

Abstract
Introduction
The mechanism behind the strong association between the ɛ2/ɛ3/ɛ4 apolipoprotein E gene (APOE) polymorphism and Alzheimer's disease is not well‐characterized. Because low plasma levels of apoE associate with risk of dementia, genetic variants altering apoE levels in general may also associate with dementia.
Methods
The APOE gene was sequenced in 10,369 individuals, and nine amino acid–changing variants with frequencies ≥2/10,000 were further genotyped in 95,228 individuals. Plasma apoE levels were measured directly.
Results
Risk of all dementia and Alzheimer's disease (AD) increased with decreasing genetically determined apoE levels (P = 5 × 10−4 and P = 1 × 10−4 after APOE ɛ2/ɛ3/ɛ4 adjustment). Hazard ratios (95% confidence intervals) for all dementia and AD were 2.76 (1.39 to 5.47) and 4.92 (2.36 to 10.29) for the group with the genetically lowest apoE versus ɛ33.
Discussion
We found that genetically low apoE levels increase and genetically high levels decrease risk, beyond ɛ2/ɛ3/ɛ4. This underscores that dementia risk more likely relates to variants affecting levels of apoE.
Keywords: Alzheimer's disease, APOE, apolipoprotein E, dementia, genetics, rare variation
1. BACKGROUND
Dementia is a major cause of disability in later life, with an increasing global prevalence. 1 , 2 , 3 , 4 , 5 Currently no curative and preventive options are available. Improved understanding of the underlying genetic background is likely to identify relevant targets for future treatment options and more precise risk prediction for targeted preventive interventions.
In genome‐wide association studies (GWASs) the apolipoprotein E (APOE) gene stands out as an impressive signal for risk of Alzheimer's disease (AD), 6 , 7 thereby confirming the identification of the common APOE ɛ4 allele as an important risk marker first reported by Corder et al., 8 and since validated in major cohorts around the globe. 9 , 10 ApoE is a central apolipoprotein in both cerebral and peripheral cholesterol metabolism and is among many functions involved in brain amyloid metabolism, blood–brain barrier integrity, and transport of various lipid species to neuronal cells, as well as in hepatic uptake of triglyceride‐rich lipoproteins. 11 , 12 , 13 , 14 , 15 , 16 Despite the impressive ɛ4 GWAS signal, the exact mechanism behind this observation is not well characterized. Of interest, low levels of plasma apoE per se were recently reported to be associated with increased risk of dementia, 10 , 17 thus suggesting that APOE variants that affect apoE protein levels in general also associate with risk of dementia. 10 , 17 , 18 , 19 Because the APOE ɛ4 allele is both common and has a large risk effect—much higher than generally observed for common variants in common diseases—the contribution from other genetic less‐frequent variants in the APOE gene to risk and their contribution to apoE levels has not been studied at the population level.
Therefore, we set out to do a systematic resequencing of the APOE gene with subsequent large‐scale genotyping and related these variants to their specific gene product—plasma levels of apoE—and to the risk of dementia. All rare variants were analyzed in the context of the well‐known APOE polymorphism, which is a combination of two genetic variants (ɛ4 rs429358 and ɛ2 rs7412), defining six common APOE genotypes (ɛ22, ɛ32, ɛ42, ɛ33, ɛ43, and ɛ44), which correspond to the isoform patterns apoE2/2, apoE3/2, apoE4/2 or 3/4+2, apoE3/3, apoE4/3, and apoE4/4. For this purpose, we used two large general population cohorts, the Copenhagen City Heart Study (CCHS) and the Copenhagen General Population Study (CGPS), and performed population‐based resequencing in 10,369 individuals from the CCHS and further genotyped nine variants with a frequency of ≥2 per 10,000 in 95,228 individuals from the CGPS.
2. METHODS
Studies were approved by institutional review boards and Danish ethical committees, and were conducted according to the Declaration of Helsinki, with written informed consent from participants. All participants were white and of Danish descent. There was no overlap of individuals between studies.
2.1. Participants
We included individuals from two similar studies of the Danish general population: The CCHS and the CGPS. Individuals were selected randomly from the national Danish Civil Registration System to reflect the adult population aged 20 through 100+ years. These studies combined included a total of 105,597 of whom 3444 developed dementia during the follow‐up period.
RESEARCH IN CONTEXT
Systematic review: We searched PubMed with the term “APOE” or “apolipoprotein E” plus “dementia”, “Alzheimer”, “sequencing”, “rare”, or “variation”. We also searched the reference lists of these articles.
Interpretation: Structural changes in apoE beyond the common apolipoprotein E gene (APOE) ε2/ε3/ε4 polymorphism contribute to risk of dementia, where genetically low apoE levels increase and genetically high apoE decrease risk. Consequently, APOE‐associated dementia risk is not a property solely related to the strong ε4 allele, but more likely relates to variants affecting levels of apoE in general.
Future directions: The study underscores apoE as an important target for future therapeutic strategies. Genetically determined low apoE levels, as seen for ɛ4 and ɛ4‐like variants, may reflect increased binding to heparan sulfate proteoglycans (HSPGs), which is necessary for development of tau pathology. Hence, inhibitors of HSPG binding may be a future path for targeted therapies in the 3% high‐risk ɛ44 group in the general population.
2.1.1. The Copenhagen City Heart Study
This prospective study of the Danish general population was initiated in 1976 to 1978 with follow‐up examinations in 1981 to 1983, 1991 to 1994, and 2001 to 2003. 10 , 20 , 21 Data collection included a questionnaire, a physical examination, and blood sampling for biochemical and DNA analyses. We included 10,369 individuals from whom blood was obtained for biochemical and DNA analyses at the 1991 to 1994 or 2001 to 2003 examinations; among these, 1093 developed dementia during follow‐up.
2.1.2. The Copenhagen General Population Study
This prospective study of the Danish general population was initiated in 2003, with the first enrollment period from 2003 to 2015, and with follow‐up examinations ongoing. 10 , 20 , 21 Participants were recruited and examined as in the CCHS. We included 95,228 individuals; among these, 2351 developed dementia during follow‐up.
2.2. Endpoints
Information on diagnoses was collected from the national Danish Patient Registry, with data on all patient contacts from all clinical hospital departments in Denmark since 1977, and from the national Danish Causes of Death Registry, with data on causes of all deaths in Denmark, as reported by hospitals and general practitioners since 1977. Alzheimer's disease was World Health Organization International Classification of Diseases (ICD) 8th Revision and 10th Revision ICD8 290.10 and ICD10 F00 and G30. All dementia included Alzheimer's disease, vascular dementia (ICD10 F01) and unspecified dementia (ICD8 290.18 and ICD10 F03). For Cox regression models for APOE variants and genetic scores, follow‐up began at birth or start of the registries (January 1, 1977), whichever came last. Follow‐up ended at occurrence of event, death, emigration, or on April 10, 2018 (last update of the registries), whichever came first. Median follow‐up from start of the registries was 40 years (range 0 to 41 years) for all dementia, with no individuals lost to follow‐up due to the completeness of the Danish nationwide registries.
2.3. Gene screening and genotyping
We screened the translated region of APOE by using five polymerase chain reaction (PCR) fragments covering exons 2, 3, and 4 and exon‐intron boundaries (APOE consensus sequence NC_000019.10) (Figure 1). Mutation screening was performed by adding a fluorescent dye (LC Green, Idaho Technology Inc, Salt Lake City, Utah) to the mixture before amplification by PCR which allows for end‐point melting (Lightscanner with Lightscanner Instrument & Analysis Software, version 2.0.0.1331, Idaho Technology Inc, Salt Lake City, Utah). 22 , 23 If two repeated Lightscanner melting analyses indicated variation, the initial PCR was followed by cycle sequencing with a dideoxy‐termination reaction 24 (Sanger sequencing) and further analysis by automated capillary electrophoresis (ABI 3730 DNA Analyzer), and finally verified by K.L.R. (Seqscape version 2.5.0, Applied Biosystems). Identified variants were confirmed by a second round of Sanger sequencing. An ABI PRISM 7900HT Sequence Detection System (Applied Biosystems Inc., Foster City, California, USA) and TaqMan‐based assays were used to genotype for p.Cys130Arg (rs429358, legacy name Cys112Arg, c.388T>C) defining the ε4 allele and p.Arg176Cys (rs7412, legacy name Arg158Cys, c.526C>T) defining the ε2 allele. Nine amino acid–changing rare variants with frequency ≥2/10,000 (allele frequency ≥0.01%) were further genotyped in the CGPS, using either PCR‐based KASP assays (LGC, Teddington, UK) (p.Thr11Ser, p.Ala23Val, p.Glu31Lys, p.Leu46Pro) or ABI PRISM 7900HT Sequence Detection System (Applied Biosystems Inc., Foster City, California, USA) and TaqMan‐based assays (p.Glu114Lys, p. Gly145Asp, p.Arg154Cys, p.Val254Glu, p. Arg269Gly). A substantial fraction of heterozygote calls and all homozygote calls for rare variants were validated by Sanger sequencing. Genotypes for three APOE promoter variants (rs449647, rs769446, and rs405509) in 74,940 individuals were determined as described previously. 18 , 19
FIGURE 1.
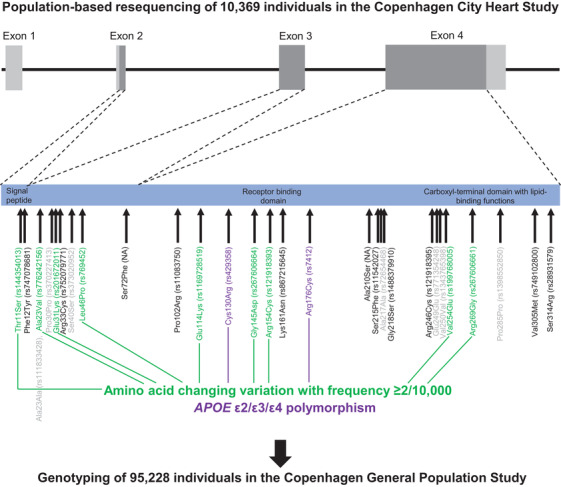
Population‐based resequencing of the translated region of the apolipoprotein E gene (APOE) in the Copenhagen City Heart Study followed by genotyping in the Copenhagen General Population Study. Gene screening and genotyping are described in the Methods Section. The dark gray regions indicate the translated regions of the APOE gene. Of the 27 rare variants found by resequencing, nine amino acid–changing variants with frequencies ≥2/10,000 are indicated in green. Amino acid–changing variants with frequencies <2/10,000 are indicated in black, and synonymous variants are indicated in light gray. In addition, the APOE ε2/ε3/ε4 polymorphism is indicated in purple. ApoE consists of two domains connected by a “hinge region” and forms a tetrameric α‐helical bundle, and in the presence of lipids, the bundle “opens” to expose the hydrophobic cores, making the protein available to interact with lipids. 28 , 42 The amino‐terminal domain (amino acids ≈19 to 209) contains the receptor binding functions (amino acids ≈148 to 168). This is preceded by a posttranslationally removed 18‐residue signal peptide (amino acids 1 to 18). 45 The carboxyl‐terminal domain (amino acids ≈243 to 317) contains the lipid‐binding functions. APOE, apolipoprotein E gene; NA, rs number not available
2.4. Biochemical analyses and other covariates
Direct measurement of plasma apoE has previously been described. 10 , 18 , 25 Biochemical analyses are described in the Appendix; other covariates are described in the legend to Table S1.
2.5. Statistical analysis
We used Stata/S.E. version 13.1 (Stata Corp., College Station, Texas, USA). P‐values <.0001 are given as powers of 10. Kruskal–Wallis equality‐of‐populations rank test and the Pearson χ2 test or Fisher exact test were used in comparisons of continuous and categorical variables, respectively. Z test evaluated plasma apoE levels for individuals with a rare variant versus individuals without this rare variant: The two‐sided P‐value was derived from the z‐value, which was calculated as z = (mean1 − mean2)/(√(standard error of the mean1 2 − standard error of the mean 2 2). Missing data (<0.4%) were imputed from age and sex in each population separately, using multinomial logistic regression for categorical variables and linear regression for continuous variables; however, if only individuals with complete data were included, results were like those reported. No genotypes, endpoints, or plasma apoE levels were imputed.
The apoE‐weighted allele score was calculated for each participant using a weighted sum of apoE‐changing variants. The weights correspond to the sum of β‐coefficients for the 11 apoE‐changing variants for each individual obtained from a linear regression for plasma levels of apoE measured directly in 103,898 individuals in CCHS and CGPS, accounting for the effect of the nine rare variants, APOE ε2/ε3/ε4, sex, age, and cohort (Table 1). By doing so, we ensured that the contribution from both the common APOE ɛ2/ɛ3/ε4 polymorphism as well as the nine rare variants were captured. Subsequently, we categorized the weighted allele score into five reasonably sized groups (given the large groups of “clean” ε33 (55%), ε32 (12%), and ε43 (25%) without rare variation) with a “clean” ɛ33 group as the reference group displaying intermediate levels of genetically determined apoE and being the most prevalent group of individuals in most populations. 15 The “clean” ɛ33 group consisted of individuals who were wild‐type for the common ε2/ε3/ε4 polymorphism and with no additional genetic variants in the coding part of APOE, thereby expressing only the apoE3 isoform. The five groups were named “very high,” “high,” ɛ33, “low,” and “very low” weighted allele score group. To illustrate the rare variants in the ε2/ε3/ε4 context we also divided the weighted allele score into 13 groups according to ε2/ε3/ε4 status with and without rare variation. Finally, as a sensitivity analysis an unweighted allele score was created by dividing individuals into 12 groups: the “clean” ε22, ε32, ε42, ε33, ε43, and ε44 genotypes, as well as the same genotypes with additional rare variation (ε22+, ε32+, ε42+, ε33+, ε43+, and ε44+).
TABLE 1.
Single per allele weights (part A) used for apolipoprotein E–weighted allele score calculation (part B)
| A: Single per allele weights for nine rare and two common amino acid changing variants in the APOE gene on plasma apoE | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of individuals (minor allele frequency) | ||||||||||
| Type of variant | Gene region | Nucleotide substitution | Variant | Rs number | CCHS Ntotal = 10,369 | CGPS Ntotal = 95,228 | Common allele | Minor allele | Single per allele weights | Contribution (%)b |
| Rare | Exon 2 | c.31A>T | p.Thr11Ser | rs144354013 | 2 (0.01) | 27 (0.01) | A | T | 0.4821579 | 3 |
| Rare | Exon 3 | c.68C>T | p.Ala23Val | rs776242156 | 2 (0.01) | 7 (0.004) | C | T | −0.6517211 | 3 |
| Rare | Exon 3 | c.91G>A | p.Glu31Lys | rs201672011 | 2 (0.01) | 6 (0.003) | G | A | −0.5511144 | 3 |
| Rare | Exon 3 | c.137T>C | p.Leu46Pro | rs769452 | 74 (0.36) | 737 (0.39)a | T | C | 0.1419617 | 3 |
| Rare | Exon 4 | c.340G>A | p.Glu114Lys | rs1169728519 | 11 (0.05) | 88 (0.05) | G | A | −1.3534370 | 3 |
| Common | Exon 4 | c.388T>C | p.Cys130Arg | rs429358 | Common (17) | Common (17) | T | C | −0.4460599 | 8 |
| Rare | Exon 4 | c.434G>A | p.Gly145Asp | rs267606664 | 4 (0.02) | 32 (0.02) | G | A | 0.0232725 | 3 |
| Rare | Exon 4 | c.460C>T | p.Arg154Cys | rs121918393 | 3 (0.01) | 19 (0.01) | C | T | 2.8210540 | 3 |
| Common | Exon 4 | c.526C>T | p.Arg176Cys | rs7412 | Common (8) | Common (8) | C | T | 1.6478710 | 26 |
| Rare | Exon 4 | c.761T>A | p.Val254Glu | rs199768005 | 12 (0.06) | 229 (0.12) | T | A | 0.1699064 | 3 |
| Rare | Exon 4 | c.805C>G | p.Arg269Gly | rs267606661 | 9 (0.04) | 102 (0.05) | C | G | −0.4358085 | 3 |
Number of individuals and rare allele frequencies in percent in brackets are shown. The single allele weights correspond to the per‐apoE allele β‐coefficients adjusted for the impact of the other variants, derived from a linear regression in 103,898 individuals in the Copenhagen City Heart Study and the Copenhagen General Population Study with plasma apoE including all 11 APOE variants, sex, age groups (20‐29, 30‐39, 40‐49, 50‐59, 60‐69, 70‐79, 80+) and cohort. aIncludes two homozygotes. bThe explanatory percentage of the interindividual variation in plasma apoE from an age, sex and cohort adjusted linear regression for each of the variants separately (R2 in percent).
TABLE 1.
(Continued)
| B: Combined single allele weights into a plasma apoE weighted allele score | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
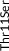
|
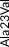
|
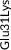
|

|
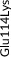
|
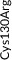
|
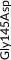
|
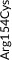
|
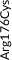
|
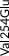
|
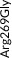
|
Weights of allele combinations | Weighted allele score in 5 groups | Weighted allele score in 13 groups | N | Percentage of population (%) |
| AA | CC | GG | TT | GG | TT | GG | CT | CT | TT | CC | 4.468925 | Very high | 3.32‐4.47 | 1 | 0.0009 |
| AA | CC | GG | TT | GG | TT | GA | CC | TT | TT | CC | 3.319015 | Very high | 3.32‐4.47 | 3 | 0.003 |
| AA | CC | GG | TT | GG | TT | GG | CC | TT | TT | CC | 3.295742 | Very high | 3.30 (ε22) | 715 | 0.7 |
| AA | CC | GG | TT | GG | TT | GG | CT | CC | TT | CC | 2.821054 | Very high | 1.67‐2.82 | 19 | 0.02 |
| AA | CC | GG | TT | GG | TC | GG | CT | CC | TT | CC | 2.374994 | Very high | 1.67‐2.82 | 2 | 0.002 |
| AA | CC | GG | TT | GG | TT | GG | CC | CT | TA | CC | 1.817777 | Very high | 1.67‐2.82 | 14 | 0.01 |
| AA | CC | GG | TC | GG | TT | GG | CC | CT | TT | CC | 1.789833 | Very high | 1.67‐2.82 | 2 | 0.002 |
| AT | CC | GG | TT | GG | TC | GG | CC | CT | TT | CC | 1.683969 | Very high | 1.67‐2.82 | 5 | 0.005 |
| AA | CC | GG | TT | GG | TT | GA | CC | CT | TT | CC | 1.671144 | Very high | 1.67‐2.82 | 27 | 0.03 |
| AA | CC | GG | TT | GG | TT | GG | CC | CT | TT | CC | 1.647871 | Very high | 1.65 (ε32) | 13,063 | 12 |
| AA | CC | GG | TC | GG | TC | GG | CC | CT | TT | CC | 1.343773 | High | 1.23‐1.34 | 76 | 0.07 |
| AA | CC | GG | TT | GG | TC | GA | CC | CT | TT | CC | 1.225084 | High | 1.23‐1.34 | 5 | 0.005 |
| AA | CC | GG | TT | GG | TC | GG | CC | CT | TT | CC | 1.201811 | High | 1.20 (ε42) | 2,953 | 3 |
| AA | CC | GA | TT | GG | TT | GG | CC | CT | TT | CC | 1.096757 | High | 0.02‐1.10 | 1 | 0.0009 |
| AA | CC | GG | TT | GG | TC | GG | CC | CT | TT | CG | 0.766003 | High | 0.02‐1.10 | 9 | 0.009 |
| AA | CC | GG | TT | GA | TT | GG | CC | CT | TT | CC | 0.294434 | High | 0.02‐1.10 | 11 | 0.01 |
| AA | CC | GG | TT | GG | TT | GG | CC | CC | TA | CC | 0.169906 | High | 0.02‐1.10 | 190 | 0.2 |
| AA | CC | GG | TC | GG | TT | GG | CC | CC | TT | CC | 0.141962 | High | 0.02‐1.10 | 13 | 0.01 |
| AT | CC | GG | TT | GG | TC | GG | CC | CC | TT | CC | 0.036098 | High | 0.02‐1.10 | 19 | 0.02 |
| AA | CC | GG | TT | GG | TT | GA | CC | CC | TT | CC | 0.023273 | High | 0.02‐1.10 | 1 | 0.0009 |
| AA | CC | GG | TT | GG | TT | GG | CC | CC | TT | CC | 0 | ε33 | 0 (ε33) | 58,428 | 55 |
| AA | CC | GG | TC | GG | TC | GG | CC | CC | TA | CC | −0.134192 | Low | −0.41‐(−0.13) | 1 | 0.0009 |
| AA | CC | GG | TT | GG | TC | GG | CC | CC | TA | CC | −0.276154 | Low | −0.41‐(−0.13) | 35 | 0.03 |
| AA | CC | GG | TC | GG | TC | GG | CC | CC | TT | CC | −0.304098 | Low | −0.41‐(−0.13) | 594 | 0.6 |
| AT | CC | GG | TT | GG | CC | GG | CC | CC | TT | CC | −0.409962 | Low | −0.41‐(−0.13) | 5 | 0.005 |
| AA | CC | GG | TT | GG | TC | GG | CC | CC | TT | CC | −0.446060 | Low | −0.45 (ε43) | 26,177 | 25 |
| AA | CC | GA | TT | GG | TT | GG | CC | CC | TT | CC | −0.551114 | Very low | −0.88‐(−0.55) | 5 | 0.005 |
| AA | CC | GG | CC | GG | CC | GG | CC | CC | TT | CC | −0.608196 | Very low | −0.88‐(−0.55) | 2 | 0.002 |
| AA | CT | GG | TT | GG | TT | GG | CC | CC | TT | CC | −0.651721 | Very low | −0.88‐(−0.55) | 8 | 0.008 |
| AA | CC | GG | TT | GG | CC | GG | CC | CC | TA | CC | −0.722213 | Very low | −0.88‐(−0.55) | 1 | 0.0009 |
| AA | CC | GG | TC | GG | CC | GG | CC | CC | TT | CC | −0.750158 | Very low | −0.88‐(−0.55) | 122 | 0.1 |
| AA | CC | GG | TT | GG | TC | GG | CC | CC | TT | CC | −0.881868 | Very low | −0.88‐(−0.55) | 80 | 0.08 |
| AA | CC | GG | TT | GG | CC | GG | CC | CC | TT | CC | −0.892120 | Very low | −0.89 (ε44) | 2898 | 3 |
| AA | CC | GA | TT | GG | TC | GG | CC | CC | TT | CC | −0.997174 | Very low | −1.80‐(−1.00) | 1 | 0.0009 |
| AA | CT | GG | TT | GG | TC | GG | CC | CC | TT | CC | −1.097781 | Very low | −1.80‐(−1.00) | 1 | 0.0009 |
| AA | CC | GG | TT | GG | CC | GG | CC | CC | TT | CG | −1.327928 | Very low | −1.80‐(−1.00) | 21 | 0.02 |
| AA | CC | GG | TT | GA | TT | GG | CC | CC | TT | CC | −1.353437 | Very low | −1.80‐(−1.00) | 73 | 0.07 |
| AA | CC | GA | TT | GG | TC | GG | CC | CC | TT | CG | −1.432983 | Very low | −1.80‐(−1.00) | 1 | 0.0009 |
| AA | CC | GG | TC | GA | TC | GG | CC | CC | TT | CC | −1.657535 | Very low | −1.80‐(−1.00) | 1 | 0.0009 |
| AA | CC | GG | TT | GA | TC | GG | CC | CC | TT | CC | −1.799497 | Very low | −1.80‐(−1.00) | 14 | 0.01 |
For all existing genotype combinations, the single allele weights were summarized into a weighted allele score. These weighted allele scores were subsequently categorized into 5 groups (“very high,” “high,” ε33, “low,” “very low”) as well as into 13 groups (3.32‐4.47, 3.30 (ε22), 1.67‐2.82, 1.65 (ε32), 1.23‐1.34, 1.20 (ε42), 0.02‐1.10, 0 (ε33), −0.41‐(−0.13), −0.45 (ε43), −0.88‐(−0.55), −0.89 (ε44), −1.80‐(−1.00)). In a sensitivity analysis, a similar score obtained from a linear regression without adjustment for sex, age and cohort resulted in identical grouping in 5 and 13 groups.
To test whether rare APOE variants were associated with increased risk of dementia, we used Cox regression models adjusted for known biologically relevant risk factors and markers of lifestyle: age (time scale), sex, body mass index, smoking, hypertension, diabetes, lipid‐lowering therapy, alcohol consumption, physical inactivity, postmenopausal status, hormonal replacement therapy, and education, and further APOE ε2/ε3/ε4 genotype. For Cox regression models, proportionality of hazards over time was assessed by plotting ‐ln(‐ln[survival]) versus ln(analysis time). There was no suspicion of nonproportionality. Interaction between apoE‐weighted allele score in five groups and sex, and between apoE weighted allele score in five groups and cohort were evaluated by the inclusion of two‐factor interaction terms in the Cox regression model, using a likelihood ratio test between models excluding and including the interaction term.
For the continuous causal, genetic estimates, we used the weighted genetic score as described above, and further we included the three common APOE promoter variants (rs449647, rs769446, and rs405509) in an alternative weighted score suited to serve as a genetic instrument for plasma apoE levels, as previously applied in Mendelian randomization studies using instrumental variable analyses. 18 , 19 For comparison with causal, genetic estimates on risk of dementia, we also studied plasma apoE on a continuous scale. These observational and causal, genetic estimates were illustrated both using restricted cubic splines and per 1 mg/dL lower plasma apoE.
3. RESULTS
Twenty‐seven rare APOE variants were identified by resequencing APOE in the CCHS, and consequently nine amino acid–changing rare variants with frequencies ≥2/10,000 were further genotyped in the CGPS (Figure 1). Characteristics of study participants are given in Tables S1, S2, and S3. Forty‐five percent of participants were ≥60 years of age at study entry corresponding to 44% of the person‐years in the genetic analyses for all dementia. The three promoter variants and the ɛ2/ɛ3/ɛ4 polymorphism are associated with expression quantitative trait loci (eQTLs) of APOE or nearby genes in linkage disequilibrium with APOE, whereas the rs769452 (Leu46Pro) is not associated with any eQTLs (Table S4). Pairwise linkage disequilibria of APOE variants are given in Table S5. ApoE‐weighted allele score in five groups and sex or cohort did not interact in predicting risk of all dementia and AD (all P‐values ≥ .10). Consequently, all further analyses were performed for sex and cohorts combined.
3.1. Individual rare APOE variants and plasma levels of apoE
Cohort‐specific age‐ and sex‐adjusted percentiles of plasma apoE for nine rare variants are given in Figure S1. Plasma apoE levels were increased for Gly145Asp and Arg154Cys (P ≤ 8 × 10−9); were decreased for Ala23Val, Leu46Pro, Glu114Lys, and Arg269Gly (P ≤ 4 × 10−5); and were unaltered from the population median for Thr11Ser, Glu31Lys, and Val254Glu (P ≥ .35) (Figure S1). Allele frequencies for the nine rare variants ranged from 0.01% to 0.36%. All nine variants were included in the weighted and in the unweighted allele scores.
3.2. Individual rare APOE variants and risk of dementia
Multifactorially adjusted Cox regression models evaluated risk of all dementia and AD for individuals with each of the rare APOE variants (Figure S2). No events were observed for heterozygote carriers of Thr11Ser, Gly145Asp, and Arg154Cys, and for homozygote carriers of Leu46Pro. Hazard ratios (HRs) (95% confidence intervals [CIs]) for heterozygotes versus non‐carriers for all dementia were 0.87 (0.12 to 6.17) for Ala23Val, 3.34 (0.47 to 23.85) for Glu31Lys, 1.87 (1.39 to 2.51) for Leu46Pro, 2.75 (1.37 to 5.51) for Glu114Lys, 0.30 (0.08 to 1.22) for Val254Glu, and 1.04 (0.34 to 3.23) for Arg269Gly. Corresponding HRs for AD were 1.88 (0.26 to 13.37), 5.79 (0.81 to 41.42), 2.36 (1.66 to 3.34), 3.64 (1.63 to 8.13), 0.57 (0.14 to 2.28), and 1.16 (0.29 to 4.64), respectively. Estimates for Glu31Lys and Glu114Lys improved after APOE ɛ2/ɛ3/ɛ4 adjustment, whereas the significant estimates for Leu46Pro heterozygotes disappeared.
3.3. ApoE‐weighted allele score, plasma apoE level, and risk of dementia
The apoE‐weighted allele score was calculated for each individual (Table 1, panel A) using a weighted sum of apoE‐changing variants and subsequently categorized into five reasonably sized groups (Table 1, panel B). The weights correspond to the sum of the individual β‐coefficients for the 11 apoE‐changing variants for each individual obtained from a linear regression accounting for the effect of the nine rare variants, APOE ɛ2/ɛ3/ɛ4, sex, age, and cohort. By doing so, we ensured that the contribution from both the common APOE ɛ2/ɛ3/ɛ4 polymorphism and the nine rare variants were captured, and at the same time ensured that linkage disequilibrium between all variants were accounted for (Table 1).
Risk of all dementia and AD increased with decreasing plasma apoE level on the continuous scale, observationally and genetically determined (Figure 2), as well as for genetically determined plasma apoE levels in five groups (P for trends 9 × 10−104 and 6 × 10−107 from the very high to high to ɛ33 to low to very low groups, and 5 × 10−4 and 1 × 10−4 after APOE ɛ2/ɛ3/ɛ4 adjustment). HRs (95% CIs) for all dementia and AD were 5.25 (4.61 to 5.98) and 7.54 (6.46 to 8.79) for the very low weighted allele score group versus ɛ33 (Figure 3). Corresponding estimates after APOE ɛ2/ɛ3/ɛ4 adjustment were 2.76 (1.39 to 5.47) and 4.92 (2.36 to 10.29).
FIGURE 2.
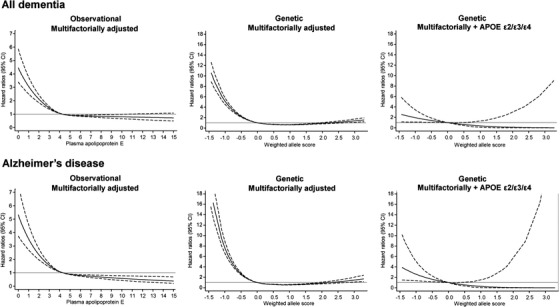
Multifactorially adjusted hazard ratios for all dementia and Alzheimer's disease according to plasma levels of apolipoprotein E (apoE) and weighted allele score in individuals in the general population. Solid lines are multifactorially adjusted hazard ratios, whereas dashed lines indicate 95% confidence intervals derived from restricted cubic splines with three knots, with the reference defined as the mean plasma level of apoE (4.3 mg/dL) or ε33 (weighted allele score = 0). For the observational estimates, follow‐up began at study entry and only if plasma apoE measurements were available. Graphs are truncated at 15 mg/dL for plasma levels of apoE and at −1.5 and 3.3 for the weighted allele score due to statistically unstable estimates outside these values, thus including 103,682 and 105,578 individuals from the Copenhagen City Heart Study and the Copenhagen General Population Study for these observational and genetic analyses. Cox regression models were adjusted for age (time scale), sex, body mass index, smoking, hypertension, diabetes, lipid‐lowering therapy, alcohol consumption, physical inactivity, postmenopausal status, hormonal replacement therapy, and education (left and middle columns), and further for APOE ε2/ε3/ε4 genotype (right column). CI, 95% confidence interval
FIGURE 3.
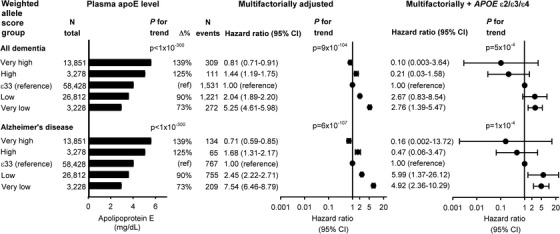
Risk of all dementia and Alzheimer's disease as a function of apolipoprotein E (apoE)‐weighted allele score in five groups. Geometric mean ± standard errors of the mean are given for plasma apolipoprotein E (left column). Cox regression models were adjusted for age (time scale), sex, body mass index, smoking, hypertension, diabetes, lipid‐lowering therapy, alcohol consumption, physical inactivity, postmenopausal status, hormonal replacement therapy, and education (middle column), and further for APOE ε2/ε3/ε4 genotype (right column). ; CI, 95% confidence interval
3.4. ApoE‐weighted allele score arranged according to APOE ɛ2/ɛ3/ɛ4 genotype and risk of dementia
For apoE‐weighted allele score in 13 groups arranged according to APOE ε2/ε3/ε4 genotype (ε22, ε32, ε42, ε33, ε43, and ε44 without rare variation), there was an overall increased risk with decreasing apoE‐weighted allele score groups for both all dementia and AD (Figure 4; P for trends ≤1 × 10−100).
FIGURE 4.
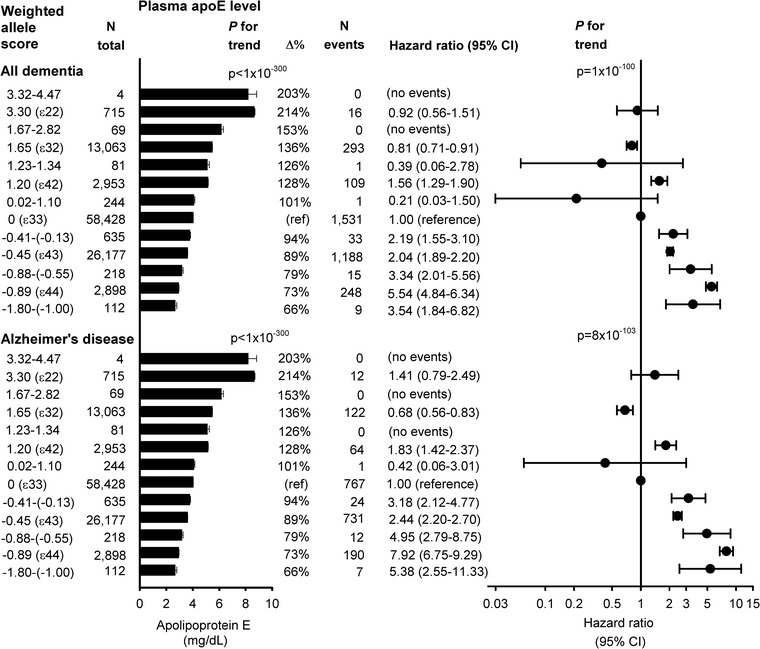
Risk of all dementia and Alzheimer's disease as a function of the weighted allele score stratified according to apolipoprotein E gene (APOE) ε2/ε3/ε4 status. The weighted allele score was divided in 13 groups according to APOE ε2/ε3/ε4 genotype (clean ε22, ε32, ε42, ε33, ε43 and ε44 genotypes without rare variation). Geometric mean ± standard errors of the mean are given for plasma apoE (left column). Cox regression models were adjusted for age (time scale), sex, body mass index, smoking, hypertension, diabetes, lipid‐lowering therapy, alcohol consumption, physical inactivity, postmenopausal status, hormonal replacement therapy, and education (middle and right column). P for trends were from 3.32‐4.47 to 3.30 (ε22) to 1.67‐2.82 to 1.65 (ε32) to 1.23‐1.34 to 1.20 (ε42) to 0.02‐1.10 to 0 (ε33) to −0.41‐(−0.13) to −0.45 (ε43) to −0.88‐(−0.55) to −0.89 (ε44) to −1.80‐(−1.00). ApoE, apolipoprotein E; CI, 95% confidence interval
HRs for a 1 mg/dL genetic decrease in plasma apoE were 2.18 (1.35 to 3.50) for all dementia and 2.66 (1.49 to 4.77) for AD for the weighted allele score after ε2/ε3/ε4 adjustment (Figure 5). Corresponding HRs for the weighted allele score in ε33 individuals only were 2.48 (1.44 to 2.25) and 3.63 (1.98 to 6.66), respectively (Figure 5). For comparison, Figure 5 also illustrates similar risk estimated for plasma apoE observationally and for genetically determined plasma apoE from APOE ɛ2/ɛ3/ɛ4 genotype and from APOE promoter variants, respectively.
FIGURE 5.
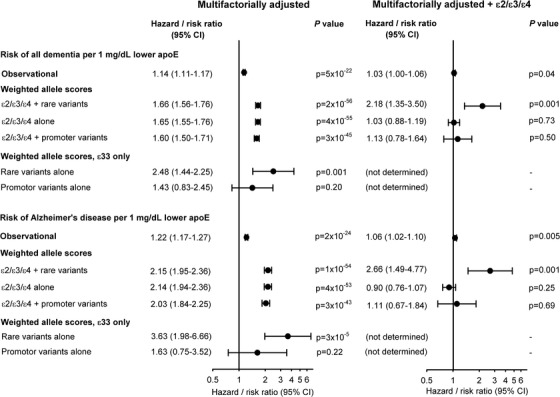
Risk for all dementia and Alzheimer's disease for a 1 mg/dL decrease in plasma apolipoprotein E (apoE) levels. Cox regression models were adjusted for age (time scale), sex, body mass index, smoking, hypertension, diabetes, lipid‐lowering therapy, alcohol consumption, physical inactivity, postmenopausal status, hormonal replacement therapy, and education (left column), and further for APOE ε2/ε3/ε4 genotype (right column). For the observational estimates, follow‐up began at study entry and only if plasma apoE measurements were available, thus including 103,744 individuals in these analyses. The first weighted allele score, ε2/ε3/ε4 + rare variants, is the score described in the Methods section (n = 105,597). For the second score, ε2/ε3/ε4 alone, the score was obtained similarly, but in a model only including the ε4 and the ε2 alleles, and not the nine rare variants (n = 105,597). The third score, ε2/ε3/ε4 + promoter variants, was obtained like the main genetic score, but included weights from ε4, ε2, and three common promoter variants (rs449647, rs769446, and rs405509) (n = 74,940). The estimates for rare variants alone and for promotor variants alone were for the same genetic scores for ε33 individuals only (n = 58,737 and 41,789). CI, 95% confidence interval
3.5. Unweighted allele score arranged according to APOE ɛ2/ɛ3/ɛ4 genotype and risk of dementia
For 12 groups of the clean ε22, ε32, ε42, ε33, ε43, and ε44 genotypes, as well as the same genotypes with additional rare variation (ε22+, ε32+, ε42+, ε33+, ε43+, and ε44+) a similar pattern as for the weighted allele score in 13 groups was observed (Figure S3; P for trends were ≤5 × 10−100).
3.6. Sensitivity analyses
Excluding three or six variants in the weighted allele score did not change estimates substantially (Figures S4 and S5). The three compound heterozygotes (Glu31Lys/Arg269Gly, Leu46Pro/Glu114Lys, and Leu46Pro/Val254Glu) and the two Leu46Pro homozygotes did not contribute to estimates with any dementia events, and risk estimates for Glu31Lys, Glu114Lys, and Val254Glu did not change after exclusion of the three compound heterozygotes (data not shown). Estimates for single rare variants and for apoE‐weighted allele score in five groups remained largely unchanged after adjustment for low‐density lipoprotein (LDL) cholesterol, high‐density lipoprotein (HDL) cholesterol, and triglycerides (data not shown and Figure S6, respectively) and after stratification by ≥60 years at study entry (Figures S7 and S8).
4. DISCUSSION
The principal findings of this study are that rare amino acid–changing variants in the APOE gene contribute to a risk of dementia beyond the strong ɛ2/ɛ3/ɛ4 polymorphism where genetically low apoE levels increase and genetically high levels decrease risk. A 1 mg/dL decrease in genetically determined plasma apoE levels increased risk of dementia two‐ to fourfold. These novel findings underscore the importance of structurally well‐functioning apoE in dementia risk and that risk increase conferred by the APOE gene is not a property solely related to the common ε2/ɛ3/ɛ4 polymorphism, but more likely relates to variants affecting levels of apoE in general.
Mechanistically, our findings can possibly be understood as follows. First, it is well established that heterozygotes for Arg176Cys (ε32) are associated with a decreased risk of dementia, likely explained by higher apoE levels caused by decreased affinity of the apoE2 isoform to LDL receptors due to the well‐established ligand‐defect of 176Cys. 15 Of interest, this is highly supported by a novel report on the APOE Arg154Ser mutation (APOE3 Christchurch, old nomenclature Arg136Ser), where the effect of a dominant presenilin 1 mutation was neutralized in a woman from a large Columbian kindred with autosomal dominant AD. 26 Functional studies have suggested that the beneficial effect is related to decreased affinity for LDL receptor family members and heparan sulfate proteoglycans (HSPGs). Consequently, no tau pathology develops because apoE binding may be necessary for neuronal uptake of extracellular tau. 27 Additional support comes from the present data, where Gly145Asp and Arg154Cys are located in the receptor binding region (amino acids ≈148 to 168) 28 and display a phenotype similar to the ɛ2‐defining Arg176Cys residue with high apoE levels and seemingly low risk of dementia (no events). Second, variation near the ε4‐defining residue Cys130Arg results in an ε4‐like increased risk of dementia. Glu114Lys is the rare variant located closest to Cys130Arg but is not in linkage disequilibrium with ε4. Of interest, this variant is associated with decreased apoE levels and increased risk of dementia, similar to ε4. HSPGs are suggested to play a role in neuronal uptake of tau and in amyloid beta metabolism, and apoE binding may play an important role here. 26 , 27 It is important to note that HSPGs binding studies have shown that apoE4 had higher affinity than apoE3, apoE2 and apoE3 Christchurch, 29 and antibodies raised against the receptor‐binding region mimicked the protective effect of apoE2 and apoE3 Christchurch, 26 thereby raising hope for apoE4‐targeted therapies and/or possibly therapies modulating apoE–HSPG interactions. 26 Functional studies of rare variation near the receptor‐ binding area—near ε4 as well as ε2—would be relevant to explore this further.
Taken together, and in light of the potential amyloid‐neutralizing effect of an apoE‐increasing mutation, 26 these ε2‐like and ε4‐like deviations of the structure of apoE have potential important biologic and therapeutic implications. Whereas brain apoE plays a major role in brain amyloid metabolism, in blood‐brain barrier integrity, and in supplying cholesterol and phospholipids to neuronal cells, 12 , 13 , 14 , 16 , 30 , 31 it is also well‐known that ε4 is associated with an adverse lipid profile and that ε22 carriers have a propensity to develop a recessive form of dysbetalipoproteinemia (formerly known as type III hyperlipoproteinemia). 15 , 18 Mutations in the LDL receptor–binding domain, as the APOE Arg154Ser Christchurch, as well as the presently identified Gly145Asp and Arg154Cys mutations, are also associated with dysbetalipoproteinemia—a condition that is, however, treatable with standard lipid medications. 32 Therefore, if receptor binding– inhibiting therapies will be developed, a natural side effect will be introduction of dysbetalipoproteinemia, unless the therapy can be limited to the brain.
This is the first time that the full spectrum of genetic variation in the coding parts of the APOE gene, corresponding effects on the direct gene product, levels of apoE, and risk of dementia are presented in a large general population cohort. The current literature is sparse on this issue and consists mostly of case reports and smaller studies. In accordance with previous studies, we found Leu46Pro in linkage disequilibrium with ε4, and whereas some studies reported Leu46Pro to add risk beyond ε4 33 , 34 through a destabilized apoE structure, 35 others found that Leu46Pro did not increase risk of AD beyond ε4. 36 , 37 Our study confirmed the latter. A resequencing study of 376 healthy individuals ≥85 years of age and 376 controls 41 to 54 years of age found no excess of rare variants in the 85+ year olds. 38 However, that study was unable to investigate the effects of the identified rare variants for development of dementia in a large prospective set‐up with an unselected population with long follow‐up. Furthermore, two case‐studies of patients with frameshift mutations and premature stop codons in exon 4 did not observe clear neurocognitive defects, and one case‐control study found a higher frequency of a combined Glu262Lys and Glu263Lys variant in patients with memory complaints (P = .08) 39 , 40 , 41 : however, these variants were not found in our population. Val254Glu and Arg269Gly are located in the lipid‐binding carboxyl‐terminal domain (amino acids ≈243 to 317). 28 , 42 We did not find risk associations, although Val254Glu was previously reported to be protective. 36 Whereas APOE ε4 is the main contributor to late‐onset AD, and has provided the clearest and most consistent signal in GWASs, 6 , 7 the risk of dementia for carriers of rare APOE variants was also substantial, with effect sizes similar to those for ɛ4 as well as other rare variants in dementia risk genes such as TREM2. 43
Collectively, the present data suggest that rare functional genetic variation in APOE, which causes lifelong changes in apoE levels, affects risk of dementia, both alone and compiled in weighted allele scores. This general statement is highly supported by the fact that the association between the weighted allele score groups and risk of dementia becomes more linear after adjusting for the ɛ2/ɛ3/ɛ4 polymorphism. Because ɛ4 is a strong risk‐increasing allele and ɛ2 is a strong apoE‐increasing allele, the ɛ42 genotype displays a higher risk than ɛ33 but also higher apoE levels than ɛ33. This results in a less linear trend for the weighted allele score group before adjustment, because the “high” weighted allele score group contains the majority of ɛ42 individuals. However, after adjustment, the weighted allele score groups reflect that genetically high apoE levels in general are associated with low risk. These findings are in accordance with our previous work on common APOE variants, where genetically low plasma apoE level was associated with increased risk of dementia and dementia‐associated mortality. 18 , 19 Hence, we now suggest that both rare and common variants in APOE contribute to plasma levels of apoE, likely mimicking brain apoE levels, and to risk of dementia in the general population. 10 , 19
A major strength of the study is the large prospective general population design with no losses to follow‐up: that is, every single individual could be followed to occurrence of event, end of follow‐up, death, or emigration. In addition, due to the large sample size, we were able to conduct survival analyses with a meaningful power despite the fact that allele frequencies for the nine rare variants were ≤0.4%. Finally, the ability to adjust for and relate estimates for rare variants to the common APOE ε2/ε3/ε4 genotype is a major advantage. One potential limitation concerns the validity of the dementia diagnoses; however, the national Danish Patient Registry includes all hospital visits, and dementia diagnoses in the Danish registries have high diagnostic validity 19 , 44 ; any unregistered events would only lead to decreased power of estimates and bias results toward the null hypothesis. Second, the generalizability of our study results is limited because we studied white individuals only, and consequently our results may not necessarily apply to other populations. In the majority of populations, the ε33 genotype is the most common; however, interestingly allelic frequencies vary in different populations, 15 which could affect the relative contribution. The impact of the APOE polymorphism is well described, however, among many ethnicities, so our results are likely to be applicable to most humans. Third, as the present large prospective cohort study is the only study with measurements of both plasma apoE and genetic variation, the weights for the 11 apoE‐changing variants were estimated from the data to which they were applied. Therefore, risk of overfitting is a potential limitation, and as the score is generated in a very homogeneous sample, it may be less likely to perform as well in other populations.
In conclusion, by resequencing the APOE gene in 10,369 individuals from the general population and genotyping selected variants in up to 105,597 individuals, we found that structural changes in apoE beyond ε2/ε3/ε4 contribute to a risk of dementia where genetically low apoE levels increase and genetically high apoE levels decrease risk. We suggest, that it is useful to genotype more than just the ε2/ε3/ε4 polymorphism, despite its strong impact. Rare genetic variation in APOE may thus add to explain missing heritability for dementia. These findings underscore the importance of structurally well‐functioning apoE in dementia risk and that this is not a property related solely to the common ε2/ɛ3/ɛ4 polymorphism, but more likely relates to variants affecting levels of apoE.
AUTHOR CONTRIBUTIONS
Katrine L. Rasmussen: Study concept and design, acquisition of data, statistical analysis, analysis and interpretation of data, drafting of the article, and critical revision of the article for important intellectual content. Anne Tybjærg‐Hansen: Acquisition of data, critical revision of the article for important intellectual content, obtained funding, and provided administrative, technical, and material support. Børge G. Nordestgaard: Acquisition of data, critical revision of the article for important intellectual content, obtained funding, and provided administrative, technical, and material support. Ruth Frikke‐Schmidt: Study concept and design, acquisition of data, statistical analysis, analysis and interpretation of data, drafting of the article, critical revision of the article for important intellectual content, obtained funding, and administrative, technical, and material support, study supervision.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We thank the staff and participants of the CGPS and the CCHS for their important contributions.
Rasmussen KL, Tybjærg‐Hansen A, Nordestgaard BG, Frikke‐Schmidt R. APOE and dementia – resequencing and genotyping in 105,597 individuals. Alzheimer's Dement. 2020;16:1624–1637. 10.1002/alz.12165
Funding information
This work was supported by the Research Council at Rigshospitalet and the Lundbeck Foundation, both to [Ruth Frikke‐Schmidt]. The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the article; and decision to submit the article for publication.
REFERENCES
- 1. Global, regional, and national age‐sex specific all‐cause and cause‐specific mortality for 240 causes of death, 1990‐2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prince M, Bryce R, Albanese E, Wimo A, Ribeiro W, Ferri CP. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement. 2013;9:63‐75. [DOI] [PubMed] [Google Scholar]
- 3. Langa KM, Larson EB, Crimmins EM, et al. A Comparison of the prevalence of dementia in the United States in 2000 and 2012. JAMA Intern Med. 2017;177:51‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Satizabal CL, Beiser AS, Chouraki V, Chene G, Dufouil S, Seshadri S. Incidence of dementia over three decades in the Framingham Heart Study. N Engl J Med. 2016;374:523‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahmadi‐Abhari S, Guzman‐Castillo M, Shipley MJ, et al. Temporal trend in dementia incidence since 2002 and projections for prevalence in England and Wales to 2040: modelling study. BMJ. 2017;358: j2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lambert JC, Ibrahim‐Verbaas CA, Naj AC, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. 2013;45:1452‐1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kunkle BW, Grenier‐Boley B, Bis JC, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corder EH, Saunders AM, Schmechel DE, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921‐923. [DOI] [PubMed] [Google Scholar]
- 9. Livingston G, Sommerlad A, Costafreda SG, et al. Dementia prevention, intervention, and care. Lancet. 2017;390:2673‐2734. [DOI] [PubMed] [Google Scholar]
- 10. Rasmussen KL, Tybjaerg‐Hansen A, Borge G, Frikke‐Schmidt R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann Neurol. 2015;77:301‐311. [DOI] [PubMed] [Google Scholar]
- 11. Cramer PE, Cirrito JRR, Lee CY, et al. ApoE‐directed therapeutics rapidly clear beta‐amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503‐1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bell RD, Winkler EA, Sagare AP, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rasmussen KL. Plasma levels of apolipoprotein E, APOE genotype and risk of dementia and ischemic heart disease: a review. Atherosclerosis. 2016;255:145‐144. [DOI] [PubMed] [Google Scholar]
- 14. Wellington CL, Frikke‐Schmidt R. Relation between plasma and brain lipids. Curr Opin Lipidol. 2016;27:225‐232. [DOI] [PubMed] [Google Scholar]
- 15. Mahley RW, Jr , Rall SC. Type III hyperlipoproteinemia (dysbetalipoproteinemia): the role of apolipoprotein E in normal and abnormal lipoprotein metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw‐Hill; 2001:2835‐2862. [Google Scholar]
- 16. Mahley RW. Central nervous system lipoproteins: ApoE and regulation of cholesterol metabolism. Arterioscler Thromb Vasc Biol. 2016;36:1305‐1315.</bib [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wolters FJ, Koudstaal PJ, Hofman A, Duijn CM, Ikram AF. Serum apolipoprotein E is associated with long‐term risk of Alzheimer's disease: The Rotterdam Study. Neurosci Lett. 2016;617:139‐142. [DOI] [PubMed] [Google Scholar]
- 18. Rasmussen KL, Tybjaerg‐Hansen A, Frikke‐Schmidt R. Plasma levels of apolipoprotein E, APOE genotype, and all‐cause and cause‐specific mortality in 105 949 individuals from a white general population cohort. Eur Heart J. 2019;40:2813‐2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rasmussen KL, Tybjaerg‐Hansen A, Nordestgaard BG, Frikke‐Schmidt R. Plasma apolipoprotein E levels and risk of dementia: a Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018;14:71‐80. [DOI] [PubMed] [Google Scholar]
- 20. Jorgensen AB, Frikke‐Schmidt R, Nordestgaard BG, Tybjaerg‐Hansen A. Loss‐of‐function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32‐41. [DOI] [PubMed] [Google Scholar]
- 21. Frikke‐Schmidt R, Nordestgaard BG, Sethi AA, et al. Association of loss‐of‐function mutations in the ABCA1 gene with high‐density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA. 2008;299:2524‐2532. [DOI] [PubMed] [Google Scholar]
- 22. Liew M, Pryor R, Meadows C, et al. Genotyping of single‐nucleotide polymorphisms by high‐resolution melting of small amplicons. Clin Chem. 2004;50:1156‐1164. [DOI] [PubMed] [Google Scholar]
- 23. Wittwer CT, Reed GH, Gundry CM, Vandersteen JG, Pryor RJ. High‐resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem. 2003;49:853‐860. [DOI] [PubMed] [Google Scholar]
- 24. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain‐terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463‐5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rasmussen KL, Tybjaerg‐Hansen A, Frikke‐Schmidt R. Plasma levels of apolipoprotein E and risk of ischemic heart disease in the general population. Atherosclerosis. 2016;246:63‐70. [DOI] [PubMed] [Google Scholar]
- 26. Arboleda‐Velasquez JF, Lopera F, Delgado‐Tirado S, et al. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019;25:1680‐1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rauch JN, Chen JJ, Miller GM, et al. Tau internalization is regulated by 6‐O sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep. 2018;8:6382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weisgraber KH. Apolipoprotein E: structure‐function relationships. Adv Protein Chem. 1994;45:249‐302. [DOI] [PubMed] [Google Scholar]
- 29. Yamauchi Y, Deguchi N, Tanaka M, et al. Role of the N‐ and C‐terminal domains in binding of apolipoprotein E isoforms to heparan sulfate and dermatan sulfate: a surface plasmon resonance study. Biochemistry. 2008;47:6702‐6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirsch‐Reinshagen V, Burgess BL, Wellington CL. Why lipids are important for Alzheimer disease? Mol Cell Biochem. 2009;326:121‐129. [DOI] [PubMed] [Google Scholar]
- 31. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hegele RA, Boren J, Arca M, et al. Rare dyslipidaemias, from phenotype to genotype to management: a European Atherosclerosis Society task force consensus statement. Lancet Diabetes Endocrinol. 2019. [DOI] [PubMed] [Google Scholar]
- 33. Scacchi R, Gambina G, Ferrari G, Corbo RM. Screening of two mutations at exon 3 of the apolipoprotein E gene (sites 28 and 42) in a sample of patients with sporadic late‐onset Alzheimer's disease. Neurobiol Aging. 2003;24:339‐343. [DOI] [PubMed] [Google Scholar]
- 34. Kamboh MI, Aston CE, Kokmen K, et al. A novel mutation in the apolipoprotein E gene (APOE*4 Pittsburgh) is associated with the risk of late‐onset Alzheimer's disease. Neurosci Lett. 1999;263:129‐132. [DOI] [PubMed] [Google Scholar]
- 35. Argyri L, Dafnis I, Gantz D, Chroni A. Molecular basis for increased risk for late‐onset Alzheimer disease due to the naturally occurring L28P mutation in apolipoprotein E4. J Biol Chem. 2014;289:12931‐12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Medway CW, Abdul‐Hay S, Zou F, et al. ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer's disease. Mol Neurodegener. 2014;9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baron M, Jimenez‐Escrig A, Simon J,et al. Apolipoprotein E Pittsburgh variant is not associated with the risk of late‐onset Alzheimer's disease in a Spanish population. Am J Med Genet B Neuropsychiatr Genet. 2003;120B:121‐124. [DOI] [PubMed] [Google Scholar]
- 38. Tindale LC, Leach S, Daley D, et al. Rare and common variants in the Apolipoprotein E gene in healthy oldest old. Neurobiol Aging. 2014;35:727‐3. [DOI] [PubMed] [Google Scholar]
- 39. Youn YC, Lim YK, Giau VV, et al. Apolipoprotein epsilon7 allele in memory complaints: insights through protein structure prediction. Clin Interv Aging. 2017;12:1095‐1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mak AC, Pullinger CR, Schwarz JM, et al. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol. 2014;71:1228‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lohse P, Brewer HB, III , Meng MS, Skarlatos SI, LaRosa JC, Brewer HB, Jr . Familial apolipoprotein E deficiency and type III hyperlipoproteinemia due to a premature stop codon in the apolipoprotein E gene. J Lipid Res. 1992;33:1583‐1590. [PubMed] [Google Scholar]
- 42. Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer's disease to AIDS. J Lipid Res. 2009;50(Suppl):S183‐S188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jonsson T, Stefansson H, Steinberg P, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013;368:107‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Phung TK, Andersen BB, Hogh P, Kessing LV, Mortensen PB, Waldemar G. Validity of dementia diagnoses in the Danish hospital registers. Dement Geriatr Cogn Disord. 2007;24:220‐228. [DOI] [PubMed] [Google Scholar]
- 45. Rall SC, Jr , Weisgraber KH, Mahley RW. Human apolipoprotein E. The complete amino acid sequence. J Biol Chem. 1982;257:4171‐4178. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information