

Abstract
A series of copolymers containing 50 mol % acrylic acid (AA) and 50 mol % butyl acrylate (BA) but with differing composition profiles ranging from an AA‐BA diblock copolymer to a linear gradient poly(AA‐grad‐BA) copolymer were synthesized and their pH‐responsive self‐assembly behavior was investigated. While assemblies of the AA‐BA diblock copolymer were kinetically frozen, the gradient‐like compositions underwent reversible changes in size and morphology in response to changes in pH. In particular, a diblock copolymer consisting of two random copolymer segments of equal length (16 mol % and 84 mol % AA content, respectively) formed spherical micelles at pH >5, a mix of spherical and wormlike micelles at pH 5 and vesicles at pH 4. These assemblies were characterized by dynamic light scattering, cryo‐transmission electron microscopy and small angle neutron scattering.
Keywords: copolymer, gradient copolymer, self-assembly, sequence control, stimuli-responsive
Copolymers of acrylic acid (AA) and butyl acrylate (BA) with constant overall composition (1:1 AA:BA) and controlled composition profiles (gradient, asymmetric diblock and asymmetric triblock) show dynamic pH‐responsive self‐assembly behavior, with reversible changes in size and formation of sphere, worm and vesicle morphologies, in contrast to the frozen micelles formed by poly(AA‐block‐BA) block copolymers.
Introduction
Natural polymers form a great variety of self‐assembled structures that are essential to their function. Remarkably, these are achieved using a very restricted alphabet of monomers: the vast natural library of proteins is made up of only 20–22 amino acids. [1] It is the arrangement of the monomers, as much as their individual properties, that determines the final structure and properties of the polymer, and allows it to fill roles ranging from structural reinforcement to precisely tuned catalysis.
In the realm of high‐volume synthetic polymers, by contrast, we dispose of a vast range of monomers. While control over polymer molar mass distribution is improving, [2] control over monomer arrangement remains limited. [3] Nature's example suggests, however, that applying even this limited degree of control could allow a wider range of properties to be obtained from copolymers of a relatively small library of monomers with controlled composition profiles.
Amphiphilic copolymers containing ionizable groups such as carboxylic acids and amines form a wide range of self‐assembled structures in response to changes in pH.[ 4 , 5 , 6 ] Transitions from unimeric solutions to spherical micelles are common,[ 7 , 8 ] while with careful control of the hydrophilic‐hydrophobic balance of the copolymer, transitions from spherical micelles to wormlike micelles[ 9 , 10 , 11 ] and/or vesicles[ 12 , 13 , 14 , 15 ] may also be observed.
Most studies of pH‐responsive polymers have focused on block copolymers comprising two or more homopolymer segments. The presence of a strongly hydrophobic segment results in the formation of kinetically trapped structures that respond slowly, if at all, to further changes in their environment. [16] Structures that can transition between different morphologies are relatively rare, and have for the most part been obtained using weakly hydrophobic monomers, such as hydroxypropyl methacrylate.[ 5 , 9 , 10 , 11 , 15 ]
More recently, amphiphilic copolymers containing mixed segments (e.g. gradient copolymers and block copolymers with hydrophilic monomers incorporated into the hydrophobic segment) have been shown to form self‐assemblies that respond dynamically to changes in their environment.[ 17 , 18 ] Gradient copolymer assemblies may exhibit reversible changes in size[ 7 , 19 , 20 ] or shape[ 20 , 21 ] in response to changes in solvent quality. Diblock and triblock copolymers containing hydrophilic polyacrylic acid and hydrophobic poly(acrylic acid‐ran‐butyl acrylate)[ 22 , 23 , 24 ] or poly(acrylic acid‐ran‐styrene)[ 25 , 26 , 27 ] segments show reversible associations across a wide pH range. Copolymers with more complex composition profiles, incorporating hydrophilic homopolymer and moderately hydrophobic gradient copolymer segments may also undergo reversible self‐assembly in response to changes in temperature[ 28 , 29 , 30 ] or pH.[ 8 , 29 ] These results indicate that the incorporation of hydrophilic groups into the hydrophobic segment of an amphiphilic copolymer can have a significant effect on its self‐assembly behavior. However, the effect of the distribution of these hydrophilic groups, whether randomly distributed in a single segment, asymmetrically distributed in a gradient throughout the polymer chain, or as part of a more complex composition profile, remains unclear.
Here we show that a single copolymer comprising equal parts of n‐butyl acrylate (BA) and acrylic acid (AA), with a controlled spatial distribution of AA groups, is capable of forming spherical micelles, wormlike micelles and vesicles in response to changes in pH. More generally, 1:1 AA‐BA copolymers with asymmetric or gradient‐like distributions of AA form dynamically responsive self‐assemblies for pH ≥5, which undergo changes in size and morphology in response to changes in pH. This is in contrast to the kinetically trapped, frozen assemblies that are formed from the block copolymer poly(AA‐block‐BA), comprising two homopolymer segments. These results demonstrate the role of the spatial distribution of hydrophilic and hydrophobic groups in amphiphilic stimuli‐responsive copolymers in modulating the size and morphology of their self‐assemblies as well as their dynamic response to changes in their environment.
Results and Discussion
Polymer precursors were synthesized from tert‐butyl acrylate (tBA) and BA via reversible addition‐fragmentation chain transfer (RAFT) polymerization (Scheme 1). These monomers displayed nearly identical reactivity, forming random copolymers without composition drift across a wide composition range (see Supporting Information, Figure S1).
Scheme 1.
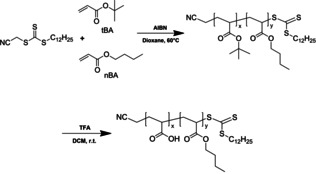
Schematic representation of the synthesis of amphiphilic block and gradient copolymers by RAFT (co)polymerizations of n‐butyl acrylate and t‐butyl acrylate followed by acidolysis of t‐butyl groups.
Four composition profiles were targeted, each containing 50 mol % BA and 50 mol % tBA (Figure 1): a poly(tBA‐block‐BA) block copolymer (B); a poly(tBA‐grad‐BA) gradient copolymer (G) of nominally linear composition profile; an asymmetric diblock copolymer (D) consisting of two poly(tBA‐ran‐BA) blocks of equal targeted number average degrees of polymerization (DP n) comprising 16 mol % and 84 mol % tBA, respectively; and an asymmetric triblock copolymer (T) consisting of a short block of poly(tBA), a longer block of poly(tBA‐ran‐BA) comprising 50 mol % tBA, and a short block of poly(BA). The targeted DP n of the component blocks of T were in the proportion 21:58:21. The gradient profile was obtained using a starved feed semibatch process with continuous addition of both monomers, [31] while the asymmetric diblock and triblock profiles were obtained via sequential polymerizations using a robotic parallel synthesizer. [32] The asymmetric structures D and T were chosen to mimic the linear gradient profile using a minimal number of steps (see Supporting Information for details on profile selection). Each composition profile was realized at overall number average molar masses (M n) of 10 and 20 kg mol−1.
Figure 1.
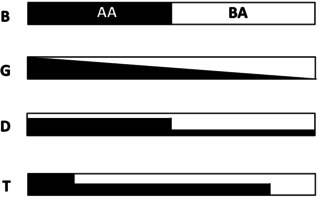
Targeted composition profiles of block copolymers (B), linear gradient copolymers (G) and asymmetric diblock (D) and triblock (T) copolymers.
The tert‐butyl groups were subsequently selectively and quantitatively acidolyzed using trifluoroacetic acid to yield amphiphilic copolymers of AA and BA, in which the distribution of AA units faithfully reproduces the distribution of tBA units in the original polymer. [33] Full details of synthesis and characterization are provided in the Supporting Information (Figures S2–S9, Tables S1 and S2); their characteristic properties are summarized in Table 1. The polymers are referred to as, for example, G20, where G represents the gradient profile, and 20 the targeted M n (before acidolysis) of 20 kg mol−1. Synthesis of the gradient copolymers with linear composition profiles was significantly more challenging than for the other structures. As a result, control over molar mass and dispersity (Ð) was less precise, with a broader range of molar mass and Ð of 1.35–1.38 for these polymers as compared to ≈1.1 for the block, asymmetric diblock and triblock copolymers.
Table 1.
Molar mass (before acidolysis) and composition data for block and gradient copolymers synthesized.
|
Polymer |
Profile |
Overall |
|
Component blocks |
|||
|---|---|---|---|---|---|---|---|
|
|
|
M n [b] (σ[a]), kg mol−1 |
Ð [b] |
%nBA[c] |
M n (σ[a]), kg mol−1 |
Ð |
%nBA[c] |
|
B10 |
Block |
11.9 (3.8) |
1.10 |
47.7 |
6.2[b] (1.8) 5.7[d] (3.3) |
1.08[b] 1.34[d] |
0 100 |
|
B20 |
Block |
23.2 (8.0) |
1.12 |
49.3 |
11.6 (3.1) 11.6[d] (7.2) |
1.07 1.38[d] |
0 100 |
|
D10 |
Asymmetric Diblock |
10.8 (3.2) |
1.09 |
49.3 |
4.9[b] (1.6) 5.9[d] (2.8) |
1.11[b] 1.23[d] |
16.0 85.4 |
|
D20 |
Asymmetric Diblock |
20.9 (6.6) |
1.10 |
45.4 |
10.0[b] (3.5) 10.9[d] (5.7) |
1.12[b] 1.27[d] |
16.1 83.9 |
|
T10 |
Asymmetric Triblock |
9.7 (2.6) |
1.07 |
47.3 |
1.7 (0.5)[b] 6.1 (2.1)[d] 2.2 (1.5)[d] |
1.10[b] 1.12[d] 1.44[d] |
0 50.0 100 |
|
T20 |
Asymmetric Triblock |
20.1 (5.3) |
1.07 |
44.2 |
4.1 (1.2)[b] 11.8 (4.1)[d] 4.2 (3.3)[d] |
1.09[b] 1.12[d] 1.60[d] |
0 49.5 100 |
|
G10 |
Gradient |
8.0 (4.9) |
1.38 |
51.4 |
|
|
|
|
G20 |
Gradient |
28.9 (17.1) |
1.35 |
43.9 |
|||
Potentiometric titration of the 20 kg mol−1 polymers in the presence of 0.1 M NaCl showed that all were essentially fully ionized at pH ≥9, 80 to 90 % ionized at pH 7, 20 to 40 % ionized at pH 5, and less than 10 % ionized at pH 4. They exhibited an apparent pKa at 50 % ionization ranging from 5.5 (B20) to 5.8 (G20). The full titration curves are reported in the Supporting Information (Figures S10–S11).
The effect of pH on the self‐assembly of the polymers in aqueous solution was first investigated by dynamic light scattering (DLS). The polymers were dissolved directly into 0.1 M NaOH solution at a concentration of 0.2 wt %, ensuring their complete ionization. The solution was then titrated first with aq. HCl until it became turbid (typically at pH ≈4), then with aq. NaOH to return to high pH. DLS correlograms were obtained at regular intervals during titration. In parallel, the polymers were directly dissolved in buffer solutions at pH of 10, 8, 7, 6, 5 and 4. At pH 4, the polymers did not dissolve spontaneously in water, and dispersions were obtained by heating with microwave irradiation. These solutions were also analyzed using DLS. The apparent particle size distributions were monomodal in nearly all cases, with polydispersity indices (PDIs) ranging from less than 0.1 in the case of D20 to a maximum of 0.46 in the case of G20 at pH 4.45 (see Supporting Information Figures S12–S23, Tables S3–S11 for details). Figure 2 shows the variation of apparent hydrodynamic diameter (D h) as a function of pH for both the titrated samples and those that were directly dispersed in buffer solution.
Figure 2.
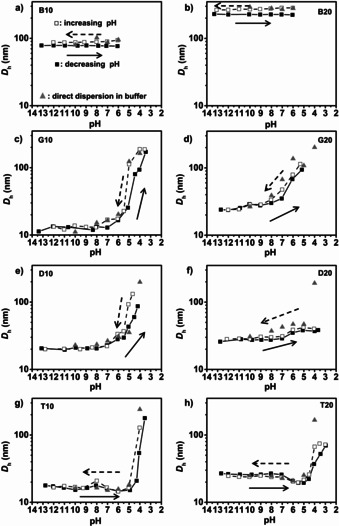
Apparent Z‐average hydrodynamic diameters (D h) obtained by DLS for different types of copolymers with molar mass of 10 and 20 kg mol−1 using two different methods (directly dispersed in different pH buffers (grey triangles) and titration study (decreasing pH: black squares; increasing pH: open squares)): a) B10 b) B20 c) G10 d) G20 e) D10 f) D20 g) T10 h) T20. The arrows indicate the direction of the titration (solid: decreasing pH; dashed: increasing pH;).
The apparent D h of self‐assemblies of the block copolymers B10 and B20 were unaffected by changes in pH for pH >5 (degree of ionization, α>20 %). At pH 5, the polymer precipitated. Significant differences in apparent D h were observed in the forward and back titrations, indicating that the self‐assemblies were kinetically trapped, non‐equilibrium species in agreement with earlier results from the literature.[ 35 , 36 , 37 ]
In contrast, the apparent D h of the gradient copolymers and asymmetric diblock copolymers were sensitive to pH, steadily increasing from 10–30 nm at pH greater than 7 to around 100 nm at pH of 4 to 5. The DLS titration curves were reversible and generally in good agreement with apparent D hs obtained by direct dissolution in buffer solution. Hysteresis was observed for pH less than ≈5, suggesting that equilibration is slow or nonexistent when the degree of ionization is low, which is consistent with previous observations of block copolymers of poly(AA) and poly(AA‐ranBA).[ 38 , 39 ] Finally, the apparent D h of the asymmetric triblock copolymers remained constant or decreased slightly as the pH decreased, before abruptly increasing at pH 4 (α<10 %). Again, the changes in size were reversible and independent of the method of preparation for pH >4.
Selected samples (B20, G20, D10 and T10, prepared by direct dispersion in buffer) were subsequently analyzed by cryo‐transmission electron microscopy (Cryo‐TEM), with representative images displayed in Figure 3 (additional Cryo‐TEM images are available in the Supporting Information, Figures S24–S40). These images were generally consistent with the trends in particle size observed by DLS, but allowed direct evaluation of particle morphology.
Figure 3.

Representative Cryo‐TEM images of the self‐assemblies of different types of copolymers directly dispersed in different pH buffers: a) B20; b) G20; c) D10; and d) T10. More images are available in the Supporting Information (Figures S24–S40).
Cryo‐TEM images of B20 displayed spherical particles with diameters of ≈40 nm. At all pHs, they are densely packed in larger clusters of bigger size, which explains the larger particle diameters obtained by DLS. The presence of these clusters in solution at pH 4 and pH 10 was confirmed by small angle neutron scattering (Figure S41). More ill‐defined, highly polydisperse spherical particles dominate at pH 4, although some larger particles including wormlike micelles and vesicles, were also observed (Figures 3 a and S27) and some macroscopic phase separation occurs.
By contrast, spherical assemblies of G20 and D10 increased in size as the pH decreased from 10 (fully ionized) to 7 (α=80–90 %). At pH 5 (α=20–40 %), D10 exhibited a mixture of wormlike and spherical structures, while at pH 4 (α<10 %) vesicles dominated. For G20, aggregates of spherical assemblies were observed at pH 5, while at pH 4 a mixture of vesicles and wormlike micelles was observed. The presence of wormlike micelles and vesicles rather than only vesicles as in the other structures may be related to the larger dispersity of the gradient copolymer, as increased dispersity is known to displace the phase diagrams of block copolymers,[ 40 , 41 , 42 , 43 , 44 ] for example pushing the cylinder/lamellar phase boundary of polystyrene‐block‐polyisoprene‐block‐polystyrene elastomers to higher volume fractions of polystyrene. [44] Wormlike structures have previously been observed in aqueous dispersions of block copolymers of BA and AA prepared at low pH; these irreversibly transform into spheres when the pH is raised.[ 35 , 45 ]
Finally, assemblies of T10 remained small (≈10 nm) and spherical from pH 10 to pH 5, while only vesicles of 100 to 300 nm in diameter were observed at pH 4.
The asymmetric diblock, triblock and gradient structures showed broad similarities in their response, forming spherical structures at high pH and vesicles at low pH, with a dynamic and reversible response to changes in pH (at least for pH ≥5) that was not observed for the block copolymers B10 and B20. While the triblock copolymers were superficially more similar in composition profile to the linear gradient copolymers, it was the shorter asymmetric diblock copolymer D10 that most closely mimicked the size and morphological transitions observed for gradient copolymers. This may be due to the presence of a segment of poly(BA) homopolymer in the triblock copolymers, which could be expected to significantly retard exchange between micelles.[ 33 , 35 , 37 ]
The observed transformations of D10 seemed particularly noteworthy, as this copolymer forms spheres of varying size, worms or vesicles in response to changes in pH, with evidence of reversibility at least for pH >5. Such a range of structures is uncommon for any single polymer composition, and in this case the effect of varying the spatial distribution of hydrophobic and hydrophilic units is particularly apparent. This sample was further investigated using small angle neutron scattering at pH 10, 7, 5 and 4.
The results, shown in Figure 4, indicated a 103‐fold increase of the molar mass of the self‐assemblies going from pH 10 to pH 4 as a consequence of the morphological transition and concomitant increase of the number of copolymer chains in each self‐assembly (see inset Figure 4). Interestingly, the scattering data at pH 10 and 7 presented a plateau at low q values indicating small quasi spherical nano‐objects. For pH 5 and 4, at low q, the scattering intensity I(q) was proportional to q−1 and q−2 respectively and indicated the presence of cylindrical and lamellar species. This qualitative observation was confirmed by the fitting of the data, which suggested that the sample comprised slightly elongated micelles at pH 10 and 7, long cylinders at pH 5 (with a possible small contribution from spherical nano‐objects, see Figure S41 for more details), and predominantly vesicles at pH 4. These conclusions are consistent with Cryo‐TEM images. Further details of the fitting procedure, including the fitted model parameters, are available in the Supporting Information (Table S12).
Figure 4.
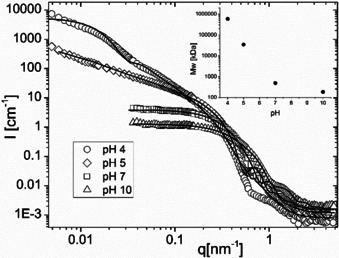
Small angle neutron scattering patterns for D10 as a function of pH for pH4, 5, 7 and 10. The lines are the fitting curves using the models detailed in Supporting Information. The shift in the scattering pattern to lower q with decreasing pH together with the increase of the forward scattering are consistent with an increase in particle size (inset), while the changing shape of the scattering patterns corresponds to a transition from slightly elongated micelles (pH 10 and 7) to long cylindrical micelles (pH 5) and vesicles (pH 4).
Conclusion
These remarkable differences in self‐assembly between block, gradient, asymmetric diblock and asymmetric triblock copolymers, despite their identical compositions and molar masses, underline the importance of controlling the distribution of functional groups within a polymer chain. Passing from a purely block‐like structure to more gradient‐like asymmetric structures that incorporate statistical copolymer segments transforms the frozen, kinetically trapped assemblies of the block copolymer into dynamically responsive assemblies that can change both their size and their shape in response to changes in their environment. By an appropriate selection of the composition profile, a single acrylic acid‐butyl acrylate copolymer (D10) can be induced to form spherical, wormlike or vesicular morphologies under different pH conditions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research was financially supported by the ASYMCOPO Project, an international collaborative research project of the Deutsche Forschungsgemeinschaft (DFG, Germany) and the Agence Nationale de la Recherche (ANR, France); DFG project: GU 1685/1‐1 (J.Z., J.U., C.G.S. and U.S.S.) and ANR project: ANR‐15‐CE08‐0039 (S.H.). C.G.S., J.U., S.H. and U.S.S. thank the Collaborative Research Center “PolyTarget” (SFB 1278, (project number 316213987), projects B02 and Z01) of the DFG for financial support. J.Z. thanks the National Natural Science Foundation of China (21901208) and the Natural Science Basic Research Plan in Shaanxi Province of China (2020JQ‐138) for financial support. B.F.M. acknowledges financial support from the Consejo Nacional de Ciencia y Tecnologia (CONACyT, Mexico) to pursue her Ph.D. Transmission electron microscopy facilities of the Jena Center of Soft Matter (JCSM) were established with a grant from the DFG and the European Funds for Regional Development (EFRE). We thank the Institut Laue‐Langevin for allocations of neutron beam time on D11, experiment no. 9‐11‐1877; dataset available at doi:10.5291/ILL‐DATA.9‐11‐1877. Open access funding enabled and organized by Projekt DEAL.
J. Zhang, B. Farias-Mancilla, I. Kulai, S. Hoeppener, B. Lonetti, S. Prévost, J. Ulbrich, M. Destarac, O. Colombani, U. S. Schubert, C. Guerrero-Sanchez, S. Harrisson, Angew. Chem. Int. Ed. 2021, 60, 4925.
Contributor Information
Prof. Ulrich S. Schubert, Email: ulrich.schubert@uni-jena.de.
Dr. Carlos Guerrero‐Sanchez, Email: carlos.guerrero.sanchez@uni-jena.de.
Dr. Simon Harrisson, Email: simon.harrisson@enscbp.fr.
References
- 1.Twenty amino acids are encoded in the standard DNA code, while in rare cases selenocysteine and pyrrolysine are encoded by the UGA and UAG stop codons, respectively. Clark D. P., Pazdernik N. J., McGehee M. R., Molecular Biology, 3 rd ed., Elsevier Academic Press, London, 2019, pp. 436–439. [Google Scholar]
- 2. Whitfield R., Parkatzidis K., Truong N. P., Junkers T., Anastasaki A., Chem 2020, 6, 1340–1352. [Google Scholar]
- 3. Gody G., Zetterlund P. B., Perrier S., Harrisson S., Nat. Commun. 2016, 7, 10514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kocak G., Tuncer C., Bütün V., Polym. Chem. 2017, 8, 144–176. [Google Scholar]
- 5. Pei Y., Lowe A. B., Roth P. J., Macromol. Rapid Commun. 2017, 38, 1600528. [DOI] [PubMed] [Google Scholar]
- 6. Grubbs R. B., Sun Z., Chem. Soc. Rev. 2013, 42, 7436–7445. [DOI] [PubMed] [Google Scholar]
- 7. Zhao Y., Luo Y.-W., Li B.-G., Zhu S., Langmuir 2011, 27, 11306–11315. [DOI] [PubMed] [Google Scholar]
- 8. Harrisson S., Ercole F., Muir B. W., Polym. Chem. 2010, 1, 326–332. [Google Scholar]
- 9. Penfold N. J. W., Lovett J. R., Warren N. J., Verstraete P., Smets J., Armes S., Polym. Chem. 2016, 7, 79–88. [Google Scholar]
- 10. Penfold N. J. W., Lovett J. R., Verstraete P., Smets J., Armes S., Polym. Chem. 2017, 8, 272–282. [Google Scholar]
- 11. Lovett J. R., Warren N. J., Ratcliffe L. P. D., Kocik M. K., Armes S. P., Angew. Chem. Int. Ed. 2015, 54, 1279–1283; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1295–1299. [Google Scholar]
- 12. Maiti C., Banerjee R., Maiti S., Dhara D., Langmuir 2015, 31, 32–41. [DOI] [PubMed] [Google Scholar]
- 13. Yildirim T., Traeger A., Sungur P., Hoeppener S., Kellner C., Yildirim I., Pretzel D., Schubert S., Schubert U. S., Biomacromolecules 2017, 18, 3280–3290. [DOI] [PubMed] [Google Scholar]
- 14. Luo C., Wei N., Luo X., Luo F., Macromol. Chem. Phys. 2018, 219, 1800124. [Google Scholar]
- 15. Lovett J. R., Warren N. J., Armes S. P., Smallridge M. J., Cracknell R. B., Macromolecules 2016, 49, 1016–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicolai T., Colombani O., Chassenieux C., Soft Matter 2010, 6, 3111–3118. [Google Scholar]
- 17. Zhang J., Farias-Mancilla B., Destarac M., Schubert U. S., Keddie D. J., Guerrero-Sanchez C., Harrisson S., Macromol. Rapid Commun. 2018, 39, 1800357. [DOI] [PubMed] [Google Scholar]
- 18. Zheng C., Soft Matter 2019, 15, 5357–5369. [DOI] [PubMed] [Google Scholar]
- 19. Okabe S., Seno K.-I., Kanaoka S., Aoshima S., Shibayama M., Macromolecules 2006, 39, 1592–1597. [Google Scholar]
- 20. Zheng C., Huang H., He T., Macromol. Rapid Commun. 2013, 34, 1654–1661. [DOI] [PubMed] [Google Scholar]
- 21. Hoogenboom R., Thijs H. M. L., Wouters D., Hoeppener S., Schubert U. S., Macromolecules 2008, 41, 1581–1583. [Google Scholar]
- 22. Lejeune E., Drechsler M., Jestin J., Müller A. H. E., Chassenieux C., Colombani O., Macromolecules 2010, 43, 2667–2671. [Google Scholar]
- 23. Charbonneau D., Lima M. D. S., Chassenieux C., Colombani O., Nicolai T., Phys. Chem. Chem. Phys. 2013, 15, 3955–3964. [DOI] [PubMed] [Google Scholar]
- 24. Lauber L., Chassenieux C., Nicolai T., Colombani O., Macromolecules 2015, 48, 7613–7619. [Google Scholar]
- 25. Bendejacq D. D., Ponsinet V., J. Phys. Chem. B 2008, 112, 7996–8009. [DOI] [PubMed] [Google Scholar]
- 26. Bendejacq D. D., Ponsinet V., Joanicot M., Langmuir 2005, 21, 1712–1718. [DOI] [PubMed] [Google Scholar]
- 27. Laruelle G., François J., Billon L., Macromol. Rapid Commun. 2004, 25, 1839–1844. [Google Scholar]
- 28. Yañez-Macias R., Kulai I., Ulbrich J., Yildirim T., Sungur P., Hoeppener S., Guerrero-Santos R., Schubert U. S., Destarac M., Guerrero-Sanchez C., Harrisson S., Polym. Chem. 2017, 8, 5023–5032. [Google Scholar]
- 29. Rabyk M., Destephen A. L., Lapp A., King S., Noirez L., Billon L., Hruby M., Borisov O., Stepanek P., Deniau E., Macromolecules 2018, 51, 5219. [Google Scholar]
- 30. Lechuga-Islas V. D., Festag G., Rosales-Guzmán M., Vega-Becerra O. E., Guerrero-Santos R., Schubert U. S., Guerrero-Sánchez C., Eur. Polym. J. 2020, 124, 109457. [Google Scholar]
- 31. D′hooge D., Van Steenberge P., Reyniers M.-F., Marin G., Polymers 2014, 6, 1074–1095. [Google Scholar]
- 32. Guerrero-Sanchez C., Harrisson S., Keddie D. J., Macromol. Symp. 2013, 325–326, 38–46. [Google Scholar]
- 33. Colombani O., Ruppel M., Schumacher M., Pergushov D., Schubert F., Müller A. H. E., Macromolecules 2007, 40, 4338–4350. [Google Scholar]
- 34. Harrisson S., Polym. Chem. 2018, 9, 1366–1370. [Google Scholar]
- 35. Jacquin M., Muller P., Talingting-Pabalan R., Cottet H., Berret J.-F., Futterer T., Theodoly O., J. Colloid Interface Sci. 2007, 316, 897–911. [DOI] [PubMed] [Google Scholar]
- 36. Johnson B. K., Prud'homme R. K., Phys. Rev. Lett. 2003, 91, 118301. [DOI] [PubMed] [Google Scholar]
- 37. Colombani O., Burkhardt M., Drechsler M., Ruppel M., Schumacher M., Gradzielski M., Schweins R., Mueller A. H. E., Macromolecules 2007, 40, 4351–4362. [Google Scholar]
- 38. Charbonneau C., Chassenieux C., Colombani O., Nicolai T., Macromolecules 2011, 44, 4487–4495. [Google Scholar]
- 39. Shedge A., Colombani O., Nicolai T., Chassenieux C., Macromolecules 2014, 47, 2439–2444. [Google Scholar]
- 40. Whitfield R., Truong N. P., Messmer D., Parkatzidis K., Rolland M., Anastasaki A., Chem. Sci. 2019, 10, 8724–8734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Listak J., Jakubowski W., Mueller L., Plichta A., Matyjaszewski K., Bockstaller M. R., Macromolecules 2008, 41, 5919–5927. [Google Scholar]
- 42. Widin J. M., Schmitt A. K., Schmitt A. L., Im K., Mahanthappa M. K., J. Am. Chem. Soc. 2012, 134, 3834–3844. [DOI] [PubMed] [Google Scholar]
- 43. Gentekos D. T., Fors B. P., ACS Macro Lett. 2018, 7, 677–682. [DOI] [PubMed] [Google Scholar]
- 44. Rosenbloom S. I., Fors B. P., Macromolecules 2020, 53, 7479–7486. [Google Scholar]
- 45. Eghbali E., Colombani O., Dreschler M., Müller A. H. E., Hoffmann H., Langmuir 2006, 22, 4766–4776. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary