

Abstract
Introduction
This phase 2b/3 trial examined the effects of plasma exchange (PE) in patients with mild‐to‐moderate Alzheimer's disease (AD).
Methods
Three hundred forty‐seven patients (496 screened) were randomized (1:1:1:1) into three PE treatment arms with different doses of albumin and intravenous immunoglobulin replacement (6‐week period of weekly conventional PE followed by a 12‐month period of monthly low‐volume PE), and placebo (sham).
Results
PE‐treated patients performed significantly better than placebo for the co‐primary endpoints: change from baseline of Alzheimer's Disease Cooperative Study–Activities of Daily Living (ADCS‐ADL; P = .03; 52% less decline) with a trend for Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS‐Cog; P = .06; 66% less decline) scores at month 14. Moderate‐AD patients (baseline Mini‐Mental State Examination [MMSE] 18‐21) scored better on ADCS‐ADL (P = .002) and ADAS‐Cog (P = .05), 61% less decline both. There were no changes in mild‐AD patients (MMSE 22‐26). PE‐treated patients scored better on the Clinical Dementia Rating Sum of Boxes (CDR‐sb) (P = .002; 71% less decline) and Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC) (P < .0001; 100% less decline) scales.
Discussion
This trial suggests that PE with albumin replacement could slow cognitive and functional decline in AD, although further studies are warranted.
Keywords: albumin, albutein, Alzheimer's disease, clinical trial, plasma exchange, plasmapheresis
1. INTRODUCTION
The initial pathological event that triggers the process that leads to Alzheimer's disease (AD) is unknown. However, autopsy studies have shown that AD is a neurodegenerative process (loss of neurons) associated with the accumulation of amyloid plaques in the brain formed from extracellular aggregates of brain amyloid beta (Aβ) protein, and the formation of intracellular neurofibrillary tangles of phosphorylated tau (P‐tau) proteins. 1 AD pathology is often accompanied by vascular disease, 2 the presence of other proteinopathies, 3 and markers of inflammation. 4
Currently there are only symptomatic treatments for AD aimed to modulate neurotransmission, such as acetylcholinesterase inhibitors (AChEI) and N‐methyl‐d‐aspartate receptor antagonists (memantine). 5 , 6 Unfortunately, none of the pharmacologic treatments available today for AD have yet been shown to inhibit or slow down the damage and neuronal death ultimately leading to morbidity and mortality associated with the disease. Despite the fact that clinical trials studying small molecule pharmacotherapy and immunotherapies to reduce brain Aβ protein have, to date, failed to demonstrate any effects on cognition and/or functional performance, 7 , 8 , 9 , 10 , 11 , 12 Aβ can still be a valid therapeutic target. 13
Plasma exchange (PE) with albumin replacement is being investigated as a new therapeutic approach for AD 14 , 15 , 16 , 17 , 18 on the basis that Aβ in the cerebrospinal fluid (CSF) is in dynamic equilibrium with plasma Aβ through the blood‐brain barrier 19 and that sequestration of Aβ in the peripheral blood would alter such balance to induce CSF Aβ to pass to plasma. 20 , 21 , 22 , 23 Hence, routine PE removal of an AD patient's plasma would favor elimination of albumin‐bound Aβ, 16 , 18 and possibly, other pathogenic elements. 24 , 25 In addition, replacement with fresh therapeutic albumin can restore the antioxidant capacity of AD patient plasma, 26 as albumin is highly oxidized and glycated. 16 , 27 , 28 , 29 Furthermore, a therapeutic action at the vascular level can have a positive impact on dementia. 30
Following this line of research, a preliminary pilot study (EudraCT#: 2005‐001616‐45) 15 conducted in 2005 and a phase 2 trial (EudraCT#: 2007‐000414‐36; ClinicalTrials.gov ID: NCT00742417) 14 , 17 conducted in 2007 showed that the decline in memory and language abilities, and in brain perfusion, was attenuated in patients treated with PE with albumin replacement. The effects on cognition persisted up to 44weeks after PE was discontinued. In this paper, we present the primary results of the Alzheimer's Management By Albumin Replacement (AMBAR) study, a phase 2b/3 trial started in 2011 to further evaluate the previously observed findings by testing PE with different replacement volumes of albumin, with or without intravenous immunoglobulin (IVIG) to ameliorate a possible decrease in endogenous immunoglobulins. 31
2. METHODS
The AMBAR trial (EudraCT#: 2011‐001598‐25; ClinicalTrials.gov ID: NCT01561053) enrolled patients at 41 sites: 19 in Spain and 22 in the United States. Institutional Review Boards or Ethics Committees from the sites and the health authorities from both countries approved the protocol. Due to the invasive nature of the study procedures, a Data Safety Monitoring Committee met when approximately half of patients were recruited. The patient and a close relative or legal representative read the patient information sheet, agreed to participate in the trial, and then signed the informed consent form.
2.1. Patients
Eligible patients were men and women between 55 and 85 years of age, had a diagnosis of probable AD dementia according to the National Institute of Neurological Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria, 32 whose baseline Mini‐Mental State Examination (MMSE) scored from 18 to 26, and were on stable dose of AChEIs and/or memantine within the previous 3 months of screening. Exclusion criteria included cerebrovascular disease and any condition in which PE is contraindicated. Full details of eligibility criteria are available elsewhere 31 and in Appendix A in supporting information.
2.2. Interventions
Enrolled patients were randomly allocated to four groups (in a 1:1:1:1 scheme): three PE treatment groups received different doses of albumin (Albutein, Grifols, Barcelona, Spain) and IVIG (Flebogamma 5% DIF, Grifols, Barcelona, Spain), and one control (placebo) group underwent a simulated PE treatment through a noninvasive procedure (sham) that mimicked PE but without any actual fluid replacement. Patients, caregivers, and raters, including investigators evaluating outcome measures and central laboratory analysis, were blinded. Details of randomization, masking, and treatment allocations are provided elsewhere 31 and in Appendix A in supporting information.
The intervention regime started with a 6‐week period of treatment with weekly sessions of conventional therapeutic PE (TPE) with replacement albumin 5% for all the active groups through a peripheral (eg, radial/cubital vein) or central access (eg, subclavian/jugular vein), followed by a second 12‐month period with monthly sessions of low volume PE (LVPE) through a peripheral line with replacement albumin 20% or IVIG. Patients received one of three treatments: (1) infusion of 20g albumin (“low‐albumin” group), (2) infusion of 20g albumin alternated with infusions of 10g IVIG 5% (“low‐albumin+IVIG” group), and (3) infusion of 40g albumin alternated with infusions of 20g IVIG 5% (“high‐albumin+IVIG” group). Further details of replaced plasma and albumin infused volumes, and procedures for TPE and LVPE, as well as of a description of TPE, LVPE, and sham PE devices are available elsewhere 31 and in Appendix A in supporting information.
2.3. Clinical and laboratory assessments
The following clinical and neuropsychological measurements were performed at baseline, plus at months 2, 6, 9, 12, and 14: Alzheimer's Disease Cooperative Study–Activities of Daily Living (ADCS‐ADL) as a functional scale; Alzheimer's Disease Assessment Scale‐Cognitive Subscale (ADAS‐Cog) as a cognitive scale; and two global assessment of change scales—Clinical Dementia Rating Sum of Boxes (CDR‐sb), and Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC). Details of the neuropsychological assessments are provided elsewhere 31 and in Appendix A in supporting information.
CSF biomarkers, Aβ40, Aβ42, total tau (T‐tau), and P‐tau were assessed. CSF samples were collected at baseline and after the TPE and LVPE treatment periods. Details of biomarker determination are provided elsewhere 31 and in Appendix A in supporting information.
Detailed physical examinations were conducted at each study visit.
RESEARCH IN CONTEXT
Systematic review: As a new approach for Alzheimer's disease (AD) treatment, it is hypothesized that sequestration through plasma exchange (PE) of amyloid beta (Aβ) bound to plasma albumin may lead to an efflux of Aβ from the central nervous system to balance the peripheral drop. Use of Aβ‐free therapeutic albumin as the fluid replacement would reinforce the mechanism.
Interpretation: This phase 2b/3, randomized, controlled clinical trial of 347 mild‐to‐moderate AD patients (496 screened) showed that PE with albumin replacement was feasible and could slow cognitive and functional decline in AD patients over a period of 14 months. This was supported by the effects observed in the co‐primary and global secondary assessment outcomes.
Future directions: These findings have the potential to offer AD patients a new modality of treatment and justify a new trial.
2.4. Efficacy and safety outcomes
There were two co‐primary efficacy variables: the ADCS‐ADL and the ADAS‐Cog scale. 31 The primary analysis assessed the change from baseline to 14 months for both of these outcomes. The global assessment of change in AD scales, CDR‐sb and ADCS‐CGIC, were secondary clinical efficacy variables. 33
The efficacy of the intervention was also assessed within severity groups based on the baseline MMSE scores; scores 22‐26 were considered mild impairment and scores 18‐21 moderate impairment.
Biomarker variables included changes in CSF levels of Aβ40 and Aβ42, T‐tau and P‐tau between baseline and the finalization of each of the two treatment periods. 31
The main safety variable was the percentage of PE procedures (including the infusion of albumin and IVIG) associated with at least one adverse event (AE) that may be related to the study procedure. 31
2.5. Statistics
A sample size of 312 patients (78 in each of the four groups) would result in over 90% power for detecting a difference in the change from baseline of 3 points on the ADAS‐Cog, between the treated and the placebo groups, with a two‐sided 5% significance level. The sample size of 312 patients provides more than 98% power for detecting a treatment difference in the change from baseline of 6.69 points on the ADCS‐ADL score between the treated and the placebo groups with a two‐sided 5% level of significance.
The primary efficacy endpoints were analyzed over time as change from baseline to month 14 using a mixed model for repeated measures (MMRM) approach. Secondary endpoints were analyzed as change from baseline to months 2, 6, 9, 12, and 14 using analysis of covariance with treatment group as a fixed effect, and the corresponding baseline score, age, and AD severity as a covariate. The differences from the placebo group were estimated using least squares (LS) means (with 95% confidence interval [CI] or ± standard error of the mean [SEM]). Details of analyses are provided in Appendix A in supporting information.
Because all treated patients had the same volume of plasma removed for PE regardless of the group allocation, it was pre‐determined that the three treatment groups were also pooled together and analyzed as the combined treatment group (“PE‐treated” group). The same analyses were performed separately within the two pre‐specified AD severity subgroups: moderate AD (baseline MMSE: 18‐21) and mild AD (baseline MMSE: 22‐26).
Effect sizes were calculated as the ratio in percentage of the change‐from‐baseline difference between placebo and treated groups over the change‐from‐baseline of the placebo group, so that if the placebo group declines, effect sizes of <100% indicate less decline of the active group while effect sizes of >100% indicate improvement over baseline. 34 For completeness, the effect sizes for primary efficacy endpoints were also determined using Cohen's d value calculated with the change from baseline standard deviation.
Additional post‐hoc sensitivity analyses were performed to assess the impact of discontinuations (ie, dropouts) on the results using a z‐score carried forward analysis (zLOCF), and to assess the consistency of results across outcomes using a global statistical test (GST). Details of post‐hoc analyses are provided in Appendix A in supporting information.
3. RESULTS
3.1. Demographics and clinical characteristics of patients
A total of 496 patients were screened between April 19, 2012 and December 16, 2016. Of this group, 347 patients were randomized into four arms (see Figure1). Twenty‐five out of the 347 randomized patients did not receive treatment. The percentage of treated patients who completed the study ranged from 65.1% to 65.4% in the low‐albumin+IVIG and high‐albumin+IVIG groups, respectively, to 78.2% and 80.0% in the half‐albumin and placebo groups, respectively.
FIGURE 1.
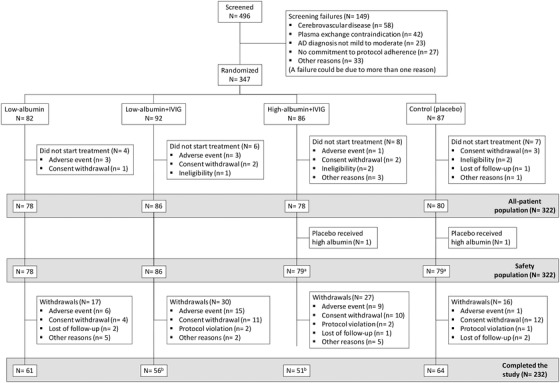
Flowchart of patients through the study. One patient randomized to placebo was implanted by error with a real central catheter and was then transferred and treated as a high‐albumin+intravenous immunoglobulin (IVIG) patient; four patients in the low‐albumin+IVIG arm and three patients in the high‐albumin+IVIG arm completed the study under a previous version of the protocol that was not blinded
Demographics and baseline characteristics of the placebo and treated groups are shown in Table1. The percentage of apolipoprotein E (APOE Ɛ4) carriers was greater in the low‐albumin arm than in the placebo arm and the other two treatment arms (63.5%vs 44.2%‐48.2%; P = .02).
TABLE 1.
Demographic and clinical characteristics at baseline by treatment arm
| Treatment modalities | ||||||
|---|---|---|---|---|---|---|
| Characteristic | Placebo (N = 80) | All PE‐treated (N = 242) | Low albumin (N = 78) | Low albumin + IVIG (N = 86) | High albumin + IVIG (N = 78) | Total (N = 322) |
| Age (years), mean (SD) | 68.4 (8.4) | 69.2 (7.4) | 68.5 (7.5) | 69.5 (6.9) | 69.5 (7.9) | 69.0 (7.7) |
| Age group (n, %) | ||||||
| < 65 | 29 (36.3) | 65 (26.9) | 26 (33.3) | 17 (19.8) | 22 (28.2) | 94 (29.2) |
| 65‐75 | 33 (41.3) | 124 (51.2) | 37 (47.4) | 52 (60.5) | 35 (44.9) | 157 (48.8) |
| > 75 | 18 (22.5) | 53 (21.9) | 15 (19.2) | 17 (19.8) | 21 (26.9) | 71 (22.0) |
| Sex (n, %) | ||||||
| Male | 44 (55.0) | 104 (43.0) | 35 (44.9) | 38 (44.2) | 31 (39.7) | 148 (46.0) |
| Female | 36 (45.0) | 138 (57.0) | 43 (55.1) | 48 (55.8) | 47 (60.3) | 174 (54.0) |
| BMI (Kg/m2), mean (SD) | 26.8 (4.3) | 26.9 (4.6) | 27.0 (4.8) | 27.1 (4.0) | 26.5 (5.0) | 26.9 (4.5) |
| Time since diagnosis of AD (years), mean (SD) | 2.5 (2.3) | 2.4 (2.4) | 2.2 (2.4) | 2.5 (2.3) | 2.4 (2.6) | 2.4 (2.4) |
| MMSE score, mean (SD) | 21.7 (2.6) | 21.7 (2.6) | 21.2 (2.4) | 22.1 (2.6) | 21.4 (2.6) | 21.6 (2.6) |
| Severity of AD (n, %) | ||||||
| Moderate (MMSE 18‐21) | 36 (45.0) | 117 (48.3) | 46 (59.0) | 37 (43.0) | 42 (53.8) | 161 (50.0) |
| Mild (MMSE 22‐26) | 44 (55.0) | 125 (51.7) | 32 (41.0) | 49 (57.0) | 36 (46.2) | 161 (50.0) |
| AD medication (n, %) | ||||||
| CEIs | 56 (70.0) | 164 (67.7) | 55 (70.5) | 50 (58.1) | 59 (75.6) | 220 (68.3) |
| Memantine | 7 (8.8) | 36 (14.9) | 13 (16.7) | 14 (16.3) | 9 (11.5) | 43 (13.4) |
| CEIs + memantine | 16 (20.0) | 40 (16.5) | 8 (10.3) | 22 (25.6) | 10 (12.8) | 56 (17.4) |
| None | 1 (1.3) | 2 (0.8) | 2 (2.6) | 0 | 0 | 3 (0.9) |
| APOE Ɛ4 | ||||||
| N | 77 | 231 | 74 | 83 | 74 | 308 |
| Carriers (n, %) | 34 (44.2) | 120 (51.9) | 47 (63.5) * | 40 (48.2) | 33 (44.6) | 154 (50.0) |
| Non‐carriers (n, %) | 43 (55.8) | 111 (49.1) | 27 (36.5) | 43 (51.8) | 41 (55.4) | 154 (50.0) |
| CSF Aβ42 | ||||||
| N | 71 | 226 | 74 | 77 | 75 | 297 |
| pg/mL, median (IQR) | 551 (380‐810) | 505 (431‐700) | 502 (436‐618) | 509 (399‐730) | 505 (407‐822) | 515 (426‐737) |
Abbreviations: AD, Alzheimer's disease; BMI, body mass index; CEI, cholinesterase inhibitor; CSF, cerebrospinal fluid; IQR, Interquartile range; IVIG, intravenous immunoglobulin; MMSE, Mini‐Mental State Examination; PE, plasma exchange; SD, standard deviation.
P = .02.
Figure B.1 of Appendix B in supporting information shows the distribution of the baseline CSF Aβ42 levels. Data separately for moderate and mild AD patients (baseline MMSE scores 18‐21 and 22‐26 respectively, are shown in Appendix B, Tables B.1 and B.2 in supporting information). Medical history of patients is shown in Appendix B, Table B.3 in supporting information.
3.2. Primary efficacy endpoints
The LS difference from baseline to month 14 for the PE‐treated group versus placebo was 3.5 points (95% CI: 0.4, 6.6; P = .03; 52% less decline) on the ADCS‐ADL scale (Figure2A), and −2.1 points (95% CI: −4.4, 0.2; P = .06; 66% less decline) on the ADAS‐Cog scale (Figure2G). The differences in the three treatment arms separately (low‐albumin, low‐albumin+IVIG, high‐albumin+IVIG) were not statistically significant (Figure2D and J). Baseline and individual visit scores are provided in Appendix B, Tables B.4 and B.5 in supporting information. Effect sizes based on the change from baseline Cohen's d values are also provided in Appendix B, Table B.6 in supporting information.
FIGURE 2.
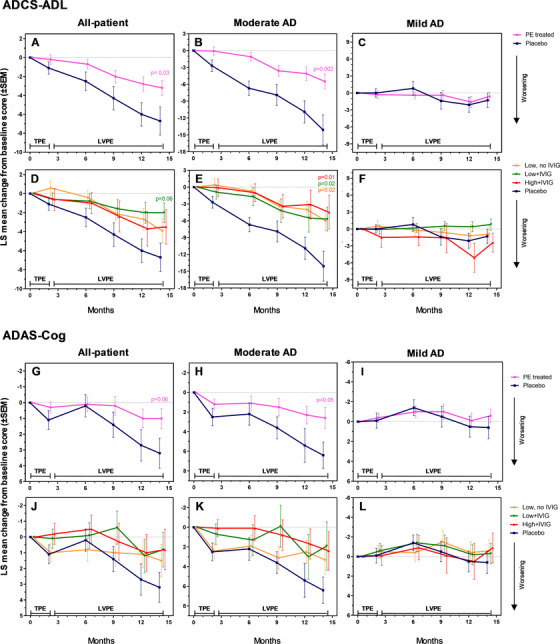
Least square (LS) mean change from baseline scores (± standard error of the mean) in the Alzheimer's Disease Cooperative Study Group–Activities of Daily Living (ADCS‐ADL) and the Alzheimer's Disease Assessment Scale–Cognitive (ADAS‐Cog) scales (co‐primary efficacy variables; panels A–F and G–L, respectively) performed on mild to moderate Alzheimer's disease (AD) patients (all‐patient [panels A, D, G, J], moderate AD [panels B, E, H, K], and mild AD [panels C, F, I, L] populations) treated with plasma exchange (PE) with albumin replacement. TPE denotes the 2‐month period of conventional therapeutic PE; LVPE denotes the period up to month 14 of low‐volume PE. The difference between the treated patient groups (PE‐treated patients combined [n = 242; panels A–C and G–I], and three active groups: low/high‐albumin dose, with/without intravenous immunoglobulin [n = 78‐86; panels D–F and J–L]) and the placebo group (n = 80) at month 14 (primary endpoint) was evaluated using a mixed model for repeated measures (MMRM) approach, with adjustment for multiple dose groups according to the Hochberg procedure for α level of 0.05. Both statistical significance (P < .05) and borderline significance (P < .1) versus placebo are indicated
3.3. Global assessment efficacy endpoints
There was significantly less decline compared to placebo in the CDR‐sb in all treatment groups (Figure3A and D), not only at month 14 (difference ranging from −1.1 [95% CI: −2.2, 0.0] to −1.2 [95% CI: −2.2, −0.2]; P values: .002 to .01; effect sizes: 65% to 71%) but at earlier time points. ADCS‐CGIC scores at month 14 showed stabilization in all treatment groups compared to placebo (difference ranging from −0.8 [95% CI: −1.44, −0.36] to −0.9 [95% CI: −1.2, −0.4]; P values: < .001 to .002; effect sizes: 100% to 113%; Figure3G and J). Baseline and individual visit scores are provided in Appendix B, Tables B.7 and B.8 in supporting information.
FIGURE 3.
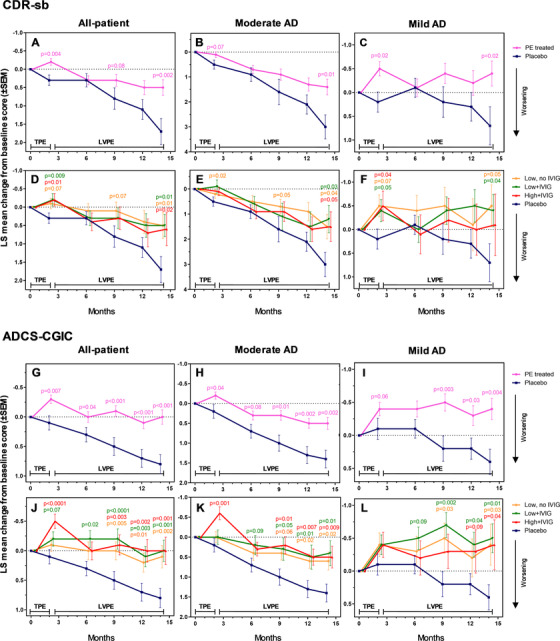
Least square (LS) mean change from baseline scores (± standard error of the mean) in the Clinical Dementia Rating Sum of Boxes (CDR‐sb), and Alzheimer's Disease Cooperative Study‐Clinical Global Impression of Change (ADCS‐CGIC) scales (global efficacy secondary variables; panels A–F and G–L, respectively) performed on mild to moderate Alzheimer's disease (AD) patients (all‐patient [panels A, D, G, J], moderate AD [panels B, E, H, K], and mild AD [panels C, F, I, L] populations) treated with plasma exchange (PE) with albumin replacement. TPE denotes the 2‐month period of conventional therapeutic PE; LVPE denotes the period up to month 14 of low‐volume PE. The difference between the treated patient groups (PE‐treated patients combined [n = 242; panels A–C and G–I], and three active groups: low/high‐albumin dose, with/without intravenous immunoglobulin [n = 78–86; panels D–F and J–L]) and the placebo group (n = 80) at months 2, 6, 9, 12, and 14 was evaluated using analysis of covariance with treatment group as a fixed effect, and the corresponding baseline value, age and AD severity, as a covariate. Both statistical significance (P < .05) and borderline significance (P < .1) versus placebo are indicated
3.4. Severity subgroup analysis
For primary endpoints, in the moderately impaired AD patients, difference between the PE‐treated group and placebo at month 14 were 8.6 for the ADCS‐ADL scale (95% CI: 3.1, 14.1; P = .002; 61% less decline; Figure2B), and −3.9 for the ADAS‐cog scale (95% CI: −7.9, 0.1; P = .05; 61% less decline; Figure2H). In the comparison of each of the three modalities of treatment versus placebo, there was a statistically significant difference in the ADCS‐ADL scale of moderate AD patients (difference ranging from 8.0 [95% CI: 2.0, 14.0] to 9.5 [95% CI: 1.6, 17.4]; P values: .01 to .02; effect sizes: 57% to 67%; Figure2E). Both ADAS‐cog and ADCS‐ADL scores remained unchanged across the study in all treatment and placebo groups for mild AD patients (Figure2C,F, I, and L).
Regarding global assessment endpoints, there was a significantly slower decline in the CDR‐sb scores versus placebo at month 14 for most treatment groups in the moderate AD patients (difference ranging ‒1.5 [95% CI: ‒3.0, 0.0] to ‒1.8 [95% CI: ‒3.2, ‒0.4]; P values: .01 to .04; effect sizes: 50% to 60%; Figure3B, E), and improvement in mild AD patients (difference ranging ‒0.8 [95% CI: ‒2.2, 0.6] to ‒1.2 [95% CI: ‒2.4, 0.0]; P values: .02 to .04; effect sizes: 157% to 171%; Figure3C, F). There was less decline in ADCS‐CGIC scores at month 14 for all treatment groups versus placebo in the moderate AD patients (difference ranging ‒0.8 [95% CI:‒1.4, ‒0.2] to ‒1.0 [95% CI: ‒1.7, ‒0.3]; P values: .002 to .02; effect sizes: 57% to 71%; Figure3H and K), and improvement in the mild AD patients (difference ranging −0.8 [95% CI: −1.3, −0.3] to −0.8 [95% CI: −1.6, −0.1]; P values: .004 to .04; effect sizes: 200%; Figure3I and L).
3.5. Complementary analysis
The results of the sensitivity analysis using zLOCF resulted in similar treatment differences and effect sizes as the primary MMRM (see Appendix B, Table B.9 in supporting information). Results were nearly identical for ADCS‐ADL, ADAS‐Cog, CDR‐sb, and ADCS‐CGIC, consistent with a missing at random assumption for the discontinuations.
When the model was adjusted for key baseline characteristics, the analysis showed statistically significant differences against placebo of most treatment groups in all relevant outcomes (Appendix B, Table B.10 in supporting information).
Consistency of results was confirmed by the calculation of effect sizes across outcomes, with slowing of progression of symptoms at 14 months of 54% for ADCS‐ADL (difference: 3.6 [95% CI: 0.6, 6.7]; P = .02), 58% for ADAS‐Cog (difference: ‒2.0 [95% CI: ‒4.01, 0.0]; P = .05), 51% for CDR‐sb (difference: ‒0.9 [95% CI: ‒1.7, ‒0.2]; P = .02), and 76% for ADCS‐CGIC (difference: ‒0.7 [95% CI: ‒1.0, ‒0.4]; P < .0001). The GST score showed slowing of progression of symptoms with statistically significant differences between all active treatments versus placebo from month 9 (difference: ‒5.6 [95% CI: ‒10.58, ‒0.69]; P = .03), month 12 (difference: ‒7.54 [95% CI: ‒12.78, ‒2.30]; P = .005), and month 14 (difference: ‒8.24 [95% CI: ‒13.52, ‒2.95]; P = .002).
3.6. Biomarkers
A total of 297 patients provided CSF at baseline. Changes from baseline CSF Aβ42 levels remained stable in all PE‐treated patients, whereas in the placebo group there was a decrease over time (Figure 4D, E, and F). This finding was particularly evident in the moderately impaired AD patients (LS means ± SEM: 1.2±9.8 vs −47.3±27.8 [P = .04] and −7.3±11.5 vs −59.8±30.2 [P = .05], at months 2 and 14, respectively; Figure 4E). With regard to T‐tau and P‐tau proteins, the placebo group showed increased values with respect to the PE‐treated patients combined in the moderate AD patients (35.5±19.8 vs 193.5±71.7 [P = .002] and 5.7±1.5 vs 13.4±4.5 [P = .04], respectively, at month 2; Figure 4H and K). Raw baseline and individual visit CSF biomarker values in the treatment groups are shown in Appendix B, Tables B.11 to B.14 in supporting information.
FIGURE 4.
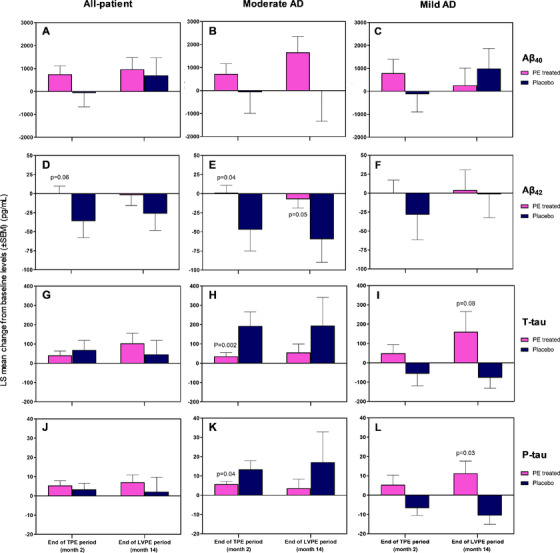
Least square (LS) mean change from baseline in cerebrospinal fluid levels (± standard error of the mean) of Aβ40 (panels A, B, C), Aβ42 (panels D, E, F), T‐tau (panels G, H, I), and P‐tau (panels J, K, L) between the finalization and beginning of each of the two plasma exchange with albumin replacement (PE) periods (TPE: conventional therapeutic PE up to month 2; LVPE: low‐volume PE up to month 14) performed on mild to moderate Alzheimer's disease (AD) patients (all‐patient [panels A, D, G, J], moderate AD [panels B, E, H, K], and mild AD [panels C, F, I, L] populations). The difference between the group of PE‐treated patients combined (n = 226 at baseline; n = 205 at end of TPE; n = 159 at end of LVPE) and the placebo group (n = 71 at baseline; n = 72 at end of TPE; n = 63 at end of LVPE) was evaluated using analysis of covariance with treatment group as a fixed effect, and the corresponding baseline value, age and AD severity, as a covariate. Both statistical significance (P < .05) and borderline significance (P < .1) versus placebo are indicated
3.7. Safety
A total of 4709 PE procedures were performed, 1223 were sham PE (placebo) and 3486 were treatment PE. Of the treatment PE, 1283 were TPE while 2203 were LVPE. All LVPE and 814 TPE (47.4%) were performed through peripheral access.
Of all 4709 PE procedures, ≈90% were uneventful, and the remaining 10.6% were associated with at least one AE. There were AEs associated with 20.1% of PE procedures with central access versus 13.1% with peripheral access. There were very few AEs related to the study product (albumin and IVIG) with respect to the number of PEs performed (0.3% to 1.4% across the PE treatment arms). The product‐related AEs were more frequent in the high‐albumin+IVIG group (17.7% of patients with at least 1 AE) than in the low‐albumin+IVIG (8.1%) or the low‐albumin (6.4%) groups.
AEs had a median duration of 1 day, the majority of them (58.5%) being transient (lasting ≤ 7 days). The most frequent AE was local catheter reactions (2.4% to 3.5% across the PE treatment arms; see Table 2). Overall AEs were more frequent among the patients in the three groups of PE treatment (87.3%‐92.3%) than in placebo (70.9%). Two patients died during the study (sepsis and suicide). As shown in Figure 1, the dropout rate due to an AE was very low in the placebo group (1.3%; 1/80 patients) whereas in the treated groups, values ranged from 7.7% (6/78) to 17.4% (15/86).
TABLE 2.
Safety of plasma exchange procedures by treatment arm
| Concept | Placebo | Low‐albumin | Low albumin + IVIG | High albumin + IVIG | Total |
|---|---|---|---|---|---|
| Total PE performed | 1223 | 1207 | 1180 | 1099 | 4709 |
| PE associated with AE, n (% of procedures) | |||||
| With procedure‐related AE | 9 (0.7) | 163 (13.5) | 148 (12.5) | 181 (16.5) | 501 (10.6) |
| With product‐related AE | 0 | 4 (0.3) | 6 (0.5) | 16 (1.4) | 26 (0.6) |
| PE associated with a specific AE, n (% of procedures) | |||||
| Catheter local reactions a | 0 | 40 (3.3) | 35 (3.0) | 38 (3.5) | 113 (2.4) |
| Hypotension | 0 | 37 (3.1) | 37 (3.1) | 29 (2.6) | 103 (2.2) |
| Muscle spasms | 0 | 15 (1.2) | 4 (0.3) | 28 (2.5) | 47 (1.0) |
| Anemia | 2 (0.2) | 12 (1.0) | 15 (1.3) | 12 (1.1) | 41 (0.9) |
| Dizziness | 0 | 8 (0.7) | 13 (1.1) | 9 (0.8) | 30 (0.6) |
| Presyncope | 1 (0.1) | 7 (0.6) | 13 (1.1) | 9 (0.8) | 30 (0.6) |
| Paresthesia | 0 | 16 (1.3) | 1 (0.1) | 11 (1.0) | 28 (0.6) |
| Nausea | 0 | 8 (0.7) | 4 (0.3) | 4 (0.4) | 16 (0.3) |
| Blood fibrinogen decreased | 0 | 1 (0.1) | 8 (0.7) | 3 (0.3) | 12 (0.3) |
| Blood/venous pressure decreased | 0 | 4 (0.3) | 3 (0.3) | 5 (0.5) | 12 (0.3) |
| Catheter/device infection b | 0 | 4 (0.3) | 4 (0.3) | 3 (0.3) | 11 (0.2) |
| Syncope | 0 | 4 (0.3) | 3 (0.3) | 4 (0.4) | 11 (0.2) |
| Contusion | 1 (0.1) | 2 (0.2) | 7 (0.6) | 1 (0.1) | 10 (0.2) |
| Anxiety | 0 | 2 (0.2) | 3 (0.3) | 5 (0.5) |
Abbreviations: AE, adverse event; IVIG, intravenous immunoglobulin; PE, plasma exchange.
AEs reported in > 5% of patients in the following categories: catheter site erythema, catheter site pain, extravasation, infusion site extravasation, vascular access complication, and poor venous access.
AEs reported in > 5% of patients with catheter site infections and device‐related infections.
Serious adverse events (SAEs) were reported more frequently in the groups receiving IVIG (1.9% to 1.7% of the PEs performed, in 20.3% to 22.1% of patients) than in the low‐albumin and the placebo group (0.7% and 0.9%, respectively, of the PE performed, in 10.3% and 10.1% of patients, respectively). The dropouts due to a SAE were: 1.3% in placebo, 1.3% in low‐albumin, 11.6% in low‐albumin+IVIG, 6.4% in high‐albumin+IVIG groups. Three patients presented four asymptomatic amyloid‐related imaging abnormalities (ARIA), two (ARIA‐E and ARIA‐H) in a patient in the low‐albumin+IVIG group and one in each of two patients in the high‐albumin+IVIG group (ARIA‐E and ARIA‐H). Details of AEs, product‐related AEs, and SAEs, are summarized in Appendix B, Tables B.15 to B.17 in supporting information.
4. DISCUSSION
This study provides encouraging results in the treatment of symptomatic AD patients. There was a reduction in the progression of symptoms in the PE‐treated patients in both co‐primary efficacy endpoints: the ADCS‐ADL showed 52% less decline in PE‐treated compared to placebo patients (P = .03) while the ADAS‐Cog showed 66% less decline (P = .06).
The effects observed in the co‐primary measures were supported by the statistically significant differences in the global assessment endpoints. PE had a significant impact on CDR‐sb and ADCS‐CGIC measures in all the treated populations, in nearly all treatment arms. Positive results on CDR‐sb and/or ADCS‐CGIC tests have not been reported in previous trials, 7 , 9 , 11 , 35 , 36 , 37 , 38 , 39 possibly due to the challenge in demonstrating treatment effects on global scales. Differences in baseline demographic characteristics did not appear to have an influence on the outcomes.
Because we wanted to examine whether the initial severity of the cognitive syndrome determined response, the cohort was dichotomized into mild and moderate AD severity. We observed that the symptom severity of our sample (baseline MMSE 18‐26) was globally milder than the one traditionally considered for mild‐to‐moderate AD (MMSE 10‐23). 40 Results by severity of the cognitive syndrome showed that the differences remained in the moderate AD group, but not in the mild AD group. Moreover, all three PE active arms showed statistically significant slower decline compared to placebo for ADCS‐ADL in the moderate group. Regarding global assessments, effects were significant in both mild and moderate patients, with moderate AD patients showing less decline, and mild AD patients showing an improvement from baseline. The better ADAS‐Cog response in moderate versus mild AD may be due to reduced sensitivity of the ADAS‐Cog in patients with better cognitive performance. 41 , 42 Consequently, cognitive benefit may be more visible with a scale that is more targeted toward early cognitive changes that are seen in these milder patients.
The percentage of dropouts (28%) over a period of 14 months (55 weeks) was similar to that reported elsewhere (13.5% to 31.3%), 7 , 8 , 9 , 10 , 12 but was higher in the two IVIG arms (34.6%‐34.9%) and lower in the other arm and placebo, both without IVIG (20.0%‐21.8%), which is consistent with the safety profile reported in patients receiving infusions of albumin and IVIG. 43 , 44 In addition, only two (0.6%) patients died during the study, which is similar to the low mortality rates reported elsewhere (1.3%‐2.5%). 8 , 9 , 10 , 12 The sensitivity analysis addressing discontinuations by imputation methods (zLOCF) suggested that bias due to discontinuations is minimal in this study. The primary MMRM results were somewhat more conservative, suggesting that lowering dropouts may increase effect sizes. The statistically stronger results after correcting for multiple baseline covariates and factors result from reduced variability and reduction of bias with these models. In addition, the overall impact of the treatment assessed with GST highlighted the consistency of the effect sizes across relevant outcomes (ADCS‐ADL, ADAS‐Cog, and CDR‐sb).
We examined CSF biomarkers of the core pathology of AD, 45 finding no clear evidence of a disease‐modifying pattern. The effect of treatment was more apparent in the moderate AD population, whose levels of CSF Aβ42 and tau protein remained stable in the PE‐treated patients across the study whereas in the placebo group decreased and increased, respectively. 46 This suggests a positive effect of PE and would be consistent with the observed clinical outcome in those patients. PE might increase clearance of CSF soluble amyloid to prevent its further deposition. 47 , 48 On the other hand, the Jack model of AD biomarkers suggests that in the moderate AD stage the amyloid deposition approaches the asymptotic part of the curve. 49 Therefore, because we observed a bigger effect in the moderate AD stage, it could be possible that the mechanism involved may be related to the clearance of deposited amyloid. Conversely, in mild AD patients the patterns of Aβ and tau levels were inconclusive or even counterintuitive. Although amyloid dynamics and their connection with tau are not well understood, our CSF biomarker results overall suggest that more than one mechanism may have been involved in the PE approach, perhaps including changes in oxidation status 28 and inflammatory mediators, 50 or could even be procedure‐related. In parallel, Aβ40 levels did not significantly change, which may be ascribed to a faster clearance compared to Aβ42, 51 as well as being less relevant for AD pathogenesis.
In our complementary analysis we examined the efficacy of the PE treatment with adjusted baseline characteristics analysis, including amyloid and APOE Ɛ4 status. Treatment effects were somewhat different between these groups, and correction for these factors resulted in more significant treatment differences. These results support the observation that all PE‐treated arms slowed progression of AD symptoms in the co‐primary and global clinical endpoints.
PE is typically used to treat a range of neurological, immunological, and metabolic disorders 52 , 53 while this study showed that PE may be a novel therapeutic approach to treat AD. Furthermore, we believe that PE treatment does not preclude the possibility of being applied in combination with current and future therapies for AD. To our knowledge, this series of 4709 apheresis procedures (including the 1223 sham PE) is the largest performed within a randomized, controlled clinical trial on a single disease. In addition, this is the first time that therapeutic apheresis was used in a phase 2b/3 trial on AD, 31 including a new form, LVPE, aimed at chronic diseases. LVPE is based on the routine plasma donation process performed millions of times a year in plasma donation centers worldwide, so its application on AD patients would be suitable on an outpatient basis.
Because AD patients are frequently in fragile health, PE treatment should be undertaken with caution due to its invasive nature. However, in our study close to 90% of the 4709 apheresis procedures were uneventful. The fact that >3500 actual PE procedures were performed in this study, most of them through peripheral access, and the high percentage (72%) of patients who completed the study, further supports that this procedure is feasible in mild‐to‐moderate AD. Peripheral access will likely be considered the only access for a future trial. Overall, the AEs reported for this study with AD patients are similar to the known safety profile of PE procedures for other indications. 54 , 55
In this study neither raters, caregivers, nor patients were aware of which treatment was being administered. The maintenance of blinding of the treatment could be a challenge as it occurs with trials that involve peripheral devices. However, there are two facts that strongly suggest that blinding was effective: first, that identical null clinical effect was observed in the primary variables of both treated and placebo groups of the mild AD subpopulation; and second, that 80.0% of the patients in the placebo group (the highest percentage of all groups) completed the whole study. It is typically expected that poor blinding causes a much higher rate of dropouts in the placebo group. 56
The findings of this study should be interpreted in the context of its limitations. Caution is advised regarding worldwide generalization of the results because the study was performed in two countries only. This study does not allow determining the specific mechanism of action, which may go beyond Aβ binding. Analysis of plasma amyloid levels, which could help determine mechanisms, was not available for this study. This study did not enroll patients based on the presence or absence of a biomarker but was based on the presence of the AD clinical syndrome. Although this will be taken into account in the design of a future trial, our distribution of baseline CSF Aβ42 levels with most patients in the lower ranges, was in line with the mean values from other studies that use similar patient population and methods of determination. 8 , 57 Moreover, because none of the three treatment arms separately showed superiority in clinical efficacy, the intervention group for the future trial will take into consideration other parameters such as dropout rate and safety profile.
In conclusion, this study showed that PE with albumin replacement could slow cognitive and functional decline in AD patients. This finding in the co‐primary outcomes was supported by a similar effect observed in key secondary global assessments. PE was shown to be feasible in the studied patient population. These findings have the potential to offer AD patients with overt dementia a new modality of treatment, although additional studies are needed to further investigate the current study areas of uncertainty.
5. THE AMBAR TRIAL GROUP
In addition to those mentioned as nominal authors, the following investigators and centers also participated in the study: Asunción Lafuente (Alzheimer Research Center and Memory Clinic, Fundació ACE Institut Català de Neurociències Aplicades ‐ Universitat Internacional de Catalunya [UIC], Barcelona, Spain. Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas [CIBERNED], Instituto de Salud Carlos III, Madrid, Spain); Juan Pablo Tartari (Hospital Universitari Mútua de Terrassa, Terrassa, Spain); Teresa Moreno (Hospital Clínico San Carlos, Madrid, Spain); Francesc Pujadas (Hospital Vall d'Hebron, Barcelona, Spain); Miguel Goñi (Hospital Universitario de Burgos, Burgos, Spain); José De La Gándara (Quantum Laboratories, Inc. Wixom, MI, USA); William A. McElveen (Bradenton Research Center, Inc., Bradenton, FL, USA); Ramon Reñé (Hospital Universitari de Bellvitge, L'Hospitalet de Llobregat, Spain); Secundino López‐Pousa (Parc Hospitalari Martí i Julià, Salt, Spain); Antonio Del Olmo (Hospital Universitario Dr. Peset, València, Spain); Douglas Young (Northern California Research, Sacramento, CA, USA); Babak Tousi (Cleveland Clinic Lou Ruvo Center for Brain Health, Las Vegas, NV, USA); Jacobo Mintzer (Roper Saint Francis Healthcare, Charleston, SC, USA); Joshua Shua‐Haim (Mid‐Atlantic Geriatric/ARC, Manchester, NJ, USA); Kimball Johnson (iResearch Atlanta, LLC, Decatur, GA, USA); Ernest Balaguer (Hospital General de Catalunya, Sant Cugat del Vallès, Spain); Sarah Berman (University of Pittsburgh Alzheimer Disease Research Center‐ADRC, Pittsburgh, PA, USA); Bridget Bellingar (DMI Research, Pinellas Park, FL, USA); Antonio Oliveros (Hospital Viamed Montecanal, Zaragoza, Spain); Norberto Rodríguez (Hospital Nuestra Señora de la Candelaria, Santa Cruz de Tenerife, Spain); Dana Kumjian (RTR Medical Group, Savannah, GA, USA); Jordi Alom (Hospital General de Elche, Elx, Spain); César García Pérez‐Cejuela (Hospital de Vinalopó, Elx, Spain); Tulio Bertorini (Neurology Clinic, P.C., Cordova, TN, USA); Bennet Machanic (Mountain View Clinical Research, Inc., Denver, CO, USA); Thomas Obisesan (Howard University College Of Medicine, Washington, DC, USA); Krzysztof Bujarski (Dartmouth‐Hitchcock Medical Center, Lebanon, NH, USA); Miquel Barceló and Natalia Afonso (Grifols, Barcelona, Spain); Paul Pinciaro (Grifols, NC, USA); Orlando Puente (Miami Dade Medical Research Institute, Miami, FL, USA); Lisa McLaughlin (American Red Cross Southern Blood Services Region, Atlanta, GA, USA), Leonardo M. Allende (L&L Research Choices, Inc., Miami, FL, USA).
CONFLICTS OF INTEREST
MB has been a consultant for Araclon, Avid, Bayer, Elan, Grifols, Janssen/Pfizer, Lilly, Neuroptix, Nutricia, Roche, Sanofi, and Servier; and received fees for lectures and funds for research from Araclon, Esteve, Grifols, Janssen, Novartis, Nutricia, Piramal, Pfizer‐Wyett, Roche, and Servier. OLL has been a consultant for Grifols and Lundbeck. JO has been a consultant for Schwabe and Grifols; and received fees for lectures and funds for research from Nutricia. ZMS has been a consultant for Grifols and Fresenius‐Kabi and participated in research supported by funds from Grifols and Fresenius‐Kabi. MPF, MPA, JL, GP‐R, JEG, FA, PO, DK, JLI, and SH received funding from Grifols to perform this study. LN, MT, CG, JB, MC, and AP are full‐time employees of Grifols.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors would like to thank Víctor Grífols‐Roura for his pioneering vision on the potential use of plasmapheresis in Alzheimer's disease, without which this project would not have been possible. Carlos Roura (Grifols) is acknowledged for providing his valuable insight and expertise in all the engineering aspects of the trial. The expert advice from Agustín Ruiz (Fundació ACE, Barcelona, Spain) to improve the manuscript is greatly acknowledged. The fruitful discussion of the study results between the authors and Prof. Bruno Vellas and Prof. Jeffrey Cummings are hereby recognized. The professionalism, caring, and dedication of the plasmapheresis nurses and the health‐care professionals who contributed greatly to the success of the study are deeply appreciated: Regina Rohe (Apheresis Care Group and Fresenius Medical Care); Cristina Prieto Fernández (Banc de Sang i Teixits); Luis Estevez, Alain Diaz, and Ruy Pardo (Allied Biomedical Research Institute, Inc.); Lori Conway, Samantha Planas, and Eddie Kubo (Fenwal); Anna Koo (Cleveland Clinic, Cleveland, OH, USA); Maria Olivera and Vivianka Lavin (L&L Research Choices, Inc.); Paul Crossley, Debbie Lang, and Klaus Freivogel (United BioSource Corporation). Last, but not least, deepest thanks from the authors to the patients and their families for their indispensable contribution.
Boada M, López OL, Olazarán J, et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer's disease: Primary results of the AMBAR Study. Alzheimer's Dement. 2020;16:1412–1425. 10.1002/alz.12137
Funding information: The AMBAR study is sponsored by Grifols, a manufacturer of therapeutic human serum albumin and intravenous immune globulin.
REFERENCES
- 1. Montine TJ, Phelps CH, Beach TG. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol (Berl). 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrovitch H, Ross GW, Steinhorn SC, et al. AD lesions and infarcts in demented and non‐demented Japanese‐American men. Ann Neurol. 2005;57:98‐103. [DOI] [PubMed] [Google Scholar]
- 3. Higashi S, Iseki E, Yamamoto R, et al. Concurrence of TDP‐43, tau and α‐synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284‐294. [DOI] [PubMed] [Google Scholar]
- 4. Eikelenboom P, Veerhuis R, van Exel E, Hoozemans JJM, Rozemuller AJM, van Gool WA. The early involvement of the innate immunity in the pathogenesis of lateonset Alzheimers disease: neuropathological, epidemiological and genetic evidence. Curr Alzheimer Res. 2011;8:142‐150. [DOI] [PubMed] [Google Scholar]
- 5. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil. JAMA. 2004;291:317. [DOI] [PubMed] [Google Scholar]
- 6. Kemp PM, Holmes C, Hoffmann S, et al. A randomised placebo controlled study to assess the effects of cholinergic treatment on muscarinic receptors in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74:1567‐1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atri A, Frölich L, Ballard C, et al. Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease. JAMA. 2018;319:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N Eng J Med. 2014;370:311‐321. [DOI] [PubMed] [Google Scholar]
- 9. Egan MF, Kost J, Tariot PN, et al. Randomized trial of verubecestat for mild‐to‐moderate Alzheimer's disease. N Eng J Med. 2018;378:1691‐1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer's disease. N Eng J Med. 2018;378:321‐330. [DOI] [PubMed] [Google Scholar]
- 11. Ostrowitzki S, Lasser RA, Dorflinger E, et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer's disease. Alzheimers Res Ther. 2017;9(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salloway S, Sperling R, Fox NC, et al. Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N Engl J Med. 2014;370:322‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tolar M, Abushakra S, Sabbagh M. The path forward in Alzheimer's disease therapeutics: reevaluating the amyloid cascade hypothesis. Alzheimers Dement. 2020. 10.1016/j.jalz.2019.09.075 [DOI] [PubMed] [Google Scholar]
- 14. Boada M, Anaya F, Ortiz P, et al. Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid‐β concentrations and cognition outcomes in Alzheimer's disease patients: a multicenter, randomized, controlled clinical trial. J Alzheimers Dis. 2017;56:129‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boada M, Ortiz P, Anaya F, et al. Amyloid‐targeted therapeutics in Alzheimer's disease: use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect. 2009;22:325‐339. [DOI] [PubMed] [Google Scholar]
- 16. Costa M, Ortiz AM, Jorquera JI. Therapeutic albumin binding to remove amyloid‐beta. J Alzheimers Dis. 2012;29:159‐170. [DOI] [PubMed] [Google Scholar]
- 17. Cuberas‐Borros G, Roca I, Boada M, et al. Longitudinal neuroimaging analysis in mild‐moderate Alzheimer's disease patients treated with plasma exchange with 5% human albumin. J Alzheimers Dis. 2018;61:321‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Milojevic J, Costa M, Ortiz AM, Jorquera JI, Melacini G. In vitro amyloid‐beta binding and inhibition of amyloid‐beta self‐association by therapeutic albumin. J Alzheimers Dis. 2014;38:753‐765. [DOI] [PubMed] [Google Scholar]
- 19. Roberts KF, Elbert DL, Kasten TP, et al. Amyloid‐beta efflux from the central nervous system into the plasma. Ann Neurol. 2014;76:837‐844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti‐A antibody alters CNS and plasma A clearance and decreases brain A burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850‐8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid‐beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264‐2267. [DOI] [PubMed] [Google Scholar]
- 22. DeMattos RB, Bales KR, Parsadanian M, et al. Plaque‐associated disruption of CSF and plasma amyloid‐beta (Abeta) equilibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002;81:229‐236. [DOI] [PubMed] [Google Scholar]
- 23. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid‐beta levels regulate amyloid‐beta clearance from the central nervous system. J Alzheimers Dis. 2009;16:325‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meca‐Lallana JE, Rodriguez‐Hilario H, Martinez‐Vidal S, et al. Plasmapheresis: its use in multiple sclerosis and other demyelinating processes of the central nervous system. An observation study. Rev Neurol. 2003;37:917‐926. [PubMed] [Google Scholar]
- 25. Weinshenker BG, O'Brien PC, Petterson TM, et al. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann Neurol. 1999;46:878‐886. [DOI] [PubMed] [Google Scholar]
- 26. Colombo G, Clerici M, Giustarini D. Redox albuminomics: oxidized albumin in human diseases. Antioxid Redox Signal. 2012;17:1515‐1527. [DOI] [PubMed] [Google Scholar]
- 27. Biere AL, Ostaszewski B, Stimson ER, Hyman BT, Maggio JE, Selkoe DJ. Amyloid beta‐peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem. 1996;271:32916‐32922. [DOI] [PubMed] [Google Scholar]
- 28. Costa M, Horrillo R, Ortiz AM, et al. Increased Albumin oxidation in cerebrospinal fluid and plasma from Alzheimer's disease patients. J Alzheimers Dis. 2018;63:1395‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramos‐Fernandez E, Tajes M, Palomer E, et al. Posttranslational nitro‐glycative modifications of albumin in Alzheimer's disease: implications in cytotoxicity and amyloid‐beta peptide aggregation. J Alzheimers Dis. 2014;40:643‐657. [DOI] [PubMed] [Google Scholar]
- 30. Hachinski V, Einhaupl K, Ganten D, et al. Preventing dementia by preventing stroke: the Berlin Manifesto. Alzheimers Dement. 2019;15:961‐984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Boada M, Lopez O, Nunez L, et al. Plasma exchange for Alzheimer's disease Management by Albumin Replacement (AMBAR) trial: study design and progress. Alzheimers Dement (N Y). 2019;5:61‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 33. European Medicines Agency (EMA) . Draft guideline on the clinical investigation of medicines for the treatment of Alzheimer's disease and other dementias. Committee for Medicinal Products for Human Use (CHMP). EMA/CHMP/539931/2014. 28 January 2016. 2016.
- 34. Hendrix SB. Requiring an amyloid‐beta1‐42 biomarker may improve the efficiency of a study, and simulations may help in planning studies. Alzheimers Res Ther. 2011;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lawlor B, Segurado R, Kennelly S, et al. Nilvadipine in mild to moderate Alzheimer disease: a randomised controlled trial. PLoS Med. 2018;15:e1002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lozano AM, Fosdick L, Chakravarty MM, et al. A phase II study of fornix deep brain stimulation in mild Alzheimer's disease. J Alzheimers Dis. 2016;54:777‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Maher‐Edwards G, Watson C, Ascher J, et al. Two randomized controlled trials of SB742457 in mild‐to‐moderate Alzheimer's disease. Alzheimers Dement (N Y). 2015;1:23‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salloway S, Honigberg LA, Cho W, et al. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti‐amyloid‐beta antibody double‐blind, placebo‐controlled, randomized phase II study in mild‐to‐moderate Alzheimer's disease (BLAZE). Alzheimers Res Ther. 2018;10:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schneider LS, Thomas RG, Hendrix S, et al. Safety and efficacy of edonerpic maleate for patients with mild to moderate Alzheimer disease: a phase 2 randomized clinical trial. JAMA Neurol. 2019;76:1330‐1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tombaugh TN, McIntyre NJ. The mini‐mental state examination: a comprehensive review. J Am Geriatr Soc. 1992;40:922‐935. [DOI] [PubMed] [Google Scholar]
- 41. Podhorna J, Krahnke T, Shear M, Harrison JE. Alzheimer's Disease Assessment Scale‐Cognitive subscale variants in mild cognitive impairment and mild Alzheimer's disease: change over time and the effect of enrichment strategies. Alzheimers Res Ther. 2016;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Skinner J, Carvalho JO, Potter GG, et al. The Alzheimer's Disease Assessment Scale‐Cognitive‐Plus (ADAS‐Cog‐Plus): an expansion of the ADAS‐Cog to improve responsiveness in MCI. Brain Imaging Behav. 2012;6:489‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alsina L, Mohr A, Montanes M, et al. Surveillance study on the tolerability and safety of Flebogamma((R)) DIF (10% and 5% intravenous immunoglobulin) in adult and pediatric patients. Pharmacol Res Perspect. 2017;5(5):e00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liumbruno GM, Bennardello F, Lattanzio A, Piccoli P, Rossettias G, et al, Italian Society of Transfusion M . Recommendations for the use of albumin and immunoglobulins. Blood Transfus. 2009;7:216‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Frank RA, Galasko D, Hampel H, et al. Biological markers for therapeutic trials in Alzheimer's disease. Proceedings of the biological markers working group; NIA initiative on neuroimaging in Alzheimer's disease. Neurobiol Aging. 2003;24:521‐536. [DOI] [PubMed] [Google Scholar]
- 46. Clark CM, Xie S, Chittams J, et al. Cerebrospinal fluid tau and beta‐amyloid: how well do these biomarkers reflect autopsy‐confirmed dementia diagnoses? Arch Neurol. 2003;60:1696‐1702. [DOI] [PubMed] [Google Scholar]
- 47. Marques MA, Kulstad JJ, Savard CE, et al. Peripheral amyloid‐beta levels regulate amyloid‐beta clearance from the central nervous system. J Alzheimers Dis. 2009;16:325‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tarasoff‐Conway JM, Carare RO, Osorio RS, et al. Clearance systems in the brain—implications for Alzheimer disease. Nat Rev Neurol. 2015;11:457‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jack CR, Jr , Wiste HJ, Lesnick TG, et al. Brain beta‐amyloid load approaches a plateau. Neurology. 2013;80:890‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int J Dev Neurosci. 2006;24:167‐176. [DOI] [PubMed] [Google Scholar]
- 51. Huang Y, Potter R, Sigurdson W, et al. Beta‐amyloid dynamics in human plasma. Arch Neurol. 2012;69:1591‐1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae‐Grant A. Evidence‐based guideline update: plasmapheresis in neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;76:294‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sachais BS, Katz J, Ross J, Rader DJ. Long‐term effects of LDL apheresis in patients with severe hypercholesterolemia. J Clin Apher. 2005;20:252‐255. [DOI] [PubMed] [Google Scholar]
- 54. Cortese I, Cornblath DR. Therapeutic plasma exchange in neurology: 2012. J Clin Apher. 2013;28:16‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mortzell Henriksson M, Newman E, Witt V, et al. Adverse events in apheresis: an update of the WAA registry data. Transfus Apher Sci. 2016;54:2‐15. [DOI] [PubMed] [Google Scholar]
- 56. Hróbjartsson A, Emanuelsson F, Skou Thomsen AS, Hilden J, Brorson S. Bias due to lack of patient blinding in clinical trials. A systematic review of trials randomizing patients to blind and nonblind sub‐studies. Int J Epidemiol. 2014;43:1272‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Willis BA, Sundell K, Lachno DR, et al. Central pharmacodynamic activity of solanezumab in mild Alzheimer's disease dementia. Alzheimers Dement (N Y). 2018;4:652‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.