

Abstract
Tucatinib is a potent tyrosine kinase inhibitor selective for human epidermal growth factor receptor 2 (HER2) approved by the US Food and Drug Administration for the treatment of HER2‐positive metastatic breast cancer and in development for other HER2‐positive solid tumors. Modest, reversible serum creatinine (SCr) elevations have been observed in tucatinib clinical trials. SCr is conveyed by the renal drug transporters organic cation transporter 2 (OCT2) and multidrug and toxin extrusion protein 1 (MATE1) and 2‐K (MATE2‐K) and can increase in the presence of inhibitors of these transporters. In vitro, tucatinib inhibited OCT2‐, MATE1‐, and MATE2‐K‐mediated transport of metformin, with IC50 values of 14.7, 0.340, and 0.135 µM, respectively. Tucatinib also inhibited OCT2‐ and MATE1‐mediated transport of creatinine, with IC50 values of 0.107 and 0.0855 µM, respectively. A phase 1 study with metformin administered orally in the absence and presence of tucatinib was conducted in 18 healthy subjects. Renal function was assessed by measuring glomerular filtration rate (GFR; based on iohexol plasma clearance) and endogenous markers (SCr, cystatin C‐based estimated glomerular filtration rate [eGFR]) with and without tucatinib. Metformin exposure increased (1.4‐fold) and renal clearance decreased (29.99‐17.64 L/h) with tucatinib, with no effect on metformin maximum concentration. Creatinine clearance transiently decreased 23% with tucatinib. GFR and eGFR, which are unaffected by OCT2 and/or MATE1/2‐K transport, were unchanged with tucatinib. These data demonstrate that tucatinib inhibits OCT2‐ and MATE1/2‐K‐mediated tubular secretion of creatinine, which may manifest as mild SCr elevations that are not indicative of renal impairment.
Keywords: tucatinib, renal function, metformin, pharmacokinetics, MATE, OCT2
Human epidermal growth factor receptor 2 (HER2) overexpression or amplification is an oncogenic driver across a range of solid tumors. 1 Reported rates of HER2 overexpression are 15% to 20% for breast cancer, 4% to 53% for gastric cancers, 2% to 11% for colorectal cancer, and 2% to 14% for lung cancer. 1 , 2 , 3 , 4 , 5 , 6 , 7 HER2‐positive status in patients with breast cancer indicates more aggressive disease with worse outcomes, but its role as a prognostic biomarker is less certain in other cancers. 1 , 5 , 8 , 9 , 10 , 11 Although advances in HER2‐targeted therapy have improved outcomes in patients with HER2‐positive breast cancer over the last 20 years, therapeutic options remain limited for patients with HER2‐positive metastatic breast cancer (MBC) who have previously received multiple courses of HER2‐targeted agents and in patients with brain metastases.
Tucatinib is a potent, HER2‐selective, small‐molecule tyrosine kinase inhibitor (TKI). In 2020, the US Food and Drug Administration approved tucatinib in combination with trastuzumab and capecitabine for the treatment of HER2‐positive MBC, including patients with brain metastases, who have received 1 or more prior anti‐HER2‐based regimens in the metastatic setting. 12 Tucatinib is also in clinical development for the treatment of other HER2‐positive solid malignancies. Tucatinib has demonstrated high selectivity for the HER2 kinase domain, with minimal inhibition of other protein targets such as epidermal growth factor receptor. 13 , 14 This may confer toxicity advantages over other available HER2‐directed TKIs that target multiple receptors (eg, lapatinib, neratinib), resulting in high rates of off‐target adverse effects, such as diarrhea and rash. 15 , 16 , 17 , 18 In a pivotal randomized study of heavily pretreated patients with HER2‐positive MBC (HER2CLIMB; NCT02614794), adding tucatinib to trastuzumab and capecitabine resulted in significant improvements in progression‐free survival and overall survival compared with trastuzumab plus capecitabine alone. 19 The efficacy and safety of tucatinib continues to be evaluated in advanced breast cancer in an ongoing randomized, double‐blind phase 3 study, in which tucatinib or placebo is combined with ado‐trastuzumab emtansine for patients with unresectable locally advanced or metastatic HER2‐positive breast cancer (HER2CLIMB‐02; NCT03975647). 20 In addition, tucatinib in combination with trastuzumab is under investigation for the treatment of patients with HER2‐positive metastatic colorectal cancer (MOUNTAINEER; NCT03043313). 21
In the pivotal HER2CLIMB study, the adverse event (AE) of elevated serum creatinine concentration was observed in a higher proportion of patients randomized to the tucatinib arm compared with those who received placebo (13.9% versus 1.5%, respectively). 19 In addition, increased creatinine as a laboratory abnormality was reported in 33% of patients in the tucatinib arm versus 6% in the placebo arm (all grade 1 or 2). 12 These increases in serum creatinine were modest, transient, reversible, clinically nonsignificant, and did not lead to renal damage or treatment discontinuation in any patient. 12 , 19
Serum creatinine is used as a clinical biomarker for glomerular filtration rate (GFR) and is routinely measured to monitor potential renal injury. However, alterations in the activity of renal uptake and efflux transporters can lead to changes in serum creatinine concentrations that do not reflect kidney damage. Inhibition of the uptake transporter organic cation transporter 2 (OCT2) and the efflux transporters multidrug and toxin extrusion protein 1 (MATE1) and 2‐K (MATE2‐K) has been associated with serum creatinine increases in the absence of renal injury with another kinase inhibitor, abemaciclib. 22
To investigate whether the elevations in serum creatinine reported with tucatinib in the HER2CLIMB study resulted from inhibition of the renal transporters or from detrimental effects on the kidney, in vitro and clinical analyses were conducted to evaluate the effects of tucatinib on renal transport. In vitro, the inhibitory effects of tucatinib on renal transport of metformin or creatinine were assessed using a transfected Madin‐Darby canine kidney (MDCK‐II) cell system expressing OCT2, MATE1, or MATE2‐K. A phase 1 clinical study in healthy subjects was conducted to investigate the impact of tucatinib on renal transport by characterizing the effects of multiple‐dose tucatinib on the single‐dose pharmacokinetics of metformin. Iohexol clearance was used as a marker of GFR. In addition to measuring serum creatinine and creatinine clearance (CrCl), cystatin C was evaluated as an alternative endogenous marker of renal injury and used to calculate estimated GFR (eGFR).
Methods
In Vitro Analysis of Transporter Interactions
Tucatinib was synthesized by Seattle Genetics (Bothell, Washington). The in vitro inhibition studies were performed employing radiolabeled 14C‐metformin and 14C‐creatinine as probe substrates, which were purchased from Moravek Biochemicals (Brea, California). Reference inhibitors used were 1000 µM of quinidine for OCT2 and 100 µM of cimetidine for MATE1 and MATE2‐K (purchased from Sigma Aldrich, St. Louis, Missouri). Other chemicals used were of analytical grade purchased from commercial suppliers. The test system for OCT2, MATE1, and MATE2‐K was a polarized monolayer of MDCK‐II cells grown on permeable supports; the cells were a purified subclone of MDCK‐II cells purchased from the University of California San Francisco cell culture facility. The MDCK‐II cells were maintained in Dulbecco's Modified Eagle Medium with low glucose and 10% fetal bovine serum. Cells were passaged and seeded at a density of 60 000 ± 10 000 cells/well on 96‐well transwell membrane plates 24 hours before being treated to express the transporter of interest or treated with a control vector. Transport experiments were conducted in Hank's Balanced Salt Solution, and the transport of each substrate was determined by radiometric detection. Experiments were performed under the same conditions for the cells expressing the transporter as for those treated with the control vector. The result from control cells is used to correct for substrate permeation by routes other than the transporter being investigated.
All in vitro inhibition studies were performed by BioIVT (Santa Clara, California). Briefly, the 96‐well plates were preincubated for 30 minutes at 37°C with orbital shaking at approximately 60 revolutions per minute (rpm) with vehicle control, reference inhibitors, or test article. After aspiration of preincubation buffer, probe substrate (10 µM of 14C‐metformin or 100 µM of 14C‐creatinine) was added, and plates were incubated for 5 minutes at 37°C with orbital shaking at approximately 60 rpm with vehicle control, reference inhibitors, or tucatinib at 6 different concentrations from 0.003 to 10 µM. After incubation, cells were washed 4 times with ice‐cold phosphate buffer solution, and 60 µL of cell extraction solution (50% acetonitrile, 50% water) was added to plates. To mix the extract, the plates were agitated for approximately 15 minutes on an orbital shaker at 120 rpm. Extract samples of 30 µL were collected, and 200 µL of scintillation fluid was added before substrate quantification with radioactivity counting on a 1450 MicroBeta (PerkinElmer, Waltham, Massachusetts).
Transport studies were conducted in triplicate in transporter‐expressing cells and control cells. Percent inhibition was calculated by dividing the transporter‐mediated transport in the presence of tucatinib or the reference inhibitor by the corresponding value for the vehicle.
For the uptake assays, half maximal inhibitory concentration (IC50) was determined applying the following equation:
where V0 is the mean transporter‐mediated flux in the absence of the test article, V is the transporter‐mediated flux in the presence of the test article throughout the concentration range tested, [I] is the inhibitor concentration, IC50 represents the value at which transport is inhibited by 50%, and n is a Hill coefficient. Inhibitory potential values also were calculated, defined as the maximal unbound plasma concentration of the interacting drug at steady state (Imax,u) over IC50.
Phase 1 Clinical Study
The study protocol and amendments were approved by a central institutional review board (Advarra, Columbia, Maryland). All subjects provided written informed consent prior to study enrollment. This study was conducted at 1 study site in the United States (PRA Health Sciences Early Development Services, Salt Lake City, Utah) in accordance with the Declaration of Helsinki, Good Clinical Practice, and other relevant regulatory guidelines.
Subjects
The study was performed in 18 healthy subjects (14 men, 4 women) aged 18‐65 years, with body weight of ≥60 kg and body mass index (BMI) of 18‐32 kg/m2. Subjects were enrolled after they were confirmed to be healthy based on their medical history, physical examination, hematology, blood chemistry, serology, and urinalysis. Female subjects were required to be of nonchildbearing potential. Male subjects who were sexually active with a woman of childbearing potential and were not surgically sterile for at least 90 days were required to agree to using a barrier method of contraception, with or without an additional method by their female partner (hormonal or intrauterine). At the time of screening, all subjects had negative results for human immunodeficiency virus, hepatitis B virus, and hepatitis C virus. Exclusion criteria included use of tobacco or other nicotine‐containing products within 21 days prior to study initiation; consumption of alcohol exceeding 7 or 14 standard drinks per week for women and men, respectively, with alcohol consumption prohibited for 48 hours prior to study initiation and throughout the study; routine or chronic use of acetaminophen at a dose of >3 g/day (limited doses were permitted); and use of prescribed or over‐the‐counter medication, health supplements, or herbal remedies within 28 days prior to study initiation through to follow‐up.
Study Design
This was a phase 1 single‐arm, open‐label, fixed‐sequence crossover study conducted in healthy subjects to evaluate the effects of tucatinib on renal transport and function by assessing the impact of tucatinib on metformin pharmacokinetics and on the plasma clearance of iohexol and cystatin C as markers of GFR and eGFR, respectively.
After a screening period of up to 20 days, eligible subjects received the following treatments: (1) metformin 850 mg administered orally in the fasted state in the morning on days 1 and 8; (2) tucatinib 300 mg orally twice daily on days 2‐8, with the morning dose on days 2 and 8 administered after an overnight fast of at least 8 hours, and the morning dose on day 8 administered immediately after metformin dosing; and (3) intravenous iohexol 5 mL (300 mg iodine per milliliter of solution), administered 10 hours after metformin dosing on days 1 and 8. Oral study drugs were administered with 8 oz of water to subjects in an upright position. Subjects were admitted to the clinical research center on the day before initiating treatment and discharged on day 9, attending a follow‐up visit on day 16.
Pharmacokinetic and Pharmacodynamic Assessments
Blood samples for metformin pharmacokinetics were obtained predose and 0.5, 1, 1.5, 2, 2.5, 3, 3.5, 4, 6, 8, 10, 12, 18, and 24 hours postdose on days 1 and 8. Metformin pharmacokinetics also were evaluated from the 24‐hour urine sampling (with collection periods of 0‐2, 2‐4, 4‐8, 8‐12, and 12‐24 hours) on days 1 and 8. Blood samples for tucatinib pharmacokinetics were obtained within 30 minutes prior to the morning dose of tucatinib on days 2‐8 and 23.5‐24 hours after the last tucatinib dose on day 9. Blood samples for the iohexol plasma clearance test were obtained predose and 1, 2, 3, and 4 hours after iohexol administration on days 1 and 8. Blood samples for evaluating serum creatinine and cystatin C concentrations were obtained on days 1, 8, and 9. Blood samples were drawn prior to and 12 hours after the metformin dose on day 1, prior to both tucatinib doses on day 8, and 24 hours after the last tucatinib dose on day 9. Urine sampling to assess concentrations of creatinine (for calculation of CrCl) and microalbumin was performed according to the aforementioned schedule.
Quantitative Assays
Analysis of tucatinib from plasma samples and metformin from plasma and urine samples was performed by Alturas Analytics, Inc. (Moscow, Idaho), and analysis of iohexol from plasma samples was performed by the bioanalytical laboratory of PRA Health Sciences (Raleigh, North Carolina). Plasma and urine drug concentrations were measured using validated high‐performance liquid chromatography (HPLC) with tandem mass spectrometry (LC‐MS/MS) methods as described below.
Reference standards for metformin and metformin‐D6 (internal standard) were purchased from MP Biomedicals, LLC (Solon, Ohio) and Clearsynth Labs Ltd. (Mississauga, Ontario, Canada), respectively. Metformin and metformin‐D6 were extracted from plasma and urine via acetonitrile precipitation.
For quantification of metformin, an aliquot of the extract was injected onto an LC‐MS/MS triple quadrupole mass spectrometer (Sciex API‐5500 or API‐6500; Applied Biosystems, Framingham, Massachusetts). A C18 HPLC column (Supelco Discovery HS‐C18, 2.1 × 50 mm, 3 µm; Sigma Aldrich, St. Louis, Missouri) was used to separate the analytes and the internal standard (metformin‐D6) from interfering compounds that may be present in the sample extract. The peak area of the product ion of the compound (metformin) was measured against the peak area of the product ion of the respective internal standard. The aqueous mobile phase A was water with 0.1% heptafluorobutyric acid (HFBA), and the organic mobile phase B was acetonitrile with 0.1% HFBA. The rinse solvents were 1:1 acetonitrile:water with 1% formic acid (metformin in plasma or urine). Flow rate was 0.7 mL/min, and the sample injection volume was 1‐40 µL, adjusted according to instrument sensitivity.
The mass spectrometers were operated with electrospray ionization (ESI) in the positive ion mode. Quantification was based on multiple‐reaction monitoring of the transitions of mass‐to‐charge ratios (m/z) of 130.0 → 60.2 for metformin and 136.2 → 60.2 for metformin‐D6. Mass spectrometer calibration functions were constructed with a linear regression model weighted by 1/x2 (where x is concentration) ranging from 0.002 to 2.0 µg/mL for metformin in plasma and 0.01 to 10.0 µg/mL for metformin in urine. The lower limit of quantification (LLOQ) for metformin in plasma and urine were 0.002 and 0.01 µg/mL, respectively. Accuracy (% bias) ranged from −2.5% to 2.2% for metformin in plasma and −2.8% to 3.3% for metformin in urine. Precision (coefficient of variation %) ranged from 3.0% to 6.4% for metformin in plasma and from 1.9% to 4.5% for metformin in urine.
Reference standards for tucatinib and ONT‐572 (internal standard) were provided by Seattle Genetics (Bothell, Washington). Tucatinib and ONT‐572 were extracted from plasma via methanol precipitation.
For quantification of tucatinib, an aliquot of the extract was injected onto an LC‐MS/MS triple quadrupole mass spectrometer (Sciex API‐5500 or API‐6500; Applied Biosystems, Framingham, Massachusetts). A C18 HPLC column (Supelco Discovery HS‐C18, 2.1 × 50 mm, 3 µm; Sigma Aldrich, St. Louis, Missouri) was used to separate the analytes and the internal standard (ONT‐572) from interfering compounds that may be present in the sample extract. The peak area of the product ion of the compound (tucatinib) was measured against the peak area of the product ion of the respective internal standard. The aqueous mobile phase A was water with 0.1% formic acid, and the organic mobile phase B was acetonitrile with 0.1% formic acid. The rinse solvent was dimethyl formamide or 1:1 methanol:water. Flow rate was 0.7 mL/min, and the sample injection volume was 1‐40 µL, adjusted according to instrument sensitivity.
The mass spectrometers were operated with ESI in the positive ion mode. Quantification was based on multiple‐reaction monitoring of the transitions of m/z of 481.3 → 274.0 for tucatinib and 487.3 → 275.0 for ONT‐572. Mass spectrometer calibration functions were constructed with a linear regression model weighted by 1/x2 (where x is concentration) ranging from 0.001 to 1.0 µg/mL for tucatinib. The LLOQ for tucatinib in plasma was 0.001 µg/mL. Accuracy (% bias) ranged from −3.4% to 3.0%, and precision (coefficient of variation %) ranged from 3.9% to 7.5% for tucatinib in plasma.
Reference materials for iohexol and Iohexol‐d5 (internal standard) were purchased from US Pharmacopeia (Rockville, Maryland) and Toronto Research Chemicals (Toronto, Ontario, Canada), respectively. Sample processing (from human K2‐ethylenediaminetetraacetic acid plasma samples) was performed by protein precipitation using a sample volume of 25.0 µL. Separation between potential metabolites and interfering endogenous compounds was achieved by ultraperformance liquid chromatography using a Waters Acquity ethylene bridged hybrid, hydrophilic interaction liquid chromatography column (2.1 × 50 mm, 1.7‐µm particles; Milford, Massachusetts) at 25°C and an isocratic elution with a postelution wash‐off using 0.1% formic acid in water as mobile phase A and 0.1% formic acid in acetonitrile as mobile phase B operating at a flow rate of 1.00 mL/min.
A triple quadrupole mass spectrometer (Triple Quad 5500, AB Sciex, Framingham, Massachusetts) equipped with a turbo ion spray source was used for detection in positive ion mode. Quantification was based on multiple‐reaction monitoring of the transitions of m/z 821.9 → 603.0 for iohexol and 826.9 → 607.9 for the internal standard. A linear calibration curve, ranging from 5.0 to 1000 µg/mL with a 1/x2 weighting factor was used. The LLOQ was 5.0 µg/mL. Accuracy (% bias) for iohexol ranged from −3.4% to 7.0%, and precision (coefficient of variation %) ranged from 0.1% to 3.1%.
Using the calibration curve for each analyte, back‐calculated concentrations were generated using the derived equation from the weighted least‐squares linear regression line of the peak area ratios. The accuracy (% bias) for the back‐calculated concentrations for each standard point needed to be within 15% (20% at the LLOQ) of the nominal value to be considered acceptable. At least 75% of the calibration standards in each batch must meet these accuracy criteria for the batch to be considered acceptable.
Pharmacokinetic and Pharmacodynamic Assessments
Tucatinib plasma trough concentrations, plasma and urine concentrations of metformin, and plasma concentrations of iohexol were determined. The following main pharmacokinetic parameters were estimated using noncompartmental analysis (Phoenix® WinNonlin® version 8.1 or higher; Certara, L.P., Princeton, New Jersey) from the plasma concentration–time data: maximum observed plasma concentration (Cmax), time to Cmax (tmax; metformin only), terminal elimination half‐life (t½), area under the plasma concentration–time curve from time 0 to the last available measurement (AUC0‐last), area under the plasma concentration–time curve from time 0 to infinity (AUC0‐inf), systemic clearance (CLsyst; iohexol only), renal clearance (CLrenal; metformin only), apparent volume of distribution (Vz/F; metformin only), and apparent oral clearance (CL/F; metformin only). Pharmacodynamic parameters (serum concentrations of creatinine and cystatin C and 24‐hour microalbumin) were summarized with changes from baseline.
Safety
Safety was assessed based on the frequency and severity of treatment‐emergent AEs (TEAEs), clinical laboratory parameters, vital signs, 12‐lead electrocardiogram, and physical examination. TEAEs were assessed and graded applying the Common Terminology Criteria for Adverse Events version 4.03 and coded according to the Medical Dictionary for Regulatory Activities version 21.1. TEAEs were summarized by preferred terms and system organ class.
Statistical Analysis
Descriptive statistics were applied to summarize demographic, pharmacokinetic, pharmacodynamic, and safety parameters.
The effect of tucatinib on metformin pharmacokinetics was assessed using the ratio and 90% confidence interval (CI) of the geometric least‐squares (LS) mean of the plasma pharmacokinetic parameters Cmax, AUC0‐inf, and AUC0‐last for metformin. A linear mixed‐effects model with treatment as fixed effect and subject as random effect was used on the log‐transformed parameters. Estimates on the original scale were obtained from point estimates on the natural log scale, and geometric LS means were provided for each treatment. There was no adjustment for multiplicity.
Iohexol plasma clearance was used to calculate GFR values using the Jødal and Brøchner Mortensen equation. 23 This calculation used CLsyst adjusted to use the elimination phase only and body surface area (BSA; calculated using the Haycock formula): 24
In this formula, denotes the 1‐pool (slope‐intercept) clearance normalized to a BSA of 1.73 m2, that is, , and where is the “missing” area under the plasma fraction curve (the 1‐pool approximation) in minutes and is plasma volume. To normalize to BSA 1.73 m2 at the level of 1‐pool clearance, , where has the unit (mL/min/1.73 m2)−1.
Based on cystatin C, eGFR values were calculated as follows, where is serum cystatin C. 25
When cystatin C was ≤0.8 mg/mL:
When cystatin C was >0.8 mg/mL:
CrCl (mL/min) was calculated as follows:
Statistical analyses were performed using SAS version 9.4 or higher (SAS Institute, Inc., Cary, North Carolina).
Results
In Vitro Analysis of Transporter Interactions
Transport of metformin mediated by OCT2, MATE1, and MATE2‐K was inhibited by tucatinib with IC50 values of 14.7, 0.340, and 0.135 µM, respectively (Table 1). Tucatinib inhibited OCT2‐ and MATE1‐mediated transport of creatinine with IC50 values of 0.107 and 0.0855 µM, respectively. Inhibitory potential values (Imax,u/IC50) were greater than 0.1 for MATE1‐ and MATE2‐K‐mediated transport of metformin and MATE1‐ and OCT2‐mediated transport of creatinine, indicating that tucatinib has the potential to inhibit these transporters in vivo. 26 However, the inhibitory potential value for OCT2 inhibition with metformin as a probe substrate was less than 0.1, suggesting that tucatinib does not inhibit OCT2‐mediated metformin transport.
Table 1.
IC50 and Inhibitory Potential Values (Imax,u/IC50) of Tucatinib on Renal Proximal Tubule Transporters OCT2, MATE1, and MATE2‐K, Determined Using Metformin or Creatinine as Probe Substrates
| Metformin | Creatinine | |||
|---|---|---|---|---|
| Transporter | IC50 (µM) | Imax,u/IC50 | IC50 (µM) | Imax,u/IC50 |
| OCT2 | 14.7 | 0.0027 | 0.107 | 0.38 |
| MATE1 | 0.340 | 0.12 | 0.0855 | 0.47 |
| MATE2‐K | 0.135 | 0.30 | ND a | ND a |
IC50, half maximal inhibitory concentration; Imax,u, maximal unbound plasma concentration of the interacting drug at steady state, determined as 0.0403 µM (Cmax of 1.39 µM and unbound fraction in plasma of 0.029, unpublished data); MATE, multidrug and toxin extrusion protein; ND, not determined; OCT2, organic cation transporter 2.
There was insufficient net creatinine uptake to accurately determine the IC50 for MATE2‐K.
Phase 1 Clinical Study
All 18 subjects enrolled in the study received at least 1 dose of tucatinib and were evaluable for safety. Of these, 17 subjects also had ≥1 post‐tucatinib dose concentration measured and were evaluable for pharmacokinetics and pharmacodynamics; 1 subject discontinued on day 3 because of an AE (moderate rash). All but 2 subjects were white (88.9%), mean ± standard deviation age was 40.6 ± 16.6 years, and mean BMI was 26.4 ± 3.6 kg/m2.
Effects of Tucatinib on the Pharmacokinetics of Metformin, a Sensitive OCT2/MATE Substrate
Coadministration of tucatinib resulted in statistically significant increases in metformin exposure measured by AUC0‐last and AUC0‐inf, which increased 1.4‐fold compared with metformin administered alone, whereas Cmax was unaffected (Table 2, Figure 1). Metformin tmax ranges were similar with (1.0–4.1 hours) and without (1.5–4.0 hours) tucatinib, but median tmax was longer with tucatinib coadministration than with metformin alone (3.0 versus 2.5 hours). In addition, t½ was longer (5.6 versus 4.5 hours), and CL/F (77.4 versus 105.4 L/h) and Vz/F (627.2 L versus 695.4 L) decreased in the presence of tucatinib (Table 1). The mean cumulative fraction of metformin excreted unchanged in urine decreased from 28.54% to 22.44%, and CLrenal decreased from 29.99 to 17.64 L/h when metformin and tucatinib were administered in combination versus metformin alone (Table 2, Figure 2).
Table 2.
Metformin Pharmacokinetics in Plasma and Urine
| Arithmetic Mean (CV%) | Geometric LSM | Geometric LSM Ratio | ||||
|---|---|---|---|---|---|---|
| Pharmacokinetic Parameter | Metformin (n = 17) | Metformin + Tucatinib (n = 17) | Metformin (n = 17) | Metformin + Tucatinib (n = 17) | Estimate | 90% CI |
| Plasma | ||||||
| AUC0‐last (µg·h/mL) | 8.4 (26.2) | 11.6 (29.9) | 8.1 | 11.1 | 1.4 | 1.2‐1.5 b |
| AUC0‐inf (µg·h/mL) | 8.6 (26.4) | 12.1 (30.5) | 8.3 | 11.6 | 1.4 | 1.3‐1.5 b |
| Cmax (µg/mL) | 1.3 (18.0) | 1.5 (27.9) | 1.3 | 1.4 | 1.1 | 1.0‐1.2 |
| Tmax (h) a | 2.5 (1.5, 4.0) | 3.0 (1.0, 4.1) | — | — | — | — |
| t½ (h) | 4.5 (11.7) | 5.6 (12.2) | — | — | — | — |
| VzF (L) | 695.4 (32.6) | 627.2 (41.5) | 664.7 | 586.8 | 0.883 | 0.763‐1.021 |
| CL/F (L/h) | 105.4 (26.7) | 77.4 (34.9) | 102.0 | 73.4 | 0.72 | 0.649‐0.799 b |
| Urine | ||||||
| Ae0‐24 (mg) | 242.6 (26.5) | 190.8 (31.7) | — | — | — | — |
| Fe0‐24 (%) | 28.5 (26.5) | 22.4 (31.7) | — | — | — | — |
| CLrenal (L/h) | 30.0 (27.0) | 17.6 (36.0) | 28.79 | 16.45 | 0.571 | 0.516‐0.632 b |
Ae0–24, amount of metformin excreted unchanged in urine; AUC0‐inf, area under the plasma concentration–time curve from time 0 to infinity; AUC0‐last, area under the plasma concentration‐time curve from time 0 to the last available measurement; CI, confidence interval; CL/F, apparent oral clearance; CLrenal, renal clearance; Cmax, maximum observed plasma concentration; CV, coefficient of variation; Fe0‐24, mean cumulative fraction of metformin excreted unchanged in urine; LSM, least‐squares mean; t½, terminal elimination half‐life; tmax, time to Cmax; VzF, apparent volume of distribution.
Median (range) is presented.
Statistically significant.
Figure 1.
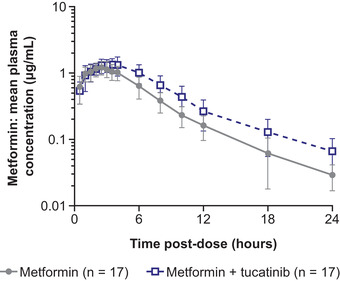
Metformin pharmacokinetics when administered alone or with tucatinib (mean ± SD). SD, standard deviation.
Figure 2.
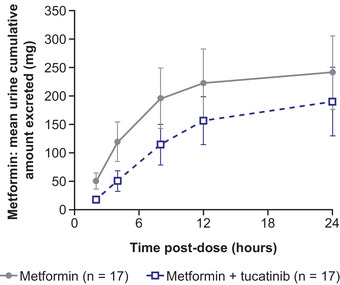
Urinary excretion of metformin when administered alone or with tucatinib.
Effects of Tucatinib on Markers of Renal Function Transported by OCT2 and MATEs
Serum creatinine remained in the normal range during the study for most subjects: 2 subjects had concentrations of 1.46 and 1.44 mg/dL, which are above the upper limit of 1.33 mg/dL, on day 8. Mean CrCl 24 hours postdose decreased by 23% in the presence of tucatinib, from 152.3 mL/min on day 2 to 117.2 mL/min on day 9 (Figure 3A). Mean urinary albumin concentrations 24 hours postdose were within the normal range (<30 mg/24 h) on day 2 (13.3 mg/24 h) and day 9 (11.1 mg/24 h); see Figure 3B.
Figure 3.
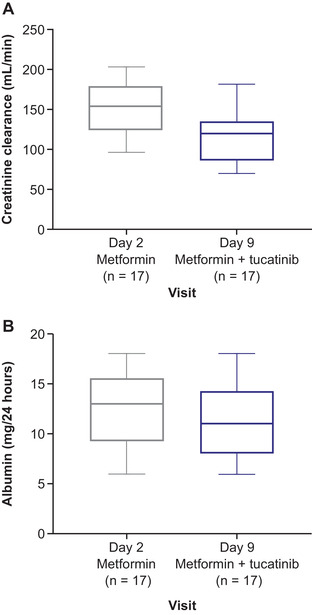
Box plots for (A) creatinine clearance and (B) urinary albumin over time.
Effects of Tucatinib on Markers of Renal Function Unaffected by Drug Transporters
Renal function evaluated using iohexol clearance as a marker of GFR was unaffected in the presence of tucatinib (Table 3, Figure 4). Mean cystatin C concentration decreased by approximately 10% when tucatinib was coadministered with metformin versus metformin alone: mean cystatin C concentration measured prior to metformin dosing was 0.846 versus 0.768 mg/L after tucatinib coadministration, and mean cystatin C concentration measured 12 hours postmetformin dosing was 0.881 versus 0.772 mg/L with metformin and tucatinib (Figure 5A). Similarly, mean eGFR derived from cystatin C concentration exhibited similar decreases with combination treatment (premetformin dose, 111.4 versus 101.4 mL/min/1.73 m2; 12 hours postdose, 110.7 versus 97.9 mL/min/1.73 m2); see Figure 5B.
Table 3.
Iohexol Pharmacokinetics in Plasma
| Arithmetic Mean (CV%) | ||
|---|---|---|
| Pharmacokinetic Parameter | Metformin (n = 17) | Metformin + Tucatinib (n = 17) |
| AUC0‐last (µg·h/mL) | 345.5 (21.2) | 346.4 (21.7) |
| AUC0‐inf (µg·h/mL) | 440.5 (27.9) | 441.9 (27.2) |
| CLsyst (mL/min) | 129.8 (22.7) | 129.0 (22.0) |
| t½ (h) | 1.762 (18.9) | 1.784 (17.5) |
| C0 (µg/mL) | 175.6 (18.0) | 178.5 (21.5) |
| GFR (mL/min/1.73 m2) | 94.99 (17.4) | 94.56 (16.9) |
AUC0‐inf, area under the plasma concentration–time curve from time 0 to infinity; AUC0‐last, area under the plasma concentration‐time curve from time 0 to the last available measurement; C0, initial concentration; CLsyst, systemic clearance; CV, coefficient of variation; GFR, glomerular filtration rate; t½, terminal elimination half‐life.
Figure 4.
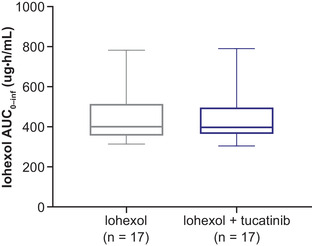
Box plot for iohexol exposure when administered alone or with tucatinib. AUC0‐inf, area under the plasma concentration‐time curve from time 0 to infinity.
Figure 5.
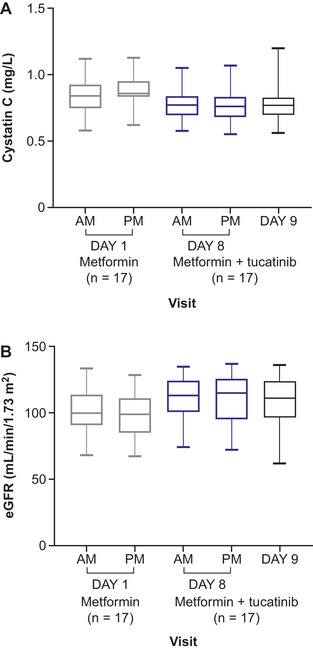
Box plots for (A) cystatin C and (B) eGFR over time. eGFR, estimated glomerular filtration rate.
Safety
Metformin and tucatinib, administered alone or in combination, were generally safe and well tolerated, and no significant clinical safety concerns were noted. Overall, 37 TEAEs were reported in 13 subjects (72.2%), all of which were mild in severity with the exception of 1 case of moderate rash. Similar proportions of subjects experienced TEAEs with metformin alone (38.9%), tucatinib alone (33.3%), and metformin plus tucatinib (35.3%). No serious AEs were reported in this study. The most commonly occurring TEAEs during the study were diarrhea (9 events in 5 subjects [27.8%]), headache (6 events in 5 subjects [27.8%]), dyspepsia (3 events in 2 subjects [11.1%]), and sinus congestion (3 events in 2 subjects [11.1%]).
All but 2 TEAEs resolved during the 10‐day study: a mild (grade 1) infusion‐site hemorrhage was reported in a subject receiving metformin plus tucatinib, considered unrelated to orally administered study treatment, and a TEAE of generalized rash occurred during tucatinib administration alone, considered by the investigator to be moderate in severity (grade 2) and related to study treatment. This second subject discontinued study participation on day 3 and was lost to follow‐up.
Discussion
Clinically nonsignificant and reversible elevations in serum creatinine have been observed in patients treated with tucatinib in a pivotal clinical trial. 19 As a biomarker for GFR, elevations in serum creatinine are typically interpreted as renal injury. Similarly, a reduction in CrCl, which is based on the amount of creatinine excreted in the urine, also may suggest renal dysfunction. However, these markers provide an approximation rather than a direct measure of renal function and may be affected by various nonrenal factors. 27
The present studies were conducted to determine whether the serum creatinine elevations reported with tucatinib in clinical trials resulted from inhibition of the renal transporters, OCT2 and MATE1/2‐K or from acute kidney injury. We found that metformin plasma exposure increased in the presence of tucatinib (1.4‐fold increase in both AUC0‐inf and AUC0‐last) compared with metformin administered alone. Consistent with the increase in exposure, reductions in metformin CL/F and CLrenal were observed when tucatinib was coadministered with metformin. A transient decrease in CrCl and increase in serum creatinine were also observed in the presence of tucatinib.
As creatinine and metformin are both transported by OCT2 and MATE transporters, further investigation was conducted to measure markers of renal function unaffected by drug transporters, and these markers were unchanged in the presence of tucatinib. Specifically, coadministration of tucatinib did not reduce GFR measured by iohexol plasma clearance or eGFR calculated using endogenous cystatin C. The stability of iohexol and cystatin C clearance in the presence of tucatinib demonstrate that the increased metformin exposure and decreased CrCl associated with tucatinib occurred in the absence of any impact on GFR. Hence, the increased metformin exposure and reduced CrCl in the presence of tucatinib are consistent with specific inhibition of renal drug transporters.
In vitro data showed that tucatinib appears to exhibit substrate‐dependent inhibition of renal transporters, with a marked difference in the inhibitory potential of OCT2‐mediated transport observed between creatinine (IC50, 0.107 µM) and metformin (IC50, 14.7 µM). Thus, it is likely that the interaction of tucatinib with metformin results from inhibition of MATE1/MATE2‐K, whereas the interaction with creatinine may result from both OCT2 and MATE1 inhibition. The less than 2‐fold increase in metformin exposure indicates that tucatinib is a weak inhibitor of the renal OCT2/MATE1/MATE2‐K pathway. 28 Given the wide therapeutic range of metformin, this increase is unlikely to be clinically meaningful and will not require dose modification of either metformin or tucatinib. Treatment with the combination of tucatinib and metformin in this study was generally safe and well tolerated with no significant TEAEs observed.
Elevated serum creatinine because of inhibition of creatinine tubular secretion has been observed with other kinase inhibitors indicated for the treatment of cancer in clinical 22 , 29 and preclinical 30 studies. In clinical trials of abemaciclib, a selective inhibitor of cyclin‐dependent kinases 4 and 6 indicated for the treatment of hormone‐receptor‐positive, HER2‐negative advanced or metastatic breast cancer, 31 reversible serum creatinine elevations of approximately 15% to 40% from baseline have been observed. 22 , 32 , 33 Abemaciclib, similar to tucatinib, has also been shown to inhibit metformin uptake by OCT2, MATE1, and MATE2‐K transporters in vitro, as well as significantly increasing metformin exposure without affecting a range of serum and urinary biomarkers of renal injury in healthy subjects, suggesting that the effects on serum creatinine may be because of renal transporter inhibition and are not associated with renal damage. 22 Increases in serum creatinine observed in clinical studies of the BRAF kinase inhibitor vemurafenib, used to treat advanced melanoma, result partly from inhibition of creatinine tubular secretion and partly from reversible renal function impairment. 29 In this study, serum creatinine increased across subjects in the presence of tucatinib (corresponding with the decreased CrCl of approximately 23%), but serum creatinine elevation was not recorded as an AE in any patient.
Together, these findings highlight the limitations of using serum creatinine as a biomarker for renal injury when patients are dosed with known OCT2 or MATE transporter inhibitors, now including tucatinib. 34 Serum creatinine elevation may be misinterpreted as renal toxicity, leading to inappropriate dose modification or discontinuation of an investigational drug during clinical trials. It is therefore important to differentiate between effects on kidney function and changes in serum creatinine resulting from inhibition of renal transporters. When clinically indicated, alternative methods of assessing renal function, such as measurement of cystatin C or GFR, may be considered to confirm whether elevated serum creatinine concentrations are indicative of kidney injury. The design of the present study enabled us to determine the effects of tucatinib on both renal transporters and renal function in vivo.
It is also important to distinguish nonpathologic transient changes in renal biomarkers from clinically relevant drug–drug interactions (DDIs) involving renal transport inhibitors. While such DDIs are less frequent than hepatic and gastrointestinal interactions associated with cytochrome P450 enzymes or drug transporters, they can involve commonly used drugs and therapies with a narrow therapeutic index. 35 Most clinically significant renal DDIs involve inhibition of transporter‐mediated tubular secretion when this is the predominant clearance pathway for the drug; the relative contribution of uptake and efflux transporters to renal drug clearance may be a key factor in such DDIs. 36 In addition, inhibition of renal transporters potentially may ameliorate the toxic effects of drugs that are substrates for the targeted transporter. 37 , 38 The possibility that interactions involving renal transporter inhibition may be substrate dependent, as reported with tucatinib, should also be considered. Hence, potential DDIs should be considered in situations in which changes in the transport of a substrate have the potential to result in a significant impact on safety and/or efficacy.
In conclusion, these data demonstrate that tucatinib inhibits tubular secretion of creatinine via OCT2 and MATE transporters, which may manifest as mild increases in serum creatinine that are not indicative of impaired renal function.
Conflicts of Interest
This study was funded by Seattle Genetics. The authors wrote the article with the assistance of a medical writer funded by the sponsor. All the authors had full access to the relevant data, vouched for the completeness and accuracy of the data and for adherence of the trial to the protocol, assumed final responsibility for the content of the article, and for the decision to submit the article for publication. A.R. Topletz‐Erickson, A.J. Lee, J.G. Mayor, E.L. Rustia, L.I. Abdulrasool, L.N. Walker, S.C. Alley, and C.J. Endres are employees of Seattle Genetics and hold stocks and shares in Seattle Genetics. B. Dailey and A.L. Wise have no conflicts of interest to disclose. S. DeChenne is an employee of Alturas Analytics Inc., with which Seattle Genetics contracts to analyze pharmacokinetic samples.
Funding
Funding for this publication was provided by Seattle Genetics, Inc. (Bothell, Washington, USA).
Data Sharing
Qualified researchers may request access to certain data and related study documents consistent with the Principles for Responsible Clinical Trial Data Sharing. Further details on data requests can be located at http://www.seattlegenetics.com/patients-healthcare-professionals/clinical-data-requests or by emailing CTDR@seagen.com. Requests to analyze anonymized patient‐level trial data used in the current study can be submitted to CTDR@seagen.com. Further details about data requests can be found at http://www.seattlegenetics.com/patients-healthcare-professionals/clinical-data-requests (see http://www.icmje.org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration.html).
Acknowledgments
The authors thank the study participants, their families, and the principal investigator of this study, Dr Shawn Searle of PRA Health Sciences, Salt Lake City, Utah. Medical writing support was provided by Tamsin Williamson, and editorial support, including formatting, proofreading, and submission, was provided by Annabel Ola, of Scion Medica, London, supported by Seattle Genetics according to Good Publication Practice guidelines (https://www.acpjournals.org/doi/10.7326/M15-0288).
References
- 1. Siena S, Sartore‐Bianchi A, Marsoni S, et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann Oncol. 2018;29(5):1108‐1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. American Cancer Society. Breast Cancer Facts & Figures 2019–2020. Atlanta: American Cancer Society, Inc. https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/breast-cancer-facts-and-figures/breast-cancer-facts-and-figures-2019-2020.pdf. Accessed May 5, 2020. [Google Scholar]
- 3. Chua TC, Merrett ND. Clinicopathologic factors associated with HER2‐positive gastric cancer and its impact on survival outcomes–a systematic review. Int J Cancer. 2012;130(12):2845‐2856. [DOI] [PubMed] [Google Scholar]
- 4. Howlader N, Altekruse SF, Li CI, et al. US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst. 2014;106(5):dju055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kim EK, Kim KA, Lee CY, Shim HS. The frequency and clinical impact of HER2 alterations in lung adenocarcinoma. PLoS One. 2017;12(2):e0171280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Owens MA, Horten BC, Da Silva MM. HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin Breast Cancer. 2004;5(1):63‐69. [DOI] [PubMed] [Google Scholar]
- 7. Richman SD, Southward K, Chambers P, et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J Pathol. 2016;238(4):562‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheang MC, Chia SK, Voduc D, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101(10):736‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chia S, Norris B, Speers C, et al. Human epidermal growth factor receptor 2 overexpression as a prognostic factor in a large tissue microarray series of node‐negative breast cancers. J Clin Oncol. 2008;26(35):5697‐5704. [DOI] [PubMed] [Google Scholar]
- 10. Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int. 2014;2014:852748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kelly CM, Janjigian YY. The genomics and therapeutics of HER2‐positive gastric cancer‐from trastuzumab and beyond. J Gastrointest Oncol. 2016;7(5):750‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Seattle Genetics Inc. Tukysa (tucatinib) US prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213411s000lbl.pdf. Accessed May 5, 2020.
- 13. Moulder SL, Borges VF, Baetz T, et al. Phase I study of ONT‐380, a HER2 inhibitor, in patients with HER2(+)‐advanced solid tumors, with an expansion cohort in HER2(+) metastatic breast cancer (MBC). Clin Cancer Res. 2017;23(14):3529‐3536. [DOI] [PubMed] [Google Scholar]
- 14. Pheneger T, Bouhana K, Anderson D, et al. Abstract #1795: in vitro and in vivo activity of ARRY‐380: a potent, small molecule inhibitor of ErbB2. Cancer Res. 2009;69(9 suppl):1795. [Google Scholar]
- 15. Capri G, Chang J, Chen SC, et al. An open‐label expanded access study of lapatinib and capecitabine in patients with HER2‐overexpressing locally advanced or metastatic breast cancer. Ann Oncol. 2010;21(3):474‐480. [DOI] [PubMed] [Google Scholar]
- 16. Mortimer J, Di Palma J, Schmid K, Ye Y, Jahanzeb M. Patterns of occurrence and implications of neratinib‐associated diarrhea in patients with HER2‐positive breast cancer: analyses from the randomized phase III ExteNET trial. Breast Cancer Res. 2019;21(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rugo HS, Di Palma JA, Tripathy D, et al. The characterization, management, and future considerations for ErbB‐family TKI‐associated diarrhea. Breast Cancer Res Treat. 2019;175(1):5‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sonnenblick A, de Azambuja E, Agbor‐Tarh D, et al. Lapatinib‐related rash and breast cancer outcome in the ALTTO phase III randomized trial. J Natl Cancer Inst. 2016;108(8):djw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murthy RK, Loi S, Okines A, et al. Tucatinib, trastuzumab, and capecitabine for HER2‐positive metastatic breast cancer. N Engl J Med. 2020;382(7):597‐609. [DOI] [PubMed] [Google Scholar]
- 20. ClinicalTrials.gov. NCT03975647: a study of tucatinib vs. placebo in combination with ado‐trastuzumab emtansine (T‐DM1) for patients with advanced or metastatic HER2+ breast cancer. https://clinicaltrials.gov/ct2/show/NCT03975647. Accessed April 3, 2020.
- 21. Strickler JH, Zemla T, Ou FS, et al. Trastuzumab and tucatinib for the treatment of HER2 amplified metastatic colorectal cancer (mCRC): initial results from the MOUNTAINEER trial. Ann Oncol. 2019;30:v200. [Google Scholar]
- 22. Chappell JC, Turner PK, Pak YA, et al. Abemaciclib inhibits renal tubular secretion without changing glomerular filtration rate. Clin Pharmacol Ther. 2019;105(5):1187‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jodal L, Brochner‐Mortensen J. Reassessment of a classical single injection 51Cr‐EDTA clearance method for determination of renal function in children and adults. Part I: Analytically correct relationship between total and one‐pool clearance. Scand J Clin Lab Invest. 2009;69(3):305‐313. [DOI] [PubMed] [Google Scholar]
- 24. Haycock GB, Schwartz GJ, Wisotsky DH. Geometric method for measuring body surface area: a height‐weight formula validated in infants, children, and adults. J Pediatr. 1978;93(1):62‐66. [DOI] [PubMed] [Google Scholar]
- 25. Inker LA, Schmid CH, Tighiouart H, et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N Engl J Med. 2012;367(1):20‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. US Food and Drug Administration. In vitro drug interaction studies—cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. Guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions. Accessed April 22, 2020. [Google Scholar]
- 27. Raman M, Middleton RJ, Kalra PA, Green D. Estimating renal function in old people: an in‐depth review. Int Urol Nephrol. 2017;49(11):1979‐1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. US Food and Drug Administration. Clinical drug interaction studies—cytochrome P450 enzyme‐ and transporter‐mediated drug interactions. Guidance for industry. https://www.fda.gov/media/134581/download. Accessed June 9, 2020.
- 29. Hurabielle C, Pillebout E, Stehle T, et al. Mechanisms underpinning increased plasma creatinine levels in patients receiving vemurafenib for advanced melanoma. PLoS One. 2016;11(3):e0149873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Omote S, Matsuoka N, Arakawa H, Nakanishi T, Tamai I. Effect of tyrosine kinase inhibitors on renal handling of creatinine by MATE1. Sci Rep. 2018;8(1):9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eli Lilly and Company. Verzenio (abemaciclib) US prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208716s004lbl.pdf. Accessed May 27, 2020.
- 32. Patnaik A, Rosen LS, Tolaney SM, et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non‐small cell lung cancer, and other solid tumors. Cancer Discov. 2016;6(7):740‐753. [DOI] [PubMed] [Google Scholar]
- 33. Sledge GW, Jr. , Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2– advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35(25):2875‐2884. [DOI] [PubMed] [Google Scholar]
- 34. Chu X, Bleasby K, Chan GH, Nunes I, Evers R. The complexities of interpreting reversible elevated serum creatinine levels in drug development: does a correlation with inhibition of renal transporters exist? Drug Metab Dispos. 2016;44(9):1498‐1509. [DOI] [PubMed] [Google Scholar]
- 35. Lepist EI, Ray AS. Renal transporter‐mediated drug‐drug interactions: are they clinically relevant? J Clin Pharmacol. 2016;56(suppl 7):S73‐S81. [DOI] [PubMed] [Google Scholar]
- 36. Feng B, Varma MV. Evaluation and quantitative prediction of renal transporter‐mediated drug‐drug interactions. J Clin Pharmacol. 2016;56(suppl 7):S110‐S121. [DOI] [PubMed] [Google Scholar]
- 37. Hucke A, Ciarimboli G. The role of transporters in the toxicity of chemotherapeutic drugs: focus on transporters for organic cations. J Clin Pharmacol. 2016;56(suppl 7):S157‐S172. [DOI] [PubMed] [Google Scholar]
- 38. Tanihara Y, Masuda S, Katsura T, Inui K. Protective effect of concomitant administration of imatinib on cisplatin‐induced nephrotoxicity focusing on renal organic cation transporter OCT2. Biochem Pharmacol. 2009;78(9):1263–271. [DOI] [PubMed] [Google Scholar]