

Abstract
Background
Histopathologic examination (HPE) of tumor tissue obtained by invasive biopsy is the standard for cancer diagnosis but is resource‐intensive and has been associated with procedural risks. The authors demonstrate that immunocytochemistry (ICC) profiling of circulating ensembles of tumor‐associated cells (C‐ETACs) can noninvasively provide diagnostic guidance in solid organ cancers.
Methods
The clinical performance of this approach was tested on blood samples from 30,060 individuals, including 9416 individuals with known cancer; 6725 symptomatic individuals with suspected cancer; and 13,919 asymptomatic individuals with no prior diagnosis of cancer. C‐ETACs were harvested from peripheral blood and profiled by ICC for organ‐specific and subtype‐specific markers relevant to the cancer type. ICC profiles were compared with HPE diagnoses to determine concordance.
Results
The presence of malignancy was confirmed by the detection of C‐ETACs in 91.8% of the 9416 individuals with previously known cancer. Of the 6725 symptomatic individuals, 6025 were diagnosed with cancer, and 700 were diagnosed with benign conditions; C‐ETACs were detected in 92.6% of samples from the 6025 individuals with cancer. In a subset of 3509 samples, ICC profiling of C‐ETACs for organ‐specific and subtype‐specific markers was concordant with HPE findings in 93.1% of cases. C‐ETACs were undetectable in 95% of samples from the 700 symptomatic individuals who had benign conditions and in 96.3% of samples from the 13,919 asymptomatic individuals.
Conclusions
C‐ETACs were ubiquitous (>90%) in various cancers and provided diagnostically relevant information in the majority (>90%) of cases. This is the first comprehensive report on the feasibility of ICC profiling of C‐ETACs to provide pan‐cancer diagnostic guidance with accuracy comparable to that of HPE.
Keywords: circulating ensembles of tumor‐associated cells (C‐ETACs), circulating tumor cells (CTCs), diagnosis, diagnostic triaging, liquid biopsy, noninvasive, solid organ cancers
Short abstract
Immunocytochemistry profiling of circulating ensembles of tumor‐associated cells conveys malignancy status as well as the organ of origin in several cancers, including brain cancer, with high sensitivity, specificity, and accuracy. This approach addresses the long‐unmet need for the noninvasive diagnostic triaging of symptomatic individuals with suspected cancers.
Introduction
Cancers are traditionally diagnosed using histologic examination (HPE) of tumor tissue obtained by invasive biopsy to identify morphologic irregularities and nuclear features. 1 Tissue biopsies, which are usually image‐guided, are specialized, invasive procedures with significant morbidity and financial implications. In addition, they necessitate patient visits to a tertiary care center with specialized facilities. Apart from the logistical aspects, there are other factors that affect tumor tissue procurement, such as inaccessibility of the tumor, proximity of the tumor to vital organs or vasculature, patients' comorbidities, and even patients' reluctance because of procedural risks. Repeat biopsies often may be desirable, such as 1) if the prior tissue sample was insufficient 2 or poorly representative, 3 2) to determine the status of therapeutically relevant biomarkers, 4 3) to characterize recurrent lesions, or 4) to identify a new lesion as a second primary or metastasis. 5 However, repeat biopsies are associated with increased procedural risks.
Noninvasive alternatives for obtaining representative tumor samples or tumor‐derived analytes can alleviate the challenges encountered with invasive procedures. 6 Circulating tumor cells 7 (CTCs) are malignant cells shed by tumors into the vasculature or lymphatics either as single cells or in clusters (eg, ≥2 cells). Because they are derived from the tumor mass itself, CTCs and their clusters are analytically equivalent to the tumor tissue. Harvesting sufficient, viable CTCs and their clusters from peripheral blood thus is comparable to obtaining a representative tissue biopsy with minimal stromal tissue or other nontumor content and may conveniently be described as oligobiopsy or microbiopsy.
Previous reports also have indicated that CTCs convey the status of diagnostic or theranostic antigens that are otherwise routinely evaluated in tumor tissue. 8 However, the application of CTCs in the clinical setting is currently confined to numerical evaluation for prognostication in a few metastatic cancers. 9 , 10 , 11 The clinical potential of CTC‐based diagnosis has not been realized because current methods and devices to harvest CTCs and their clusters from peripheral blood principally rely on immunomagnetic enrichment or microfluidic separation, neither of which yields sufficient numbers for meaningful applications. 12 , 13 , 14 We previously described the ubiquity of circulating ensembles of tumor‐associated cells (C‐ETACs) in solid organ tumors; C‐ETACs include CTCs, which are CD45‐negative cells, as well as CD45‐positive and CD8‐positive cells, such as tumor‐associated macrophages and tumor‐associated leucocytes, in addition to cancer stem cells (CD44‐positive). 15 We previously described a novel approach for the negative enrichment of C‐ETACs (and CTCs) from peripheral blood samples based on the apoptosis resistance of malignant cells of tumorigenic origin. 15 We used this approach to achieve high detection and harvest rates of C‐ETACs in a large cohort of patients who had prior diagnoses of various cancers and in symptomatic individuals who had results that were suspicious for cancer. In a subset analysis, C‐ETACs were characterized by immunocytochemistry (ICC) profiling for organ‐specific and subtype‐specific (OSS) antigens, which are routinely evaluated in HPE and ICC, to determine the tissue of origin. Here, we describe the suitability of this approach for adoption in clinical practice because it noninvasively provides diagnostically relevant information not inferior to that obtained by HPE of tumor tissue.
Materials and Methods
Study Design
The data presented in this report were derived from patient samples obtained during the course of 4 observational studies: the TrueBlood study (Tissue Biopsy Replacement With Unique Evaluation of Circulating Biomarkers for Morphological Evaluation and Clinically Relevant Molecular Typing of Malignancies From Blood Samples; Clinical Trials Registry‐India [CTRI]/2019/03/017918); the ProState study (Utility of ProState, the Liquid Biopsy Platform, in Distinguishing Prostate Malignancies From Benign Prostatic Hyperplasia; CTRI/2019/02/017863), the GlioLENS study (Utility of Gliotrack, the Liquid Biopsy Platform for Gliomas, in Distinguishing Glioblastoma From Other Central Nervous System Lesions With Equivocal Findings on Neuroimaging; registered on the World Health Organization International Clinical Trials Registry Platform; CTRI/2019/02/017663), and the RESOLUTE study (Realtime Enrichment Screen for Outright Detection of Latent Undiagnosed Malignant Tumors in Asymptomatic Individuals Efficiently; CTRI/2019/01/017219). All studies were evaluated by the institutional review boards and approved by the ethics committees of the study sponsor (Datar Cancer Genetics) and of the respective participating institutions. All trials were conducted in accordance with existing ethical guidelines and regulations, such as those of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use‐Good Clinical Practice (ICH‐GCP), as well as the Declaration of Helsinki.
The TrueBlood study enrolled adult men and women (aged ≥18 years) who had a histopathologically confirmed diagnosis of a solid organ cancer irrespective of the extent of disease or therapy status. The ProState study enrolled adult men (aged ≥18 years) who had a confirmed diagnosis of either prostate cancer or benign prostate enlargement as well as individuals who had results that were suspicious for prostate cancer. The GlioLENS study enrolled adults (aged ≥18 years) who presented with radiologic intracranial space‐occupying lesions that were suspicious for central nervous system (CNS) malignancies. The RESOLUTE study enrolled asymptomatic adult men and women who had an age‐associated elevated risk of cancer. Details of all studies are available online by querying for the respective trial identification numbers (https://apps.who.int/trialsearch/, last accessed on 29‐Aug‐2020).
Study Participants and Samples
For the current study, we primarily evaluated 9416 patients who had prior diagnoses of various cancers and 6725 who had suspected cancers, among whom 6025 were subsequently diagnosed with cancer and 700 were diagnosed with benign or inflammatory conditions of various organs. Clinical details of these patients' cancers were determined from the most recent clinical reports. Finally, the study evaluated 13,919 asymptomatic individuals who had an age‐associated elevated risk of cancer but had negative (normal) findings on screening investigations for cancer, including low‐dose computed tomography, mammography, Papanicolaou smears, as well as serum antigens (cancer antigen 125 [CA125], prostate‐specific antigen [PSA], carbohydrate antigen 19‐9 [CA19‐9], α‐fetoprotein [AFP], and carcinoembryonic antigen [CEA]). Demographic data from study participants are provided in Table 1. Details of various observational trials from which our study cohort was populated are provided in Supporting Table 1. Details of cancer types and benign conditions are provided in Supporting Tables 2 and 3, respectively. All study participants were counselled regarding the aims and scope of each study, after which they provided signed, written informed consent. From 15 to 20 mL of peripheral blood was collected into ethylenediaminetetraacetic acid vacutainer tubes from all study participants. For the 6725 individuals with suspected cancer, blood was collected before undergoing a biopsy or any other invasive procedure. Blood samples were transported to the laboratory at between 2 °C and 8 °C within 48 hours. All samples were processed at the facility of the study sponsor, which is accredited by the College of American Pathologists under the Clinical Laboratory Improvement Amendments and by the International Organization for Standardization number 15189:2012 (National Accreditation Board for Testing and Calibration Laboratories‐International Laboratory Accreditation Cooperation, NABL‐ILAC).
TABLE 1.
Patient Demographics a
| Characteristic | Cancer | Benign | Asymptomatic |
|---|---|---|---|
| Sex | |||
| Men | 6773 | 434 | 5807 |
| Women | 8668 | 266 | 8112 |
| Total | 15,441 | 700 | 13,919 |
| Age: Median (range), y | 57 (18‐102) | 55 (18‐90) | 53 (40‐75) |
| Therapy status | |||
| Naive | 6025 | — | — |
| Treated | 9416 | — | — |
| Metastatic status | |||
| Nonmetastatic | 3947 | — | — |
| Metastatic | 9675 | — | — |
| Unavailable | 1819 | — | — |
The study cohort included 9416 previously diagnosed and treated cases of cancer, 6025 recently diagnosed therapy‐naive cases of cancer, 700 individuals with benign conditions, and 13,919 asymptomatic individuals.
Enrichment, Harvesting, and Detection of C‐ETACs
Peripheral blood mononuclear cells were obtained from 15 mL of whole blood using RBC Lysis Buffer (Thermo Fisher Scientific USA) according to the manufacturer's instructions, and aliquots were transferred into multiwell plates for treatment with epigenetically activating media, as described previously. 15 Processed samples were observed by phase contrast microscopy on the fifth day. Viable apoptosis‐resistant (malignant) tumorigenic cells and their clusters were harvested by aspiration for further processing. Harvested single cells and clusters were gently transferred to 96‐well, imaging‐compatible plates for the identification of C‐ETACs and CTCs by ICC (see Immunocytochemistry Workflow, below). C‐ETACs were defined as epithelial cell adhesion molecule (EpCAM)‐positive, pan‐cytokeratin (PanCK)‐positive, and irrespective of CD45 status for all epithelial malignancies (carcinomas); as cell‐surface vimentin ‐positive and smooth muscle actin ‐positive/S100‐positive, irrespective of CD45 status for all sarcomas; and as glial fibrillary acidic protein (GFAP)‐positive, S100‐positive/Nestin‐positive and CD45‐negative for all glial CNS malignancies. To differentiate active C‐ETACs from random/transient associations of cells, C‐ETACs were defined as clusters of ≥3 cells that stained positive for the indicated markers, irrespective of CD45 status. 15 CTCs were defined as single cells that stained positive for the indicated markers and negative for CD45.
Immunocytochemistry Workflow
Dissociated C‐ETACs (single cells) were fixed on slides with 4% paraformaldehyde, pH 6.9, for 20 minutes. Cell permeabilization was achieved with 0.3% Triton X‐100 (15 minutes) followed by blocking with 3% bovine serum albumin (30 minutes). Cells were immunostained with primary antibodies (60 minutes), washed with phosphate‐buffered saline, pH 7.4, incubated with secondary antibodies (60 minutes), washed with phosphate‐buffered saline, and then incubated with 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) in the dark (15 minutes). All incubations were at ambient temperature (range, from 20 °C to 25 °C). Positive and negative cell line controls were also processed with each batch of samples. ICC slides were scanned by using the Cell Insight CX7 High‐Content Screening platform (Thermo Fisher Scientific USA), which enables nuclear size filters and calibration of intensity thresholds for individual fluorophore‐conjugated antibodies. The intensity of each antigen expression was compared with that of batch controls (reference cell lines) (Supporting Table 4). These precautions avoid or eliminate crosstalk in multiplexed analysis with different fluorophore‐conjugated antibodies.
TABLE 2.
Organ‐Specific and Subtype‐Specific Antibodies a
| Cancer Type | Marker 1 | Marker 2 | Marker 3 | Marker 4 |
|---|---|---|---|---|
| Bladder | Uroplakin‐II | GATA3 | CK20 | CK7 |
| Breast | GCDFP‐15 | GATA3 | EMA | CK7 |
| CNS | GFAP | S100 | Nestin | Olig‐2 |
| Cervix | p63 | p16 | CEA | CK7 |
| Colorectum | CDX2 | MUC2 | CK20 | — |
| Gallbladder | CEA | Maspin | CK19 | CK7 |
| Head and neck | p63 | HMWCK | CK5/CK6 | — |
| Kidney | CA‐IX | RCC | CD10 | Pax‐8 |
| Liver | Glypican 3 | Hep Par‐1 | AFP | Arginase |
| Lung | Napsin‐A | TTF‐1 | p40 | CK7 |
| Esophagus | p63 | CK5/CK6 | MUC2 | CK7 |
| Ovary | CA125 | WT‐1 | Pax‐8 | CK7 |
| Pancreas | CA19.9 | CK19 | Maspin | CK7 |
| Prostate | AMACR | PSMA | p63 | PSCA |
| Sarcomas | SMA | S100 | CSV | — |
| Stomach | CDX2 | CEA | CK7 | — |
| Thyroid | TTF‐1 | Thyroglobulin | Calcitonin | CK19 |
| Uterine | CK19 | Pax‐8 | CEA | CK7 |
Abbreviation: CNS, central nervous system.
The listed organ‐specific and subtype‐specific markers were evaluated by immunocytochemistry profiling for each cancer type.
Design of Organ‐Specific and Subtype‐Specific Immunocytochemistry Panels
Where EpCAM‐positive, PanCK‐positive clusters (C‐ETACs) were detected, these clusters were gently dissociated into single cells for ICC profiling to determine the status of OSS markers. We observed that the detection of OSS markers was more efficient and sensitive in single cells than in clusters. Cancer‐specific prescreening panels of OSS markers were designed based on publicly available information on antigen markers used in routine HPE or ICC analysis (Table 2). ICC methods were initially developed and optimized on respective control cell lines, which also were used for analytical validation. Details of the control cell lines, antibodies (primary and secondary), and fluorophores are provided in Supporting Table 4. All control cell lines used in this study were procured in the last 3 years. All cell lines were mycoplasma‐free.
TABLE 3.
Organ and Subtype‐Specific Antibody Panels to Discern Primary From Metastatic Deposits
| Primary | Metastasis | Primary | Metastasis | ||
|---|---|---|---|---|---|
| Marker 1 | Marker 2 | Marker 1 | Marker 2 | ||
| Bladder | Brain | Uroplakin‐II | GATA3 | GFAP | S100 |
| Breast | Brain | GCDFP15 | GATA3 | GFAP | S100 |
| Lung | GCDFP15 | GATA3 | Napsin‐A | TTF1 | |
| Liver | GCDFP15 | GATA3 | Glypican3 | HepPar1 | |
| Cervix | Brain | p63 | CK7 | GFAP | S100 |
| Colon | Brain | CDX2 | MUC2 | GFAP | S100 |
| Lung | CDX2 | MUC2 | Napsin‐A | TTF1 | |
| Liver | CDX2 | MUC2 | HepPar1 | Glypican3 | |
| Head and neck | Brain | p63 | HMWCK | GFAP | S100 |
| Kidney | Brain | CA‐IX | RCC | GFAP | S100 |
| Liver | Lung | Glypican3 | HepPar1 | Napsin‐A | TTF1 |
| Lung | Brain | Napsin‐A | TTF1 | GFAP | S100 |
| Liver | Napsin‐A | TTF1 | Glypican3 | HepPar1 | |
| Esophagus | Brain | p63 | CK5/6 | GFAP | S100 |
| Lung | p63 | CK5/6 | Napsin‐A | TTF1 | |
| Ovary | Brain | WT1 | PAX8 | GFAP | S100 |
| Liver | WT1 | PAX8 | Glypican3 | HepPar1 | |
| Pancreas | Lung | CA19.9 | Maspin | Napsin‐A | — |
| Liver | CA19.9 | Maspin | Glypican3 | — | |
| Stomach | Brain | CDX2 | CK7 | GFAP | S100 |
| Lung | CDX2 | CK7 | Napsin‐A | TTF1 | |
Combined Prospective and Retrospective Evaluation of Concordance
In a subset of 3509 samples (see Supporting Table 5), C‐ETACs were profiled with respective cancer‐specific OSS‐ICC panels. This subset included 2281 previously diagnosed and pretreated cases in which OSS‐ICC findings were retrospectively evaluated for concordance with HPE findings on a foundational biopsy during primary diagnostic workup. Concordance (%) was determined as the proportion of samples in which OSS‐ICC findings agreed with prior HPE findings. The remaining 1228 samples formed the prospective, double‐blinded evaluation cohort in which OSS‐ICC profiling of C‐ETACs and HPE of a biopsied tumor tissue sample were conducted concurrently. For all 1228 samples, OSS‐ICC panels were selected on the basis of clinician's recommendation of a suspected primary. Findings of HPE and OSS‐ICC profiling were masked from each other until all samples had been evaluated. After unblinding, concordance (%) was determined as the proportion of samples in which OSS‐ICC findings agreed with recent HPE findings.
In a subset of 229 samples (Supporting Table 6) from metastatic cancers, including samples from 163 previously diagnosed and pretreated patients and from 66 recently diagnosed therapy‐naive patients, the ICC profile of C‐ETACs was evaluated to determine fidelity in representing the primary organ versus the commonly observed organ(s) of metastases, such as lung, liver, and brain. C‐ETACs in this subset of samples were profiled with OSS‐ICC markers of the primary organs as well as the organ of metastasis (Table 3) to determine whether the approach accurately discerns the primary organ.
Results
Detection of C‐ETACs
C‐ETACs were detected in 14,221 of 15,441 samples (sensitivity, 92.1%) of various cancers. C‐ETACs were detected in 3623 of 3947 (91.8%) nonmetastatic cases, in 8959 of 9675 (92.6%) metastatic cases, in 5578 of 6025 (92.6%) recently diagnosed therapy‐naive cases, and in 8643 of 9416 (91.8%) previously diagnosed pretreated cases. C‐ETAC detection rates for each cancer type are provided in Table 4. Figure 1 depicts representative sets of images for ICC profiling of C‐ETACs and CTCs for EpCAM, PanCK, and CD45 status. Representative images depicting ICC profiling of C‐ETACs for these markers have also been published previously. 15 Among the 700 patients who were diagnosed with benign or other nonmalignant conditions, C‐ETACs were detected in 35 individuals (5%). Among the 13,919 asymptomatic individuals who had negative findings on all screening investigations, C‐ETACs were undetectable in 13,408 individuals (specificity, 96.3%).
TABLE 4.
Circulating Tumor Cell Detection Rates (Sensitivity) and Concordance of Organ‐Specific and Subtype‐Specific Panels With Histopathologic Examination Data (Accuracy)
| Cancer Type | CTC Detection Rate, % | OSS Marker Concordance Rate, % | ||||
|---|---|---|---|---|---|---|
| Prospective | Retrospective | Overall | Prospective | Retrospective | Overall | |
| Bladder | 91.0 | 96.2 | 94.7 | 100.0 | 98.1 | 98.5 |
| Breast | 92.4 | 92.5 | 92.5 | 95.4 | 93.4 | 94.0 |
| CNS | 90.0 | — | 90.0 | 90.0 | — | 90.0 |
| Cervix | 96.0 | 86.7 | 89.8 | 87.7 | 88.6 | 88.3 |
| Colorectum | 89.8 | 93.4 | 92.4 | 91.8 | 92.8 | 92.6 |
| Gallbladder | 97.3 | 90.1 | 93.0 | 100.0 | 87.9 | 91.3 |
| Head and neck | 92.2 | 92.8 | 92.5 | 97.9 | 98.5 | 98.3 |
| Kidney | 92.2 | 95.0 | 93.5 | 100.0 | 100.0 | 100.0 |
| Liver | 91.8 | 91.8 | 91.8 | 94.3 | 83.3 | 91.5 |
| Lung | 95.8 | 94.1 | 95.0 | 91.8 | 86.3 | 89.2 |
| Esophagus | 96.8 | 92.3 | 94.7 | 88.4 | 85.1 | 86.3 |
| Ovary | 86.8 | 85.6 | 85.9 | 96.2 | 86.2 | 87.7 |
| Pancreas | 96.6 | 91.8 | 94.1 | 100.0 | 93.0 | 96.0 |
| Prostate | 93.7 | 97.9 | 96.0 | 91.3 | 96.3 | 93.0 |
| Sarcoma | 94.1 | 95.3 | 95.1 | 100.0 | 100.0 | 100.0 |
| Stomach | 92.6 | 95.3 | 93.8 | 96.0 | 97.8 | 96.8 |
| Thyroid | 100.0 | 94.5 | 97.0 | 100.0 | 100.0 | 100.0 |
| Unknown primary | 89.3 | 81.5 | 88.9 | — | — | — |
| Uterine | 88.4 | 88.1 | 88.2 | 85.7 | 77.6 | 79.7 |
| Other | 89.4 | 85.0 | 89.3 | — | — | — |
| Overall | 92.6 | 91.8 | 92.1 | 93.6 | 92.8 | 93.1 |
Abbreviations: CNS, central nervous system; CTC, circulating tumor cells; OSS, organ and subtype specific.
Figure 1.
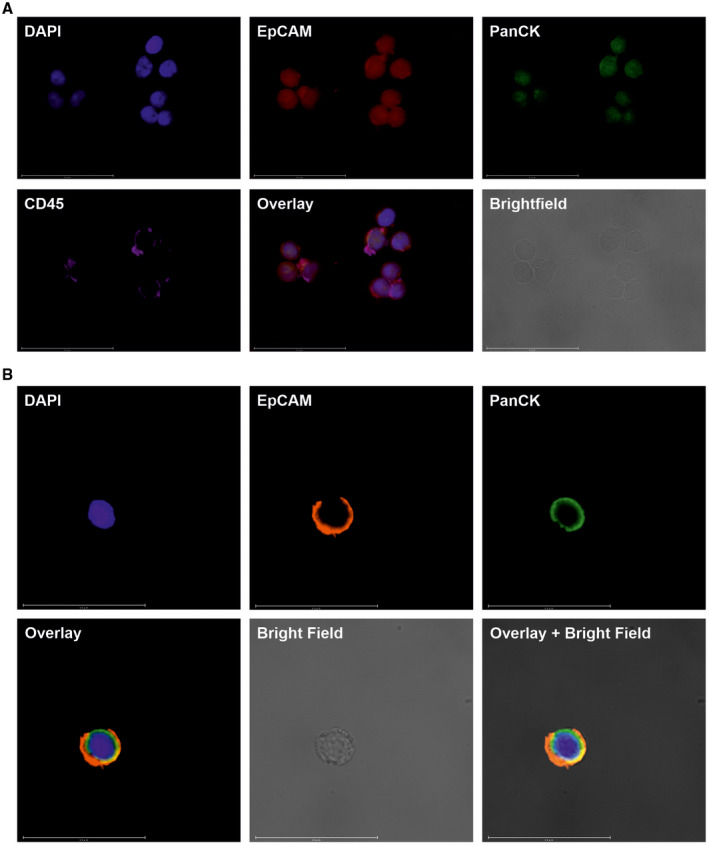
Images depict the identification of circulating ensembles of tumor‐associated cells (C‐ETACs) and circulating tumor cells (CTCs) by immunocytochemistry profiling. (A) C‐ETACs are defined as clusters of ≥3 cells that are positive for epithelial cell adhesion molecule (EpCAM), positive for pan‐cytokeratin (PanCK), and irrespective of CD45 status. (B) CTCs are defined as single cells that are positive for EpCAM, positive for PanCK, and negative for CD45. Representative images of C‐ETACs and CTCs are shown for 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI), EpCAM, and PanCK staining along with a fluorescence overlay, a brightfield image, and a brightfield image with fluorescence overlay.
Concordance of OSS‐ICC Profiling With HPE Findings
To determine whether ICC profiling can provide accurate representation of histologically relevant information, such as the organ of origin and subtype, we evaluated C‐ETACs from a subset of 3509 patient samples. Among the 1228 recently diagnosed and therapy‐naive individuals who formed the prospective cohort, OSS‐ICC profiling was accurate in 1150 cases (93.6%) and negative or aberrant in 78 cases (6.4%). Among the 2281 previously diagnosed and pretreated patients who formed the retrospective cohort, OSS‐ICC profiling was accurate in 2116 cases (92.8%) and negative or aberrant in 164 cases (7.2%). Overall, among the 3509 samples, OSS markers were accurate in 3266 cases (93.1%) and negative or aberrant in 243 cases (6.9%). Cancer‐specific concordance of OSS markers is detailed in Table 4. Also among the 3509 samples, OSS marker positivity rates were comparable in CTCs from metastatic (92.4%) and nonmetastatic (94%) samples irrespective of prior treatment status.
In the subset of 229 samples in which ICC profiling of C‐ETACs was evaluated for fidelity in determining primary deposits and in discerning primary from metastatic deposits, an overall 96.9% accuracy was determined based on 95.5% accuracy in 66 therapy‐naive cases and 97.5% accuracy in 163 pretreated cases (see Supporting Table 6).
Among the 35 C‐ETAC positive benign cases (out of a total of 700), C‐ETACs from 5 samples (0.71% out of 700) were positive for ≥1 OSS marker associated with the organ of suspicion, indicating a possible risk of malignancy. The low OSS positivity rate in benign indicates high specificity of the approach to discern malignant and benign conditions in a particular organ.
C‐ETACs detected in asymptomatic individuals were not profiled by ICC.
C‐ETAC positivity in individuals with benign conditions and in asymptomatic individuals were conveyed to the referring clinicians for further surveillance, the results of which will be communicated later. Figure 2 and Figure 3 depict representative images of CTCs profiled for OSS markers. Additional images of CTCs profiled for OSS markers are provided in the online Supporting Information (see Supporting Figs. 1‐10). Images of C‐ETACs profiled for OSS markers have been published previously.15
Figure 2.

Images depict immunohistochemistry (ICC) profiling of circulating tumor cells (CTCs) for organ‐specific and subtype‐specific (OSS) markers of carcinomas. Representative images show ICC profiles of CTCs from (A) lung cancers, (B) breast cancers, and (C) prostate cancers. (A) In lung cancer, (i) Napsin‐A and (ii) thyroid transcription factor‐1 (TTF‐1) are specific for adenocarcinoma (AD), whereas (iii) p40 is specific for squamous cell carcinoma (SCC). (B) In breast cancer, (iv) GATA‐binding protein 3 (GATA3) and (v) gross cystic disease fluid protein 15 (GCDFP15) are specific for ductal and lobular breast carcinomas. (C) In prostate cancer, (vi) prostate‐specific membrane antigen (PSMA) and (vii) α‐methylacyl coenzyme A racemase (AMACR) are specific for prostate adenocarcinoma (AD). Each row of images (i‐vii) shows 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) staining, an OSS marker, a fluorescence overlay, a brightfield image, and a brightfield image with fluorescence overlay. All CTCs were negative for CD45 (not depicted). Additional representative ICC profiling images of CTCs from other cancers are provided in the online Supporting Information.
Discussion
With approximately 18 million new cases diagnosed annually,16 cancer contributes significantly to the global disease burden. HPE of malignant tissue obtained by invasive biopsy is the current standard to determine malignant status in suspected cancer cases as well as for morphologic characterization of subtype, aggressiveness, and grade. Invasive biopsies not only cause pain, discomfort, and anxiety to patients but are also associated with procedural risks, such as hemorrhage, sepsis, and tumor seeding.17,18 Organ‐specific risks pose additional challenges to invasive biopsies. In the lung, percutaneous computed tomography‐guided transthoracic needle biopsy is associated with a risk of pneumothorax, leading to lung collapse, pneumonia, and systemic air embolism.19‐22 In the kidney, the risks of biopsy include dysuria, hematuria, hematoma, and arteriovenous fistula. 23 , 24 Biopsies of the liver and gallbladder are known to be associated with risks such as pneumothorax, hemothorax, bile peritonitis, hemobilia, intrahepatic arteriovenous fistula, and neuralgia. 25 Percutaneous biopsies of the pancreas are associated with risks such as macrohematuria, pancreatitis, exocrine leak, and inadvertent biopsy of other organs. 26 , 27 Risks associated with prostate biopsies include hematuria, hematospermia, rectal bleeding, vasovagal episodes, urosepsis, and acute urinary retention. 28 Among all biopsies, brain biopsies in individuals who present with intracranial space‐occupying lesions are perhaps most daunting because these are associated with risks of intracranial hemorrhage, morbidity, and mortality. 29 In addition to these risks, invasive biopsies may not be possible because of inaccessibility of the tumor or comorbidities. 2 , 3 , 5 , 30
The RESOLUTE, TrueBlood, GLIOLens and ProState studies were designed to evaluate the feasibility of detection and in vitro ICC profiling of C‐ETACs and/or CTCs for noninvasively screening and obtaining diagnostically relevant information in various cancers. On the basis of an evaluation of blood samples from an initial 10,625 samples in the RESOLUTE study, we previously demonstrated that C‐ETACs are rare (3.7%) in asymptomatic populations, 15 and detection rates were lower (3%) in individuals who formed the baseline‐risk subgroup (no aberrant findings in cancer markers or on low‐dose computed tomography, mammography, or Papanicolaou smear). Subsequently, 12,009 additional individuals were enrolled and, among the total 22,634 individuals, 13,919 formed the baseline‐risk subgroup, whereas 8715 who had ≥1 aberrant finding formed the elevated‐risk subgroup. C‐ETACs were detected in 4.5% of the entire population (n = 22,634), which included a 3.7% detection rate in the baseline‐risk subgroup and a 5.8% detection rate in the elevated‐risk subgroup.
Figure 3.
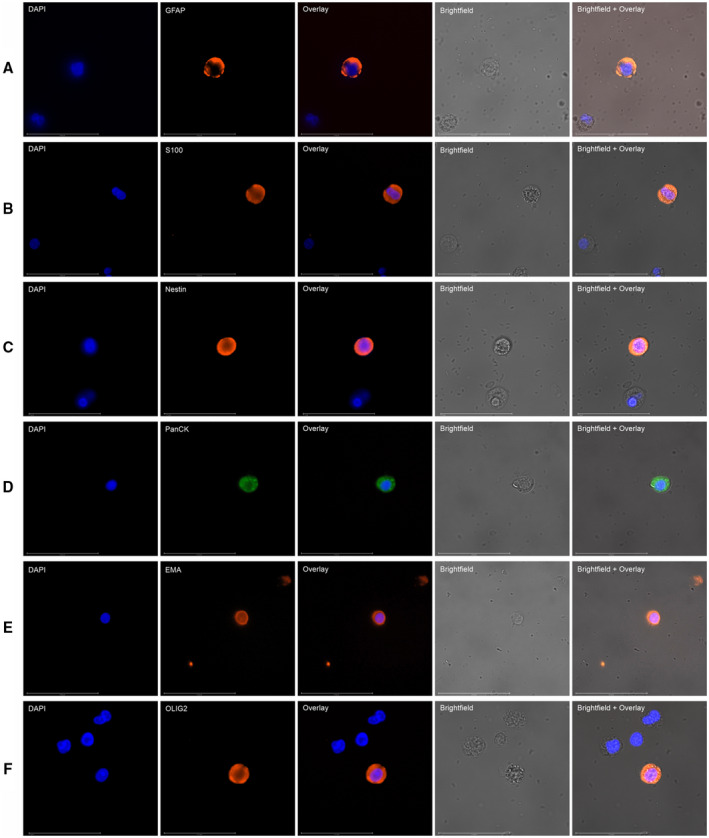
Images depict immunohistochemistry profiling of circulating tumor cells for central nervous system (CNS)‐specific markers. The markers used for CNS malignancy were (A) glial fibrillary acidic protein (GFAP), (B) S100, (C) Nestin, (D) pan‐cytokeratin (PanCK), (E) epithelial membrane antigen (EMA), and (F) oligodendrocyte transcription factor (OLIG2). Each row of 5 images shows 4′,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) staining, a CNS‐specific marker, a fluorescence overlay, a brightfield image, and a brightfield image with fluorescence overlay. All samples were negative for CD45 (not depicted).
The negative‐enrichment approach described previously 15 yielded sufficient C‐ETACs to permit meaningful downstream applications. The high detection rates of C‐ETACs across the entire cancer cohort were consistent with confirmed diagnoses of cancer, and the baseline detection rates in individuals with benign conditions and in asymptomatic individuals indicated high specificity. Because the detection and yield of C‐ETACs were not affected by therapy status, we found this approach suitable for longitudinal evaluations during treatment. The objective of the current study was not to evaluate a quantitative change in C‐ETACs based on extent of disease or in response to treatment.
The current approach is primarily intended for symptomatic individuals who have been referred for a biopsy but have not yet undergone the biopsy. Therefore, it is imperative to evaluate the performance characteristics of this approach on blood samples from a similarly biopsy‐naive population rather than a postbiopsy population to accurately ascertain its sensitivity. Hence the study population had sufficient representation from suspected cases in which the blood samples were collected before a biopsy. We observed no significant differences in C‐ETAC detection rates between biopsy‐naive individuals and patients who had undergone a previous diagnostic biopsy or between individuals with metastatic and nonmetastatic disease.
The detection of C‐ETACs (or CTCs) offers direct, visual evidence of malignancy; it is comparable to a positive finding of malignancy in HPE on a tumor tissue sample and is effectively an oligobiopsy/microbiopsy without stromal content, necrotic content, or normal tissue. C‐ETACs are viable malignant cells shed from a tumor and hence contribute to and retain (a subset of or in total) the overall molecular and functional imprint of the parent tumor. 31 C‐ETACs are a source of tumor analytes (proteins, DNA, RNA) as well as CTCs that may be evaluated for diagnostic inference. 32 In the current study, CTCs were present in all C‐ETAC–positive cases. A few prior reports have indicated the feasibility of evaluating CTCs for organ or origin markers. 8 , 33 , 34 In a subset analysis of 3,509 cancer samples, we observed significant concordance between OSS‐ICC profiles of C‐ETAC samples and HPE diagnoses in both the prospective and retrospective settings. Likewise, in another subset analysis of 229 samples from patients with metastatic solid organ cancers, we observed that ICC profiles of C‐ETACs accurately conveyed the primary cancer type/organ without any interference from OSS markers specific to the organ of metastasis. Thus we observed that C‐ETACs retained and faithfully conveyed the molecular and functional characteristics of the tumor tissue of origin, irrespective of metastatic status or prior therapy status, and had minimal intermarker interference.
Prior reports have indicated lower expression of OSS markers in poorly differentiated or undifferentiated tumors as well as in CTCs undergoing epithelial‐to‐mesenchymal transition. 35 In the current study, OSS markers were negative (undetectable) in C‐ETACs from a limited number of samples; this false negativity may be speculatively ascribed to dedifferentiation or epithelial‐to‐mesenchymal transition. Similarly, detection rates of OSS markers are currently restricted to classical CTCs (CD45‐negative). During analysis of our samples, OSS marker positivity was also observed in CD45‐positive subpopulations of C‐ETACs.
The standard for diagnosis in the current study is HPE of biopsied tumor tissue, the verdict of which (malignant vs benign) determined C‐ETAC findings as true‐positive or false‐positive. Any error in HPE would result in a conflict in diagnosis. Therefore, although the 5% detection rate of C‐ETACs in HPE‐determined benign cases is undesirable, it falls in the realm of inherent methodological limitations. No morphologic differences were observed between the C‐ETACs detected in malignant and benign cases. The 3.7% detection rate of C‐ETACs in asymptomatic individuals may represent a risk of malignancy that is currently without clinical or symptomatic manifestation. Individuals in both cases have been advised surveillance.
Image‐guided biopsies require specialized infrastructure and highly trained staff, which are generally unavailable at primary care centers and hence necessitate the patient's visit to a secondary or tertiary care facility, which can lead to increased time to diagnosis. By comparison, the current noninvasive approach requires a simple blood draw, which can be fulfilled at any primary health care clinic or even at the patient's home.
It is acknowledged that the total number of biopsies performed every year exceeds the actual number of diagnosed cases; the additional biopsies account for negative (benign cases and false‐negatives) and inconclusive findings on subsequent HPE. For example, it has been estimated that benign fibroadenomas account for the majority of all breast masses as well as biopsied lesions, thus adding up to a significantly high rate of negative findings. 36 , 37 , 38 Similarly, it has been estimated that the majority of all enlarged prostate cases are benign enlargements or inflammatory conditions. 39 In the ProState study, we evaluated 140 known cases of prostate cancer, 71 known cases of benign prostate hyperplasia/prostatitis, and 347 symptomatic cases with enlarged prostate suspicious for prostate cancer; of the latter, 111 were eventually diagnosed with prostate cancer, and 236 were diagnosed with benign conditions based on HPE of biopsied tissue. In our analysis of the 347 samples, in which the operator was initially blinded to the findings of HPE, we observed 98.9% overall accuracy for detection of prostate cancer in samples that were positive for at least 1 marker (α‐methylacyl coenzyme A racemase/prostate‐specific membrane antigen) and 93.1% overall accuracy in discerning prostate cancer from benign conditions. Currently, the only limitation of this approach for prostate cancers is that it has not been validated for concordance with the Gleason score. Among the 236 benign cases, 228 (96.6%) were accurately identified based on the absence of C‐ETACs. In a real‐world scenario, this represents the number of individuals with benign conditions for whom an unnecessary biopsy can be avoided. We foresee this analysis being used in conjunction with multiparametric magnetic resonance imaging for assessment, to identify patients who have a high probability of cancer and to achieve diagnostic triaging for individuals in whom the diagnosis still needs confirmation by tissue biopsy.
Malignancies of the CNS (brain tumors) are especially challenging because these are associated with significant limitations to biopsy and postbiopsy complications. Brain biopsies are considered especially challenging compared with biopsies of other organs because of the greater risks of morbidity and mortality associated with procedural complications, such as intracranial hemorrhage. 29 Unarguably, a noninvasive diagnostic approach would be most appreciated for CNS malignancies. In the subset of samples from the GlioLENS study, ICC profiling of C‐ETACs (with GFAP, S100, Nestin, PanCK, epithelial membrane antigen, and oligodendrocyte transcription factor) helped differentiate CNS malignancies from benign conditions and metastasis from primary carcinoma with 90% and 100% specificity, respectively, and it also ascertained the glial lineage with 90% accuracy.
Often a repeated biopsy may be desirable after suspected false‐negative or inconclusive findings on HPE or when progression of disease is suspected in the case of CNS malignancies. However, this may not be advisable or immediately viable because of health risks, expenses, logistical considerations, and delayed diagnosis and treatment. Conversely, inconclusive findings in C‐ETAC–based diagnosis merely necessitate another blood draw. Noninvasive approaches that can reduce dependence on invasive biopsies or defer the immediate need for a biopsy could alleviate infrastructural burdens on the health care system.
The objective of the current study was to raise and answer 3 analytical questions with regard to the clinical utility of a C‐ETAC–based diagnostic approach for symptomatic individuals presenting at a tertiary cancer care center and who have been advised to undergo an invasive biopsy: 1) whether it is possible to provide a noninvasive diagnosis of cancer with accuracy that is not inferior to that of conventional tissue‐based procedures, 2) whether C‐ETACs can be used for immunopathologic characterization of the tumor according to the tissue of origin, and 3) whether this approach is suitable and robust for the real‐time assessment of tumor dynamics in patients with pretreated cancer. All of these questions are answered affirmatively in light of the study findings. We demonstrate that viable C‐ETACs can be obtained in most patients with cancer and that ICC profiling of these C‐ETACs can provide diagnostically relevant information. The strength of the study lies in demonstrating: 1) the ability to detect and harvest C‐ETACs in a significant proportion of a large cohort of patients, 2) the ability to detect OSS markers in a majority of samples covering diverse cancer types, and 3) that the approach is feasible in all patients irrespective of extent of disease (metastatic status) and therapeutic status.
Furthermore, and because C‐ETACs are probably derived from the leading edge or tumor‐budding elements of a growing cancer and have their own evolving transcriptome, future research on molecular profiling of C‐ETACs may help unravel the metastatic potential and inherent aggressive nature of the evolving cancer and would be an intuitive addition to existing approaches for the molecular profiling of circulating tumor nucleic acids for diagnostic and treatment purposes.
The current study is based on existing antigen markers that are approved for use in the diagnosis of various solid organ cancers by HPE. Like HPE, the success of C‐ETAC–based diagnostic approaches are affected by the inherent limitations of these markers, including detection rates and cross‐reactivity. We have not evaluated the interference of ongoing chemotherapy on C‐ETAC yields or ICC; a gap of 21 days was ensured as a washout period for patients on systemic therapy before blood collection. Currently, this study is unable to report on melanoma because of lower prevalence rates and an insufficient sample size. Further evaluation of C‐ETACs for determining additional parameters, such as Ki‐67, grade, and status of therapeutically relevant markers (eg, estrogen receptor, human epidermal growth factor receptor , androgen receptor, programmed death‐ligand 1, and neurotrophic tyrosine receptor kinase), is expected to add to the value of this noninvasive approach.
In conclusion, in a large cohort study, we demonstrate for the first time the clinical potential of using C‐ETACs for noninvasive diagnostic triaging of suspected cancer cases, particularly in cases unfit for biopsy or in which biopsy is difficult for any reason, and for clinical decision making. The current study goes some way toward that Holy Grail of a simple blood test to detect cancer.
Funding Support
This entire study was self‐funded by Datar Cancer Genetics, and no external funding was received for the study.
Conflict of Interest Disclosures
Revati Patil, Pradip Fulmali, Vineet Datta, Prashant Kumar, Darshana Patil, and Dadasaheb Akolkar are full‐time employees of the study sponsor (Datar Cancer Genetics) and report personal fees from the company during the conduct of the study. The remaining authors made no disclosures.
Author Contributions
Darshana Patil,Prashant Kumar, and Dadasaheb Akolkar: Study design, supervision, data review, advised on results interpretation, and wrote the article. Revati Patil: Sample analysis, data review, and advised on results interpretation. Pradip Fulmali: Sample analysis and data review. Tim Crook: Study design and reviewed the article. Andrew Gaya: Study design and reviewed the article. Nicholas Plowman: Study design and reviewed the article. Raymond Page: Study design and reviewed the article. Vineet Datta: Study design and reviewed the article. Sewanti Limaye: Study design and reviewed the article. Anantabhushan Ranade: Study design and reviewed the article. Amit Bhatt: Study design and reviewed the article. All authors read and approved the final version.
Supporting information
Fig S1‐S10
Table s1‐6
Supplementary Material
Gaya A, Crook T, Plowman N, Ranade A, Limaye S, Bhatt A, Page R, Patil R, Fulmali P, Datta V, Kumar P, Patil D, Akolkar D. Evaluation of circulating tumor cell clusters for pan‐cancer noninvasive diagnostic triaging. Cancer Cytopathol 2021. 10.1002/cncy.22366
We acknowledge all patients and asymptomatic individuals who consented to participate in this study and provided blood samples. Samples from asymptomatic individuals were obtained from Medall Spark Diagnostics (multiple pan‐India locations), and samples from patients with cancer were obtained from HCG Manavata Cancer Center (Nasik), NueClear Healthcare (Mumbai), Chandak Cancer Care (Jalgaon), and HCG Cancer Center (Bengaluru). We also acknowledge the contributions of all Datar Cancer Genetics staff and scientists in managing various operational aspects of the study.
References
- 1. Mills SE. Histology for Pathologists. 4th ed. Lippincott Williams & Wilkins; 2012. [Google Scholar]
- 2. Hiley CT, Le Quesne J, Santis G, et al. Challenges in molecular testing in non‐small‐cell lung cancer patients with advanced disease. Lancet. 2016;388:1002‐1011. [DOI] [PubMed] [Google Scholar]
- 3. Abraham NE, Mendhiratta N, Taneja SS. Patterns of repeat prostate biopsy in contemporary clinical practice. J Urol. 2015;193:1178‐1184. [DOI] [PubMed] [Google Scholar]
- 4. Walk EE, Yohe SL, Beckman A, et al; College of American Pathologists Personalized Health Care Committee . The cancer immunotherapy biomarker testing landscape. Arch Pathol Lab Med. 2020;144:706‐724. [DOI] [PubMed] [Google Scholar]
- 5. Shachar SS, Fried G, Drumea K, Shafran N, Bar‐Sela G. Physicians' considerations for repeat biopsy in patients with recurrent metastatic breast cancer. Clin Breast Cancer. 2016;16:e43‐e48. [DOI] [PubMed] [Google Scholar]
- 6. De Rubis G, Rajeev Krishnan S, Bebawy M. Liquid biopsies in cancer diagnosis, monitoring, and prognosis. Trends Pharmacol Sci. 2019;40:172‐186. [DOI] [PubMed] [Google Scholar]
- 7. Krebs MG, Hou JM, Ward TH, Blackhall FH, Dive C. Circulating tumour cells: their utility in cancer management and predicting outcomes. Ther Adv Med Oncol. 2010;2:351‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cummings J, Sloane R, Morris K, et al. Optimisation of an immunohistochemistry method for the determination of androgen receptor expression levels in circulating tumour cells. BMC Cancer. 2014;14:226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cristofanilli M, Hayes DF, Budd GT, et al. Circulating tumor cells: a novel prognostic factor for newly diagnosed metastatic breast cancer. J Clin Oncol. 2005;23:1420‐1430. [DOI] [PubMed] [Google Scholar]
- 10. Danila DC, Heller G, Gignac GA, et al. Circulating tumor cell number and prognosis in progressive castration‐resistant prostate cancer. Clin Cancer Res. 2007;13:7053‐7058. [DOI] [PubMed] [Google Scholar]
- 11. Cohen SJ, Punt CJA, Iannotti N, et al. Relationship of circulating tumor cells to tumor response, progression‐free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:3213‐3221. [DOI] [PubMed] [Google Scholar]
- 12. Banko P, Lee SY, Nagygyorgy V, et al. Technologies for circulating tumor cell separation from whole blood. J Hematol Oncol. 2019;12:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gwak H, Kim J, Kashefi‐Kheyrabadi L, Kwak B, Hyun KA, Jung HI. Progress in circulating tumor cell research using microfluidic devices. Micromachines. 2018;9:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hong B, Zu Y. Detecting circulating tumor cells: current challenges and new trends. Theranostics. 2013;3:377‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akolkar D, Patil D, Crook T, et al. Circulating ensembles of tumor‐associated cells: a redoubtable new systemic hallmark of cancer. Int J Cancer. 2020;146:3485‐3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu CC, Maher MM, Shepard JA. Complications of CT‐guided percutaneous needle biopsy of the chest: prevention and management. Am J Roentgenol. 2011;196:W678‐W682. [DOI] [PubMed] [Google Scholar]
- 17. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 18. Sennerstam RB, Franzen BSH, Wiksell HOT, Auer GU. Core‐needle biopsy of breast cancer is associated with a higher rate of distant metastases 5 to 15 years after diagnosis than FNA biopsy. Cancer Cytopathol. 2017;125:748‐756. [DOI] [PubMed] [Google Scholar]
- 19. Robertson EG, Baxter G. Tumour seeding following percutaneous needle biopsy: the real story! Clin Radiol. 2011;66:1007‐1014. [DOI] [PubMed] [Google Scholar]
- 20. Khan MF, Straub R, Moghaddam SR, et al. Variables affecting the risk of pneumothorax and intrapulmonal hemorrhage in CT‐guided transthoracic biopsy. Eur Radiol. 2008;18:1356‐1363. [DOI] [PubMed] [Google Scholar]
- 21. Freund MC, Petersen J, Goder KC, Bunse T, Wiedermann F, Glodny B. Systemic air embolism during percutaneous core needle biopsy of the lung: frequency and risk factors. BMC Pulm Med. 2012;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lang D, Reinelt V, Horner A, et al. Complications of CT‐guided transthoracic lung biopsy: a short report on current literature and a case of systemic air embolism. Wien Klin Wochenschr. 2018;130(7‐8):288‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trajceska L, Severova‐Andreevska G, Dzekova‐Vidimliski P, et al. Complications and risks of percutaneous renal biopsy. Open Access Maced J Med Sci. 2019;7:992‐995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lubomirova M, Krasteva R, Bogov B, Paskalev E. Incidence of A‐V fistulas after renal biopsy of native and transplanted kidney—two centers experience. Open Access Maced J Med Sci. 2015;3:241‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Machado NO. Complications of liver biopsy—risk factors, management and recommendations. In: Takahashi H, ed. Liver Biopsy. IntechOpen; 2011. Accessed August 29, 2020. https://www.intechopen.com/books/liver‐biopsy/complications‐of‐liver‐biopsy‐risk‐factors‐management‐and‐recommendations [Google Scholar]
- 26. Atwell TD, Gorman B, Larson TS, Charboneau JW, Ingalls Hanson BM, Stegall MD. Pancreas transplants: experience with 232 percutaneous US‐guided biopsy procedures in 88 patients. Radiology. 2004;231:845‐849. [DOI] [PubMed] [Google Scholar]
- 27. Klassen DK, Weir MR, Cangro CB, Bartlett ST, Papadimitriou JC, Drachenberg CB. Pancreas allograft biopsy: safety of percutaneous biopsy‐results of a large experience. Transplantation. 2002;73:553‐555. [DOI] [PubMed] [Google Scholar]
- 28. Efesoy O, Bozlu M, Çayan S, Akbay E. Complications of transrectal ultrasound‐guided 12‐core prostate biopsy: a single center experience with 2049 patients. Turk J Urol. 2013;39:6‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malone H, Yang J, Hershman DL, Wright JD, Bruce JN, Neugut AI. Complications following stereotactic needle biopsy of intracranial tumors. World Neurosurg. 2015;84:1084‐1089. [DOI] [PubMed] [Google Scholar]
- 30. Amir E, Miller N, Geddie W, et al. Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J Clin Oncol. 2012;30:587‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Germano G, Mauri G, Siravegna G, et al. Parallel evaluation of circulating tumor DNA and circulating tumor cells in metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17:80‐83. [DOI] [PubMed] [Google Scholar]
- 32. Batth IS, Mitra A, Manier S, et al. Circulating tumor markers: harmonizing the yin and yang of CTCs and ctDNA for precision medicine [published correction appears in Ann Oncol. 2019;30:1845]. Ann Oncol. 2017;28:468‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paoletti C, Muniz MC, Thomas DG, et al. Development of circulating tumor cell‐endocrine therapy index in patients with hormone receptor‐positive breast cancer. Clin Cancer Res. 2015;21:2487‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chou J, Provot S, Werb Z. GATA3 in development and cancer differentiation: cells GATA have it! J Cell Physiol. 2010;222:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qi LN, Xiang BD, Wu FX, et al. Circulating tumor cells undergoing EMT provide a metric for diagnosis and prognosis of patients with hepatocellular carcinoma. Cancer Res. 2018;78:4731‐4744. [DOI] [PubMed] [Google Scholar]
- 36. Cerrato F, Labow BI. Diagnosis and management of fibroadenomas in the adolescent breast. Semin Plast Surg. 2013;27:23‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chang DS, McGrath MH. Management of benign tumors of the adolescent breast. Plast Reconstr Surg. 2007;120:13e‐19e. [DOI] [PubMed] [Google Scholar]
- 38. Lee M, Soltanian HT. Breast fibroadenomas in adolescents: current perspectives. Adolesc Health Med Ther. 2015;6:159‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miah S, Catto J. BPH and prostate cancer risk. Indian J Urol. 2014;30:214‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S10
Table s1‐6
Supplementary Material