

Abstract
The ability of metal ionophores to induce cellular metal hyperaccumulation endows them with potent antimicrobial activity; however, the targets and mechanisms behind these outcomes are not well understood. This work describes the first utilization of proteome-wide measurements of protein folding stability in combination with protein expression level analysis to identify protein targets of copper, thereby providing new insight into ionophore-induced copper toxicity in E. coli. The protein folding stability analysis employed a one-pot protocol for pulse proteolysis (PP) in combination with a semi-tryptic peptide enrichment strategy for proteolysis procedures (STEPP) to generate stability profiles for proteins in cell lysates derived from E. coli exposed to copper with and without two ionophores, the antimicrobial agent pyrithione and its β-lactamase-activated prodrug, PcephPT. As part of this work, the above cell lysates were also subject to protein expression level analysis using conventional quantitative bottom-up proteomic methods. The protein folding stability and expression level profiles generated here enabled the effects of ionophore vs. copper to be distinguished and revealed copper-driven stability changes in proteins involved in processes spanning metabolism, translation, and cell redox homeostasis. The 159 differentially stabilized proteins identified in this analysis was significantly more (~3x) than the 53 proteins identified with differential expression levels. These results illustrate the unique information protein stability measurements can provide to decipher metal-dependent processes in drug mode of action studies.
Graphical Abstract
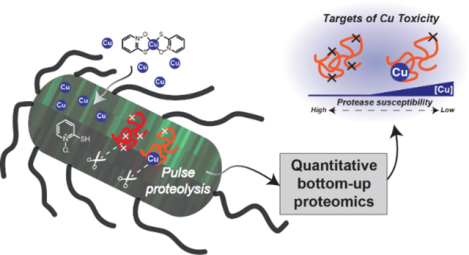
INTRODUCTION
The conformational properties of proteins are closely tied to their function. A protein’s folding stability, and consequently its function, is altered upon interaction with a ligand, be it a cofactor, substrate, inhibitor, another protein, or metal ion. Metal-protein interactions in particular are known both to stabilize and destabilize protein folded states, with stark ramifications for function.1–3 To avoid malfunction due to insufficient, excess, or misplaced metals, cells regulate protein metalation by concerted metal trafficking and homeostasis processes. Among the d-block biometals, Cu is the most stringently regulated, with labile levels kept in check at attomolar levels.4 Such extremely tight regulation alludes to an inherent and unique peril of “free” Cu, with dire consequences for cells that fail to maintain Cu homeostasis.
While cytotoxicity associated with Cu has long been recognized for its antimicrobial benefits,5,6 the mechanisms and targets through which Cu elicits damage are not fully understood.7,8 A widespread presumption has long held that oxidative stress promoted by Cu+/2+ redox cycling is to blame, although evidence to support these claims is often circumstantial.7,9 Cu is toxic even in anaerobic conditions,10 and has been shown to occur without inducing a strong cellular antioxidant response,11 indicating that production of reactive oxygen species cannot be the sole mechanism of its toxicity. The ability of Cu ions to displace metals such as Zn2+ and Fe2+ from metalloenzymes and generally impair protein folding are prevailing mechanisms that have garnered compelling experimental support.4,10–15
The exquisite sensitivity of cells to excess Cu has generated interest in developing compounds to leverage its antimicrobial and cytotoxic activity in a targeted way.8,16 Metal ionophores that form neutral, lipophilic, and kinetically labile metal complexes can hyperaccumulate and redistribute cellular metal ions in ways that distinguish them from conventional chelating agents that sequester metals in high-affinity or kinetically trapped complexes.17 We and others have shown that ionophores provide chemical pathways that bypass cellular Cu homeostasis processes to effect disease-related outcomes.18–22 However, the cellular targets of excess Cu and the small molecule ionophores have yet to be identified, which is an important step in understanding the mechanisms that ultimately lead to cellular malfunction induced by Cu.
The kinetic lability of metal-ionophore complexes allows both the metal and the ionophore to interact with different biological targets via metal-ligand exchange reactions. The released metal could bind proteins in productive or destructive ways, while the released ionophore could bind and redistribute other metal ions, react with biomolecules, or inhibit metalloenzymes by scavenging metal cofactors or blocking active sites. As protein stability measurements are a general probe potentially sensitive to all of these outcomes, we sought to identify the protein targets of metals and metal-ionophore complexes by measuring protein stability changes on the proteomic scale as a function of Cu delivered pharmacologically by small molecule ionophores.
A suite of mass spectrometry-based proteomics techniques has been developed to identify the protein targets of ligands such as small molecule drugs and enzyme cofactors. These techniques include the stability of proteins from rates of oxidation (SPROX),23–25 thermal proteome profiling,26,27 pulse proteolysis,28–31 chemical denaturation and protein precipitation,32 and limited proteolysis33 techniques. Here, we describe the application of a previously established pulse proteolysis protocol, termed one-pot STEPP-PP (semi-tryptic peptide enrichment strategy for proteolysis procedures with pulse proteolysis),34,35 to identify bacterial protein targets of Cu by exposing E. coli to Cu in the absence and presence of one of two small molecules, the antimicrobial ionophore pyrithione (PT) and a prodrug of pyrithione which is activated by β-lactamase, PcephPT (Scheme 1). We have shown previously that both compounds lead to increased levels of cellular Cu in E. coli.36 The STEPP-PP approach is especially advantageous for this study with Cu as it involves neither a protein precipitation step (as in thermal proteome profiling and chemical denaturation and protein precipitation) or a chemical reagent (as in SPROX). The presence of Cu has been shown to impact protein precipitation in unpredictable ways,11 and the hydrogen peroxide used in SPROX is likely to perturb the oxidation state of Cu.
Scheme 1. PcephPT cleavage releases PT.
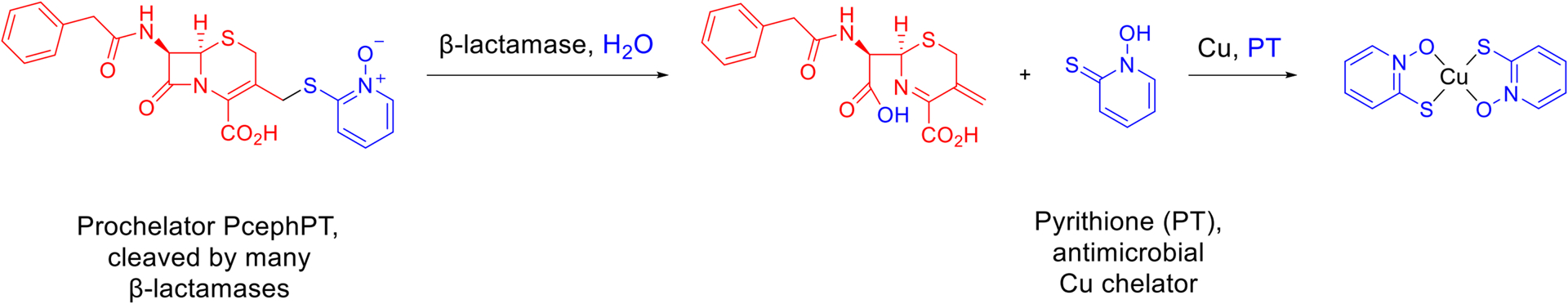
As a prodrug that releases a metal-binding agent, PcephPT consists of a β-lactam core (red) that is recognized by β-lactamase enzymes and a masked leaving group (blue) that prevents metal binding by the prochelator form. When cleaved, PcephPT releases the antimicrobial pyrithione (PT, blue), a bidentate metal-binding compound that forms a lipophilic 2:1 complex with Cu2+.
As part of the current work, we identified protein targets of Cu in E. coli by analyzing changes in both protein folding stability and protein expression levels. While protein expression level studies can provide some information about global cellular adaptations induced in response to a stressor, the functional significance of a protein’s altered expression level can be dubious. Functionally relevant proteins with expression levels that are unchanged by treatment can also go undiscovered in conventional protein expression level studies.37 Indeed, in this study we observed relatively few expression level changes under the treatment conditions investigated, whereas the one-pot STEPP-PP methodology identified over 150 proteins with treatment-induced stability changes. These proteins spanned many biological processes including metal detoxification, iron-sulfur cluster biogenesis, metabolism (glycolysis and the tricarboxylic acid cycle), and translation (the ribosome). Further analysis of the data revealed distinct drivers of these stability changes and separated the consequences of increased Cu levels from those of the small molecule ionophores. Enzyme activity assays on two of the protein hits, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and isocitrate dehydrogenase (IDH), revealed that the ligand-induced stability changes indeed correlated with ligand-induced functional changes in these enzymes. This work describes the first utilization of mass spectrometry-based proteomics methods for protein folding and stability measurements to interrogate metal-protein interactions on the proteomic scale.
RESULTS
E. coli accumulates Cu+ after cotreatment of Cu with PT.
E. coli strain K-12 MG1655 expressing the CTX-M-1 β-lactamase were grown in the absence and presence of various combinations of CuCl2, PT, or PcephPT at concentrations below their minimum inhibitory concentrations (MICs), which were found to be consistent with our previous report (4 mM for CuCl2, 17.5 μM for PT and PcephPT, and 8.8 μM for PT and PcephPT cotreated with 10 μM Cu).36 Cell-associated Cu levels were quantified using inductively coupled plasma mass spectrometry (ICP-MS). Consistent with the results observed in our previous work,36 treatment of 10 μM Cu, 4 μM PT, or 4 μM PcephPT alone did not substantially increase (less than two-fold change) cell-associated Cu compared to untreated cells (Figure 1a). However, cotreatment of E. coli cells with 10 μM Cu and 4 μM PT resulted in a nearly ten-fold increase in cellular Cu compared to treatment with 10 μM Cu alone. Cotreatment of the cells with 10 μM Cu and 4 μM PcephPT resulted in a five-fold increase compared to 10 μM Cu alone. Even though the concentration of Cu in the growth medium was low, cotreatments of the cells with 10 μM Cu and either PT or PcephPT caused the cells to hyperaccumulate Cu to levels that rivaled the ten-fold increase induced by treatment with 2 mM Cu.
Figure 1. PT and PcephPT increase cellular Cu+ levels.
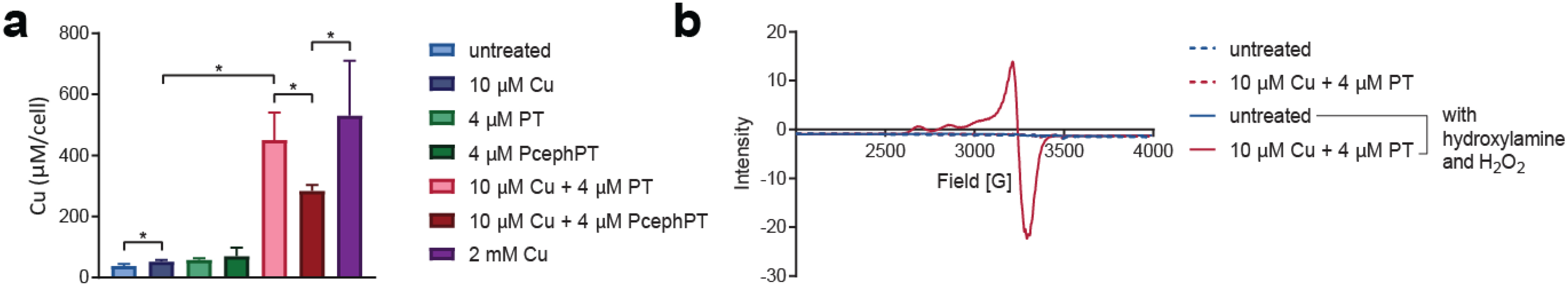
a, ICP-MS data comparing cellular Cu content of E. coli MG1655 CTX-M-1 grown to OD600 of 0.1–0.2 and treated for 15 min. * indicates p < 0.05 using a one-tailed Student’s t test. b, Whole cell EPR spectra of E. coli MG1655 CTX-M-1 grown to OD600 of 0.8–1 and treated for 15 min with Cu and PT as indicated. Treated cell pellets subsequently oxidized with H2O2 (1 M) plus catalase inhibitor hydroxylamine (1 mM) displayed a 4-line signal (g∥ = 2.28, g⊥ = 2.06) attributable to Cu2+. Spectra obtained at 77 K.
Intracellular Cu is generally assumed to be in its reduced Cu+ state. To assess the oxidation state of Cu that hyperaccumulates in cells upon cotreatment of Cu with PT, we measured the electron paramagnetic resonance (EPR) spectra of whole cell E. coli that had been treated with or without Cu and PT. No Cu-associated EPR signal was observed in any of our cell samples initially, indicating that there was either not enough Cu in the sample to detect or that the Cu was in an EPR-silent form. To test these scenarios, a large excess of hydrogen peroxide (1 M) was added before spectra acquisition to one set of samples to force the oxidation of any Cu present and compared with its matched, unoxidized sample. The untreated cells did not show a signal regardless of hydrogen peroxide addition, indicating the low amount of Cu present under normal growth conditions, consistent with our ICP-MS results. However, when hydrogen peroxide was added to the cells treated with Cu and PT, a prominent signal (g∥ = 2.28, g⊥ = 2.06) with a hyperfine splitting pattern attributable to Cu2+ appeared, which was distinctly different from the control spectra of Cu2+ coordinated by PT (Figure 1b and Supplementary Figure 1). These results confirm that PT mediates hyperaccumulation of Cu and, importantly, verifies that Cu delivered to cells in this artificial manner exists in an EPR-silent speciation. We attribute the EPR silence of samples containing sufficient Cu to arise from reduced Cu+ species, although antiferromagnetically-coupled Cu2+ species cannot be ruled out. Regardless, these data indicate that any Cu(PT)2 complexes that may exist extracellularly dissociate inside the cell, leaving the individual Cu and PT components free to interact with intracellular targets.
Stability reveals more treatment effects than expression.
Based on results from the Cu accumulation studies (Figure 1a), five treatment conditions that yielded a range of cellular Cu levels (35–450 μM/cell) were selected for proteomics experiments (no treatment, 10 μM Cu, 4 μM PT, 10 μM Cu + 10 μM PcephPT, and 10 μM Cu + 10 μM PT). Such experiments were designed to reveal changes in protein expression level and folding stability as a function of Cu and PT or PcephPT (Figure 2, Supplemental Figures 2–3). The one-pot STEPP-PP protocol employed here enables the relative stability of the assayed proteins to be assessed across the treatment conditions. In pulse proteolysis, the denaturant dependence of a proteolytic digestion reaction involving a protease with broad specificity (i.e., thermolysin) is evaluated using a sample that spans a range of denaturant concentrations. As proteins unfold, they become more susceptible to cleavage by thermolysin, which retains its activity in high concentration of denaturant. A protein stabilized by treatment requires a higher concentration of denaturant to unfold compared to the concentration of denaturant required to unfold the same protein in untreated cells. Conversely, a destabilized protein would require a lower concentration of denaturant to unfold relative to the same protein in untreated cells. In the one-pot protocol, individual protein unfolding curves are not generated because the samples from all the denaturant concentrations that would be used to construct the protein unfolding curve are pooled. The resulting proteomics measurement instead produces the average signal from all the data points (i.e., the Favg value as defined in Figure 2). When a specific protein’s Favg value is unchanged across treatment conditions, its stability is revealed to be unchanged. However, if the Favg value is significantly different across treatment conditions, it indicates that protein’s stability has been perturbed.
Figure 2. Experimental workflows for protein expression level (left box) and protein stability (right box) analyses.
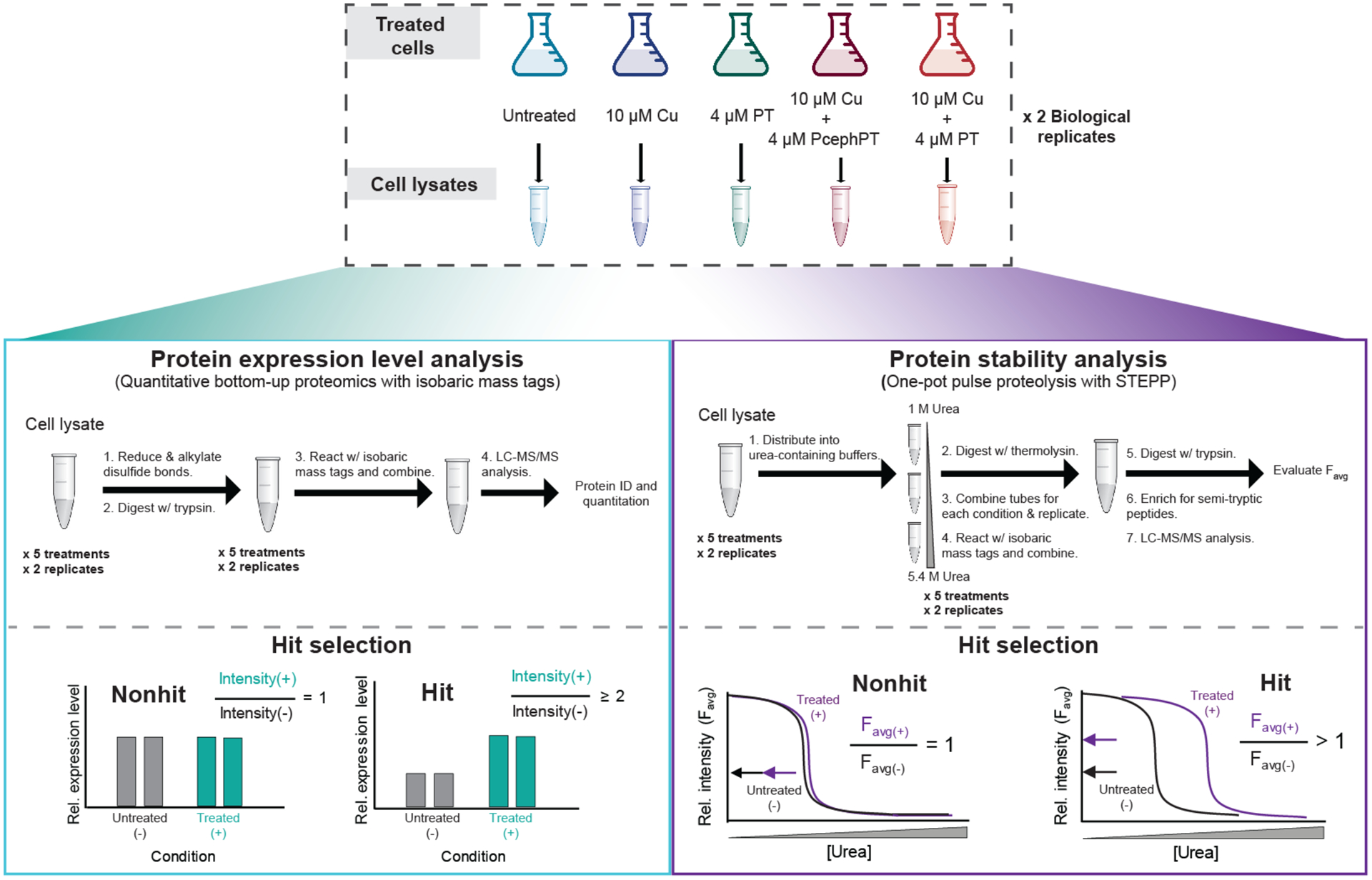
Top, E. coli MG1655 CTX-M-1 were grown to OD600 of 0.1–0.2, treated as indicated for 15 min, and harvested. Clarified lysate from the pellets was utilized in the proteomics workflows. Left side, A total of four biological replicates of the protein expression level analysis were performed using a traditional bottom-up proteomics workflow with an isobaric mass tagging strategy. Right side, A total of four biological replicates of the STEPP-PP technique were performed using a “one-pot” strategy, a technique we have previously reported.35 For each workflow, a total of two biological replicates per treatment condition were analyzed in one LC-MS/MS sample using the isobaric mass tagging strategy employed here. Each workflow was performed twice, resulting in four biological replicates per treatment condition. See Supplementary Figures 2 and 3 for detailed descriptions of the isobaric mass tagging schemes.
The proteomic data search output for each protein expression level sample and protein stability sample from Proteome Discoverer is summarized in Supplementary Datasets 1–4. Summarized in Supplementary Table 1 are the proteomic coverages, which were close to 900 and 600 proteins in the expression level and stability analyses, respectively, and the numbers of protein “hits”, which ranged from 8 to 84 depending on the type of analysis and the experimental condition. Volcano plots of the data from these analyses are shown in Figure 3.
Figure 3.
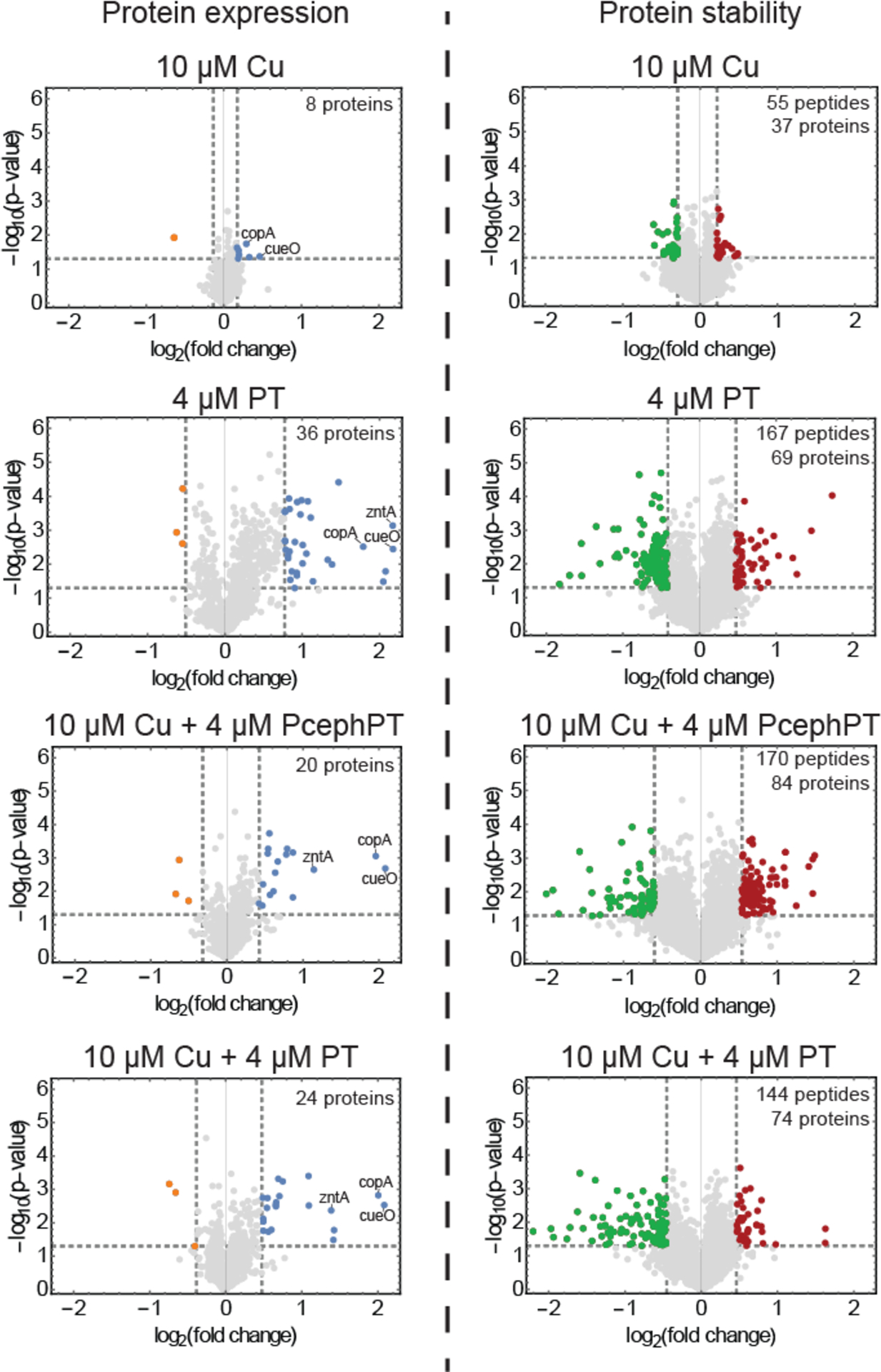
Protein expression level analysis (left) and protein stability analysis (right) of E. coli cells treated under different conditions. Gray lines represent criteria used for selection of differentially expressed and differentially stabilized proteins (|protein or peptide log2(fold change)| ≥ 2 STDEV from average log2(fold change), p ≤ 0.05). Blue and orange dots represent hit proteins that passed the selection criteria for overexpression and underexpression, respectively. Green (stabilized) or red (destabilized) dots represent hit peptides that passed the selection criteria for significantly altered stability changes. Known metal detoxification proteins are labeled in the protein expression volcano plots.
The protein expression level analysis identified a relatively small number of hits across all conditions (<4% of the proteins assayed) (Supplementary Table 2, Supplementary Dataset 5). Notably, most of the protein expression hits involved an increase, not decrease, in protein expression level. Under the Cu-only treatment condition, only eight proteins had significant changes in protein expression (Figure 3, left side, and Supplementary Figure 4). Overexpressed proteins in this condition included metal detoxification and homeostasis proteins (copA, cueO, fiu), and a number of proteins involved in metabolic and biosynthetic processes. The treatment of PT alone resulted in the most protein hits, with 14 of its 36 hits also being identified in the Cu + PT treatment. In addition to three metal detoxification proteins (copA, cueO, and zntA), these shared hits included iron-sulfur cluster biogenesis proteins (iscR, iscS, iscU), heat shock proteins (ibpA, ibpB, dnaK, groS) and luciferase-like monooxygenase yhbw. The 22 hits identified exclusively for PT alone included amino acid biosynthesis proteins (e.g. serA, cysK), lipid A biosynthesis protein lpxC, tricarboxylic acid (TCA) cycle protein fumC, and chaperone protein htpG. The three metal detoxification proteins and iron-sulfur cluster biogenesis proteins found as hits in the PT-containing conditions were also identified as hits for the Cu + PcephPT treatment condition.
The protein stability analysis included a slightly smaller set of assayed proteins (Supplementary Table 1, Supplementary Dataset 6). However, it is remarkable that the protein folding stability analysis generated significantly more (~3x) hit proteins than the protein expression level analysis (a total of 159 unique proteins compared to 53 unique proteins) (Supplementary Table 3). The hit proteins in the protein folding stability analysis spanned many biological processes including but not limited to metal detoxification proteins (copA, cueO, zntA), iron-sulfur cluster proteins or those involved in iron-sulfur cluster biogenesis and its transcriptional activation (e.g. iscR, iscS), metabolic enzymes including enzymes involved in glycolysis and the TCA cycle (e.g. gapA, acnB, icd), enzymes involved in biosynthesis, protein chaperones or proteases (e.g. dnaK, hslV, hslU, groL), and ribosomal proteins or other proteins involved in translation (Supplementary Table 4 and Supplementary Figure 5). Nearly half of the protein expression level hits also had protein folding stability changes. However, it is noteworthy that most (86%) of the protein stability changes detected here were not identified as protein expression level hits, even though all the stability hits were assayed in the expression experiment. This finding illustrates the unique molecular level information protein stability measurements can provide compared to conventional protein expression level analysis in the context of drug mode of action studies.
Cluster analysis reveals drivers of stability changes.
A fuzzy c-means cluster analysis38 was employed to identify common hit behaviors across all 407 peptide stability hits. Using this analysis, four main clusters of hit behaviors were identified including those showing trends in: a) Cu-driven stabilization, b) PcephPT-driven destabilization, c) PT-driven stabilization, and d) PT-driven destabilization (Figure 4, Supplementary Dataset 7). Membership scores for each of these clusters were assigned to all peptide hits, and lower confidence classifications (scores < 0.7) were ignored in subsequent analyses. Peptides that had low membership scores most likely have complex stability changes that cannot be explained by the four main clusters identified. Hits with high confidence classifications in the four clusters above (~60% peptide hits mapping to ~100 proteins) were considered as targets of Cu-, PT-, or PcephPT-related activities.
Figure 4. Hit peptides clustered according to their stability change trends.
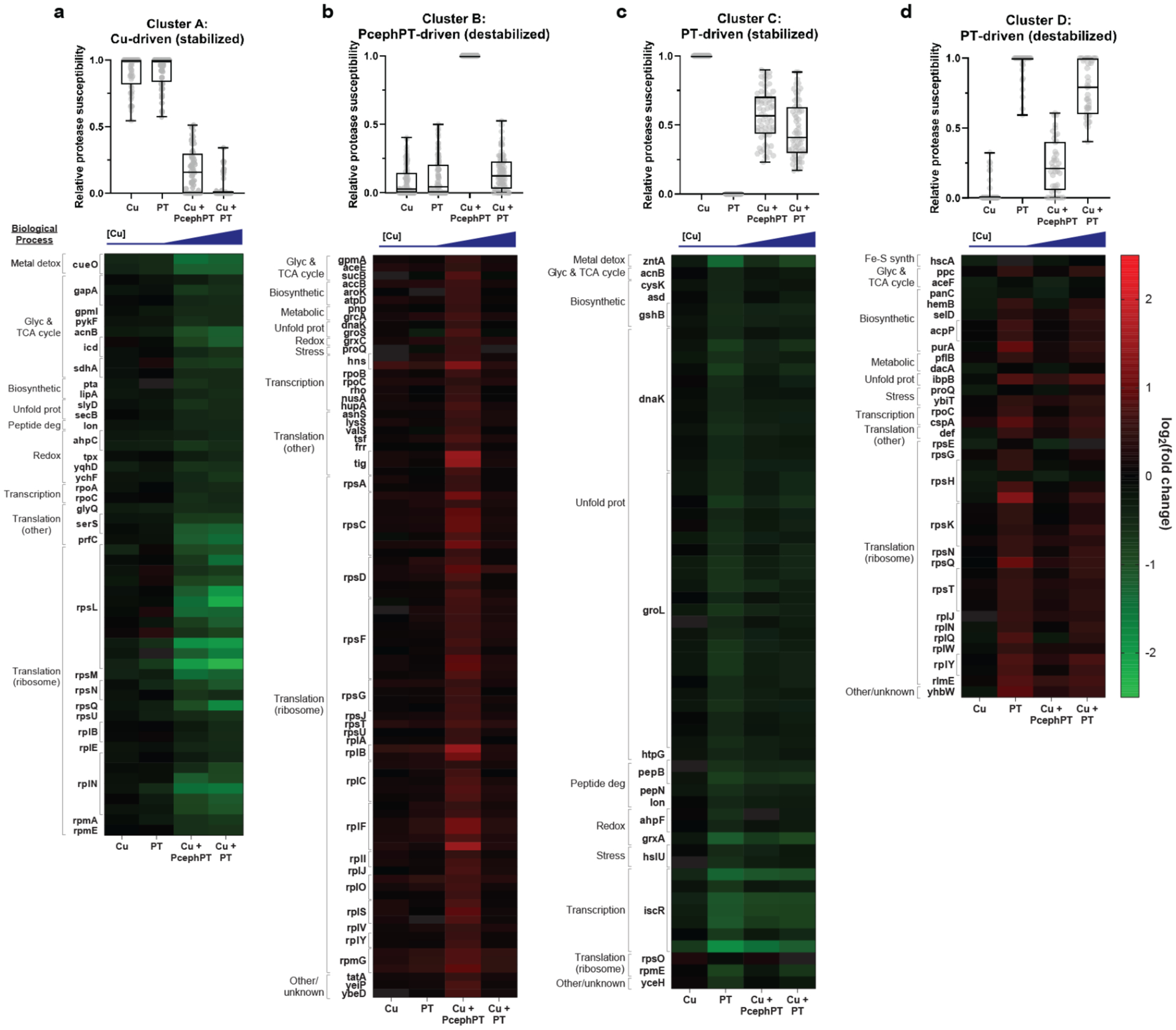
Fuzzy c-means clustering identified four clusters that described the behavior of ~60% of peptide hits mapping to ~100 proteins. For each cluster, a graph indicating the relative protease susceptibility of member peptides is shown above a heatmap displaying the raw log2(fold change) of member peptides. Genes codes corresponding to member proteins are shown to the left of each heatmap. a, Peptides in cluster A exhibit decreased protease susceptibility (indicating protein stabilization) as a consequence of increased cellular Cu. b, Peptides in cluster B exhibit increased protease susceptibility (protein destabilization) in the treatment condition that includes PcephPT. c, Peptides in cluster C exhibit decreased protease susceptibility (protein stabilization) most significantly in the PT alone condition, and partially in conditions containing PT and PcephPT in the presence of Cu. d, Peptides in cluster D exhibit increased protease susceptibility (protein destabilization) most significantly in the PT alone condition, and partially in conditions containing PT and PcephPT in the presence of Cu. Within each heatmap, peptides are grouped according to their biological process GO terms (see also Supplementary Table 5).
The peptide hits in each cluster were sorted into groups based on the biological process GO terms associated with the proteins to which they mapped to better understand what cellular processes were impacted by the different treatment types (Supplementary Table 5, Supplementary Dataset 8). The cluster with the most hits (91 peptides) was composed of peptides exhibiting PcephPT-driven destabilization. A PANTHER bioinformatic analysis39,40 of this cluster revealed that it is significantly enriched (4-fold) in peptides mapping to ribosomal proteins and other proteins involved in translation (e.g tig, tsf, frr). The second largest cluster contains peptides exhibiting PT-driven stabilization. Many peptides in this cluster mapped to chaperone proteins (e.g. dnaK, groL) as well as iron-sulfur cluster transcription regulator iscR and zinc export protein zntA. Another similar cluster included peptides that had PT-driven effects that led to destabilization. Peptides from many ribosomal proteins as well as peptides from proteins involved in metabolic processes (e.g. pflB, aceF, ppc) were found in this category. The final cluster includes peptides exhibiting Cu-driven stabilization, a behavior independent of the small molecule chelators and purely a consequence of the increasing Cu concentrations imparted by the treatment conditions. Peptides in this category include those mapping to copper detoxification protein CueO, to key enzymes in glycolysis and the TCA cycle (e.g. gapA, icd, sdhA), to proteins involved in translation (e.g. ribosomal proteins, serS, prfC), to proteins involved in cell redox homeostasis (e.g. tpx, ahpC, ychF), and to proteins containing iron-sulfur clusters (lipA, acnB). The breadth of this list shows that Cu disrupts a multitude of cellular pathways when it is delivered artificially by metal ionophores such as PT.
Protein stability changes correlate with protein function.
As part of this work we correlated the Cu-induced stability changes observed for two enzymes, glyceraldehyde-3-phosphate dehydrogenase (GAPDH, gene code gapA) and isocitrate dehydrogenase (IDH, gene code icd), with changes in function. These assays were performed in unpurified cell lysates and purified protein samples.
Treating cells with the concentrations of Cu and PT used in the STEPP-PP experiment did not induce measurable changes in GAPDH activity. However, doubling these concentrations to 20 μM Cu + 8 μM PT resulted in 85% inhibition, with more PT resulting in further inhibition (Figure 5a). Treating with 20 μM Cu alone had no effect on GAPDH activity, whereas treatment of cells with 2 mM Cu led to significant inhibition. It is also noteworthy that treating the cells with just PT (up to 20 μM) produced no significant change in GAPDH activity (Supplementary Figure 6a). This differential response of GAPDH activity to the level of supplemental Cu is an indicator that E. coli’s Cu-resistance mechanisms are sufficient to safely handle low micromolar amounts of Cu but can be broached by elevated concentrations in its growth environment. Inclusion of the ionophore clearly lowers that threshold, as shown by the similar inhibition of GAPDH activity by 2 mM Cu vs. 20 μM Cu + 8 μM PT.
Figure 5. GAPDH and IDH activity changes correlate with stability and conformational changes.
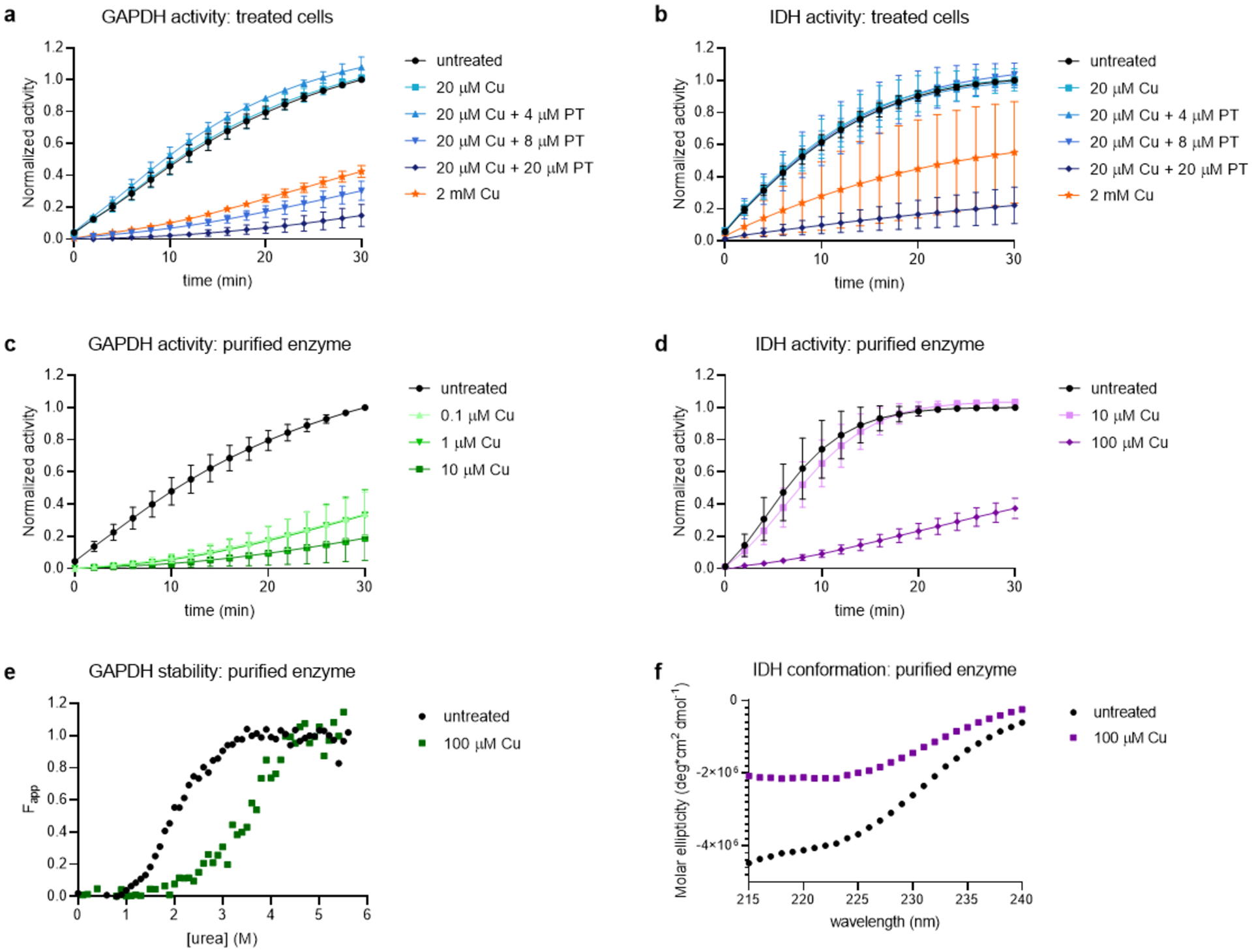
Activity of GAPDH and IDH as monitored by absorbance at 340 nm for the conversion of their cofactors NAD+ and NADP+ to NADH and NADPH, respectively. a, Activity of GAPDH measured in cell lysates of E. coli treated for 15 min as indicated; b, activity of IDH measured in cell lysates of E. coli treated for 15 min as indicated; c, activity of 0.25 μM purified GAPDH exposed to increasing [Cu] as indicated; d, activity of 32 μM purified IDH exposed to increasing [Cu] as indicated; e, CD denaturation curves of 2.8 μM GAPDH showing stabilization in the presence of Cu to urea-induced unfolding. f, CD spectra of 2.2 μM IDH showing significant change in molar ellipticity and decreased alpha helicity (222 nm).
In order to test the hypothesis that the GAPDH inhibition observed in whole cell lysates is the result of direct inhibition of the enzyme by Cu, we measured the catalytic activity of purified GAPDH and indeed found it to be inhibited by Cu (Figure 5c). To gain further insight into the effect of Cu on the stability of this enzyme, we determined the chemical denaturant-induced equilibrium unfolding properties of the protein both in the presence and absence of Cu using circular dichroism (CD) spectroscopy (Figure 5e). The shift of the unfolding curve to higher urea concentrations indicates that the presence of Cu stabilizes this protein against denaturation, a result that is consistent with the Cu-induced stabilization detected in the STEPP-PP experiment.
IDH activity in treated cells was evaluated under the same treatment conditions described for GAPDH (Figure 5b). According to the STEPP-PP results, IDH exhibited significant ligand-induced stability changes in all four treatment conditions. In the cell lysate assay, IDH activity was 85% inhibited by cotreatment of 20 μM Cu and 20 μM PT. IDH activity was also ~50% inhibited in the 2 mM Cu condition, although the error associated with the replicate measurements was relatively large (Figure 5b). As with GAPDH, treating the cells with just PT (up to 20 μM) produced no significant change in IDH activity (Supplementary Figure 6b).
A purified IDH sample was also inhibited by Cu (Figure 5d). However, CD spectra collected on IDH in the absence and presence of Cu were not similar. Notably, the molar ellipticity values were significantly reduced at a range of different wavelengths in the Cu-treated sample (Figure 5f). While the differences in spectra precluded the ability to obtain meaningful protein denaturation curves, this result provides evidence that Cu interacts directly with the purified protein and alters its secondary structure.
DISCUSSION
E. coli relies on multiple systems to maintain metal homeostasis, including copper resistance. It is notable that only a small number of protein expression changes occurred in the 10 μM Cu condition (Figure 3), as it indicates that the Cu resistance and detoxification systems do not need to be upregulated to the same degree as in the ionophore-containing conditions to cope with exposure to low micromolar Cu during growth. This result is not surprising considering this low concentration of supplemental Cu has little consequence on total cellular Cu levels compared to untreated controls (Figure 1a). However, it is striking that the presence of the small molecule ionophores, PT and PcephPT, when paired with only 10 μM Cu, caused similar cellular Cu accumulation as treatment with 2 mM Cu. Predictably, the common protein expression hits across treatment conditions that include PT or PcephPT are proteins involved in metal resistance: copA, cueO, and zntA. CopA, a P-type ATPase that transports Cu(I) from the cytoplasm into the periplasm, and CueO, a periplasmic copper oxidase, compose the primary aerobic system for copper detoxification in E. coli.41 Similarly, ZntA is a P-type ATPase that transports Zn2+.42 These results, combined with the small changes in expression observed for the Cu-only treatment, indicate that these small molecules induce E. coli to significantly upregulate metal resistance proteins in response to otherwise inconsequential levels of Cu, an outcome that is consistent with the recognized ionophore activity of PT. The observation that PcephPT treatment caused significant upregulation of the same metal detoxification proteins is a strong indicator that these treated cells experience metal-induced stress as a primary mode of action when PT is released in situ upon cleavage of its prochelator version PcephPT. One key observation from the expression level analysis is that cells do not need to hyperaccumulate Cu to sense Cu-induced stress. This is clearly illustrated in the PT-alone treatment condition in which CopA, CueO, and ZntA are significantly overexpressed, despite the absence of Cu hyperaccumulation as measured by ICP-MS (Figure 1a). One hypothesis for this behavior is that cells experience less stress when Cu hyperaccumulation is managed by the Cu homeostasis system; however, when this system is bypassed by metal ionophores that artificially deliver Cu, the cells experience uncontrollable levels of metal-induced stress. Future studies will be aimed at comparing the targets of artificially-delivered Cu vs. Cu overload from high Cu content in the media to better understand why cells are experiencing Cu stress, even if the measurable levels are not elevated.
The identification of proteins involved in iron-sulfur cluster biogenesis as protein expression level hits for the PT-containing conditions is in line with previous work showing that iron-sulfur clusters are targets of excess Cu and that the ionophore activity of PT may help mediate this effect.43–46 In fungi, PT has been shown to act as a Cu ionophore to import Cu into the cell, leading to Cu toxicity and damage to iron-sulfur clusters in metabolic enzymes.43 In addition, PT was observed to impact the iron-sulfur cluster assembly system, particularly the transfer of the cluster from its scaffold to the target protein, as well as proteins involved in metabolic processes such as the citric acid cycle and amino acid synthesis.43–46 The unique upregulation of several proteins in the PT-alone condition suggests that this treatment may influence other pathways in the cell aside from iron-sulfur cluster maturation and may elicit distinct responses in biological processes such as amino acid and lipid biosynthesis.
Although the protein expression level analysis provided some insight into the E. coli response to pharmacologically induced Cu stress, they identified relatively few hits (<1–4% of the assayed proteins). In contrast, a significantly larger fraction (6–15%) of the assayed proteins in the folding stability analysis were identified as hits. The stability changes observed for the protein hits identified in the pulse proteolysis analysis could in principle result from direct protein-ligand binding interactions, where the ligand in these cases could be Cu, PT, a Cu-PT complex, or PcephPT. Alternatively, a hit could come from a protein that was indirectly stabilized or destabilized by a partner protein that was directly impacted by binding to one of the treatment components. We aimed to better understand the underlying effects driving the peptide hit behaviors by looking for consistent trends. We reasoned that protein stability hits resulting from direct Cu-protein interactions are likely to correlate with the cellular Cu concentration, which our ICP-MS results showed covers a range based on treatment conditions. However, we could not rule out the impact of PT or PcephPT on protein stability, which would be independent of Cu concentration. The deconvolution of the separate effects of Cu and small molecules was facilitated by the cluster analysis that revealed four main clusters of hit behaviors driving stability changes for different groups of proteins.
Our data provide evidence that unregulated Cu may target central carbon metabolism in E. coli, similar to the findings of recent work exploring the response to copper stress in Staphylococcus aureus.47 Of the 31 proteins in the Cu-driven category, six are involved in glycolysis or the TCA cycle. We showed that two of these enzymes, GAPDH and IDH, had catalytic activities directly inhibited by Cu in purified form and by the combination of extracellular Cu and PT in whole cell lysates. GAPDH was also found to be a target of silver toxicity in E. coli.48 A striking number of peptides from ribosomal proteins (28 peptides, ten proteins) and tRNA ligases (two peptides, two proteins) were also found in the Cu-driven category, suggesting the peril of increasing intracellular Cu may target the ribosome and protein biosynthesis.47,49 Predictably, peptides from proteins involved in cell redox homeostasis were shown to have Cu-driven stability changes, suggesting that Cu’s ability to catalyze reactive oxygen species or otherwise incite oxidative stress may contribute to its mode of action, in addition to its other discovered effects on metabolism and translation. By separating the effects of Cu from the effects of its small molecule carriers, this analysis reveals both established and novel mechanisms of action for Cu-induced cell death. Moreover, we now have evidence that these effects can be amplified by the presence of a small molecule ionophore with the ability to functionally overwhelm Cu homeostasis.
Proteins not classified as having stability changes driven by Cu were classified as being driven by either PT or PcephPT. Such stability changes were independent of Cu concentration and suggest unique modes of action mediated by the presence of the small molecule chelator and prochelator. Of the 61 peptides classified in the PT-driven stabilization cluster, 36 (59%) are from proteins involved in the unfolded protein response, although most of these peptides correspond to two main proteins, dnaK and groL, and many other peptides are from protein chaperones or proteases. These results implicate PT-driven activities in having a direct effect on protein quality control. Interestingly, iron-sulfur cluster biogenesis transcriptional regulator iscR is clustered into the PT-driven cluster and not into the Cu-driven cluster, implying that the presence of the small molecule chelator impacts this protein’s stability more than increasing cellular Cu alone. As noted above, past studies have shown that the antifungal effect of PT is due to its disruption of enzymes containing iron-sulfur clusters,43,44 while other studies have shown that excess Cu could be the primary interactor.12,13,50,51 Our results suggest PT alone has a strong effect on inducing the transcriptional regulation of iron-sulfur cluster biogenesis, while Cu and PT each may directly target iron-sulfur cluster containing enzymes.
Despite their different outcomes regarding intracellular Cu levels in β-lactamase-expressing E. coli, PcephPT and PT were shown in our previous work to have similar minimum inhibitory concentrations (MICs) against E. coli that expressed a β-lactamase enzyme capable of cleaving PcephPT.36 Since PcephPT functions by releasing PT, it is reasonable to expect the two molecules to share some aspects of their modes of action, while also having distinct targets due to their different structures. Indeed, the cluster analysis revealed 91 peptides mapping to 49 proteins for which the stability changes were driven uniquely by PcephPT. This set of proteins was enriched 4-fold in ribosomal proteins and proteins involved in translation, which is in line with a thermal proteome profiling study in E. coli that uncovered targets of ampicillin, an antibiotic that shares structural similarity with the β-lactam core of PcephPT.52 Overall, our analysis indicates that small molecules such as PT and PcephPT have both a metal-driven mode of action and modes of action independent of the metals they bind.
One advantage of using protein folding stability measurements to understand the biological activities of small molecules is that protein folding stability is closely connected to protein function. However, it can be challenging to connect a detected protein folding stability change to a specific functional change in complex biological systems due to the limited availability of appropriate functional assays. Fortunately, two of the protein hits identified in this work, GAPDH and IDH, are enzymes with well-established assays47,53 that can be performed on both the purified protein and the protein in unpurified cell lysates obtained from treated E. coli. Our enzyme activity data on these proteins demonstrated that changes in enzyme stability (GAPDH) or enzyme conformation (IDH) led to changes in enzyme function in whole cell E. coli. Low micromolar concentrations of PT and Cu were sufficient to reduce GAPDH and IDH activity by at least 85%. Interestingly, while 2 mM Cu consistently inhibited GAPDH, IDH was not as reliably inhibited in treated cells. Our ICP-MS results show high variation in the amount of cell-associated Cu after treatment with 2 mM Cu (Figure 1a), which could explain the variable effects of this treatment on enzyme inhibition for IDH if the amount of Cu imported is close to its IC50. This result highlights the importance of the small molecule PT as a delivery vehicle that allows even low concentrations of Cu to consistently affect enzyme stability and activity. As showcased in our work with these key metabolic enzymes, it is likely that other proteins exhibiting significant ligand-induced stability changes also experience changes in their functions.
In this work, we show that increasing intracellular Cu can affect a wide range of protein targets. Although protein expression levels alone failed to identify the direct targets of PT and Cu, measuring changes in protein stability revealed proteins involved in multiple biological processes that are directly disrupted by treatment with PT, Cu, and a prodrug of PT. Furthermore, we observed a clear link between changes in protein stability and changes in protein function, thereby elucidating new insights into long-standing questions about the specific targets and functional consequences of cellular toxicity induced by Cu. More generally, these findings point to the utility of protein stability-based methods such as one-pot STEPP-PP as powerful tools for interrogating protein-metal and protein-drug binding interactions on the proteomic scale to gain insights into the complex mechanisms behind a drug’s mode of action.
Supplementary Material
Supplementary Text | Description of Materials and Methods
Supplementary Table 1 | Proteomic coverage from protein expression level and protein stability analyses.
Supplementary Table 2 | Complete list of protein expression level hits.
Supplementary Table 3 | Complete list of protein stability hits.
Supplementary Table 4 | Biological process classification of protein stability hits.
Supplementary Table 5 | Biological process classification of protein stability hits within each cluster.
Supplementary Figure 1 | Electron paramagnetic resonance spectroscopy of Cu titrated with PT in 10% glycerol.
Supplementary Figure 2 | Experimental workflow and isobaric mass tagging scheme for protein expression analysis.
Supplementary Figure 3 | Experimental workflow and isobaric mass tagging scheme for protein stability analysis.
Supplementary Figure 4 | Heat map visualizing magnitude of expression changes for hit proteins grouped by biological process.
Supplementary Figure 5 | Heat maps visualizing magnitude of stability changes for hit peptides grouped by biological process.
Supplementary Figure 6 | GAPDH and IDH activity in cells treated with PT.
Supplementary Dataset 1 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the first TMT10-plex labeled sample generated in the protein expression level analyses (i.e., biological replicates 1 and 2).
Supplementary Dataset 2 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the second TMT10-plex labeled sample generated in the protein expression level analyses (i.e., biological replicates 3 and 4).
Supplementary Dataset 3 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the first TMT10-plex labeled sample generated in the protein stability analyses (i.e., biological replicates 1 and 2).
Supplementary Dataset 4 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the first TMT10-plex labeled sample generated in the protein stability analyses (i.e., biological replicates 3 and 4).
Supplementary Dataset 5 | Hit analysis of overlapping proteins identified in the two TMT10-plex labeled samples generated in the protein expression level analyses (i.e., biological replicates 1–4).
Supplementary Dataset 7 | Fuzzy c-means cluster analysis of peptide stability hits.
Supplementary Dataset 8 | DAVID functional analysis and resulting GO biological process classifications of protein expression level hits and proteins corresponding to peptide stability hits.
Supplementary Dataset 6 | Hit analysis of overlapping proteins identified in the two TMT10-plex samples generated in the protein stability analyses (i.e., biological replicates 1–4).
ACKNOWLEDGMENTS
This project was supported with funds from the National Institutes of Health (R01GM084176 to K.J.F., R01GM134716 to M.C.F.), N.W. and J.M.Z.-B. acknowledge fellowship support from the Duke Pharmacological Sciences Training Program (T32 GM007105). We thank Dr. Abigail Jackson and Elena Puccio at Duke University for synthesizing PcephPT, Dr. Bradford Becken at Duke University for providing the E. coli MG1655 strain expressing the CTX-M-1 β-lactamase, and the Duke Proteomics Facility for collecting the LC-MS/MS data.
Footnotes
SUPPLEMENTARY INFORMATION
The supplementary information accompanying this manuscript includes Supplementary Text, 5 Supplementary Tables, 6 Supplementary Figures, and 8 Supplementary Datasets as summarized below:
DATA DEPOSITION
The mass spectrometry data generated in this work have been uploaded to the ProteomeXchange Consortium via the PRIDE partner repository (PXD021198). The raw MS data are available for confidential review using the account, reviewer79333@ebi.ac.uk, and the password, bU1driKV.
REFERENCES
- 1.Palm-Espling ME, Niemiec MS & Wittung-Stafshede P Role of metal in folding and stability of copper proteins in vitro. Biochim. Biophys. Acta BBA - Mol. Cell Res 1823, 1594–1603 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Chang CJ Searching for harmony in transition-metal signaling. Nat. Chem. Biol 11, 744–747 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Leal SS, Botelho HM & Gomes CM Metal ions as modulators of protein conformation and misfolding in neurodegeneration. Coord. Chem. Rev 256, 2253–2270 (2012). [Google Scholar]
- 4.Osman D et al. Bacterial sensors define intracellular free energies for correct enzyme metalation. Nat. Chem. Biol 15, 241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borkow G & Gabbay J Copper, An Ancient Remedy Returning to Fight Microbial, Fungal and Viral Infections. Curr. Chem. Biol 3, 272–278 (2009). [Google Scholar]
- 6.Grass G, Rensing C & Solioz M Metallic Copper as an Antimicrobial Surface. Appl. Environ. Microbiol 77, 1541–1547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macomber L, Rensing C & Imlay JA Intracellular Copper Does Not Catalyze the Formation of Oxidative DNA Damage in Escherichia coli. J. Bacteriol 189, 1616–1626 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemire JA, Harrison JJ & Turner RJ Antimicrobial activity of metals: mechanisms, molecular targets and applications. Nat. Rev. Microbiol 11, 371–384 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Imlay JA Diagnosing oxidative stress in bacteria: not as easy as you might think. Curr. Opin. Microbiol 24, 124–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan G et al. Anaerobic Copper Toxicity and Iron-Sulfur Cluster Biogenesis in Escherichia coli. Appl. Environ. Microbiol 83, e00867–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saporito-Magriñá CM et al. Copper-induced cell death and the protective role of glutathione: the implication of impaired protein folding rather than oxidative stress. Metallomics (2018) doi: 10.1039/C8MT00182K. [DOI] [PubMed] [Google Scholar]
- 12.Macomber L & Imlay JA The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci 106, 8344–8349 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan G et al. Copper binding in IscA inhibits iron-sulphur cluster assembly in Escherichia coli. Mol. Microbiol 93, 629–644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Djoko KY & McEwan AG Antimicrobial Action of Copper Is Amplified via Inhibition of Heme Biosynthesis. ACS Chem. Biol 8, 2217–2223 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Johnson MDL, Kehl-Fie TE & Rosch JW Copper intoxication inhibits aerobic nucleotide synthesis in Streptococcus pneumoniae. Metallomics 7, 786–794 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Besold AN, Culbertson EM & Culotta VC The Yin and Yang of Copper During Infection. J. Biol. Inorg. Chem 21, 137–144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding W-Q & Lind SE Metal ionophores – An emerging class of anticancer drugs. IUBMB Life 61, 1013–1018 (2009). [DOI] [PubMed] [Google Scholar]
- 18.Oliveri V Biomedical applications of copper ionophores. Coord. Chem. Rev 422, 213474 (2020). [Google Scholar]
- 19.Steinbrueck A et al. Transition metal chelators, pro-chelators, and ionophores as small molecule cancer chemotherapeutic agents. Chem. Soc. Rev (2020) doi: 10.1039/C9CS00373H. [DOI] [PubMed] [Google Scholar]
- 20.Hunsaker EW & Franz KJ Emerging Opportunities To Manipulate Metal Trafficking for Therapeutic Benefit. Inorg. Chem 58, 13528–13545 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helsel ME & Franz KJ Pharmacological activity of metal binding agents that alter copper bioavailability. Dalton Trans. 44, 8760–8770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soma S et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc. Natl. Acad. Sci 115, 8161–8166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeArmond PD, Xu Y, Strickland EC, Daniels KG & Fitzgerald MC Thermodynamic Analysis of Protein–Ligand Interactions in Complex Biological Mixtures using a Shotgun Proteomics Approach. J. Proteome Res 10, 4948–4958 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran DT, Adhikari J & Fitzgerald MC Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC)-Based Strategy for Proteome-Wide Thermodynamic Analysis of Protein-Ligand Binding Interactions. Mol. Cell. Proteomics 13, 1800–1813 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West GM et al. Quantitative proteomics approach for identifying protein–drug interactions in complex mixtures using protein stability measurements. Proc. Natl. Acad. Sci 107, 9078–9082 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Savitski MM et al. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346, 1255784 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Lu K-Y et al. Plasmodium chaperonin TRiC/CCT identified as a target of the antihistamine clemastine using parallel chemoproteomic strategy. Proc. Natl. Acad. Sci 117, 5810–5817 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adhikari J & Fitzgerald MC SILAC-Pulse Proteolysis: A Mass Spectrometry-Based Method for Discovery and Cross-Validation in Proteome-Wide Studies of Ligand Binding. J. Am. Soc. Mass Spectrom 25, 2073–2083 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Chang Y, Schlebach JP, VerHeul RA & Park C Simplified proteomics approach to discover protein–ligand interactions. Protein Sci. Publ. Protein Soc 21, 1280–1287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu P-F, Kihara D & Park C Energetics-based Discovery of Protein–Ligand Interactions on a Proteomic Scale. J. Mol. Biol 408, 147–162 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park C & Marqusee S Pulse proteolysis: A simple method for quantitative determination of protein stability and ligand binding. Nat. Methods 2, 207–212 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Meng H, Ma R & Fitzgerald MC Chemical Denaturation and Protein Precipitation Approach for Discovery and Quantitation of Protein–Drug Interactions. Anal. Chem 90, 9249–9255 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Feng Y et al. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol 32, 1036–1044 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Ma R, Meng H, Wiebelhaus N & Fitzgerald MC Chemo-Selection Strategy for Limited Proteolysis Experiments on the Proteomic Scale. Anal. Chem 90, 14039–14047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cabrera A et al. Comparative Analysis of Mass-Spectrometry-Based Proteomic Methods for Protein Target Discovery Using a One-Pot Approach. J. Am. Soc. Mass Spectrom 31, 217–226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zaengle-Barone JM et al. Copper Influences the Antibacterial Outcomes of a β-Lactamase-Activated Prochelator against Drug-Resistant Bacteria. ACS Infect. Dis 4, 1019–1029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ruprecht B et al. A mass spectrometry-based proteome map of drug action in lung cancer cell lines. Nat. Chem. Biol 1–9 (2020) doi: 10.1038/s41589-020-0572-3. [DOI] [PubMed] [Google Scholar]
- 38.Dunn JC Well-Separated Clusters and Optimal Fuzzy Partitions. J. Cybern 4, 95–104 (1974). [Google Scholar]
- 39.Mi H, Muruganujan A, Casagrande JT & Thomas PD Large-scale gene function analysis with PANTHER Classification System. Nat. Protoc 8, 1551–1566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas PD et al. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Res. 13, 2129–2141 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rensing C & Grass G Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev 27, 197–213 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Rensing C, Mitra B & Rosen BP The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc. Natl. Acad. Sci 94, 14326–14331 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reeder NL et al. Zinc Pyrithione Inhibits Yeast Growth through Copper Influx and Inactivation of Iron-Sulfur Proteins. Antimicrob. Agents Chemother 55, 5753–5760 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park M, Cho Y-J, Lee YW & Jung WH Understanding the Mechanism of Action of the Anti-Dandruff Agent Zinc Pyrithione against Malassezia restricta. Sci. Rep 8, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yasokawa D et al. DNA microarray analysis suggests that zinc pyrithione causes iron starvation to the yeast Saccharomyces cerevisiae. J. Biosci. Bioeng 109, 479–486 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Garcia-Santamarina S, Uzarska MA, Festa RA, Lill R & Thiele DJ Cryptococcus neoformans Iron-Sulfur Protein Biogenesis Machinery Is a Novel Layer of Protection against Cu Stress. mBio 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarrant E et al. Copper stress in Staphylococcus aureus leads to adaptive changes in central carbon metabolism. Metallomics 11, 183–200 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H et al. Antimicrobial silver targets glyceraldehyde-3-phosphate dehydrogenase in glycolysis of E. coli. Chem. Sci 10, 7193–7199 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker J et al. Copper Stress Induces a Global Stress Response in Staphylococcus aureus and Represses sae and agr Expression and Biofilm Formation. Appl. Environ. Microbiol 76, 150–160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chillappagari S et al. Copper Stress Affects Iron Homeostasis by Destabilizing Iron-Sulfur Cluster Formation in Bacillus subtilis. J. Bacteriol 192, 2512–2524 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brancaccio D et al. [4Fe-4S] Cluster Assembly in Mitochondria and Its Impairment by Copper. J. Am. Chem. Soc 139, 719–730 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Mateus A et al. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol. Syst. Biol 14, e8242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purves J, Cockayne A, Moody PCE & Morrissey JA Comparison of the Regulation, Metabolic Functions, and Roles in Virulence of the Glyceraldehyde-3-Phosphate Dehydrogenase Homologues gapA and gapB in Staphylococcus aureus. Infect. Immun 78, 5223–5232 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Text | Description of Materials and Methods
Supplementary Table 1 | Proteomic coverage from protein expression level and protein stability analyses.
Supplementary Table 2 | Complete list of protein expression level hits.
Supplementary Table 3 | Complete list of protein stability hits.
Supplementary Table 4 | Biological process classification of protein stability hits.
Supplementary Table 5 | Biological process classification of protein stability hits within each cluster.
Supplementary Figure 1 | Electron paramagnetic resonance spectroscopy of Cu titrated with PT in 10% glycerol.
Supplementary Figure 2 | Experimental workflow and isobaric mass tagging scheme for protein expression analysis.
Supplementary Figure 3 | Experimental workflow and isobaric mass tagging scheme for protein stability analysis.
Supplementary Figure 4 | Heat map visualizing magnitude of expression changes for hit proteins grouped by biological process.
Supplementary Figure 5 | Heat maps visualizing magnitude of stability changes for hit peptides grouped by biological process.
Supplementary Figure 6 | GAPDH and IDH activity in cells treated with PT.
Supplementary Dataset 1 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the first TMT10-plex labeled sample generated in the protein expression level analyses (i.e., biological replicates 1 and 2).
Supplementary Dataset 2 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the second TMT10-plex labeled sample generated in the protein expression level analyses (i.e., biological replicates 3 and 4).
Supplementary Dataset 3 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the first TMT10-plex labeled sample generated in the protein stability analyses (i.e., biological replicates 1 and 2).
Supplementary Dataset 4 | Search output and log2(fold change) calculations from LC-MS/MS analyses of the first TMT10-plex labeled sample generated in the protein stability analyses (i.e., biological replicates 3 and 4).
Supplementary Dataset 5 | Hit analysis of overlapping proteins identified in the two TMT10-plex labeled samples generated in the protein expression level analyses (i.e., biological replicates 1–4).
Supplementary Dataset 7 | Fuzzy c-means cluster analysis of peptide stability hits.
Supplementary Dataset 8 | DAVID functional analysis and resulting GO biological process classifications of protein expression level hits and proteins corresponding to peptide stability hits.
Supplementary Dataset 6 | Hit analysis of overlapping proteins identified in the two TMT10-plex samples generated in the protein stability analyses (i.e., biological replicates 1–4).