

Abstract
The repeated failure of clinical trials targeting the amyloid beta (Aβ) protein has challenged the amyloid cascade hypothesis. In this perspective, I discuss the biogenesis and biology of Aβ, from the arrangement of its atoms to its effects on the human brain. I hope that this analysis will help guide future attempts to home in on this elusive therapeutic target.
1. INTRODUCTION
Are amyloid beta protein (Aβ) therapies for Alzheimer's disease (AD) dead?
Genetics and neuropathology inextricably link AD to the amyloid precursor protein (APP), of which one breakdown product—the amyloid beta Aβ—has been virtually the sole focus. After two decades of effort, antibodies targeting Aβ and drugs reducing the production of Aβ have consistently failed to improve cognition or reduce rates of cognitive decline. Aβ is released from the proteolysis of APP by beta‐site APP cleavage enzyme (BACE1) 1 and the γ‐secretase complex. 2 , 3 Recently, reports on the failure of two more BACE1 inhibitors 4 , 5 tipped the scale of an influential opinion leader away from Aβ as a therapeutic target. 6
Here, I argue that experimental treatments aimed at Aβ are still viable, provided they target the specific forms that cause neurological dysfunction. The forms of Aβ found in amyloid plaques, which several of the failed experimental therapies target, are probably not the most important ones causing neurological dysfunction. Some researchers believe that in the initial stages of disease, Aβ triggers pathological changes in tau, and that at later stages tau becomes unyoked from Aβ. This idea forms the rationale for applying Aβ treatments early on, even before symptoms are present. 7 To develop effective therapies, it is important to be clear about what the different forms of Aβ do to the brain.
2. WHAT FORMS OF Aβ CAUSE NEUROLOGICAL DYSFUNCTION?
Type 1 oligomers, specifically in mice with modest or no Aβ plaques; type 2 oligomers, specifically in mice with plaque loads that exceed levels typically seen in humans with Alzheimer's disease (AD). Type 1 oligomers cause more large‐scale harm to brain function than type 2 oligomers.
Aβ oligomers, rather than Aβ monomers or Aβ fibrils, are the most pathogenic Aβ species. 8 , 9 , 10 , 11 Therefore, delineating the biogenesis and self‐assembly of Aβ oligomers in vivo is paramount for understanding how and why Aβ causes neurological dysfunction. Aβ oligomers form in the brain when Aβ monomers merge. It is likely that not all Aβ oligomers accumulate, however, and that the ones that accumulate are difficult to break down due to a lattice of non‐covalent bonds linking the monomers and stabilizing the oligomers. Stable Aβ oligomers that cause neurological dysfunction have specific, quaternary structures, bind different conformation‐specific antibodies, and probably arise via distinct nucleation processes.
2.1. Quaternary structures
The quaternary structure of an Aβ oligomer refers to the arrangement of its constituent monomers. Aβ derives its name from the β‐sheet quaternary structure of its most abundant pathological form—the amyloid fibril. The β‐sheet, first described in 1951 by Pauling and Corey, 12 consists of rows of polypeptides (not identical in protein structures, but usually identical in amyloid fibrils) with β‐strand secondary structure. Lining up the rows evenly (in‐register) produces β‐sheets that stack side‐by‐side, forming sheaves that bundle together to form fibrils. There are two types of β‐sheet fibrils, which differ in the orientation of their polypeptide chains. In many, naturally occurring, non‐pathological fibrils, such as those found in silk and feathers, the polypeptide chains run parallel to the length of the fibril. In amyloid fibrils, most of which are pathological, the polypeptides—typically segments of 10 to 100 amino acids—run perpendicular to the fibril, as was first observed in boiled egg white. 13 The structural term for this arrangement is “cross‐β.” The amyloid structure of Aβ fibrils was known before its amino acid sequence was deciphered from brain proteins purified from patients with AD. 14
When the rows of polypeptide chains are staggered (out‐of‐register), the resulting β‐sheet structures are stubby—they lack topological tails—not fibrillary, in shape. An excellent review of the amyloid structure of proteins was written by Eisenberg and Jucker. 15
Despite knowledge of the basic quaternary structure and amino acid sequence of Aβ fibrils, the arrangement of the actual atoms remained mysterious for two decades. Thanks to advances in an assortment of biophysical techniques, several structures—fibrils and oligomers—have now been resolved at high levels of resolution. All but one of the deduced structures contain β‐sheets of one variety or another. These structures include in‐register parallel β‐sheets, 16 , 17 in‐register anti‐parallel β‐sheets, 18 out‐of‐register β‐barrels, 19 , 20 and β‐hairpin assemblies. 21 Intriguingly—although terminologically counterintuitive—Aβ can also form α‐sheets, 22 which do not exist as fibrils but may comprise oligomers. α‐Sheets, first described in 1951 by Pauling and Corey, 23 differ from β‐sheets in the orientation of the amide bonds in the polypeptide strands; in α‐sheets the carboxyl groups in the α‐strands face the same direction, whereas in β‐sheets, the carboxyl groups in the β‐strands alternate. In both α‐ and β‐sheets, the amino‐acid side chains alternate. However, in β‐sheets the side‐chains may be in‐ or out‐of‐register, while in α‐sheets they are always out‐of‐register.
2.2. Nucleation processes define type 1 and type 2 oligomers
The nucleation of Aβ assemblies refers to the mechanism by which Aβ monomers come together. Primary nucleation refers to two or more monomers simultaneously forming a long‐lived cluster. Secondary nucleation is when a previously formed assembly, usually a fibril, acts as a catalyst bringing monomers together. In 2015, we hypothesized that the biogenesis of Aβ oligomers in the brain likely involves both primary and secondary nucleation processes, and named Aβ oligomers formed by primary nucleation type 1 oligomers, and Aβ oligomers formed by secondary nucleation type 2 oligomers 24 (Figure 1).
FIGURE 1.
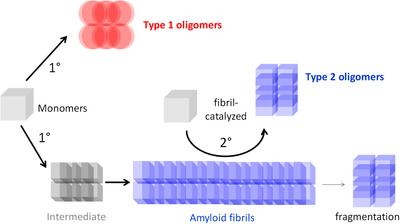
Primary and secondary nucleation of Aβ oligomers. Adapted, with permission, from Liu et al., “Quaternary structure defines a large class of Aβ oligomers neutralized by sequestration” Cell Reports, 2015. Type 1 oligomers arise via primary nucleation. Type 2 oligomers arise via fibril‐catalyzed secondary nucleation. Both types of oligomers compete for a common pool of monomers
Secondary nucleation is the favored reaction process, as was shown in vitro by measuring the levels of Aβ oligomers under different experimental conditions 25 (Figure 2). Adding fibrils to monomers increased the production of Aβ oligomers by ≈ 20‐fold, convincingly demonstrating that the fibrils catalyzed a secondary nucleation reaction that is far more efficient than primary nucleation.
FIGURE 2.
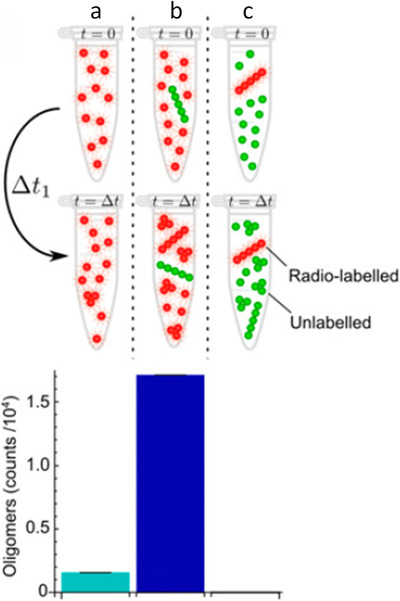
Fibril‐catalyzed secondary nucleation is the favored reaction process. Adapted, with permission, from Cohen et al., “Proliferation of amyloid‐β42 aggregates occurs through a secondary nucleation mechanism” PNAS, 2013. Radio‐labeled monomers (reaction a), radiolabelled monomers plus unlabeled fibrils (reaction b), or unlabeled monomers plus radio‐labeled fibrils (reaction c) were incubated for a period of time, Δt. The resultant oligomers in the reaction mixture were isolated by size‐exclusion chromatography, and the amount of radioactivity in the oligomer fractions was measured. The quantity of oligomers generated by secondary nucleation (reaction b) greatly exceeds that generated by primary nucleation (reaction a) or by fragmentation (reaction c)
2.3. Antibodies binding type 1 and type 2 oligomers
Aβ oligomers in the brain are hard to purify for biophysical studies. Therefore, their quaternary structures are inferred, using conformation‐specific antibodies. Developed by Kayed and Glabe, the conformation‐specific antibody prototypes are polyclonal antisera, OC 26 and A11. 27 OC preferentially binds in‐register parallel β‐sheets. 24 , 26 A11 recognizes a variety of out‐of‐register β‐sheet structures 28 and α‐sheets. 22
Type 1 oligomers bind A11, but not OC. 24 Type 2 oligomers bind OC, but not A11. 24 Type 1 oligomers do not contain in‐register β‐sheets, and probably consist of out‐of‐register α‐ or β‐sheets. In contrast, type 2 oligomers do not contain out‐of‐register sheets, and probably consist of in‐register β‐sheets.
2.4. The biology of Aβ oligomers in the brain
The biological effects of Aβ oligomers are related directly to their biogenesis in the brain. It is important to distinguish between experimental effects observed for Aβ oligomers studied in situ versus those applied ex situ or ex vivo. The former are situated where they are generated, the latter injected into places where the oligomers are not necessarily found in the intact brain.
Type 1 oligomers, which form by primary nucleation, independently of Aβ fibrils, do not contain structural features of amyloid fibrils (ie, do not contain in‐register β‐sheets), appear before amyloid plaques, and are not concentrated around amyloid plaque cores, which are dense knots of fibrils stained by Thioflavin S and other amyloid dyes. 24 Type 2 oligomers, which form by secondary nucleation, catalyzed by Aβ fibrils, share the basic structural features of amyloid fibrils (ie, in‐register parallel β‐sheets), only appear after amyloid plaques, and are found only in the immediate vicinity of amyloid plaque cores 24 (Figure 3). To be fully relevant to AD, these studies, conducted in mice, must be confirmed in humans.
FIGURE 3.
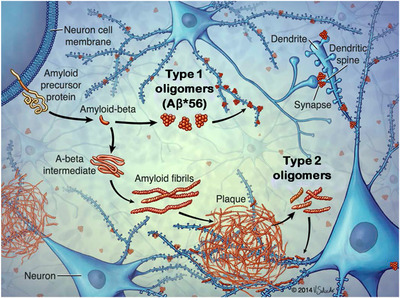
Biogenesis of types 1 and 2 Aβ oligomers in the brain. Adapted, with permission, from Liu et al., “Quaternary structure defines a large class of Aβ oligomers neutralized by sequestration” Cell Reports, 2015. Type 1 oligomers form by primary nucleation, do not have basic the structural feature of amyloid fibrils (ie, do not contain in‐register β‐sheets), are not concentrated around amyloid plaque cores, and appear before amyloid plaques. 24 Aβ*56 10 is a type 1 oligomer. 24 Type 2 oligomers form by secondary nucleation, contain in‐register β‐sheet structure, are concentrated around amyloid plaque cores, and appear after amyloid plaques. 24 Both types of oligomers affect neuronal signaling pathways through interactions with dendritic spines
Both type 1 and type 2 oligomers alter neuronal signaling pathways, although probably through different sets of molecules, and lead to varying degrees of synaptic dysfunction, synaptic loss, and neuron death. 29 , 30 , 31 , 32 , 33 Although both types of oligomers are toxic, what matters most for how they affect brain function is the way they are distributed in the brain. 24 Type 1 oligomers cause neurological dysfunction, even in tiny amounts, because they are dispersed. 24 Type 2 oligomers may go unnoticed, even in relatively large amounts, because the oligomers remain sequestered. 24
3. DO TYPE 2 Aβ OLIGOMERS AFFECT NEUROLOGICAL FUNCTION IN PERSONS WITH ALZHEIMER'S DISEASE?
Type 2 oligomers do not cause large‐scale neurological dysfunction. They might interfere with neural activity in some patients, in particular, those with high plaque density, perhaps in the default‐mode network nodes. However, any potentially beneficial effects of removing them are probably due to a temporary offset in the neurological deterioration that is due to a separate, distinct pathogenic process.
Knowing how type 2 oligomers cause toxicity in mice may help interpret clinical trial results in humans. In mice, type 2 oligomers reside in a “toxic halo” extending ≈50 μm from the outer surface of amyloid plaque cores, 24 , 34 within which neurites are dystrophic 35 and dysfunctional. 36 Type 2 oligomers remain sequestered within these toxic spheres and cause neurological dysfunction depending on the amount of brain the spheres occupy. Adjacent toxic spheres coalesce when the plaque core load, obtained using Thioflavin S to estimate the volume of brain tissue containing dense cores, is ≈ 0.5% to 4% (Figure 4).
FIGURE 4.
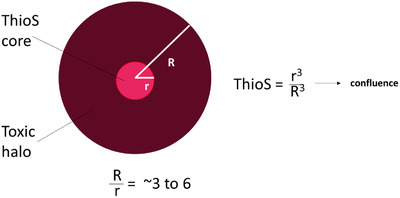
Type 2 oligomers reach confluence when the Thioflavin S plaque load is ≈ 0.5% to 4%. The Thioflavin S (ThioS) core (red) contains densely packed amyloid fibrils. The toxic halo (brown) contains Type 2 oligomers and extends ≈ 50 microns from the edge of the ThioS core. 34 The radius of ThioS cores (r) ranges from ≈ 10 to 25 microns. The ratio of R/r ranges from ≈ 3 to 6. When the ThioS plaque load equals r3/R3, the toxic halos in the brain become confluent. This occurs when the ThioS plaque load is 1/63 to 1/33 = ≈ 0.5 to 4%. In humans with Alzheimer's disease, the ThioS plaque load ranges from ≈ 0.1% to 0.4% 37
Neurological dysfunction in some, but not all, mouse models of Aβ amyloidosis is caused by type 2 oligomers. In mouse models that develop amyloid plaques, the Thioflavin S plaque load increases with age, and ranges from < 0.2% to 6% in 10‐ to 13‐month‐old mice. 37 In 5xFAD mice, the Thioflavin S plaque load reaches ≈ 4% by 6 to 9 months (at the upper limit of the range of Thioflavin S load, 0.5% to 4%, when type 2 oligomers coalesce), corresponding to the onset of memory dysfunction in 5xFAD mice, 38 suggesting that their deficits result from the coalescence of type 2 oligomers. In APPPS1ΔE9 mice, memory deficits appear at 6 to 9 months, 39 when the Thioflavin S plaque load is ≈ 1%. Based on the estimated radius of plaques in these mice, ≈ 14 microns, 37 the type 2 oligomers are probably confluent at this age, suggesting that type 2 oligomers are the major cause of deficits in APPPS1ΔE9, also. Type 2 oligomers do not cause memory deficits in all mouse models, however. For example, 7‐ to 11‐month‐old Tg2576 mice lack neuritic plaques but are nonetheless impaired, 40 indicating other causes of neurological dysfunction at work.
In humans, as in mice, a toxic halo defined by the presence of dystrophic neurites extends from the outer surface of Thioflavin S plaque cores. The distance between the edge of the core and the perimeter of the halo in humans is probably similar to that in mice, based on qualitative comparisons between mouse and human neuritic plaques. The distribution of type 2 oligomers around plaque cores in humans has not been measured as accurately as in mice, which was achieved by biochemical analyses of microdissected cores, halos, and surrounding tissue. 24 If the yet‐to‐be‐defined mechanisms that keep the oligomers sequestered within neuritic plaques in mice are defective in AD, then it is possible that type 2 oligomers spill out beyond the 50‐micron perimeter, and may impair the function, if not the structure, of cells beyond the neuritic plaque itself. Barring this possibility, the overall Thioflavin S plaque load in AD, which ranges from ≈ 0.1% to 0.4%, 37 is not sufficiently high for toxic spheres of type 2 oligomers to coalesce except, perhaps, in brain regions where the density of plaque cores is exceptionally high, such as in the default‐mode network nodes. 41 Whether the Thioflavin S plaque load in some patients with AD reaches the threshold for coalescence, ≈ 0.5% to 4% assuming human and mouse plaques are similar, is currently unknown.
The failure of Aβ antibodies in clinical trials, even when they effectively erase amyloid plaques, may be due to the limited number of neurons adversely affected by type 2 oligomers in AD. The monoclonal antibody aducanumab does not bind monomers and probably does not bind type 1 oligomers, but binds type 2 oligomers and fibrils. 42 The antibody BAN2401 exhibits similar binding properties. 43 Both antibodies can completely clear plaques in patients. Two large‐scale clinical trials of aducanumab were halted for lack of efficacy, 44 a result that is consistent with the neurological silence of type 2 oligomers in mice with plaque loads comparable to those in AD.
Negative results notwithstanding, clinical decline was slower in some persons with mild cognitive impairment or very mild AD treated with aducanumab or BAN2401, first observed in small‐scale trials, and subsequently found in a large‐scale trial, and may potentially be related to erasing type 2 oligomers from brain regions laden with dense clusters of neuritic plaques. In a small clinical trial of aducanumab, the group of subjects receiving 10 mg/kg showed significantly less decline on the Mini Mental Status Examination (MMSE) cognitive test from weeks 52 to 132, but statistical significance was lost at week 164. 45 Similarly, subjects receiving 10 mg/kg BAN2401 also showed slower rates of decline. 46 In two large clinical trials, subjects receiving 10 mg/kg aducanumab showed significant slowing in one trial, but a slight hastening in the other. 47
In further analyses, it was possible to define, in both large trials, a subset of subjects receiving 10 mg/kg aducanumab that declined more slowly than a matched subset of subjects receiving placebo. 47 The effect sizes in these trials are small and highly sensitive to parsing, raising the possibility that a true signal, if present, is derived from particular subjects. A testable hypothesis is that the positive signals may be an indication that plaque removal, possibly from default network nodes, temporarily offsets the neurological deterioration that is due to a separate, distinct pathogenic process. A direct correlation between the amyloid load at the onset of treatment and a beneficial cognitive response would support the hypothesis that excessive amounts of amyloid are harmful. Even a small and short‐lived effect would be an important discovery from a scientific perspective, and could potentially help identify patients more likely to benefit from plaque‐clearing therapies.
4. DO TYPE 1 Aβ OLIGOMERS AFFECT NEUROLOGICAL FUNCTION IN PERSONS WITH ALZHEIMER'S DISEASE?
Type 1 oligomers impair neurological function in mice, but whether they cause neurological dysfunction in humans is unknown.
Memory dysfunction is associated with type 1 oligomers, specifically in mouse models of Aβ amyloidosis in which the Thioflavin S load does not exceed 1% to 2%. 24 , 33 , 37 Neurological dysfunction in mice with type 1 oligomers is reversed and memory function is fully restored by blocking Aβ with antibodies. 48 This dysfunction, which does not kill neurons or cause substantial synaptic loss, may result from specific changes in neuronal signaling triggered when type 1 oligomers, such as Aβ*56, bind N‐methyl‐d‐aspartate (NMDA) receptors. 32 The reversal of memory loss has not yet been observed in patients with mild cognitive impairment or mild AD treated with anti‐Aβ with antibodies, indicating that neurological dysfunction caused by type 1 oligomers in mouse models is not the same as dementia in humans. Sporadic late‐onset AD is preceded by decades of elevated Aβ, implying a much more insidious and slowly progressing assault on synaptic function and neuronal integrity than what occurs in the mice.
This difference notwithstanding, some recent findings raise the possibility that type 1 oligomers may indirectly trigger dementia, through their interaction with tau proteins. Aβ oligomers binding A11 induce tau to form toxic oligomers, but Aβ fibrils do not have this effect. 49 Because type 1 oligomers also bind A11, it is tempting to speculate that they amplify their adverse effects by transforming tau into neurotoxic species. Such a process could explain why clinical deterioration correlates better with tau pathology than amyloid plaques. Type 1 oligomers may set the stage for tau to inflict neurotoxic damage leading to dementia.
The relevance of type 1 oligomers ultimately depends on resolving some crucial questions, the most important of which is how well the temporal and spatial patterns of type 1 oligomers correspond to the neurological and cognitive statuses of persons in the preclinical and clinical stages of AD. The involvement of type 1 oligomers critically depends on showing that the temporal profile of the appearance of neurological abnormalities and type 1 oligomers is linked in time much more closely than the Aβ detected by amyloid positron emission tomography (PET). A specific type 1 oligomer, the putative dodecamer, Aβ*56, has been measured in denaturing gels. In the inferior temporal gyrus in human autopsy specimens, Aβ*56 is highest in elderly, cognitively intact persons, and lowest in persons with AD. 50 This observation argues against a direct causal relationship between type 1 oligomers and brain dysfunction, but supports the idea that they cause tau to become pathogenic prior to the appearance of clinical symptoms. An additional consideration is the possibility that there are species of type 1 oligomers that denature easily, and therefore disintegrate using conventional, denaturing sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis. Type 1 oligomers in humans need to be characterized more definitively, using alternative, complementary techniques.
5. SINCE BACE1 INHIBITORS AND SOLANEZUMAB SHOULD DISPROPORTIONATELY LOWER TYPE 1 OLIGOMERS, DOESN'T THE FAILURE OF THESE TREATMENTS ARGUE AGAINST TYPE 1 OLIGOMERS DRIVING NEUROLOGICAL DETERIORATION?
This is a good point. In the case of BACE1 inhibitors, there is an explanation unrelated to Aβ. One of the physiological roles of BACE1‐mediated cleavage of APP may be to facilitate memory and synaptic plasticity. Therefore, inhibiting BACE1 may impair neurological function. Regarding solanezumab, it may be an issue of dosage or timing.
Because secondary nucleation is the favored reaction process, lowering the production of Aβ monomers, a common pool that feeds both reactions, should disproportionately reduce type 1 Aβ oligomers. This prediction is supported by experimental data, involving suppressing the expression of APP in transgenic mice producing both types of oligomers. 24 Antibodies binding monomers and small molecules inhibiting BACE1 reduce the pool of Aβ monomers, and would therefore be expected to lower type 1 oligomer levels.
Solanezumab is one such antibody. The prediction is that it reduces type 1 oligomers, but actual data supporting this conclusion have not been reported. Although solanezumab did not slow cognitive decline sufficiently to warrant continuing its development as a viable therapy for persons with AD symptoms, its ability to prevent the conversion from asymptomatic to symptomatic disease were held promise. 51 Unfortunately, hopes were recently dashed when solanezumab did not appear to benefit subjects with dominantly inherited AD (https://www.alzforum.org/news/research‐news/topline‐result‐first‐dian‐tu‐clinical‐trial‐negative‐primary). It is unclear whether the antibody lowered the monomer pool enough to reduce type 1 oligomers. If solanezumab indeed prevents or delays the conversion to AD in future studies, then it is likely that reducing type 1 Aβ oligomers is involved in the underlying mechanism.
Data from mice and humans support the possibility that cleavage of APP by BACE1 may underlie normal memory function. In mice, the loss of even one BACE1 allele attenuated APP‐induced enhanced spatial memory function and synaptic plasticity, and was associated with a reduction in the levels of an APP intracellular domain (AICD), 52 which preserves memory processes in a mouse model of Aβ amyloidosis. 53 BACE1 inhibitors worsen cognition in patients with AD 4 , 5 , 54 ; that 10 distinct inhibitors caused patients to decline argues against off‐target effects causing their deterioration. Lowering Aβ by inhibiting BACE1 may not only lower type 1 oligomers, but may also reduce the memory‐preserving effects of AICD.
6. CONCLUSION
The Aβ hypothesis of AD is not dead. Removing type 2 oligomers may ameliorate neurological decline, but in whom and for how long we do not know. Targeting type 1 oligomers remains a viable treatment strategy, but requires additional investigation. In practice, it is easier to study type 2 than type 1 oligomers, because type 2 oligomers are more abundant and easier to measure. The polyclonal and monoclonal antibodies binding type 2 oligomers are reliable. Unfortunately, the currently available polyclonal antibodies binding type 1 oligomers are not entirely reliable, and there are no specific monoclonal antibodies. Monoclonal antibodies to type 1 oligomers can probably be successfully developed, however, since various A11 antisera contain specific antibodies that presumably arise from single clones. Identifying and harvesting such clones is a feasible strategy for generating these valuable antibodies. It would be worthwhile to develop monoclonal antibodies, as well as other capture reagents, targeting type 1 oligomers.
Future research will yield exciting discoveries. The development of reagents that selectively bind type 1 oligomers will permit testing of the effects of neutralizing type 1 oligomers, and enable the purification of type 1 oligomers to decipher their precise biophysical structures. These studies will lay the foundation for testing the ability of compounds that target type 1 oligomers to halt or reverse the progression of AD. Finally, it is likely that other types of Aβ oligomers await discovery, followed by the elucidation of their biogenesis and biology in the brain.
CONFLICTS OF INTEREST
No conflicts of interest
ACKNOWLEDGMENTS
I thank David Knopman and Rob Tycko for provocative discussions and insightful comments about this manuscript, and Peng Liu for his vital contributions to the delineation and characterization of type 1 and type 2 oligomers in the brain. Funding: NIH R01‐NS/AG33249, B. Grossman, T. and P. Grossman Family Foundation.
Ashe KH. The biogenesis and biology of amyloid β oligomers in the brain. Alzheimer's Dement. 2020;16:1561–1567. 10.1002/alz.12084
REFERENCES
- 1. Vassar R, Bennett BD, Babu‐Khan S, et al. Beta‐secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999; 286(5440): 735‐741. [DOI] [PubMed] [Google Scholar]
- 2. De Strooper B, Saftig P, Craessaerts K, et al. Deficiency of presenilin‐1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998; 391(6665): 387‐390. [DOI] [PubMed] [Google Scholar]
- 3. Takasugi N, Tomita T, Hayashi I, et al. The role of presenilin cofactors in the gamma‐secretase complex. Nature. 2003; 422(6930): 438‐441. [DOI] [PubMed] [Google Scholar]
- 4. Egan MF, Kost J, Voss T, et al. Randomized trial of verubecestat for prodromal Alzheimer's disease. N Engl J Med. 2019; 380(15): 1408‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henley D, Raghavan N, Sperling R, Aisen P, Raman R, Romano G. Preliminary results of a trial of atabecestat in preclinical Alzheimer's disease. N Engl J Med. 2019; 380(15): 1483‐1485. [DOI] [PubMed] [Google Scholar]
- 6. Knopman DS. Lowering of amyloid‐beta by beta‐secretase inhibitors—Some informative failures. N Engl J Med. 2019; 380(15): 1476‐1478. [DOI] [PubMed] [Google Scholar]
- 7. Gauthier S, Alam J, Fillit H, et al. Combination therapy for Alzheimer's disease: perspectives of the EU/US CTAD Task Force. J Prev Alzheimers Dis. 2019; 6(3): 164‐168. [DOI] [PubMed] [Google Scholar]
- 8. Roher AE, Chaney MO, Kuo YM, et al. Morphology and toxicity of Abeta‐(1‐42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J Biol Chem. 1996; 271(34): 20631‐20635. [DOI] [PubMed] [Google Scholar]
- 9. Lambert MP, Barlow AK, Chromy BA, et al. Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998; 95(11): 6448‐6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lesne S, Koh MT, Kotilinek L, et al. A specific amyloid‐beta protein assembly in the brain impairs memory. Nature. 2006; 440(7082): 352‐357. [DOI] [PubMed] [Google Scholar]
- 11. Kim J, Chakrabarty P, Hanna A, et al. Normal cognition in transgenic BRI2‐Abeta mice. Mol Neurodegener. 2013; 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pauling L, Corey RB. The structure of feather rachis keratin. Proc Natl Acad Sci U S A. 1951; 37(5): 256‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Astbury WT, Dickinson S, Bailey K. The X‐ray interpretation of denaturation and the structure of the seed globulins. Biochem J. 1935; 29(10): 2351‐2360.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Glenner GG, WongCW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984; 120(3): 885‐890. [DOI] [PubMed] [Google Scholar]
- 15. Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012; 148(6): 1188‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Multiple quantum solid‐state NMR indicates a parallel, not antiparallel, organization of beta‐sheets in Alzheimer's beta‐amyloid fibrils. Proc Natl Acad Sci U S A. 2000; 97(24): 13045‐13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Petkova AT, Ishii Y, Balbach JJ, et al. A structural model for Alzheimer's beta ‐amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002; 99(26): 16742‐16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiang W, Yau WM, Luo Y, Mattson MP, Tycko R. Antiparallel beta‐sheet architecture in Iowa‐mutant beta‐amyloid fibrils. Proc Natl Acad Sci U S A. 2012; 109(12): 4443‐4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serra‐Batiste M, Ninot‐Pedrosa M, Bayoumi M, Gairi M, Maglia G, Carulla N. Abeta42 assembles into specific beta‐barrel pore‐forming oligomers in membrane‐mimicking environments. Proc Natl Acad Sci U S A. 2016; 113(39): 10866‐10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Do TD, LaPointe NE, Nelson R, et al. Amyloid beta‐Protein C‐Terminal Fragments: formation of Cylindrins and beta‐Barrels. J Am Chem Soc. 2016; 138(2): 549‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kreutzer AG, Hamza IL, Spencer RK, Nowick JS. X‐ray crystallographic structures of a trimer, dodecamer, and annular pore formed by an Abeta17‐36 beta‐hairpin. J Am Chem Soc. 2016; 138(13): 4634‐4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shea D, Hsu CC, Bi TM, et al. Alpha‐sheet secondary structure in amyloid beta‐peptide drives aggregation and toxicity in Alzheimer's disease. Proc Natl Acad Sci U S A. 2019; 116(18): 8895‐8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pauling L, Corey RB. The pleated sheet, a new layer configuration of polypeptide chains. Proc Natl Acad Sci U S A. 1951; 37(5): 251‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu P, Reed MN, Kotilinek LA, et al. Quaternary structure defines a large class of amyloid‐beta oligomers neutralized by sequestration. Cell Rep. 2015; 11(11): 1760‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cohen SI, Linse S, Luheshi LM, et al. Proliferation of amyloid‐beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci U S A. 2013; 110(24): 9758‐9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kayed R, Head E, Sarsoza F, et al. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007; 2: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003; 300(5618): 486‐489. [DOI] [PubMed] [Google Scholar]
- 28. Liu C, Zhao M, Jiang L, et al. Out‐of‐register beta‐sheets suggest a pathway to toxic amyloid aggregates. Proc Natl Acad Sci U S A. 2012; 109(51): 20913‐20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature. 2009; 457(7233): 1128‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cisse M, Sanchez PE, Kim DH, Ho K, Yu GQ, Mucke L. Ablation of cellular prion protein does not ameliorate abnormal neural network activity or cognitive dysfunction in the J20 line of human amyloid precursor protein transgenic mice. J Neurosci. 2011; 31(29): 10427‐10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Larson M, Sherman MA, Amar F, et al. The complex PrP(c)‐Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci. 2012; 32(47): 16857‐16871a. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 32. Amar F, Sherman MA, Rush T, et al. The amyloid‐beta oligomer Abeta*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation. Sci Signal. 2017; 10(478). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chiang ACA, Fowler SW, Reddy R, et al. Discrete pools of oligomeric amyloid‐beta track with spatial learning deficits in a mouse model of Alzheimer amyloidosis. Am J Pathol. 2018; 188(3): 739‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. D'Amore JD, Kajdasz ST, McLellan ME, Bacskai BJ, Stern EA, Hyman BT. In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine‐S‐associated alterations in neurite trajectories. J Neuropathol Exp Neurol. 2003; 62(2): 137‐145. [DOI] [PubMed] [Google Scholar]
- 35. Knowles RB, Wyart C, Buldyrev SV, et al. Plaque‐induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci U S A. 1999; 96(9): 5274‐5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stern EA, Bacskai BJ, Hickey GA, Attenello FJ, Lombardo JA, Hyman BT. Cortical synaptic integration in vivo is disrupted by amyloid‐beta plaques. J Neurosci. 2004; 24(19): 4535‐4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu P, Reichl JH, Rao ER, et al. Quantitative comparison of dense‐core amyloid plaque accumulation in amyloid‐beta protein precursor transgenic mice. J Alzheimers Dis. 2017; 56(2): 743‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kimura R, Ohno M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis. 2009; 33(2): 229‐235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jankowsky JL, Melnikova T, Fadale DJ, et al. Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer's disease. J Neurosci. 2005; 25(21): 5217‐5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsiao K, Chapman P, Nilsen S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996; 274(5284): 99‐102. [DOI] [PubMed] [Google Scholar]
- 41. Buckner RL, Snyder AZ, Shannon BJ, et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005; 25(34): 7709‐7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016; 537(7618): 50‐56. [DOI] [PubMed] [Google Scholar]
- 43. Tucker S, Moller C, Tegerstedt K, et al. The murine version of BAN2401 (mAb158) selectively reduces amyloid‐beta protofibrils in brain and cerebrospinal fluid of tg‐ArcSwe mice. J Alzheimers Dis. 2015; 43(2): 575‐588. [DOI] [PubMed] [Google Scholar]
- 44. Available at : https://www.alzforum.org/therapeutics/aducanumab. Accessed March 30, 2020.
- 45. Available at : https://investors.biogen.com/static-files/90eae2d3-532c-49c3-aa06-14a32b80d16a. Accessed March 30, 2020.
- 46. Available at : https://www.alzforum.org/therapeutics/ban2401. Accessed March 30, 2020.
- 47.Available at: https://investors.biogen.com/static-files/ddd45672-9c7e-4c99-8a06-3b557697c06f. Accessed March 30, 2020.
- 48. Kotilinek LA, Bacskai B, Westerman M, et al. Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci. 2002; 22(15): 6331‐6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lasagna‐Reeves CA, Castillo‐Carranza DL, Guerrero‐Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010; 49(47): 10039‐10041. [DOI] [PubMed] [Google Scholar]
- 50. Lesne SE, Sherman MA, Grant M, et al. Brain amyloid‐beta oligomers in ageing and Alzheimer's disease. Brain. 2013; 136(Pt 5): 1383‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. AlzForum s. Available from: https://www.alzforum.org/therapeutics/solanezumab.
- 52. Ma H, Lesne S, Kotilinek L, et al. Involvement of beta‐site APP cleaving enzyme 1 (BACE1) in amyloid precursor protein‐mediated enhancement of memory and activity‐dependent synaptic plasticity. Proc Natl Acad Sci U S A. 2007; 104(19): 8167‐8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Deyts C, Clutter M, Pierce N, et al. APP‐Mediated Signaling Prevents Memory Decline in Alzheimer's Disease Mouse Model. Cell Rep. 2019; 27(5): 1345‐1355.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Available from: https://www.alzforum.org/therapeutics/search?fda_statuses=&target_types=&therapy_types%5B%5D=166&conditions=&keywords-entry=&keywords=BACE%2CBACE+inhibitor. Accessed March 30, 2020.