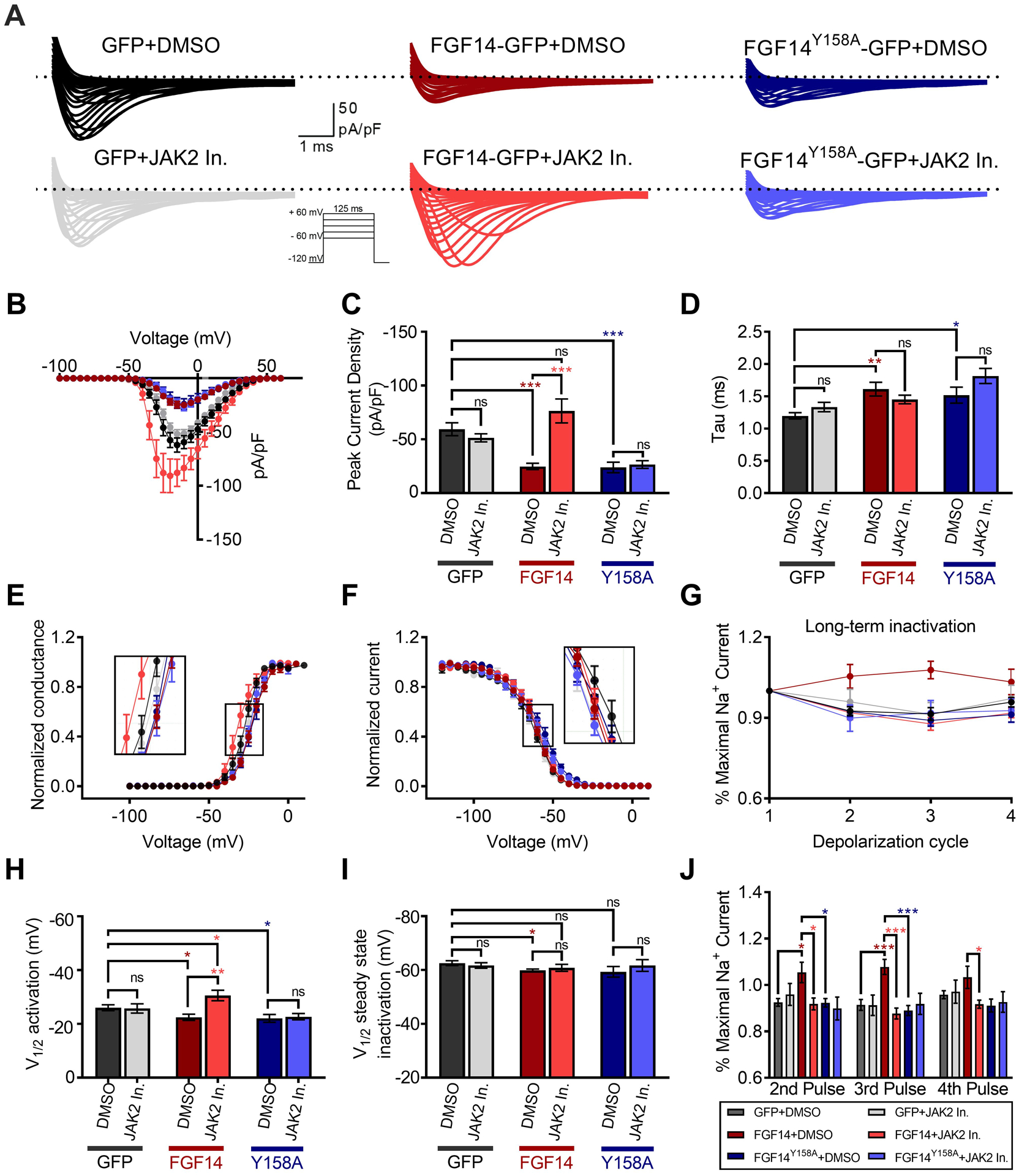
(A) Representative traces of Na+ transient currents (INa) in response to depolarizing voltage steps recorded from HEK-Nav1.6 cells transiently expressing GFP, FGF14-GFP, or the FGF14Y158A-GFP mutant. Cells were treated with either the JAK2 inhibitor Fedratinib (20 μM) or vehicle (0.05% DMSO) alone. (B) Current-voltage relationships of INa from experimental groups described in (A). (C) Peak-current densities at −10 mV derived from A and B. (D) Tau of fast inactivation was calculated at the peak current density (−10 mV) from experimental groups described in A. Normalized conductance (E) and current (F) are plotted as a function of the membrane potential (mV) and were used to extrapolate the voltage-dependence of INa activation and steady-state inactivation, respectively. (G) Summary of percent maximal sodium current following four depolarizing pulses used to study long-term inactivation (LTI). Significance for each pulse is shown in panel J. Note that either inhibition of JAK2 or the FGF14Y158A mutation was sufficient to abolish the FGF14-dependent change in Nav1.6 LTI. Voltage of half-maximal (V1⁄2) of activation (H) and steady-state inactivation (I). (J) Mean normalized sodium current following depolarizing pulses represented in panel G. Data in B-J are mean ± SEM. Statistical significance between groups was assessed using Student’s t-test; ***, p<0.0001, **, p<0.001, *, p<0.05.
To confirm that this effect was mediated through Y158, as suggested by LCA and mass spectrometry, we next assessed the effect of Fedratinib on Nav1.6 currents in the presence of the FGF14Y158A mutant (HEK-Nav1.6 cells transfected with FGF14Y158A-GFP, hereafter referred to as HEK-Nav1.6 FGF14Y158A). In these cells, the peak current density of Nav1.6-mediated transient INa+ was similar to that observed in the presence of the wild-type FGF14, and not statistically different when treated with Fedratinib (−26.65 ± 3.5 pA/pF, n = 13) compared to DMSO (−23.97 ± 4.8 pA/pF, n = 12, p < 0.6567; Figure 7A–C). Hence, neither the Y158A mutation alone, nor in combination with Fedratinib treatment, was capable of rescuing the FGF14-mediated suppression of Nav1.6 currents.
Further analysis revealed that for HEK-Nav1.6 GFP, the decay time constant (τ) of INa+ was not significantly affected by Fedratinib (1.3 ± 0.07 ms, n = 13) compared to DMSO (1.2 ± 0.05 ms, n = 10), confirming that JAK2 does not affect τ in the absence of FGF14. However, τ was significantly slower in the presence of FGF14 (1.6 ± 0.1 ms, n = 14, p < 0.0052), similar to as observed previously[28,33]), as well as in the presence of FGF14Y158A (1.5 ± 0.12 ms, n = 12, p < 0.0268) compared to GFP alone. Although Fedratinib partially abolished this effect of FGF14 (1.4 ± 0.06 ms, n = 12, Figure 7D and Suppl. Table 3), τ was significantly slower in the FGF14Y158A group treated with Fedratinib (1.8 ± 0.11 ms, n = 12, p < 0.0147) compared to FGF14-GFP.
Similar to our previous reports[20,28,33], we observed a depolarizing shift in the V½ of activation of Nav1.6 in the presence of FGF14-GFP (−22.4 ± 1.1 mV, n = 12) compared to controls (GFP+DMSO; −26.03 ± 1.1 mV, n = 14, p < 0.0358, Figure 7E,H and Suppl. Table 3). Crucially, pretreatment with Fedratinib abolished this shift for the wild-type FGF14 (−30.56 ± 1.9 mV, n =13, p < 0.0019) compared to GFP+DMSO control, but had no effect on the channel alone (GFP+Fedratinib, −25.7 ± 1.7 mV, n = 13). In contrast, the FGF14-induced depolarizing shift in the V½ of activation was neither abolished by the Y158A point mutation in itself (i.e., DMSO, −22.06 ± 1.4 mV, n = 12), nor was Fedratinib capable of rescuing this phenotype (−22.69 ± 1.2 mV, n = 13).
Likewise, expression of wild-type FGF14 caused a depolarizing shift in V½ of steady-state inactivation relative to the HEK-Nav1.6 GFP control group when treated with DMSO alone (−59.8 ± 0.5 mV, n = 12 vs. −62.6 ± 0.9 mV, n = 12, p < 0.0019; Figure 7F,I), and the presence of the Y158A mutation did not rescue this phenotype mutation (−59.3 ± 2.0 mV, n = 12). Fedratinib had no significant effect on the channel alone (i.e., GFP, −61.7 ± 1.0 mV, n = 15) relative to HEK-Nav1.6 GFP treated with DMSO. However, pretreatment of HEK-Nav1.6 FGF14 cells with Fedratinib partially abolished this phenotype (−60.8 ± 1.2 mV, n = 16), as it resulted in no significant change compared to DMSO for both HEK-Nav1.6 GFP and HEK-Nav1.6 FGF14. Treatment of HEK-Nav1.6 FGF14Y158A cells with Fedratinib behaved similarly, with no significant change compared to DMSO for any condition. Altogether, these results indicate that in the presence of the Y158A point mutation, FGF14 remains functionally active toward Nav1.6, but that JAK2 is incapable of modulating this interaction. This suggests that Y158 is the key node of JAK2-dependent regulation of FGF14.
Intracellular FGFs expressing the 1a N-terminal tail have been shown to induce Nav channel long-term inactivation (LTI), a slow inactivation process that controls channel availability over repetitive stimulation[50–52]. Following a 30 min incubation with either DMSO or Fedratinib, LTI in all experimental groups was evaluated using depolarizing steps (from −90 mV to −10 mV) spaced by 40 ms, a time interval that allows Nav channels to fully recover from fast inactivation. In HEK-Nav1.6 GFP cells, Nav1.6 channels recovered nearly completely from LTI for both controls (DMSO) as well as for Fedratinib treatment (Figure 7G,J). In the presence of FGF14, the fraction of Nav1.6 channels that underwent LTI was significantly reduced, resulting in potentiated INa over the course of the depolarization cycles (FGF14+DMSO (maroon), 1.08 ± 0.03%, n = 15 vs. GFP+DMSO control (dark grey), 0.91 ± 0.02%, n = 14, p < 0.0001 for pulse 3). Crucially, pretreatment with Fedratinib rescued this FGF14-dependent phenotype (0.89 ± 0.02%, n = 14 for pulse 3) back to the level of the GFP control, suggesting that phosphorylation of FGF14 by JAK2 is required for regulation of slow inactivation of Nav1.6. However, unlike all other phenotypes described (Figure 7A–F), the Y158A mutation abrogated the effect of FGF14 on slow inactivation (Figure 7G,J, dark blue), although Fedratinib similarly had no effect in the presence of the Y158A mutation. These findings suggest that Y158 has an additional role in mediating FGF14-dependent modulation of slow inactivation, independent of phosphorylation status.
3.8. JAK2 regulates FGF14-dependent firing of hippocampal CA1 pyramidal neurons
Based on the conclusive evidence demonstrating that JAK2 controls FGF14-dependent modulation of Nav1.6, we next confirmed these findings ex vivo by examining the effect of the JAK2 inhibitor Fedratinib on intrinsic excitability of hippocampal CA1 pyramidal neurons (Figure 8). Previous studies have shown that these neurons abundantly express FGF14 and Nav1.6 channels, especially at the axonal initial segment, as well as exhibit a firing pattern consistent with Nav1.6 activity[20,30,53].
Figure 8. Inhibition of JAK2 increases hippocampal CA1 pyramidal neuron firing in an FGF14-dependent manner.
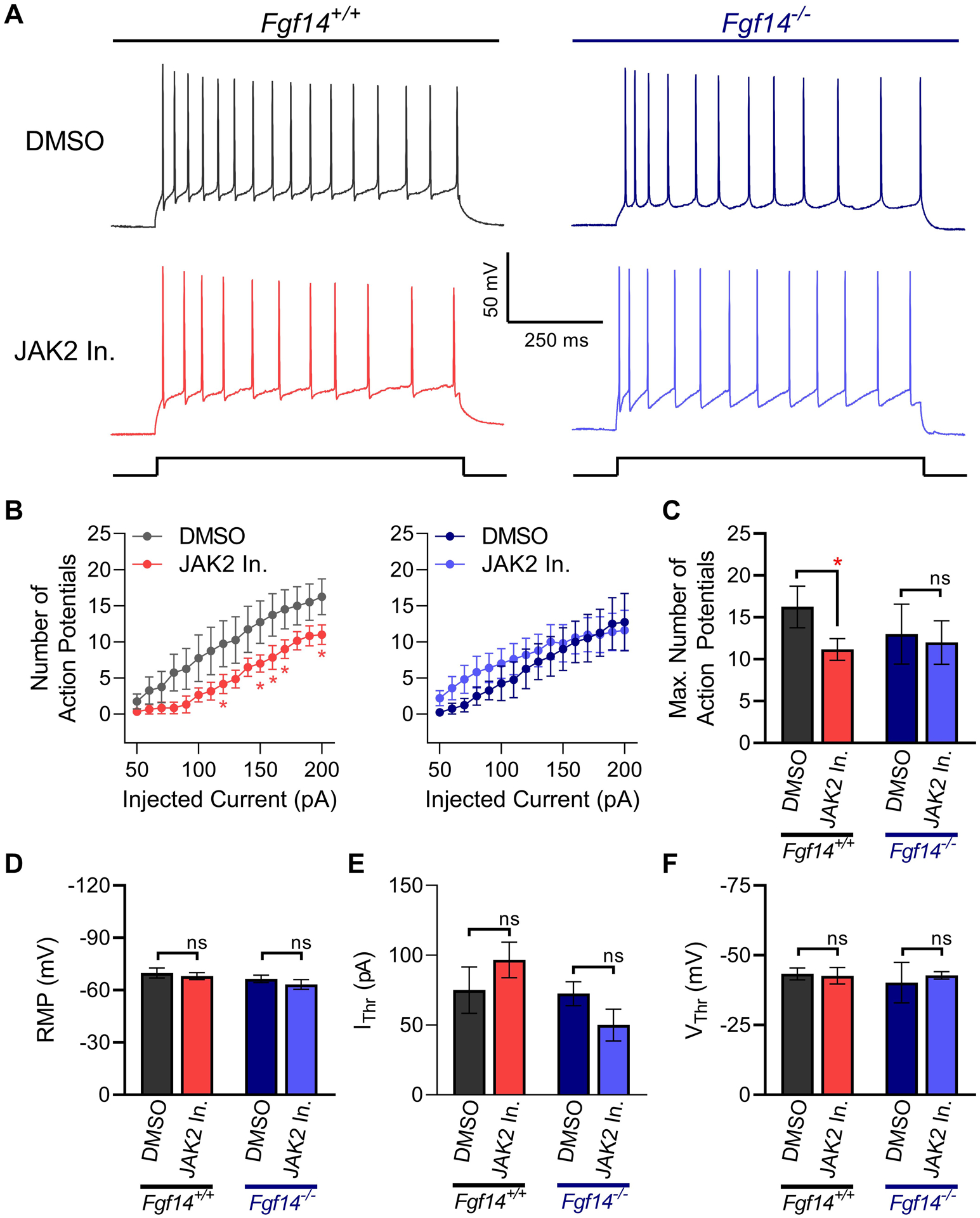
(A) Representative traces of maximal evoked action potentials in response to current injection in CA1 pyramidal neurons of Fgf14+/+ and Fgf14−/− mice following 1 hr incubation of coronal brain slices in DMSO (0.02%) or Fedratinib (20 μM). (B) Input-output curves of average number of evoked action potentials recorded in CA1 pyramidal neurons from Fgf14+/+ (left) or Fgf14−/− (right) mice in response to varying injected current stimuli. (C) Maximum number of action potentials fired from CA1 pyramidal neurons. (D) Resting membrane potential (RMP) (E) Current threshold (IThr) and (F) voltage threshold (VThr) for action potential firing in response to injected current in CA1 pyramidal neurons. Data in B-F are mean ± SEM. Statistical significance between groups was assessed using Student’s t-test with Welch Correction; *, p<0.05.
Using whole-cell patch clamp electrophysiology of acutely treated hippocampal slices from Fgf14+/+ mice, we observed that Fedratinib (20 μM) significantly decreases the maximum number of evoked action potentials (AP) (11.3 ± 1.4, n = 6) relative to DMSO controls (16.3 ± 2.5, n = 4, p < 0.05; t-test with Welch Correction) as seen in Figure 8B. This phenotype is likely driven via a reduction in Nav channel function as no significant changes were found in resting membrane potential (RMP; Figure 8D), current threshold and voltage threshold for action potential firing (Figure 8E,F) or other active or passive properties (Suppl. Table 4) that could likely drive the increase in firing.
To determine whether the mechanism of Fedratinib required the presence of FGF14 as expected from in vitro and in cell studies, similar patch-clamp experiments were conducted in Fgf14−/− mice. We found that in the knockout mouse model, Fedratinib did not significantly affect evoked action potentials (11.8 ± 2.7, n = 5) compared to DMSO controls (13 ± 3.9, n = 4, p = 0.82), in direct contrast to what was observed in Fgf14+/+ animals. Additionally, no significant changes were found in other active or passive properties (Suppl. Table 4) of hippocampal CA1 pyramidal neurons from Fgf14−/− mice, indicating that inhibition of JAK2 does not substantially modify these neuronal properties in the absence of FGF14.
Discussion
Our results build on previous studies showing that the Nav1.6 channel complex, a fundamental determinant of neuronal firing, is regulated by phosphorylation. We have previously conducted smaller scale screening campaigns [20,21,25] that identified multiple Ser/Thr kinases, including Akt/PI3K, PKC, and GSK3β, as regulators of the FGF14:Nav1.6 complex. Follow-up studies supported these results and demonstrated extensive regulation of Nav complexes by GSK3β, controlling neuronal excitability in both diseased and healthy states [43], in part through direct phosphorylation of FGF14 at S226, as well as both the Nav1.2 [17] and Nav1.6 [18] channel isoforms. While these initial discoveries laid the groundwork for the present studies, prior campaigns suffered from the following limitations: i) small scale screening library (~385 kinase inhibitors), resulting in lack of significant target overlap between inhibitors (i.e, only 1–2 compounds/target); ii) kinase target representation was neither complete nor fully distributed (i.e., preference toward Ser/Thr over Tyr kinase inhibitors; not all known kinases were represented, such as multiple tyrosine kinases); and iii) lack of initial rapid counter-screening studies to explore potential mechanisms, such as comparison of target effects on the FGF14:FGF14 vs FGF14:Nav1.6 complexes.
Here, we screened ~3,000 well-characterized compounds and FDA-approved kinase inhibitors with diverse mechanisms and an extremely wide range of targets covering the majority of the known “kinome.” The high degree of target overlap between screened libraries, in addition to a small degree of compound overlap (i.e., identical compound from different sources), was used as a measure of reproducibility to increase confidence in results. For instance, multiple hits targeting a common kinase, combined with similar effects being observed for the same compound across libraries, lends strong support toward that kinase being a true regulator rather than artefacts arising from inhibitor promiscuity. We selected hits through a serial selection pipeline that combined both unbiased and hypothesis-based criteria. Following exclusion of toxic compounds, “hits” were initially selected using binary Z-score and % luminescence cut-offs. Hits were then clustered by primary target, and we identified those targets with the highest proportion of hits to total number of screened compounds (Table 1), resulting in the identification of JAK2 and Src tyrosine kinases as the highest-ranking candidates. Additional high-ranking targets also corroborated findings of our previous studies that demonstrated a role of the GSK3, Akt/PI3K, NF-kB, and PKC pathways as regulators of the Nav channel [21]. Based upon these rankings, in addition to the identified phospho- and binding-motifs in the FGF14 sequence (Table 2) for corresponding kinases such as p38 MAPK, MEK, JAK2, STAT3, and Src, we selected hits for initial concentration-dependency studies (Figure 2 and Suppl. Fig 3). This important step identified true “hits” demonstrating ideal concentration dependency behavior, separating these from compounds yielding linear dependency (often indicative of promiscuity), and we observed numerous structurally diverse JAK2 and Src inhibitors.
Following target analysis and initial concentration-dependency studies, hits were counter-screened to determine the effect of these pathways on FGF14:FGF14 dimerization. Similar to secreted FGFs, intracellular FGFs retain the ability to dimerize [28,29]. While dimerization of secreted FGFs is the essential molecular step for activation of transmembrane FGF receptors [54], the biological role of iFGF dimerization is unknown. Homodimerization of iFGFs is supported by structural evidence and homology model-guided mutations in cells, with FGF13 and FGF12 homodimers having been resolved using X-ray crystallography [29,55]. These crystallography studies also demonstrated a significant overlap between the PPI interfaces of FGFs with a variety of Nav channel isoforms mediated by the iFGF core domain, but did not investigate points of structure-function divergence between the iFGF:iFGF homodimer and iFGF:Nav complex interfaces. We have recently begun to investigate this area using a combination of in-cell assays, site directed mutagenesis and electrophysiology [27,28], and the present study builds on these findings by demonstrating the importance of a single residue in dynamically regulating the equilibrium between protein:protein complexes.
During the initial FGF14:FGF14 homodimer counter-screening, differential regulation of the homodimer vs FGF14:Nav1.6 complex species was only observed for inhibitors targeting JAK2. We hypothesize that in the cell milieu an increase in FGF14 dimerization would “sequester” monomeric FGF14, such that less monomeric FGF14 is available to bind the Nav1.6 C-terminal tail. While the two assays presented separately measure FGF14:Nav1.6 or FGF14:FGF14 binding, the FGF14 dimer is predicted to exist in both systems. For the FGF14:Nav1.6 assay, it would exist as a dimer of CLuc-FGF14:CLuc-FGF14, yielding no luminescence in the presence of luciferin; increases in this dimer would reduce the FGF14 available for binding to CD4-Nav1.6-NLuc, thereby reducing reconstituted luciferase enzyme capable of generating luminescence. Although this hypothesis cannot be directly tested using the specific LCA constructs presented here, the high-throughput nature of said assays makes them ideal tools for hypothesis generation and guiding subsequent validation studies.
While JAK2 inhibitors resulted in increased stability of the FGF14:FGF14 dimer and inhibition of the FGF14:Nav1.6 complex, Src kinase inhibitors were largely only effective on the FGF14:Nav1.6 complex, resulting in only moderate inhibition of the FGF14 dimer, and only at high concentrations. However, phosphomotif scanning and prediction algorithms identified potential JAK2 and Src phosphorylation sites at two adjacent residues, Y158 and Y162, within an overlapping SH2 domain (Table 2 and Figure 5A), and thus we hypothesized that both of these kinases could play important, albeit distinct, regulatory roles. Both Y158 and Y162 are at the β−9 sheet of FGF14 that mediates dimerization and interaction with the Nav1.6 channel. However, while Y158 is a bona fide hot-spot at the interface of both complexes [28], Y162 is buried in the β−9 sheet at a position more distal from the PPI interface, largely sequestered toward the FGF14 core by hydrophobic interactions, and is not within the 8 Å distance ( > 12 Å) required for defining the residue as a hot spot (Suppl. Fig. 2). In addition, Y158, along with the adjacent site V160, was previously shown to mediate structure-function properties of the FGF14:FGF14 dimer and FGF14:Nav1.6 complex PPI interface [28]. Results from expanded concentration-dependency studies with repurchased JAK2 and Src inhibitors supported the findings of the counter-screening, and SPR demonstrated that phosphorylation by JAK2 altered FGF14 homodimerization kinetics to a much greater extent than phosphorylation by Src. Thus, both Src and JAK2 kinases might transduce physiologically distinct mechanisms with variable effects on FGF14 species depending on upstream signaling stimulus. Although the mechanisms of Src-mediated changes were not further studied here, the importance of Y158 in JAK2-dependent regulation of FGF14 was subsequently confirmed by observing phosphorylation of this site in vitro using mass-spectrometry, as well as Y158A mutagenesis studies (Figures 5 and 6).
By providing evidence for differential effects of Y158 modification on the two FGF14 complexes, this study strengthens the hypothesis of two structurally related, but distinct PPI interfaces at the FGF14 surface, and provides new insight into how dimeric vs monomeric iFGFs might differentially function in cells. The FGF14:FGF14 homodimer and FGF14:Nav1.6 complexes were previously thought to largely share a common PPI interface. However, we show initial evidence for a single JAK2-dependent phosphorylation event that destabilizes the prior while promoting the latter. Thus, one plausible prediction is that if in close proximity to its targets, activation of JAK2 could shift the FGF14 dimer equilibrium toward free monomeric FGF14 available to bind the Nav1.6 C-terminal tail, resulting in drastic changes to Nav channel function. This mechanism could enable rapid and efficient modulation of Nav channel function in response to extracellular signals transduced through transmembrane receptors.
The proximal 2/3rd of the C-terminal tail (residues 1767–1912) of Nav channels is the primary binding site for iFGFs and other interacting proteins such as calmodulin, while the distal 1/3rd is predicted to be disordered and not well conserved [28,29,39,56]. SCN8A mutations leading to epileptic encephalopathies are largely found in the more conserved protein domains, including two missense mutations (causing R1872W and R1871Q) in the proximal portion of the C-terminus [56] that would occur in relative proximity to the FGF14 binding region shown in Suppl. Fig 2. However, evidence for modulation of Nav channel inactivation by iFGFs have spurred the idea that other contacts also exist outside those between the channel C-terminal tail and the iFGFs core domain. The prevailing hypothesis is that the flexible N-terminal tail of the iFGFs-1a isoforms and of FGF14–1b (which, unlike other iFGF isoforms, possesses a unique 60 aa N-terminal tail) might protrude into channel domains distal to the C-terminal tail and more proximal to the plasma membrane. The Nav channel intracellular III-IV loop, located in close proximity to the channel pore and referred to as the inactivation loop, is a postulated interacting site for the N-terminal tail of iFGFs [52]. However, whether such putative interaction with the inactivation loop is mediated by an iFGF monomer or heterodimer (i.e., FGF14:Nav1.6 complex) is unclear. In addition, studies have shown that pore-forming Nav α subunits assemble and function as dimers [57], which could potentially accommodate two iFGF molecules per channel C-terminal tail forming a larger macromolecular complex beneath the plasma membrane.
When applied to cells expressing the Nav1.6 channel and FGF14, the JAK2 inhibitor Fedratinib normalized previously described FGF14-dependent phenotypes of Nav1.6 properties [28,33] (Figure 6). Crucially, FGF14-induced suppression of transient Na+ currents was abolished in the presence of Fedratinib. However, FGF14 was also found to slow channel entry into fast inactivation (tau), as well as cause a depolarizing shift in both voltage-dependence of activation and steady-state inactivation, phenotypes which were similarly abolished by inhibition of JAK2. The effect on FGF14 on Nav1.6 long-term inactivation, which leads to potentiation of Na+ currents during repetitive stimulation, was also fully reversed by Fedratinib. Furthermore, electrophysiology studies with the FGF14Y158A mutant indicate that in the presence of the Y158A point mutation, FGF14 remains functionally active and presumably capable of binding Nav1.6, but that JAK2 is incapable of modulating this interaction. Of all phenotypes examined, only FGF14-dependent modulation of long-term inactivation was abolished by the presence of Y158A, suggesting an additional role for Y158 in mediating FGF14-dependent modulation of slow inactivation, independent of phosphorylation.
On the basis of in vitro and in-cell studies we sought to determine the extent to which the FGF14/JAK2 axis could impact intrinsic firing of CA1 pyramidal neurons, a subtype of hippocampal neurons where both FGF14 and Nav1.6 are highly expressed [20,30,53]. In these cells, Fedratinib caused a decrease in intrinsic firing, a phenotype that was absent in FGF14−/− animals (Figure 8). Other passive properties that could account for a change in firing were not significantly affected in either FGF14+/+ or FGF14−/− animals, suggesting that the observed phenotype is likely induced by attenuation of Na+ currents and is dependent upon the presence of FGF14. As previous studies have demonstrated that the role of FGF14 in the native system is to stimulate Nav channel function [33,51,58,59], this phenotype suggests that native JAK2 may increase neuronal excitability through potentiating binding of FGF14 to Nav1.6. Therefore, in summary, our study presents three key findings to support this novel mechanism: i) inhibition of JAK2 limits FGF14:Nav1.6 complex assembly, potentiates FGF14 homodimerization, and abolishes FGF14-dependent modulation of Nav1.6 currents, contingent upon the presence of Y158; ii) JAK2 phosphorylates FGF14 at Y158; and iii) in native brain slices, inhibition of JAK2 reduces neuronal excitability, contingent upon the presence of FGF14.
JAK2 is a non-receptor (protein) tyrosine kinase and is largely studied as part of the JAK/STAT pathway. Like other kinases in the same family, JAK2 serves as the catalytic signaling component for a wide range of transmembrane receptors, including those for interleukins, interferons, growth hormone, erythropoietin, and leptin[61], to contribute to physiological processes ranging from cell survival to inflammation. Additionally, there is emerging evidence for a role of JAK2 in neuronal function, as well as for the potential of therapeutically targeting the JAK/STAT pathway for neuropsychiatric disorders, including depression [26,62]. The wide spectrum of signal transduction pathways associated with JAK2 in neurons support the idea that FGF14 phosphorylation by JAK2 could be involved in a variety of physiological processes ranging from BDNF-dependent synaptic plasticity to neuroinflammatory signaling. Additionally, our study has identified a new role of JAK2 in the hippocampus, a brain region involved in learning and memory.
Previous studies have shown that JAK2 is functionally relevant in hippocampal neurons [63–67], as well as in the nucleus accumbens [68]. For instance, through activation of STAT3, JAK2 plays a role in regulating apoptosis following white matter injury [67], as well as in leptin-mediated neuroprotective effects following cerebral ischemia [69]. In hippocampal CA3 neurons, inactivation of JAK2-STAT5 signaling has also been linked with elevation of brain-derived neurotrophic factor (BDNF) and reduced apoptosis in experimental models of depression [65]. Extending on the role of BDNF in synaptic plasticity, recent evidence has demonstrated a role of JAK-STAT signaling in activity-dependent long-term depression (LTD) of hippocampal CA1 synapses [63]. Interestingly, unlike synaptic plasticity elicited by stimulation of the CA3 Schaffer collaterals, LTD induced in CA1 neurons by stimulation of extrahippocampal temporoammonic inputs requires rapid gene transcription and is dependent upon glutamate receptor internalization mediated by JAK2 activation and subsequent STAT3-driven gene transcription. In another recent study, BDNF/TrkB reduction was associated with upregulation of gene transcription, δ-secretase activity, and Aβ and Tau alterations in murine brains through activation of neuroinflammatory pathways in a JAK2/STAT3-dependent manner [71]. BDNF-dependent synaptic plasticity and neuronal homeostasis are intimately linked to neuronal excitability [72], so it is plausible that by regulating the equilibrium between FGF14 species, JAK2 might enable neurons to dynamically adjust firing in response to transmembrane receptor signaling as part of a homeostatic regulatory loop.
Overall, this study highlights that protein tyrosine kinases (PTKs) play a role in regulating the Nav1.6 channel complex. We observed a high proportion of screening hits targeting PTKs including Lck, Syk, Fyn, FLT3, Src, and JAK2, and validation studies demonstrated clear regulation of the complex by Src and JAK2. These results build on previous investigations of the relationship between PTKs and Nav channels. Nav1.2 channels in rat brain have been found to directly interact with Fyn [23], a tyrosine kinase closely related to Src. The interaction was found to be functionally relevant, affecting Na+ current amplitudes and channel availability through Tyr phosphorylation induced by activation of TrkB/p75 signaling via BDNF [73]. A number of Fyn kinase phosphorylation sites have been found on Nav channels, including Y1497 and Y1498 on Nav1.2 close to the inactivation gate [73] and others on the cardiac Nav1.5 N-terminal and C-terminal tails[74]. Additionally, both FGF14 and Nav1.6 are phosphorylated by GSK3β and thereby controlled via the IP3K/Akt/GSK3β pathway of Ser/Thr kinases downstream of receptor tyrosine kinases [18,20,75,76], adding layers of complexity to the FGF14 role in kinase signaling. Under basal conditions, FGF14 and Nav1.6 are highly clustered at the AIS, the site of action potential initiation. Thus, it is plausible that both Ser/Thr and Tyr kinases also cluster in this compartment, forming a structurally organized signaling complex of which FGF14, the Nav1.6 channel, kinases and the transmembrane receptor are all components. In this manner, specific post-translational modifications could confer functional specificity to the Nav channel complex in distinct subcellular compartments of the neuron, contributing to specialized signaling important for firing and synaptic plasticity[22,24,77].
Conclusions
Based on these results, we concluded that activation of JAK2 results in FGF14 phosphorylation at Y158, dimer dissociation, and subsequent increases in binding of monomeric FGF14 to the Nav1.6 C-terminal tail. Functionally, JAK2 is thereby capable of controlling FGF14-dependent modulation of Nav1.6 channel currents, as well as firing of CA1 pyramidal neurons in the hippocampus. Future studies will determine whether this novel signaling mechanism could enable neurons to dynamically adjust firing in response to receptor-mediated JAK2 signaling, as well as implications for synaptic plasticity and neuroinflammation.
Supplementary Material
Highlights:
The voltage-gated Na+ channel (Nav1.6) is controlled by protein:protein interactions with FGF14
JAK2 differentially regulates FGF14:Nav1.6 and FGF14:FGF14 dimer assembly through phosphorylation of FGF14Y158
Functionally, JAK2 thereby controls FGF14-dependent modulation of Nav1.6 currents and firing of hippocampal CA1 pyramidal neurons
By regulating the equilibrium of FGF14 homodimerization, activation of JAK2 might enable neurons to dynamically adjust firing in response to receptor signaling.[60, 70]
Funding:
This work was supported by the National Institutes of Health (NIH) [grant numbers: T32 AG051131 (P.A.W.) from the NIA; T32 ES007254 from the NIEHS (C.M.T); NIH S10OD023576 (F.L.); and R01 MH095995 (F.L.) and R01 MH111107 (F.L.) from the NIMH]; Pharmaceutical Research and Manufacturers of America (PhRMA) Foundation Pre-doctoral Fellowship in Pharmacology/Toxicology (P.A.W.); Cancer Prevention & Research Institute of Texas (CPRIT) [grant number RP150578 (C.S.)]; Sealy and Smith foundation grant to the Sealy Center for Structural Biology and Molecular Biophysics; and the UTMB Technology Commercialization Program (F.L.).
Footnotes
Competing interests
Dr. Fernanda Laezza is the founder and President of IonTx Inc. and she owns stock in the company. All other authors declare that they have no competing interests.
Supplementary information accompanies this paper in a separate document. All data generated or analyzed during this study are included in this article (and its Supplementary Information files), and any additional data may be provided upon reasonable request to the corresponding author.
References
- [1].Ahern CA, Payandeh J, Bosmans F, Chanda B, The hitchhiker’s guide to the voltage-gated sodium channel galaxy, J. Gen. Physiol, 147 (2016) 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Catterall WA, Signaling complexes of voltage-gated sodium and calcium channels, Neurosci. Lett, 486 (2010) 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hsu WCJ, Nilsson CL, Laezza F, Role of the axonal initial segment in psychiatric disorders: Function, dysfunction, and intervention, Front. Psychiatry, 5 (2014) 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Di Re J, Wadsworth PA, Laezza F, Intracellular fibroblast growth factor 14: Emerging risk factor for brain disorders, Front. Cell. Neurosci, 11 (2017) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Abriel H, Cardiac sodium channel Nav15 and its associated proteins, Arch. Mal. Coeur Vaiss, 100 (2007) 787–793. [PubMed] [Google Scholar]
- [6].Buffington SA, Rasband MN, The axon initial segment in nervous system disease and injury, Eur. J. Neurosci, 34 (2011) 1609–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Emmett MR, Kroes RA, Moskal JR, Conrad CA, Priebe W, Laezza F, Meyer-Baese A, Nilsson CL, Integrative biological analysis for neuropsychopharmacology, Neuropsychopharmacology, 39 (2014) 5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kovacs DM, Gersbacher MT, Kim DY, Alzheimer’s secretases regulate voltage-gated sodium channels, Neurosci. Lett, 486 (2010) 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gasparini S, Magee JC, Phosphorylation-dependent differences in the activation properties of distal and proximal dendritic Na+ channels in rat CA1 hippocampal neurons, J. Physiol, 541 (2002) 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Loftis JL, King DD, Colbert CM, Kinase-dependent loss of Na+ channel slow-inactivation in rat CA1 hippocampal pyramidal cell dendrites after brief exposure to convulsants, Eur. J. Neurosci, 18 (2003) 1029–1032. [DOI] [PubMed] [Google Scholar]
- [11].Colbert CM, Johnston D, Protein kinase C activation decreases activity-dependent attenuation of dendritic Na+ current in hippocampal CA1 pyramidal neurons, J. Neurophysiol, 79 (1998) 491–495. [DOI] [PubMed] [Google Scholar]
- [12].Cantrell AR, Catterall WA, Neuromodulation of Na+ channels: an unexpected form of cellular plasticity, Nat. Rev. Neurosci, 2 (2001) 397–407. [DOI] [PubMed] [Google Scholar]
- [13].Scheuer T, Regulation of sodium channel activity by phosphorylation, Semin. Cell Dev. Biol, 22 (2011) 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Aromolaran AS, Chahine M, Boutjdir M, Regulation of cardiac voltage-gated sodium channel by kinases: Roles of protein kinases A and C, Handb. Exp. Pharmacol, 246 (2018) 161–184. [DOI] [PubMed] [Google Scholar]
- [15].Berendt FJ, Park K-S, Trimmer JS, Multisite phosphorylation of voltage-gated sodium channel alpha subunits from rat brain, J. Proteome Res, 9 (2010) 1976–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bréchet A, Fache MP, Brachet A, Ferracci G, Baude A, Irondelle M, Pereira S, Leterrier C, Dargent B, Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G, J. Cell Biol, 183 (2008) 1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].James TF, Nenov MN, Wildburger NC, Lichti CF, Luisi J, Vergara F, Panova-Electronova NI, Nilsson CL, Rudra JS, Green TA, Labate D, Laezza F, The Nav12 channel is regulated by GSK3, Biochim. Biophys. Acta - Gen. Subj, 1850 (2015) 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Scala F, Nenov MN, Crofton EJ, Singh AK, Folorunso O, Zhang Y, Chesson BC, Wildburger NC, James TF, Alshammari MA, Alshammari TK, Elfrink H, Grassi C, Kasper JM, Smith AE, Hommel JD, Lichti CF, Rudra JS, D’Ascenzo M, Green TA, et al. , Environmental Enrichment and Social Isolation Mediate Neuroplasticity of Medium Spiny Neurons through the GSK3 Pathway, Cell Rep, 23 (2018) 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hien YE, Montersino A, Castets F, Leterrier C, Filhol O, Vacher H, Dargent B, CK2 accumulation at the axon initial segment depends on sodium channel Nav1, FEBS Lett, 588 (2014) 3403–3408. [DOI] [PubMed] [Google Scholar]
- [20].Shavkunov AS, Wildburger NC, Nenov MN, James TF, Buzhdygan TP, Panova-Elektronova NI, Green TA, Veselenak RL, Bourne N, Laezza F, The fibroblast growth factor 14-voltage-gated sodium channel complex is a new target of glycogen synthase kinase 3 (GSK3), J. Biol. Chem, 288 (2013) 19370–19385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hsu WC, Nenov MN, Shavkunov A, Panova N, Zhan M, Laezza F, Identifying a kinase network regulating FGF14:Nav16 complex assembly using split-luciferase complementation, PLoS One, 10 (2015) 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Francis KR, Wei L, Yu SP, Src Tyrosine Kinases Regulate Neuronal Differentiation of Mouse Embryonic Stem Cells Via Modulation of Voltage-Gated Sodium Channel Activity, Neurochem. Res, 40 (2015) 674–687. [DOI] [PubMed] [Google Scholar]
- [23].Beacham D, Ahn M, Catterall WA, Scheuer T, Sites and molecular mechanisms of modulation of NaV12 channels by Fyn tyrosine kinase, J. Neurosci, 27 (2007) 11543–11551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Li Y, Zhu T, Yang H, Dib-Hajj SD, Waxman SG, Yu Y, Le Xu T, Cheng X, Nav17 is phosphorylated by Fyn tyrosine kinase which modulates channel expression and gating in a cell type-dependent manner, Mol. Pain, 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wadsworth PA, Folorunso O, Nguyen N, Singh AK, D’Amico D, Powell RT, Brunell D, Allen J, Stephan C, Laezza F, High-throughput screening against protein:protein interaction interfaces reveals anti-cancer therapeutics as potent modulators of the voltage-gated Na+ channel complex, Sci. Rep, 9 (2019) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nicolas CS, Amici M, a Bortolotto Z, Doherty A, Csaba Z, Fafouri A, Dournaud P, Gressens P, Collingridge GL, Peineau S, The role of JAK-STAT signaling within the CNS, Jak-Stat, 2 (2013) e22925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ali S, Shavkunov A, Panova N, Stoilova-McPhie S, Laezza F, Modulation of the FGF14:FGF14 Homodimer Interaction Through Short Peptide Fragments, CNS Neurol. Disord. - Drug Targets, 13 (2014) 1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ali SR, Singh AK, Laezza F, Identification of amino acid residues in fibroblast growth factor 14 (FGF14) required for structure-function interactions with voltage-gated sodium channel Nav16, J. Biol. Chem, 291 (2016) 11268–11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goetz R, Dover K, Laezza F, Shtraizent N, Huang X, Tchetchik D, Eliseenkova AV, Xu CF, Neubert TA, Ornitz DM, Goldfarb M, Mohammadi M, Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels, J. Biol. Chem, 284 (2009) 17883–17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Alshammari MA, Alshammari TK, Laezza F, Improved Methods for Fluorescence Microscopy Detection of Macromolecules at the Axon Initial Segment, Front. Cell. Neurosci, 10 (2016) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shavkunov A, Panova N, Prasai A, Veselenak R, Bourne N, Stoilova-McPhie S, Laezza F, Bioluminescence Methodology for the Detection of Protein–Protein Interactions Within the Voltage-Gated Sodium Channel Macromolecular Complex, Assay Drug Dev. Technol, 10 (2012) 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shavkunov AS, Ali SR, Panova-elektronova NI, Laezza F, Split-Luciferase Complementation Assay to Detect Protein-Protein Interactions in Live Cells, in: Protein-Protein Interact. Methods Appl. Methods Mol. Biol, 2015: pp. 497–514. [DOI] [PubMed] [Google Scholar]
- [33].Ali SR, Liu Z, Nenov MN, Folorunso O, Singh A, Scala F, Chen H, James TF, Alshammari M, Panova-Elektronova NI, White MA, Zhou J, Laezza F, Functional Modulation of Voltage-Gated Sodium Channels by a FGF14-Based Peptidomimetic, ACS Chem. Neurosci, 9 (2018) 976–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sittampalam NP Coussens K Brimacombe, et al. , Assay Guidance Manual, Eli Lilly Co. Natl. Cent. Adv. Transl. Sci, (2019). [PubMed] [Google Scholar]
- [35].Seashore-Ludlow B, Rees MG, Cheah JH, Coko M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, Alexander B, Li A, Montgomery P, Wawer MJ, Kuru N, Kotz JD, Suk-Yee Hon C, Munoz B, Liefeld T, Dančik V, et al. , Harnessing connectivity in a large-scale small-molecule sensitivity dataset, Cancer Discov, 5 (2015) 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A, A curated compendium of phosphorylation motifs, Nat. Biotechnol, 25 (2007) 285–286. [DOI] [PubMed] [Google Scholar]
- [37].Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S, Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence, Proteomics, 4 (2004) 1633–1649. [DOI] [PubMed] [Google Scholar]
- [38].Horn H, Schoof EM, Kim J, Robin X, Miller ML, Diella F, Palma A, Cesareni G, Jensen LJ, Linding R, KinomeXplorer: An integrated platform for kinome biology studies, Nat. Methods, 11 (2014) 603–604. [DOI] [PubMed] [Google Scholar]
- [39].Wang C, Chung BC, Yan H, Lee SY, Pitt GS, Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin, Structure, 20 (2012) 1167–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Remy I, Michnick SW, A highly sensitive protein-protein interaction assay based on Gaussia luciferase, Nat. Methods, 3 (2006) 977–979. [DOI] [PubMed] [Google Scholar]
- [41].Hsu WCJ, Scala F, Nenov MN, Wildburger NC, Elferink H, Singh AK, Chesson CB, Buzhdygan T, Sohail M, Shavkunov AS, Panova NI, Nilsson CL, Rudra JS, Lichti CF, Laezza F, CK2 activity is required for the interaction of FGF14 with voltage-gated sodium channels and neuronal excitability, FASEB J, 30 (2016) 2171–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Park SY, Kim YH, Park G, Cucurbitacins attenuate microglial activation and protect from neuroinflammatory injury through Nrf2/ARE activation and STAT/NF-κB inhibition, Neurosci. Lett, 609 (2015) 129–136. [DOI] [PubMed] [Google Scholar]
- [43].Hsu WCJ, Wildburger NC, Haidacher SJ, Nenov MN, Folorunso O, Singh AK, Chesson BC, Franklin WF, Cortez I, Sadygov RG, Dineley KT, Rudra JS, Taglialatela G, Lichti CF, Denner L, Laezza F, PPARgamma agonists rescue increased phosphorylation of FGF14 at S226 in the Tg2576 mouse model of Alzheimer’s disease, Exp. Neurol, 295 (2017) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Singer JW, Al-Fayoumi S, Taylor J, Velichko S, O’Mahony A, Comparative phenotypic profiling of the JAK2 inhibitors ruxolitinib, fedratinib, momelotinib, and pacritinib reveals distinct mechanistic signatures, PLoS One, 14 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Singer JW, Al-Fayoumi S, Ma H, Komrokji RS, Mesa R, Verstovsek S, Comprehensive kinase profile of pacritinib, a nonmyelosuppressive janus kinase 2 inhibitor, J. Exp. Pharmacol, 8 (2016) 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Aittomäki S, Pesu M, Therapeutic targeting of the JAK/STAT pathway, Basic Clin. Pharmacol. Toxicol, 114 (2014) 18–23. [DOI] [PubMed] [Google Scholar]
- [47].Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E, Tefferi A, CYT387, a selective JAK1/JAK2 inhibitor: In vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients, Leukemia, 23 (2009) 1441–1445. [DOI] [PubMed] [Google Scholar]
- [48].Uitdehaag JCM, Verkaar F, Alwan H, De Man J, Buijsman RC, Zaman GJR, A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets, Br. J. Pharmacol, 166 (2012) 858–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Smolinski MP, Bu Y, Clements J, Gelman IH, Hegab T, Cutler DL, Fang JWS, Fetterly G, Kwan R, Barnett A, Lau JYN, Hangauer DG, Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2–391 and KX2–361), J. Med. Chem, 61 (2018) 4704–4719. [DOI] [PubMed] [Google Scholar]
- [50].Venkatesan K, Liu Y, Goldfarb M, Fast-onset long-term open-state block of sodium channels by A-type FHFs mediates classical spike accommodation in hippocampal pyramidal neurons, J. Neurosci, 34 (2014) 16126–16139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].White HV, Brown ST, Bozza TC, Raman IM, Effects of FGF14 and NaVβ4 deletion on transient and resurgent Na current in cerebellar Purkinje neurons, J. Gen. Physiol, 151 (2019) 1300–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Dover K, Solinas S, D’Angelo E, Goldfarb M, Long-term inactivation particle for voltage-gated sodium channels, J. Physiol, 588 (2010) 3695–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lorincz A, Nusser Z, Cell-type-dependent molecular composition of the axon initial segment, J. Neurosci, 28 (2008) 14329–14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sarabipour S, Hristova K, Mechanism of FGF receptor dimerization and activation, Nat. Commun, 7 (2016) 10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Olsen SK, Garbi M, Zampieri N, Eliseenkova AV, Ornitz DM, Goldfarb M, Mohammadi M, Fibroblast Growth Factor (FGF) Homologous Factors Share Structural but Not Functional Homology with FGFs, J. Biol. Chem, 278 (2003) 34226–34236. [DOI] [PubMed] [Google Scholar]
- [56].Wagnon JL, Meisler MH, Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy, Front. Neurol, 6 (2015) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Clatot J, Hoshi M, Wan X, Liu H, Jain A, Shinlapawittayatorn K, Marionneau C, Ficker E, Ha T, Deschênes I, Voltage-gated sodium channels assemble and gate as dimers, Nat. Commun, 8 (2017) 2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Laezza F, Gerber BR, Lou J-YJ-Y, Kozel MA, Hartman H, Marie Craig A, Ornitz DM, Nerbonne JM, Craig AM, Ornitz DM, Nerbonne JM, The FGF14F145S Mutation Disrupts the Interaction of FGF14 with Voltage-Gated Na+ Channels and Impairs Neuronal Excitability, J. Neurosci, 27 (2007) 12033–12044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, Giza J, Tchetchik D, Kelley K, Vega A, Matthews G, Rossi P, Ornitz DM, D’Angelo E, Fibroblast Growth Factor Homologous Factors Control Neuronal Excitability through Modulation of Voltage-Gated Sodium Channels, Neuron, 55 (2007) 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sopjani M, Konjufca V, Rinnerthaler M, Rexhepaj R, Dërmaku-Sopjani M, The Relevance of JAK2 in the Regulation of Cellular Transport, Curr. Med. Chem, 23 (2016) 578–588. [DOI] [PubMed] [Google Scholar]
- [61].Hubbard SR, Mechanistic insights into regulation of JAK2 tyrosine kinase, Front. Endocrinol. (Lausanne), 8 (2018) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Shariq AS, Brietzke E, Rosenblat JD, Pan Z, Rong C, Ragguett RM, Park C, McIntyre RS, Therapeutic potential of JAK/STAT pathway modulation in mood disorders, Rev. Neurosci, 30 (2019) 1–7. [DOI] [PubMed] [Google Scholar]
- [63].Nicolas CS, Peineau S, Amici M, Csaba Z, Fafouri A, Javalet C, Collett VJ, Hildebrandt L, Seaton G, Choi SL, Sim SE, Bradley C, Lee K, Zhuo M, Kaang BK, Gressens P, Dournaud P, Fitzjohn SM, Bortolotto ZA, Cho K, et al. , The JAK/STAT Pathway Is Involved in Synaptic Plasticity, Neuron, 73 (2012) 374–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chiba T, Yamada M, Sasabe J, Terashita K, Shimoda M, Matsuoka M, Aiso S, Amyloid-β causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons, Mol. Psychiatry, 14 (2009) 206–222. [DOI] [PubMed] [Google Scholar]
- [65].Tian RH, Bai Y, Li JY, Guo KM, Reducing PRLR expression and JAK2 activity results in an increase in BDNF expression and inhibits the apoptosis of CA3 hippocampal neurons in a chronic mild stress model of depression, Brain Res, 1725 (2019) 146472. [DOI] [PubMed] [Google Scholar]
- [66].Jia L, Wang F, Gu X, Weng Y, Sheng M, Wang G, Li S, Du H, Yu W, Propofol postconditioning attenuates hippocampus ischemia-reperfusion injury via modulating JAK2/STAT3 pathway in rats after autogenous orthotropic liver transplantation, Brain Res, 1657 (2017) 202–207. [DOI] [PubMed] [Google Scholar]
- [67].Chen XM, Yu YH, Wang L, Zhao XY, Li JR, Effect of the JAK2/STAT3 signaling pathway on nerve cell apoptosis in rats with white matter injury, Eur. Rev. Med. Pharmacol. Sci, 23 (2019) 321–327. [DOI] [PubMed] [Google Scholar]
- [68].Liu NY, Zhang L, Zhang L, Wang XN, Mediating effect of dopamine D3 receptors on Jak2 and GABAAα1 expression in mouse brains induced by cocaine, Chin. Med. J. (Engl), 120 (2007) 910–914. [PubMed] [Google Scholar]
- [69].Zhang W, Jin Y, Wang D, Cui J, Neuroprotective effects of leptin on cerebral ischemia through JAK2/STAT3/PGC-1-mediated mitochondrial function modulation, Brain Res. Bull, 156 (2020) 118–130. [DOI] [PubMed] [Google Scholar]
- [70].Crofton EJ, Nenov MN, Zhang Y, Scala F, Page SA, McCue DL, Li D, Hommel JD, Laezza F, Green TA, Glycogen synthase kinase 3 beta alters anxiety-, depression-, and addiction-related behaviors and neuronal activity in the nucleus accumbens shell, Neuropharmacology, 117 (2017) 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Wang ZH, Xiang J, Liu X, Yu SP, Manfredsson FP, Sandoval IM, Wu S, Wang JZ, Ye K, Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease, Cell Rep, 28 (2019) 655–669.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Turrigiano G, Homeostatic signaling: the positive side of negative feedback, Curr. Opin. Neurobiol, 17 (2007) 318–324. [DOI] [PubMed] [Google Scholar]
- [73].Ahn M, Beacham D, Westenbroek RE, Scheuer T, Catterall WA, Regulation of NaV12 channels by brain-derived neurotrophic factor, TrkB, and associated Fyn kinase, J. Neurosci, 27 (2007) 11533–11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].ju Liu C, Dib-Hajj SD, Renganathan M, Cummins TR, Waxman SG, Modulation of the cardiac sodium channel Nav15 by fibroblast growth factor homologous factor 1B, J. Biol. Chem, 278 (2003) 1029–1036. [DOI] [PubMed] [Google Scholar]
- [75].Minichiello L, TrkB signalling pathways in LTP and learning, Nat. Rev. Neurosci, 10 (2009) 850–60. [DOI] [PubMed] [Google Scholar]
- [76].Noorolyai S, Shajari N, Baghbani E, Sadreddini S, Baradaran B, The relation between PI3K/AKT signalling pathway and cancer, Gene, 698 (2019) 120–128. [DOI] [PubMed] [Google Scholar]
- [77].Wang ZH, Xiang J, Liu X, Yu SP, Manfredsson FP, Sandoval IM, Wu S, Wang JZ, Ye K, Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease, Cell Rep, 28 (2019) 655–669.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.