

Abstract
We and other groups have demonstrated that exposure to nickel nanoparticles (Nano-Ni) results in severe and persistent lung inflammation and fibrosis, but the underlying mechanisms remain unclear. Here, we propose that miR-21 may play an important role in Nano-Ni-induced lung inflammation, injury, and fibrosis. Our dose- and time-response studies demonstrated that exposure of C57BL/6J (WT) mice to Nano-Ni resulted in upregulation of miR-21, proinflammatory cytokines, and profibrotic mediators. Histologically, exposure to Nano-Ni caused severe pulmonary inflammation and fibrosis. Based on the dose- and time-response studies, we chose a dose of 50 μg of Nano-Ni per mouse to compare the effects of Nano-Ni on WT with those on miR-21 KO mouse lungs. At day 3 post-exposure, Nano-Ni caused severe acute lung inflammation and injury that were reflected by increased neutrophil count, CXCL1/KC level, LDH activity, total protein concentration, MMP-2/9 protein levels and activities, and proinflammatory cytokines in the BALF or lung tissues from WT mice, which were confirmed histologically. Although Nano-Ni had similar effects on miR-21 KO mice, the above mentioned levels were significantly lower than those in WT mice. Histologically, lungs from WT mice exposed to Nano-Ni had infiltration of a large number of polymorphonuclear (PMN) cells and macrophages in the alveolar space and interstitial tissues. However, exposure of miR-21 KO mice to Nano-Ni only caused mild acute lung inflammation and injury. At day 42 post-exposure, Nano-Ni caused extensive pulmonary fibrosis and chronic inflammation in the WT mouse lungs. However, exposure of miR-21 KO mice to Nano-Ni only caused mild lung fibrosis and chronic lung inflammation. Our results also showed that exposure to Nano-Ni caused upregulation of TGF-β1, phospho-Smad2, COL1A1, and COL3A1 in both WT and miR-21 KO mouse lungs. However, levels were significantly lower in miR-21 KO mice than in WT mice, except TGF-β1, which was similar in both kinds of mice. Decreased expression of Smad7 was observed in WT mouse lungs, but not in miR-21 KO mice. Our results demonstrated that knocking out miR-21 ameliorated Nano-Ni-induced pulmonary inflammation, injury, and fibrosis, suggesting the important role of miR-21 in Nano-Ni-induced pulmonary toxicity.
Keywords: Nickel nanoparticles (Nano-Ni), miR-21, lung injury, lung fibrosis, Smad7
Introduction
ISO/TS 80004 has defined the nanoscale as a “length range approximately from 1 nm to 100 nm” and a nanomaterial as a “material with any external dimension in the nanoscale or having internal structure or surface structure in the nanoscale” (ISO 2015). The unique properties that manifest in the nanoscale has led to the development, manufacture, and use of metal nanoparticles in many applications, such as plastics, colorants, coatings, cosmetics, semiconductors, drug delivery systems, and more (Colvin 2003; Niska et al. 2018; Stankic et al. 2016). Increased demand for manufactured nanomaterials has accompanied the development of nanotechnology, and rising prevalence of such materials in everyday life inevitably calls for a greater understanding of their effects on human health and the environment.
Nickel nanoparticles have characteristics such as high surface area, high surface energy, high magnetism, low melting point, and low burning point, which lends itself to use in printing inks, rechargeable batteries, ceramics and catalysts, and in the electronics industry for their magnetic and optical properties (Bajpai et al. 2012; Lei et al. 2016). Novel applications that utilize the nanoscale properties of nickel continue to be developed, such as in homogeneous nickel catalysis or in materials requiring high compressive strength (Seo et al. 2018; Sharma et al. 2018; Tasker et al. 2014). The intentional production and increased use of nickel nanoparticles has increased environmental and occupational exposure, which is associated with an elevated risk for skin allergies, the development of pulmonary inflammation, fibrosis and cancer, and so on (Anon 1990; IARC 1990; Journeay and Goldman 2014; Morgan and Usher 1994; Phillips et al. 2010). Two case reports demonstrate some of the health effects of nickel nanoparticles on humans. In one case report, a previously healthy man inhaled ~1g nickel nanoparticles while using a metal arc process to spray nickel nanoparticles onto bushes for turbine bearings. The worker died from adult respiratory distress syndrome (ARDS) at day 13 post-exposure (Phillips et al. 2010). Inhalation of 1 g nickel nanoparticles is equivalent to ~1.43 μg/cm2 epithelium, assuming a total alveolar surface area of a human lung of 70 m2 (Frohlich et al. 2016). In another case report, a laboratory technician developed nickel sensitization while working with nickel nanoparticles without special respiratory protection or control measures (Journeay and Goldman 2014). Properties of metal nanoparticles often drastically differ from that of bulk materials, suggesting unique mechanisms of toxicity. Our current understanding of these mechanisms lags behind the development of nanomaterials, so studying the potential health effects of nickel nanoparticles is urgent to safeguard the health of the public and the environment.
The skin, lungs, and gastrointestinal tract are constantly in contact with the environment, hence they are the most likely portals of entry for natural and anthropogenic nanoparticles (Oberdorster et al. 2005). While the skin is generally an effective barrier to foreign substances, the lungs and gastrointestinal tract are more vulnerable. The lungs are the major portal of entry for various particles and, as such, a key organ for whole-body defense by clearing most inhaled particles. The pulmonary toxicity of metal particles in both occupational and non-occupational settings is not only proportional to the concentration of metal exposure, but also to the particle size. Metal nanoparticles have wide range of physical and chemical properties that can alter their in vivo activity (Aillon et al. 2009; Shin et al. 2015). Our previous studies have demonstrated that nickel nanoparticles (Nano-Ni) with mean diameter of 20 nm instilled into rat lungs caused a greater inflammatory response than that of standard-sized nickel particles (Zhang et al. 2003). The small diameter and subsequent high particle number of Nano-Ni appear to play an important role in Nano-Ni-induced toxicity (Dick et al. 2003; Zhang et al. 1998; Zhang et al. 2003). We and other previous studies have demonstrated that exposure to Nano-Ni caused severe and persistent lung inflammation, injury, and fibrosis (Dick et al. 2003; Mo et al. 2019; Zhang et al. 1998; Zhang et al. 2003), and that pro-inflammatory cytokines were involved in these effects (Capasso et al. 2014; Glista-Baker et al. 2014; Zhang et al. 2003). Immune cells and inflammatory cytokines play essential roles in regulating the early inflammatory response and lung injury after Nano-Ni exposure. Although Nano-Ni-induced inflammatory responses were previously studied, the underlying mechanisms of acute and subacute biological responses and even chronic effects of Nano-Ni, such as lung fibrosis, still require further investigation.
MicroRNAs (miRNAs), noncoding endogenous RNAs of ~22 nucleotides, modulate the expression of multiple downstream genes by transcript degradation and/or translation inhibition (Lewis et al. 2005). Previous studies have demonstrated that miRNAs are dynamically regulated in various human diseases, including pulmonary and cardiovascular diseases (Brown et al. 2014; Ha 2011a, 2011b; Latronico and Condorelli 2009; Port and Sucharov 2010; Sessa and Hata 2013) and tumorigenesis (Chen et al. 2014; Ha 2011a). Certain studies have indicated that miRNAs are extensively involved in inflammation and fibrosis (Liu et al. 2010; O’Connell et al. 2012; Roggli et al. 2010). microRNA-21 (miR-21), one of the most extensively studied miRNAs, is highly expressed in many cell types and has been shown to play an important role in lung development and in the pathogenesis of various pulmonary diseases (Brown et al. 2014; Jiang et al. 2019). Notably, previous studies showed that exposure to particles, such as diesel exhaust particles and particulate matter, caused up-regulation of miR-21 (Bollati et al. 2010; Zhou et al. 2015). miR-21 was reported to contribute to pulmonary and cardiac fibrosis by repressing Spry1 and PTEN (Bronnum et al. 2013; Thum et al. 2008).
The aim of this study was to examine whether miR-21 was involved in Nano-Ni-induced lung inflammation, injury, and fibrosis in vivo. We proposed that exposure to Nano-Ni may upregulate miR-21 expression that may further cause lung inflammation, injury, and fibrosis; knocking out miR-21 may ameliorate these Nano-Ni-induced pulmonary effects. We also explored potential mechanisms that may be involved in miR-21-regulated Nano-Ni-induced pulmonary toxicity.
Materials and methods
Nickel nanoparticles and their characterization
Nano-Ni used in this study was provided by Inabata & Co., Ltd., Vacuum Metallurgical Co., Ltd., Japan, which is composed of Ni (85–90%) and NiO (10–15%) (Mo et al. 2008; Mo et al. 2019; Wan et al. 2011). The mean diameter of Nano-Ni in the powder was 20 nm, which was determined by transmission electron microscopy (TEM), and the average hydrodynamic size of Nano-Ni was about 250 nm (240~280 nm), measured by dynamic light scattering (DLS) (Mo et al. 2019; Wan et al. 2011). The specific surface area was 43.8 m2/g, and the Zeta potential was 2.0 ± 1.4 mV (Mo et al. 2019; Wan et al. 2011). Nano-Ni was dispersed in physiological saline, ultrasonicated for 10 min, and vibrated thoroughly prior to each experiment.
Animals
The miR-21 knock-out (KO) mice (B6;129S6-Mir21atm1YoLi/J) were originally obtained from Dr. Yong Li at University of Louisville, in which the 93 bp precursor to the miR-21 (pre-miR-21) sequence in the 3’ UTR of the vacuole membrane protein 1 (Vmp1 or Tmem49) transcript was replaced with a pGK-gb2 loxPIFRT-flanked neomycin resistance cassette (Ma et al. 2011). The mice were bred in the animal facility of University of Louisville. Eight-week-old male C57BL/6J mice, weighing about 22–28 g, were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were housed in an air-conditioned room (temperature of 20 ± 2°C, relative humidity of 60 ± 10%) with a 12-h light and 12-h dark cycle environment and with free access to food and water. The mice were allowed to acclimatize for 1–2 weeks before starting the experimental protocol and were monitored daily for general health. Animal use was reviewed and approved by the University of Louisville Institutional Animal Care and Use Committee.
Exposure of mice to nickel nanoparticles
Mice were exposed to Nano-Ni by intratracheal instillation as described previously (Mo et al. 2019; Wan et al. 2017; Zhang et al. 1998; Zhang et al. 2003). The intratracheal instillation model is an easy and reliable method compared with an inhalation study, and has been widely used to identify particle toxicity and to compare responses to different particle types (Driscoll et al. 2000). The C57BL/6J (WT) and miR-21 KO mice with similar age and weight were randomly assigned to physiological saline-instilled (control) or Nano-Ni-instilled groups. Under anesthesia, mouse neck skin was opened by a small midline incision and the trachea was isolated. Then, 10, 20, 50, or 100 μg of Nano-Ni in 50 μL physiological saline were instilled intratracheally by a syringe with a 28G1/2 needle under anesthesia (Mo et al. 2019; Wan et al. 2017). Control mice were instilled with 50 μL physiological saline. Immediately after instillation, the skin was closed with a monofilament nylon suture. Sutures were removed in 7–10 days. The mice were sacrificed at multiple time points following instillation of Nano-Ni.
BAL and BALF analysis
BALF was collected to assess lung inflammatory responses after Nano-Ni instillation as described previously (Mo et al. 2019; Wan et al. 2017; Zhang et al. 1998; Zhang et al. 2003). Briefly, mice were anesthetized by an overdose injection of tribromoethanol into the abdominal cavity, and the abdominal aorta was severed. The trachea was exposed and cannulated, allowing the lungs to be lavaged with 0.5 mL of ice-cold sterile 1x PBS containing 0.4 mM EDTA. The lavage fluid was retrieved by gentle massage. The procedure was repeated another five times. The first and second collected lavage fluid samples were pooled together and centrifuged at 200 g for 5 min at 4°C. The supernatant was collected and stored at −80°C for subsequent analysis of biochemical markers, proinflammatory cytokines, and MMP-2/9 protein levels and activities. The cells in the BALF were pooled together. The total number of cells in BALF was calculated based on the number of cells counted under microscope using a hemocytometer. The cell differential was evaluated on a cytospin slide stained with Giemsa and May-Grünwald stains (Sigma-Aldrich, St. Louis, MO, USA).
The levels of lactate dehydrogenase (LDH) activity and CXCL1/KC in the BALF were measured by an LDH Cytotoxicity Detection Kit (TaKaRa Bio Inc., Shiga, Japan) and a Mouse CXCL1/KC PicoKine™ ELISA Kit (Boster Biological, Pleasanton, CA, USA). The concentration of total protein in the BALF was determined via the Bradford method using Bio-Rad Protein Assay (Hercules, CA, USA). The MMP-2 and MMP-9 protein levels in BALF were determined by Mouse MMP-2 or MMP-9 PicoKine™ ELISA Kit (Boster Biological, Pleasanton, CA, USA). The protein contents of proinflammatory cytokines IL-6 and TNFα in BALF were determined by IL-6 or TNF alpha Mouse ELISA Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s instructions.
Gelatin zymography assay
MMP-2 and MMP-9 activities in the BALF and lung tissues were determined by a gelatin zymography assay as described previously (Mo et al. 2009b; Mo et al. 2019; Wan et al. 2008; Wan et al. 2011; Zhang et al. 2016; Zhang et al. 2019). A total of 60 μL of BALF mixture (48 μL BALF + 12 μL 5x loading buffer) was loaded into each lane of 10% SDS-PAGE copolymerized with 0.5 mg/mL gelatin, which was used as the substrate under non-reducing conditions. Lung tissues were homogenized on ice in 20 volumes per tissue mass of RIPA lysis buffer supplemented with PMSF, protease inhibitor cocktail, and sodium orthovanadate (Santa Cruz Biotechnology, Santa Cruz, CA) using a Tissue-Tearor homogenizer (BioSpec Products, Bartlesville, OK). After the homogenate was centrifuged at 10,000 g for 15 min at 4°C, the supernatant was collected and the protein concentration was determined by Bio-Rad Protein Assay (Hercules, CA). 30 μg protein per lane was subjected to electrophoresis on 10% SDS-PAGE copolymerized with 0.5 mg/mL gelatin. After electrophoresis, the gels were washed at room temperature in 50 mM Tris-HCl buffer (pH 7.5) containing 2.5% Triton X-100 (Sigma, St. Louis, MO) for 1 h, changing the solution every 15 min. The gels were then incubated at 37°C overnight in 50 mM Tris-HCl buffer (pH 7.5) containing 0.2 M NaCl, 7.55 mM CaCl2, 1 μM ZnCl2, and 1% Triton X-100 with gentle shaking to develop the enzyme activity bands. After washing with distilled water twice, 5 min each time, the gels were stained with 0.1% Coomassie Brilliant Blue R-250 (Bio-Rad, Hercules, CA) and destained with 10% acetic acid until clear bands were observed against the background of Coomassie blue-stained gel.
Lung histology and trichrome staining
Mouse lung tissues were fixed in 10% neutral buffered formalin at an inflation pressure of 20 cm H2O for 5 min through a catheter inserted into the trachea, collected, and placed in 10% neutral buffered formalin overnight at 4°C. Lung tissues were then dehydrated stepwise through an ascending series of alcohol solutions and finally degreased in xylene. The tissues were embedded in paraffin and sectioned at 5 μm by a microtome (Thermo Scientific, Rockford, IL).
To examine lung histology changes after Nano-Ni exposure, lung sections were stained with hematoxylin and eosin (HE) stain. Images were captured and the volume fraction of lung parenchyma with chronic inflammation after Nano-Ni exposure was quantified by morphometric analysis using ImageJ (http://imagej.nih.gov/ij/) as described previously (Mo et al. 2013; Mo et al. 2015; Mo et al. 2019).
To observe and compare the extent of pulmonary fibrosis in WT and miR-21 KO mice after Nano-Ni exposure, Masson’s Trichrome for Connective Tissue kit (Electron Microscopy Sciences, Hatfield, PA) was used to stain the mouse lung tissues according to the manufacturer’s instructions with minor modifications as described previously (Mo et al. 2019; Wan et al. 2017).
Dual immunofluorescent staining
Mouse lung sections were deparaffinized with xylene and rehydrated in water through graded ethanol. Antigen retrieval was performed in 10 mM sodium citrate solution (pH 6.0) with 0.05% Tween-20 added freshly and incubated in 95°C water bath for 30 min. The nonspecific protein binding was blocked by incubating the sections with 3% BSA, 5% normal goat serum, and 0.3% Triton X-100 at room temperature for 30 min. The primary antibodies used were TTF1 (thyroid transcription factor 1, 1:250, cat. no. ab76013, Abcam, Cambridge, MA) and α-SMA (α-smooth muscle actin, 1:400, cat. no. A5228, Sigma-Aldrich, St. Louis, MO). TTF1 stains lung type II pneumocytes and club (Clara) cells positive, while activated fibrogenic cells, myofibroblasts, contains α-SMA. After incubation with primary antibodies at room temperature for 2h and washing with 1x PBS three times, the lung sections were incubated with donkey anti-rabbit IgG (Alexa Fluor 594, 1:500, Abcam, Cambridge, MA) and goat anti-mouse IgG (Alexa Fluor 488, 1:500, Invitrogen, Carlsbad, CA) at room temperature for 1 h. After washing, the sections were mounted with Prolong Gold Antifade Reagent with DAPI (Invitrogen, Carlsbad, CA) and examined using fluorescence microscopy.
Hydroxyproline assay
Measurement of hydroxyproline content in mouse left lungs was used to determine the amount of collagen in mouse lungs after Nano-Ni exposure. This method is based on the acid hydrolysis of the tissue and subsequent determination of the free hydroxyproline in hydrolysate. The hydroxyproline content in mouse left lungs was measured as previously described (Mo et al. 2013; Mo et al. 2019). The amount of hydroxyproline was determined against a standard curve generated using known concentrations of hydroxyproline (Sigma-Aldrich, St. Louis, MO).
Total RNA isolation and real-time PCR
To determine the expression levels of miR-21, proinflammatory cytokines, and fibrosis-related genes in lung tissues after Nano-Ni exposure, total RNA was isolated from lung tissues using mirVana miRNA Isolation Kit (Abcam, Cambridge, MA) according to the manufacturer’s instructions. Lung tissues were weighed and homogenized in 10 volumes per tissue mass of Lysis/Binging Buffer on ice using a Tissue-Tearor homogenizer (BioSpec Products, Bartlesville, OK). At the end of the procedure, total RNA was eluted in nuclease-free water and its concentration was measured by absorbance at 260 nm with a DU 730 Spectrophotometer (Beckman Coulter, Fullerton, CA).
To measure the level of miR-21, TaqMan® MicroRNA Assay was used (Applied Biosystems, Assay ID 000397). There were two-steps: (1) Reverse transcription using TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems) and (2) Real-Time PCR. Briefly, 10 ng total RNA was reverse transcribed in a total volume of 15 μL which contained 0.15 μL of 100 mM dNTPs, 1 μL MultiScribe™ Reverse Transcriptase (50 U/μL), 1.5 μL of 10x Reverse Transcription Buffer, 0.19 μL RNase Inhibitor (20 U/μL), and 3 μL of 5x RT primer. Then, 2 μL RT product from each sample was used to perform real‐time PCR in a total volume of 20 μL which contained 10 μL of TaqMan® Universal PCR Master Mix and 1 μL of TaqMan® Small RNA Assay (20x) (Applied Biosystems). Values of miR-21 expression in mouse lungs was normalized to the endogenous control U6 snRNA (Applied Biosystems, Assay ID 001973), and reported as fold increase as compared to the control without Nano-Ni exposure.
To determine the mRNA expression levels of proinflammatory cytokines and fibrosis-related genes, 2 μg total RNA was reverse‐transcribed into cDNA in a total volume of 25 μL, which included 1 μL of M‐MLV reverse transcriptase (Promega, Madison, WI), 2 μL of 0.5 μg/ μL oligo(dT)18 primer, 1.25 μL of 10 mM dNTP, 0.75 μL RNasin Plus RNase Inhibitor, and 5 μL of 5x M‐MLV reaction buffer. Real‐time PCR was performed using a Bio‐Rad iQ5 iCycler as described previously (Mo et al. 2009a; Mo et al. 2012). Briefly, 1 μL cDNA solution from each sample was used to perform real‐time PCR in a total volume of 20 μL using 10 μL of iTaq Universal SYBR Green Supermix (Bio‐Rad) and 1 μL of 5 μM of each primer. The experimental protocol consisted of four programs: (i) Denaturation of the cDNA/RNA hybrid at 95°C for 3 min; (ii) Amplification of cDNA for 40–50 cycles, each cycle sequentially 95°C for 10 s, 58°C for 45 s, and 72°C for 45 s; (iii) Analysis of the melting curve to confirm the single product amplification during the PCR assay; and (iv) Cooling the rotor and thermal chamber to 25°C. The specific primers for each gene are listed in Table 1. The relative expression level of each gene was calculated using the 2−ΔΔCT (Livak) method (Livak and Schmittgen 2001) and normalized to the expression of a reference gene β-actin in the same sample, and then expressed as the fold increase of the gene in the Nano-Ni-exposed group relative to the control without Nano-Ni exposure.
Table 1.
Mouse primers used for real-time PCR.
| Gene | Forward (5’ → 3’) | Reverse (5’ → 3’) | Product size (bp) |
|---|---|---|---|
| IL-6 | TTG GGA CTG ATG CTG GTG ACA | TTG GAA ATT GGG GTA GGA AGG A | 470 |
| TNFα | GCC CAC GTC GTA GCA AAC CAC CAA | ACA CCC ATT CCC TTC ACA GAG CAA T | 445 |
| TGF-β1 | ACC CCC ACT GAT ACG CCT GA | AGC AGT GAG CGC TGA ATC GAA | 109 |
| COL1A1 | GTG CTC CTG GTA TTG CTG GT | GGC TCC TCG TTT TCC TTC TT | 212 |
| COL3A1 | GGG TTT CCC TGG TCC TAA AG | CCT GGT TTC CCA TTT TCT CC | 237 |
| β-actin | GGC ATT GTT ACC AAC TGG GAC | ACC AGA GGC ATA CAG GGA CAG | 219 |
Protein isolation and Western blot
Protein was extracted according to our previous studies (Feng et al. 2015; Long et al. 2019; Mo et al. 2009a; Mo et al. 2012; Yu et al. 2010; Zhang et al. 2016). Mouse lung tissues were homogenized on ice in 20 volumes per tissue mass of RIPA lysis buffer supplemented with PMSF, protease inhibitor cocktail, and sodium orthovanadate (Santa Cruz Biotechnology, Santa Cruz, CA) using a Tissue-Tearor homogenizer (BioSpec Products, Bartlesville, OK). After setting on ice for 40 min and centrifuging at 10,000 g and 4°C for 15 min, the supernatant was collected. The protein concentration in the supernatant was determined by Bradford method with a DU730 Spectrophotometer (Beckman Coulter, Fullerton, CA). Western blot was performed as previously described (Feng et al. 2015; Long et al. 2019; Mo et al. 2009a; Mo et al. 2012; Yu et al. 2010; Zhang et al. 2016). The primary antibody for Smad7 (1:200, cat. no. PA1–41506) was obtained from Invitrogen (Carlsbad, CA), for COL1A1 (1:1000, cat. no. 84336), TGF-β (1:1000, cat. no. 3711), phospho-Smad2 (1:1000, cat. no. 12747), and Smad2 (1:1000, cat. no. 5339) from Cell Signaling Technology (Beverly, MA), and for β-actin (1:4000, cat. no. A1978) from Sigma (Saint Louis, MO). HRP-conjugated goat anti-rabbit IgG (1:2000, for TGF-β, phospho-Smad2, Smad2, Smad7, and COL1A1) was from CHEMICON (Temecula, CA), while HRP-conjugated horse anti-mouse IgG (1:2000, for β-actin) was from Cell Signaling Technology (Beverly, MA). Immunoreactive bands were detected using ECL™ Western Blotting Detection Reagents (GE Healthcare, Amersham™, Buckinghamshire, UK) followed by exposure to CL-XPosure™ film (Thermo Scientific, Rockford, IL) and quantified using NIH ImageJ software (http://imagej.nih.gov/ij/).
Statistical analysis
Statistical analyses were carried out using SigmaPlot 13.0 software (Systat Software, Inc., San Jose, CA, USA). Data were expressed as the mean ± SEM. Differences among groups were evaluated by one-way or two-way analysis of variance (ANOVA). If necessary, transformation of data was used to achieve normally distributed data before ANOVA analysis. If a p-value was less than 0.05, a difference was considered statistically significant.
Results
Nano-Ni exposure upregulated miR-21
Dose-response and time-response studies were performed to obtain an overall view of the effects of Nano-Ni exposure on miR-21 expression. In the dose-response studies, C57BL/6J mice were instilled intratracheally with 10, 20, 50, or 100 μg per mouse of Nano-Ni and sacrificed at day 3 after exposure. Nano-Ni exposure caused miR-21 upregulation in the mouse lungs, and 50 or 100 μg per mouse of Nano-Ni instillation caused a statistically significant increase in miR-21 expression (Figure 1A). A dose of 50 μg of Nano-Ni per mouse was selected for the following studies. In the time-response studies, C57BL/6J mice were intratracheally instilled with 50 μg per mouse of Nano-Ni and sacrificed at days 1, 3, 7, 14, 28, or 42 after exposure. Control mice instilled with physiological saline were sacrificed at days 1 and 42 after instillation. miR-21 expression was significantly increased as early as day 3 after 50 μg per mouse of Nano-Ni exposure, peaked at day 14, and then declined but still remained significantly higher than that of the control mice at days 28 and 42 after Nano-Ni exposure (Figure 1A), suggesting that Nano-Ni can cause miR-21 upregulation in mouse lungs.
Figure 1.
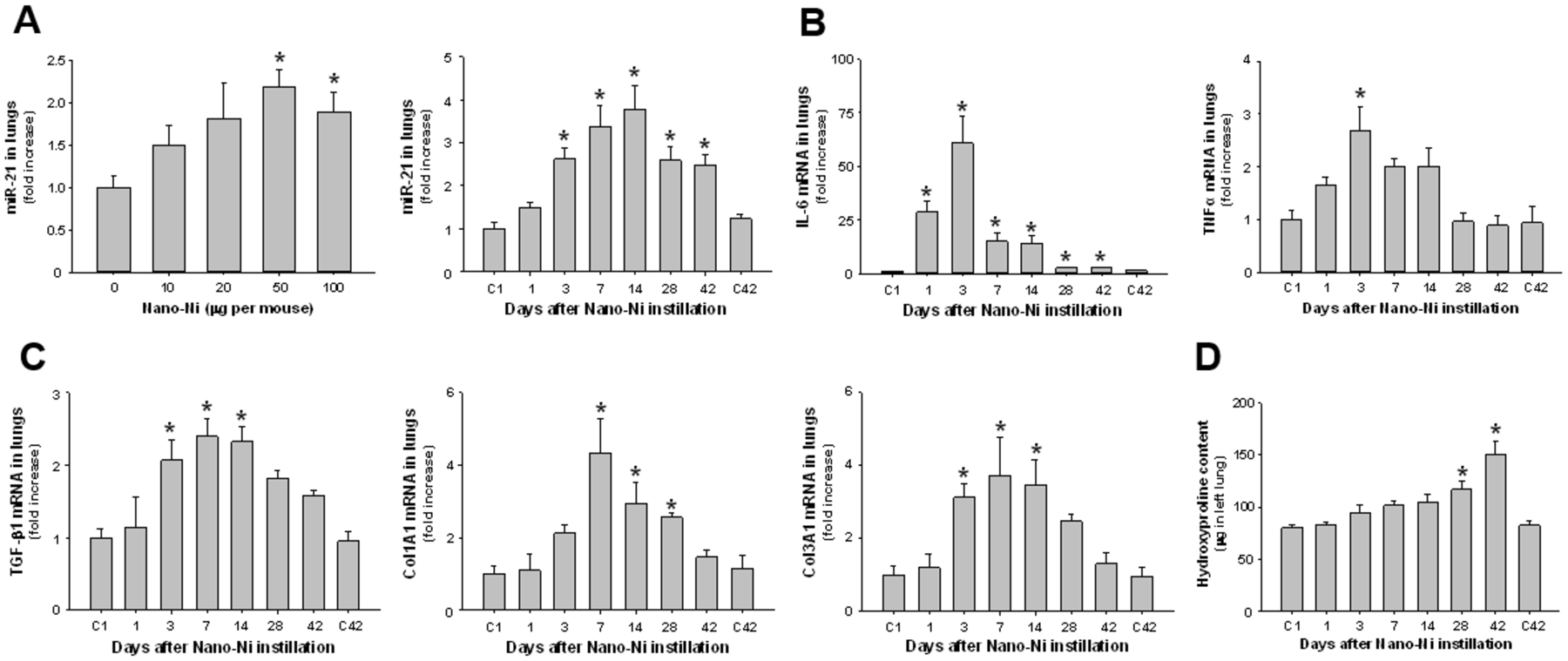
Upregulation of miR-21 (A), proinflammatory cytokines (B), and fibrosis-related genes (C), and increased hydroxyproline content (D) in mouse lungs after Nano-Ni exposure. In the dose-response study, C57BL/6J mice were instilled intratracheally with 0, 10, 20, 50, or 100 μg per mouse of Nano-Ni, and lung tissues were collected at day 3 after exposure. In the time-response study, C57BL/6J mice were instilled intratracheally with 50 μg per mouse of Nano-Ni, and lung tissues were collected at multiple times after exposure. Control mice were instilled with physiological saline and sacrificed at day 1 (C1) and day 42 (C42) after exposure. The expression of miR-21 (A), proinflammatory cytokines (B), and fibrosis-related genes (C) was analyzed by real-time PCR, normalized to the endogenous control U6 snRNA (A) or β-actin (B-C), and reported as fold increase as compared to the control. The hydroxyproline content was measured in the left lung (D). Data are shown as mean ± SEM (n=4~5). * p<0.05 vs. control.
Nano-Ni exposure caused upregulation of proinflammatory cytokines and fibrosis-related genes
Our previous study showed that exposure of mice to Nano-Ni caused severe and persistent lung inflammation and fibrosis (Mo et al. 2019). Dysregulation of proinflammatory cytokines such as IL-6 and TNFα was observed in pulmonary inflammation and fibrosis (Howrylak and Nakahira 2017; Le et al. 2014; Zhang et al. 2003), and the TGF-β1/Smad signaling pathway is critical for the fibrotic response (Upagupta et al. 2018). A time-response study was performed to assess changes in proinflammatory cytokines such as IL-6 and TNFα and in fibrosis-related genes such as TGF-β1, COL1A1, and COL3A1 following Nano-Ni exposure. Mice were instilled intratracheally with 50 μg per mouse of Nano-Ni, and lung tissues were collected at days 1, 3, 7, 14, 28 and 42 after exposure. Lung tissues from control mice were collected at days 1 and 42 after physiological saline instillation. The expression of proinflammatory cytokines and fibrosis-related genes was determined by real-time PCR. Our results showed that the mRNA expression level of proinflammatory cytokine IL-6 increased as early as day 1 after exposure, peaked at day 3, and decreased thereafter from day 7 to day 42 post-instillation (Figure 1B). However, even at day 42 after exposure, its expression level was still significantly higher than that of the control (Figure 1B). Nano-Ni exposure caused slight TNFα upregulation at day 1 after exposure, and the increase in TNFα expression became statistically significant at day 3 after Nano-Ni exposure (Figure 1B). The expression levels of fibrosis-related genes such as TGF-β1, COL1A1, and COL3A1 increased over time and peaked at day 7 after Nano-Ni exposure, although TGF-β1 and COL3A1 expression levels were already significantly increased at day 3 after exposure (Figure 1C). Then their expression levels gradually decreased over time, but never reaching the baseline levels of the controls even at day 42 after exposure (Figure 1C). The increased and continued expression of proinflammatory cytokines and fibrosis-related genes in mouse lungs after Nano-Ni exposure suggest that Nano-Ni can induce persistent inflammatory and profibrotic responses in mouse lungs.
Nano-Ni exposure caused pulmonary inflammation and fibrosis
To confirm the impact of Nano-Ni on pulmonary inflammation and fibrosis, mice were intratracheally instilled with 50 μg per mouse of Nano-Ni, and lung tissues were collected at days 1, 3, 7, 14, 28, or 42 after exposure. H&E staining was performed on the lung sections to observe histopathological changes after Nano-Ni exposure, while trichrome staining, which stains collagen blue, and hydroxyproline assay, which measures the content of hydroxyproline in lungs, were used to confirm lung fibrosis after Nano-Ni exposure. In the control mice sacrificed at day 1 and day 42 after physiological saline instillation, normal structure of lung parenchyma was observed (Figure 2 A&H). Nano-Ni exposure caused lung inflammation with a tendency for increased involvement of lung tissues and multiple lobes with time after exposure. As early as day 1 after exposure, leukocytes, including neutrophils, macrophages, and eosinophils, were observed to infiltrate the lung parenchyma, mainly at the perivascular, peribronchial, and peribronchiolar areas, although some were observed in the alveolar spaces (Figure 2B). At day 1 after exposure, nearly 20% of the cells in the bronchoalveolar lavage fluid (BALF) were neutrophils (Mo et al. 2019). At day 3 after exposure, a large number of leukocytes infiltrated into the lung parenchyma (Figure 2C), in which more than 30% of the cells were neutrophils (Mo et al. 2019). Enlarged macrophages containing Nano-Ni agglomerates were also observed in the alveolar space (Figure 2C). No fibrosis was observed in the controls (Figure 2 I&P) and at days 1 and 3 after Nano-Ni exposure (Figure 2J–K). At day 7 after exposure, solitary and small aggregates of enlarged macrophages were found in the lung parenchyma (Figure 2D). Only in these macrophage aggregates, focal increase in collagen deposition (blue staining) was observed by Masson’s trichrome staining (Figure 2L), indicating focal pulmonary fibrosis. Increasingly large macrophage aggregates were observed, correlating with time after exposure (Figure 2E–G). In the lumen of some bronchi and bronchioles, granulomatous lesions were observed at day 28 after exposure and beyond (Figure 2F–G). Necrosis was observed at the center of some granulomatous lesions at day 42 after exposure (Figure 2G, inserted image). In some areas of the lungs, alveolar space and septa were infiltrated primarily by a large number of foamy macrophages, enlarged macrophages with a larger nuclei and prominent nucleoli, and some were mixed with polymorphonuclear cells (PMNs) (Figure 2F–G). Lymphocyte infiltration in some areas of lungs and thickening of subepithelial areas of bronchi and bronchioles and alveolar septa were also observed at days 28 and 42 after Nano-Ni exposure (Figure 2F–G). Increased extent of fibrosis from day 14 to day 42 after Nano-Ni exposure was confirmed by trichrome staining, which showed collagen deposition in the granulomatous lesions, subepithelial areas, and alveolar septa (Figure 2M–O). The pulmonary fibrosis was quantified by measuring the hydroxyproline content in the mouse left lungs, which is a significant biomarker of collagen deposition (Mo et al. 2013; Mo et al. 2019). Our results demonstrated that exposure to Nano-Ni caused a time-dependent increase in the hydroxyproline content in the left lungs, and this increase was significant at days 28 and 42 after Nano-Ni exposure (Figure 1D). Our lung histopathological examination confirmed that Nano-Ni could cause severe and persistent lung inflammation, injury, and fibrosis.
Figure 2.
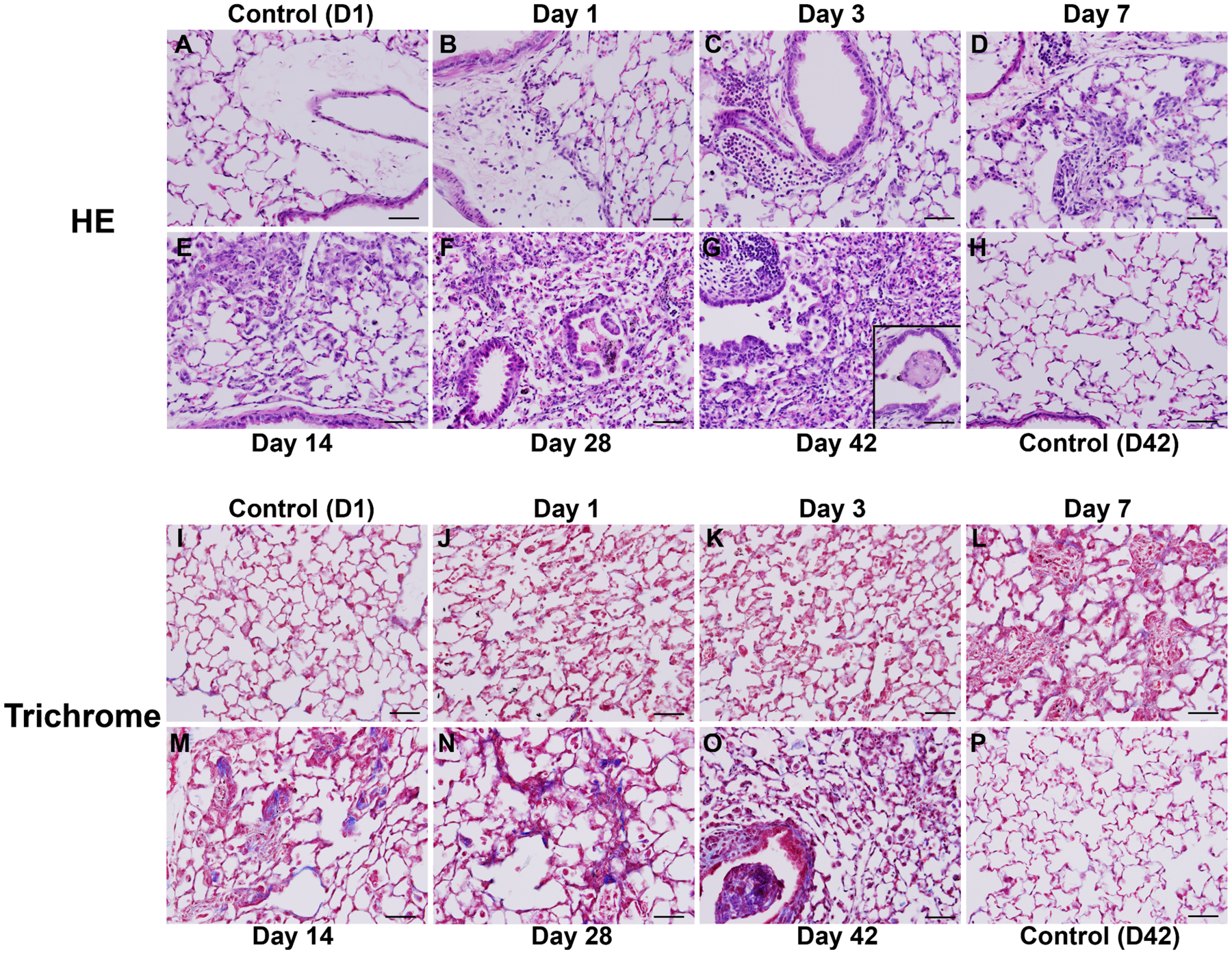
Histology of mouse lungs after Nano-Ni exposure (time-course). Mice were instilled intratracheally with 50 μg per mouse of Nano-Ni, and lung tissues were collected at multiple times after exposure. Control mice were instilled with physiological saline and sacrificed at day 1 (D1) and day 42 (D42) after exposure. Lung tissues were subjected to HE (A-H) or trichrome (I-P) staining. A, H, I, and P show the normal structure of lung parenchyma in the control mice. Nano-Ni exposure caused acute pulmonary inflammation indicated by large numbers of polymorphonuclear (PMN) cells and macrophages infiltrating into the lung parenchyma (B-C), which followed by subacute and chronic inflammation and fibrosis with time after exposure (D-G). The inserted image in G (right lower corner) shows a granulomatous lesion with necrosis. Pulmonary fibrosis was confirmed by trichrome staining (I-P). No fibrosis was observed at Day 1 (J) and Day 3 (K) after Nano-Ni exposure. Small amount of collagen deposition (blue staining) was observed at Day 7 (L) after Nano-Ni exposure and the collagen deposition gradually increased with time after exposure (M-O), indicating pulmonary fibrosis. Scale bar represents 50 μm in all panels.
Knocking out miR-21 ameliorated Nano-Ni-induced acute pulmonary inflammation and injury in mice
To compare the acute pulmonary effects of Nano-Ni on wild-type (WT) and miR-21 knock-out (KO) mice, mice were intratracheally instilled with 50 μg per mouse of Nano-Ni and sacrificed at day 3 post-exposure. The choice of a dose of 50 μg per mouse and a time point of day 3 post-exposure was based on our previous (Mo et al. 2019) and current dose-response and time-response studies (Figure 1A–B) and observations that a significant increase in the expression of miR-21 was observed at day 3 after 50 μg of Nano-Ni exposure (Figure 1A). No mice died in the control groups and Nano-Ni-exposed groups during the 3-day experimental period.
(A). BALF profiles
To compare Nano-Ni-induced acute pulmonary inflammation and injury in WT mice with that in miR-21 KO mice, mice underwent bronchoalveolar lavage at day 3 after 50 μg per mouse of Nano-Ni exposure, and the cellular and biochemical constituents in BALF were analyzed. Our results showed that intratracheal instillation of Nano-Ni caused severe acute pulmonary inflammation and lung injury in WT mice, manifesting as a marked increase in neutrophil count, CXCL1/KC level, LDH activity, and concentration of total protein in BALF obtained from mice at day 3 after Nano-Ni exposure (Figure 3A). Eosinophil count in BALF from WT mice exposed to Nano-Ni was also increased (data not shown). Although Nano-Ni exposure also caused a statistically significant increase in these cellular and biochemical parameters in BALF obtained from miR-21 KO mice, the increases were significantly lower than those of the WT mice (Figure 3A).
Figure 3.
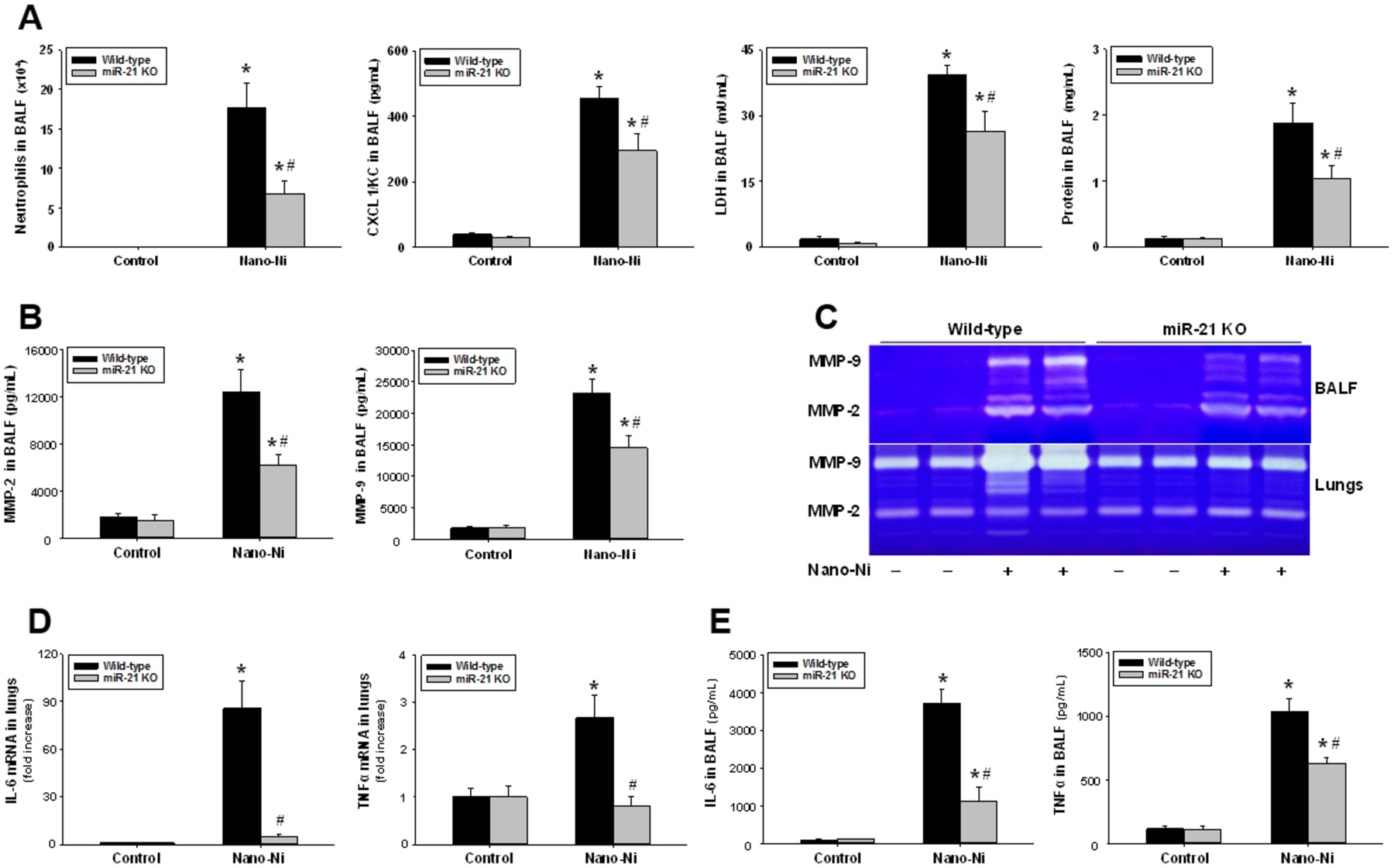
Cellular and biochemical parameters (A), MMP-2/9 (B-C), and proinflammatory cytokines (D-E) in BALF and lungs from mice at day 3 after Nano-Ni exposure. Wild-type and miR-21 KO mice were intratracheally instilled with 50 μg per mouse of Nano-Ni. Control mice were instilled with physiological saline. (A) Cellular and biochemical parameters in BALF. The level of LDH activity was measured by an LDH Cytotoxicity Detection Kit. The concentration of total protein was determined via the Bradford method. The protein level of CXCL1/KC was determined by an ELISA kit. (B) The protein levels of MMP-2 and MMP-9 in BALF were determined by ELISA kits. (C) MMP-2 and MMP-9 activities in BALF and lungs were determined by gelatin zymography. (D) mRNA expression of IL-6 and TNFα in lungs was determined by real-time PCR. (E) The protein levels of IL-6 and TNFα in BALF were determined by ELISA kits. Data are shown as mean ± SEM (n=5~9). * p<0.05 vs. the control group of the same genotype; # p<0.05 vs. wild-type Nano-Ni group.
(B). MMP-2 and MMP-9 protein levels and activities in BALF and lungs
MMP-2 (gelatinase A) and MMP-9 (gelatinase B) are zinc metalloendopeptidases in the matrix metalloproteinase (MMP) family, which are capable of degrading type IV collagen and gelatin (Hannocks et al. 2019). MMP-2 and MMP-9 have been implicated in lung inflammation and injury. Increased MMP-2 and MMP-9 in BALF or tracheal aspirates have been observed in patients with ARDS (Gushima et al. 2001; Hsu et al. 2015). MMP-2 and/or MMP-9 have also been implicated in the development of experimental acute lung injury (Blazquez-Prieto et al. 2018; Mo et al. 2019). In addition, exposure to Nano-Ni has been reported to cause ARDS (Phillips et al. 2010). To determine whether the presence of MMP-2 and MMP-9 was associated with Nano-Ni-induced lung inflammation, injury, and fibrosis, MMP-2 and MMP-9 protein levels in BALF were determined by commercially available ELISA kits, while their activities in both BALF and lung tissue homogenates were analyzed by gelatin zymography assay.
Our results demonstrated that exposure to Nano-Ni led to significant increases in MMP-2 and MMP-9 protein levels in the BALF obtained from WT mice as compared to those of control mice (Figure 3B). Although exposure of miR-21 KO mice to Nano-Ni also caused a significant increase in MMP-2 and MMP-9 protein levels in BALF, their levels were significantly lower than those of the Nano-Ni-exposed WT mice (Figure 3B). The MMP-2 and MMP-9 activities in BALF and lung tissue homogenates analyzed by gelatin zymography assay were consistent with their protein levels in BALF (Figure 3C). Although Nano-Ni exposure caused significant increases in MMP-2 and MMP-9 activities in BALF and lung tissues from both WT and miR-21 KO mice, their activities in miR-21 KO mice were much lower than those of WT mice (Figure 3C).
(C). Proinflammatory cytokines in BALF and lungs
The mRNA expression of proinflammatory cytokines IL-6 and TNFα in lung tissues collected from WT and miR-21 KO mice were compared by real-time PCR. Our results showed that Nano-Ni caused significant increases in IL-6 and TNFα mRNA levels in WT mice, but not in miR-21 KO mice (Figure 3D). The protein levels of IL-6 and TNFα in BALF were determined by ELISA. The results showed that Nano-Ni caused significant increases in IL-6 and TNFα protein levels in the BALF obtained from both WT and miR-21 KO mice. However, their levels were significantly lower in miR-21 KO mice as compared to those of WT mice (Figure 3E).
(D). Lung histology
H&E staining was performed on lung sections to compare and examine the histopathological changes in WT and miR-21 KO mice after Nano-Ni exposure. In the control mice with physiological saline instillation, normal structure of lung parenchyma was observed (Figure 4 A&C). Instillation of Nano-Ni in WT mice caused severe acute lung inflammation evidenced by infiltration of a large number of polymorphonuclear (PMN) cells and macrophages into the perivascular, peribronchial, and peribronchiolar areas and alveolar spaces and septa (Figure 4B). Macrophages containing cytoplasmic particles were also observed (Figure 4B arrow). Some mice exhibited intra-alveolar hemorrhage and exudation of serum proteins into the alveolar spaces. However, instillation of Nano-Ni to miR-21 KO mice only induced mild lung inflammation (Figure 4D). Although some macrophages, neutrophils, and eosinophils infiltrated into the lung parenchyma of Nano-Ni-exposed miR-21 KO mice, their numbers were much less than that observed in the lungs of Nano-Ni-exposed WT mice (Figure 4D).
Figure 4.
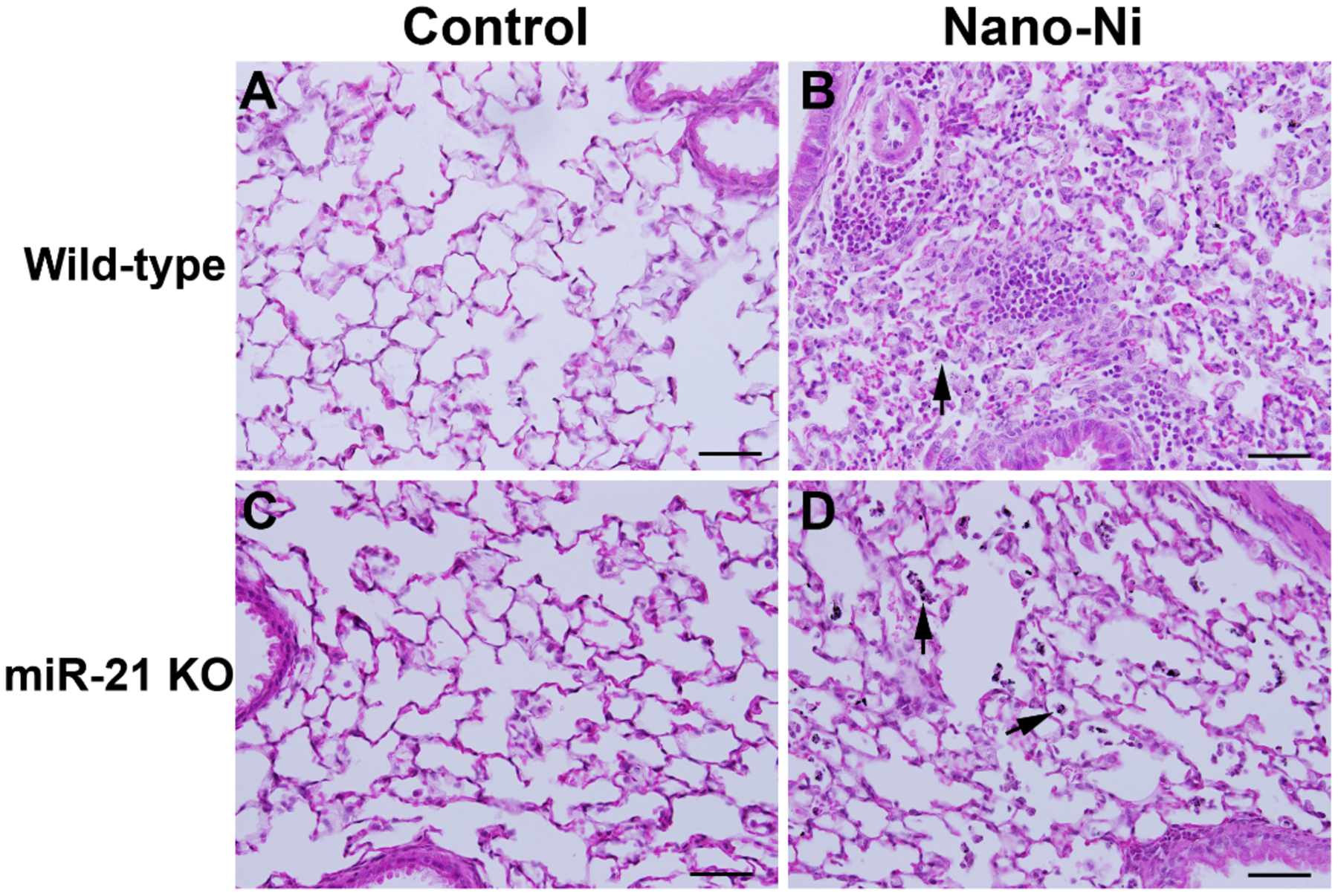
Acute pulmonary inflammation in mouse lungs at day 3 after Nano-Ni exposure. Wild-type and miR-21 KO mice were instilled intratracheally with 50 μg per mouse of Nano-Ni. Control mice were instilled with physiological saline. Lung tissues collected from mice at day 3 after exposure were analyzed by H&E staining. A and C show the normal structure of alveoli and peribronchiolar areas in the control mice. B shows acute inflammation in the lungs of a wild-type mouse with Nano-Ni exposure, evidenced by large numbers of polymorphonuclear (PMN) cells and macrophages infiltrating into the lung parenchyma. Acute pulmonary inflammation was also observed in the lungs of Nano-Ni-exposed miR-21 KO mice, but to a much lesser extent (D). Arrows: Nano-Ni-phagocytized macrophages. Scale bar represents 50 μm in all panels.
Knocking out miR-21 reduced Nano-Ni-induced chronic pulmonary inflammation and fibrosis in mice
To examine whether miR-21 was involved in Nano-Ni-induced chronic and persistent pulmonary inflammation and fibrosis, lung histopathological changes after exposure of WT and miR-21 KO mice to Nano-Ni were examined and compared. A dose of 50 μg per mouse of Nano-Ni and a time point of day 42 post-exposure were selected, as our time-response results demonstrated extensive pulmonary fibrosis in WT mice at that time.
(A). Lung histology
WT and miR-21 KO mice were instilled intratracheally with 50 μg per mouse of Nano-Ni. At day 42 after exposure, mice were sacrificed, and the lung tissues were collected for histological examination by H&E staining. Normal structure of lung parenchyma was observed in WT and miR-21 KO control mice with physiological saline instillation (Figure 5A&C). In WT mice, Nano-Ni instillation caused extensive chronic pulmonary inflammation and fibrosis (Figure 5B). A large number of enlarged and foamy macrophages with/without a mixed infiltrate with PMNs were observed in the alveolar space, alveolar septa, and the lumen of bronchi and bronchioles. In some areas of pneumonitis, granulomatous lesions (some with central necrosis), bronchiolization of alveoli, lymphocyte aggregates, deposition of proteinaceous materials in the alveoli, and increased number of pulmonary intravascular macrophages were also observed. Nano-Ni-induced lung fibrosis was reflected by thickening of alveolar septa and subepithelial areas of bronchi and bronchioles. Fibrosis also developed in the granulomatous lesions. Although chronic inflammation and fibrosis were observed in some areas of Nano-Ni-exposed miR-21 KO lungs, they were much milder and focal (Figure 5D). Morphometric analysis was used to quantify the extent of chronic inflammation in the right three lobes, showing that the volume fraction inflamed in the lungs of WT or miR-21 KO mice was significantly increased compared to that of the controls (Figure 5M). Although there was no statistically significant difference in involvement of pneumonitis between Nano-Ni-exposed WT and miR-21 KO mice, chronic inflammation was much milder in the lungs of miR-21 KO mice (Figure 5D&M).
Figure 5.
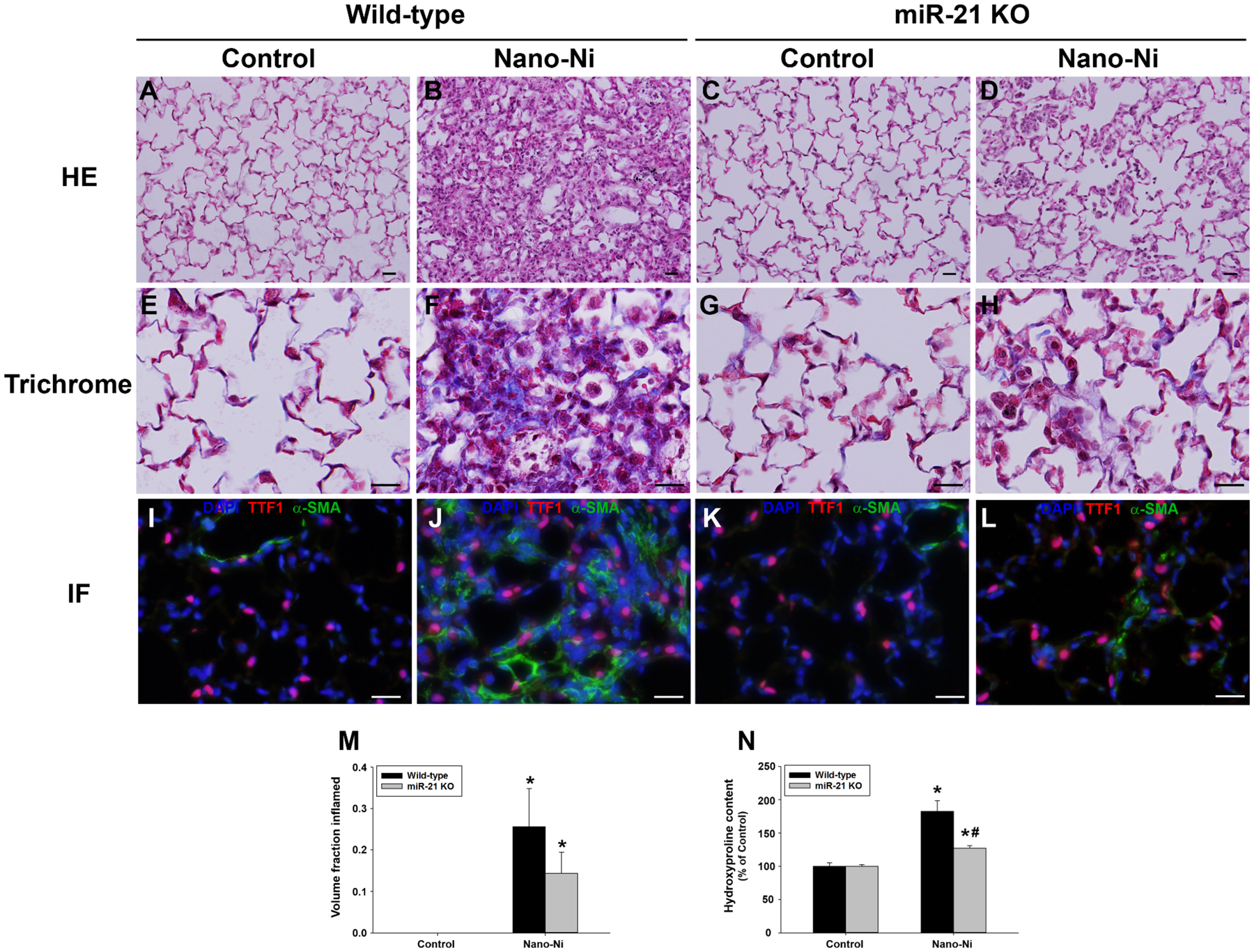
Chronic inflammation and pulmonary fibrosis in mouse lungs at day 42 after Nano-Ni exposure. Wild-type and miR-21 KO mice were instilled intratracheally with 50 μg per mouse of Nano-Ni. Control mice were instilled with physiological saline. A and C show the normal alveolar structure in the control mice by HE staining. B shows extensive chronic inflammation and fibrosis in the lungs of wild-type mice with Nano-Ni exposure, which is to a much lesser extent in the miR-21 KO lungs (D). Pulmonary fibrosis was further confirmed by trichrome staining (E-H, collagen deposition, blue staining) and dual immunofluorescent staining (I-L, α-SMA positive, green staining). Lung type II pneumocytes are TTF1-positive (red staining), and DAPI (blue staining) stains cell nuclei. Scale bar represents 20 μm for all panels. Extent of pneumonitis was also calculated by the volume fraction inflamed on H&E stained sections (M) and hydroxyproline content was determined in the left lungs (N). Data are shown as mean ± SEM (n=5~6). * p<0.05 vs. the control group of the same genotype; # p<0.05 vs. wild-type Nano-Ni group.
(B). Lung fibrosis
Exposure of WT mice to Nano-Ni revealed extensive lung fibrosis at day 42 post-exposure by H&E staining, which was further confirmed by Masson’s trichrome staining and measurement of hydroxyproline content in the mouse left lungs. Collagen deposition in the alveolar septa and in subepithelial areas of bronchi and bronchioles and proliferation of interstitial cells in the lung tissues were observed in WT mice exposed to Nano-Ni (Figure 5F). Fibrosis was also observed in some areas of the lungs from miR-21 KO mice exposed to Nano-Ni, but to a much lesser extent than that of the WT mice (Figure 5H). Dual immunofluorescent staining showed more α-SMA staining (green staining) in the WT mouse lungs (Figure 5J) than that of the miR-21 KO mouse lungs (Figure 5L), indicating more myofibroblasts developed in the lungs of WT mice after Nano-Ni exposure. Hydroxyproline is a major component of the protein collagen, hence measuring its content in lung tissues is commonly used to quantify pulmonary fibrosis (Mo et al. 2013; Mo et al. 2019; Taylor et al. 2002). Exposure to Nano-Ni resulted in a significant increase in hydroxyproline content in the left lungs of WT mice compared to that of control mice (Figure 5N). Although Nano-Ni caused a significant increase in hydroxyproline content in the left lungs of miR-21 KO mice relative to the controls, the level was significantly lower than that of WT mice (Figure 5N).
(C). The mechanism involved in Nano-Ni-induced pulmonary fibrosis
The lungs of WT and miR-21 KO mice at day 7 after Nano-Ni exposure were used to examine the role of miR-21 in Nano-Ni-induced pulmonary fibrosis. mRNA expression of TGF-β1, COL1A1, and COL3A1 was determined by real-time PCR, and protein expression of TGF-β, phosphorylated Smad2, Smad2, Smad7, and COL1A1 was determined by Western blot. Our results showed that Nano-Ni exposure caused a significant increase in mRNA expression of TGF-β1 in both WT and miR-21 KO mouse lungs (Figure 6A). Although TGF-β1 expression was slightly lower in miR-21 KO than in WT mice, there was no statistically significant difference between them (Figure 6A). Nano-Ni exposure caused significant upregulation of COL1A1 and COL3A1 mRNA expression in WT mouse lungs compared to that of the controls, but not in the miR-21 KO mouse lungs (Figure 6A). Our Western blot results of TGF-β and COL1A1 expression were consistent with the real-time PCR results (Figure 6B–D). Nano-Ni exposure caused significant increase in phosphorylated Smad2 and decrease in Smad7 expression in WT lungs compared to that of the controls, but not in miR-21 KO lungs (Figure 6B&D). These results suggest that TGF-β1 may be involved in the regulation of miR-21 and that Smad2 and Smad7 are the downstream targets of miR-21, which are involved in Nano-Ni-induced fibrosis.
Figure 6.
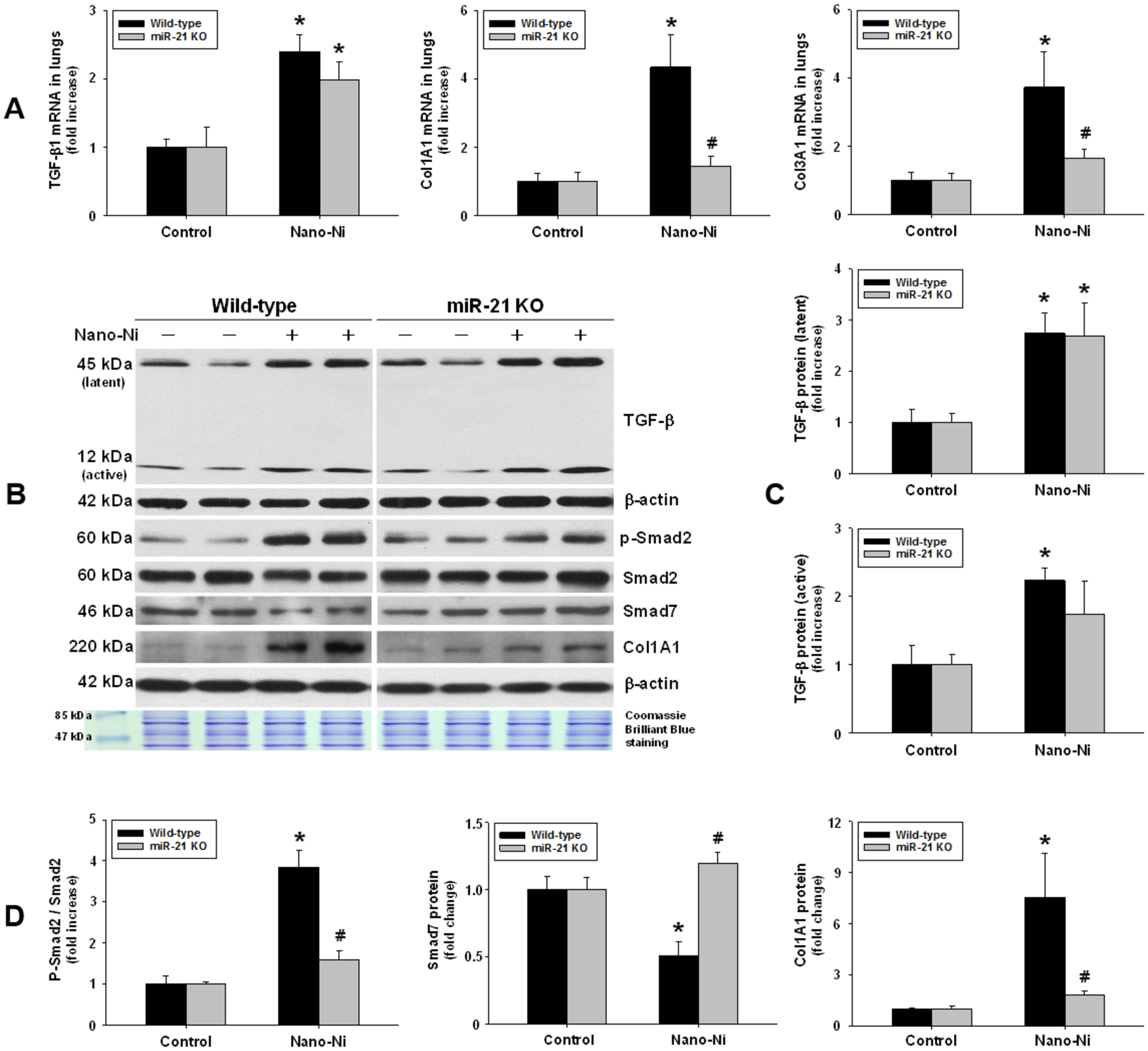
Fibrosis-related genes and TGF-β/Smad signaling in mouse lungs after Nano-Ni exposure. Wild-type and miR-21 KO mice were instilled intratracheally with 50 μg per mouse of Nano-Ni. Control mice were instilled with physiological saline. At day 7 after exposure, mouse lung tissues were collected for total RNA and protein isolation. (A) The mRNA expression of fibrosis-related genes was determined by real-time PCR. B shows representative Western blot results, while C and D are normalized band densitometry readings. β-actin and Coomassie Brilliant Blue stained gels served as loading controls. p-Smad2: phospho-Smad2. Data are shown as mean ± SEM (n=4~5). * p<0.05 vs. the control group of the same genotype; # p<0.05 vs. wild-type Nano-Ni group.
Discussion and conclusion
Nickel is widely used in industry as nickel alloys, electroplating, batteries, coins, industrial plumbing, spark plugs, machinery parts, stainless-steel, nickel-chrome resistance wires, and catalysts (Bajpai et al. 2012). Exposure to nickel and its compounds (salts, oxides, and alloys) has caused respiratory tract irritation, inflammation, emphysema, varying degrees of pulmonary cell hyperplasia, fibrosis, and even cancer (Lei et al. 2016). Nickel compounds are Group 1 carcinogens (carcinogenic to humans), while metallic nickel and nickel alloys are Group 2B (possibly carcinogenic to humans) according to the International Agency for Research on Cancer (IARC) (IARC 1990). Nanoparticles refer to particles with a diameter less than 100 nm (Anon 1990); vastly different properties manifest on the nanoscale. This suggests that metal nanoparticles may have different mechanisms of toxicity than the bulk material. There are case reports demonstrating serious complications resulting from nickel nanoparticle exposure, such as sensitization in a chemist handling nickel nanoparticles without protective measures (Journeay and Goldman 2014) and death of a previously healthy man from adult respiratory distress syndrome (ARDS) after spraying nickel nanoparticles using a metal arc process (Phillips et al. 2010). The use of nickel nanoparticles will continue to increase, requiring the scientific community access their potential toxicological, genotoxic, and carcinogenic effects.
Our previous studies have shown that exposure to Nano-Ni caused pulmonary inflammation and injury, reflected by increased neutrophil count, CXCL1/KC level, LDH activity, and total protein concentration in the BALF, which appeared as early as day 1, peaked at day 3, and then attenuated at day 7 and beyond post-instillation (Mo et al. 2019). Although these parameters in the BALF dramatically decreased over time, their levels were still higher than those of the control, even at day 42 post-exposure (Mo et al. 2019). In this study, we examined expression of proinflammatory cytokines and fibrosis-related genes in the lungs of mice exposed to Nano-Ni. The results showed that exposure to Nano-Ni caused upregulation of proinflammatory cytokines such as IL-6 and TNF-α, which reached peak at day 3 after exposure, and fibrosis-related genes such as TGF-β1, COL1A1, and COL3A1 in mouse lungs. Lung histopathological examination also demonstrated that Nano-Ni caused severe and persistent lung inflammation, injury, and fibrosis. The pulmonary fibrosis was confirmed by trichrome staining and by determination of lung hydroxyproline content. Based on our current and previous (Mo et al. 2019) dose-response and time-response studies, the dose of 50 μg per mouse of Nano-Ni and the time points of day 3 (acute) and day 42 (chronic) after Nano-Ni exposure were chosen for comparative studies.
Many miRNAs are involved in lung inflammation. Among them, miR-21 has been shown to be strongly implicated in pulmonary inflammation (Brown et al. 2014; Ha 2011b; Sessa and Hata 2013) and in the repair, regeneration, and remodeling of the injured respiratory system (Jiang et al. 2019; Liu et al. 2010). In this study, we demonstrated a dose-dependent upregulation of miR-21 in mouse lungs after Nano-Ni exposure. Following exposure of mice to 50 μg per mouse of Nano-Ni, a significant increase was first observed at day 3, peaked at day 14, and then declined but remaining significantly higher than that of the controls at days 28 and 42 post-exposure. Although miR-21 upregulation had been widely found in many pulmonary inflammatory diseases and fibrosis, the role of miR-21 in Nano-Ni-induced lung inflammation, injury, and fibrosis is still lacking. Our results raised the intriguing possibility that miR-21 may be involved in these processes, hence we used miR-21 KO mice to investigate its functional roles in Nano-Ni-induced lung injury.
Analysis of the cellular and biochemical profile of BALF, such as of the type and number of differentiated immune cells and of the soluble factors, is widely used to evaluate particle-induced lung inflammation and injury. Accumulation of neutrophils and macrophages in the lungs is a key inflammatory response to various particle exposures, including metal nanoparticles (Mo et al. 2019; Wan et al. 2017). CXCL1/KC is considered an important neutrophil chemoattractant released by the lungs in many animal models of airway inflammation after exposure to various particles (Mo et al. 2019; Wan et al. 2017). Evaluation of LDH activity and total protein concentration in BALF may provide a quantitative assessment of cell and tissue damage (Mo et al. 2019; Wan et al. 2017). Our short-term study results demonstrated that exposure to Nano-Ni caused severe acute lung inflammation and injury in WT mice, which were supported by increased neutrophil count, CXCL1/KC level, total protein concentration, and LDH activity in BALF. Although exposure of miR-21 KO mice to Nano-Ni also caused a significant increase in these BALF parameters, their levels were significantly lower than those of the WT mice. The milder pulmonary inflammation in miR-21 KO mice compared to WT mice after Nano-Ni exposure was further confirmed by histopathological examination. Our proinflammatory cytokine expression results were also consistent with the above findings; knock-out of miR-21 ameliorated Nano-Ni-induced upregulation of IL-6 and TNFα. All these results suggest that miR-21 may play a crucial role in regulating pulmonary inflammatory response and injury after Nano-Ni exposure. Although it remains unclear how deletion of miR-21 ameliorates Nano-Ni-induced lung inflammation, miR-21 has been shown to be upregulated in many inflammatory diseases, including lipopolysaccharide (LPS)-stimulated lung inflammation (Qi et al. 2015), allergic airway inflammation (Lu et al. 2009), cigarette smoke-induced airway inflammation (Pace et al. 2019), osteoarthritis (Tsezou 2014), psoriasis and atopic eczema (Guinea-Viniegra et al. 2014; Vennegaard et al. 2012), colitis (Shi et al. 2013), alcoholic hepatitis (Wu et al. 2018), and many others. Increased miR-21 may be a consequence of activation of the IL-6/Stat3 pathway (Guinea-Viniegra et al. 2014; Loffler et al. 2007). Previous studies have suggested a therapeutic role of miR-21 in renal injury in a mouse model of type 2 diabetes (Zhong et al. 2013), in patient-derived psoriatic skin xenotransplants in mice (Guinea-Viniegra et al. 2014), and in a psoriasis-like mouse model (Guinea-Viniegra et al. 2014). Anti-miR-21 treatment caused downregulation of TNFα, IL-17, IL-21, and IL-23 (Guinea-Viniegra et al. 2014). Furthermore, miR-21 may regulate immune-related target genes, including the confirmed targets IL-12p25 (Lu et al. 2009) and transforming growth factor-β receptor 2 (TGFBR2) (Mishra et al. 2014). Interestingly, other studies demonstrated anti-inflammatory properties of miR-21 in macrophages by targeting the proinflammatory programmed cell death 4 gene (PDCD4) (Barnett et al. 2016; Merline et al. 2011; Sheedy 2015). The role of miR-21 in inflammation may manifest in a time-dependent and/or cell-type-specific manner, which requires further investigation.
MMP-2 (72 kDa gelatinase A) and MMP-9 (92 kDa gelatinase B) belong to the matrix metalloproteinases (MMPs) family, which includes 25 secreted and cell surface-bound neutral proteinases (Hannocks et al. 2019). Matrix-degrading MMP enzymes are not only directly responsible for airway and pulmonary injury and inflammation, but also play an important role in the repair process (Greenlee et al. 2007; Murphy et al. 1994; Parks 2003). Activation of MMP-2 and MMP-9 plays an important role in many pulmonary diseases (Parks 2003). Our previous in vitro and in vivo studies demonstrated that Nano-Ni caused increased expression and activity of MMP-2 and MMP-9 (Mo et al. 2019; Wan et al. 2011). Here, we demonstrated that exposure of mice to Nano-Ni caused significant increase in protein levels and activities of MMP-2 and MMP-9 in BALF and lung tissues collected from either WT or miR-21 KO mice. However, their levels in miR-21 KO mice were much lower than that of the WT mice, suggesting the involvement of miR-21 in Nano-Ni-induced upregulation of MMP-2 and MMP-9. Both membrane-anchored MMP inhibitor RECK and tissue inhibitor of metalloproteinases-3 (TIMP3) were confirmed to be targets of miR-21 (Gabriely et al. 2008; Jiang et al. 2019; Reis et al. 2012). RECK has the ability to regulate at least three members of the MMPs family: MMP-2, MMP-9, and MT1-MMP (Oh et al. 2001; Takahashi et al. 1998). Nano-Ni-induced miR-21 upregulation may cause a loss of inhibitory effects of RECK on MMP-2 and MMP-9, resulting in their upregulation. MMP-2 and MMP-9 are believed to be the critical enzymes for degrading type IV collagen, a major component of basement membrane (Greenlee et al. 2007; Murphy et al. 1994; Parks and Shapiro 2001; Parks 2003). The lung has a rich vascular network and is in continuous and direct communication with the outside environment. In addition to gas exchange, the lung also functions in host defense. Nano-Ni-induced activation of MMP-2 and MMP-9 may cause increased degradation of type IV collagen and dysfunction of basement membrane, leading to a loss of barrier protection, an increase in pulmonary permeability, a loss of lung homeostasis, exacerbation of lung inflammation, and triggering lung remodeling.
The hallmark of fibrosis is tissue remodeling with excess deposition of extracellular matrix (ECM) components, predominantly collagens. Accumulating ECM components replace functional tissue and disrupts organ architecture (Bonnans et al. 2014). A variety of cell types, cytokines, growth factors, and enzymes are orchestrated together in complex pathogenic networks, converging on the activation and/or recruitment of fibroblasts and their differentiation into myofibroblasts, which are α-smooth muscle actin (α-SMA)-enriched cells overproducing ECM (Baum and Duffy 2011; Bonnans et al. 2014; Sun et al. 2016). Several studies have demonstrated that miRNAs directly regulate ECM synthesis and that the TGF-β signaling is a key profibrotic pathway (Liu et al. 2010; Xie et al. 2011; Yang et al. 2012). For example, downregulation of miR-200 was reported in mouse lungs with bleomycin-induced fibrosis; restoration of miR-200 expression reversed lung fibrosis via inhibiting TGF-β, suggesting its antifibrotic role (Yang et al. 2012). Notably, miR-21 was found upregulated in bleomycin-induced lung fibrosis and in the lungs of patients with idiopathic pulmonary fibrosis (IPF) (Liu et al. 2010). Even the delayed administration of antisense nucleotides blocking miR-21 was able to attenuate the profibrotic effect exerted by bleomycin (Liu et al. 2010). Our results showed that Nano-Ni caused miR-21 upregulation and fibrosis in WT mouse lungs. At day 42 after exposure, Nano-Ni caused extensive pulmonary fibrosis and proliferation of interstitial cells in WT mice, which was consistent with our previous study (Mo et al. 2019). However, Nano-Ni only caused slight to mild lung fibrosis in miR-21 KO mice that was confirmed by Masson’s trichrome staining, α-SMA immunofluorescent staining, and measurement of the hydroxyproline content in the left lungs. Exposure to Nano-Ni also caused TGF-β1 upregulation in the lungs of both WT and miR-21 KO mice. TGF-β1 has been shown to mediate a post-transcriptional activation of miR-21 by processing pri-miR-21 to pre-miR-21 in the cell nucleus (Davis et al. 2008). Addition of TGF-β1 to primary pulmonary fibroblast culture enhanced miR-21 expression (Liu et al. 2010). Thus Nano-Ni-induced miR-21 upregulation may be through Nano-Ni-induced TGF-β1 activation.
TGF-β/Smad signaling plays a vital role in pulmonary fibrosis. The Smads are TGF-β type I receptor-activated signaling effectors. Smad2 and Smad3 are R-Smads and can be phosphorylated and activated by TGF-β type I receptor. The phosphorylated Smad2/3 form a complex with a co-Smad, Smad4, which is translocated into the nucleus, where it regulates transcription (Derynck et al. 1998). Smad7 is an I-Smad, which suppresses the activity of R-Smads and is a general TGF-β signal inhibitor (Yan et al. 2016). Exposure to Nano-Ni caused downregulation of Smad7 in the lungs of WT mice, but not in the lungs of miR-21 KO mice, suggesting the involvement of miR-21 upregulation in Nano-Ni-induced pulmonary fibrosis may be through suppressing Smad7. Prior studies clearly demonstrated a 3’ untranslated region (UTR) binding site for miR-21 on Smad7 mRNA (Li et al. 2013; Lin et al. 2014; Liu et al. 2010). Down-regulation of Smad7 upregulated phospho-Smad2 and α-SMA, thus resulting in lung fibrosis (Shukla et al. 2009), and overexpression of Smad7 in gingival fibroblasts caused down-regulation of phospho-Smad2 and α-SMA and significant reduction of type I collagen production (Sobral et al. 2011). Our results demonstrate that the level of phospho-Smad2 was higher in WT than in miR-21 KO lungs after Nano-Ni exposure. It appears miR-21 functions in a vicious amplifying circuit to enhance TGF-β1/Smad signaling events and finally to promote lung fibrosis. Taken together, Nano-Ni-induced pulmonary fibrosis may be through Nano-Ni-induced upregulation of TGF-β1 and miR-21 and down-regulation of Smad7, thus resulting in increased collagen secretion and lung fibrosis. Since miR-21 is known to have multiple targets, other pathways that may be influenced by miR-21 knock-out cannot be completely excluded and need to be further studied.
Up to 85% of the connective tissue mass of a normal human lung is collagen, of which type I and type III collagen are the predominant types, with type I approximately twice as abundant as type III (Hance and Crystal 1975; Seyer et al. 1976). In the normal lung, continuous collagen synthesis and collagen degradation are precisely balanced to maintain normal tissue architecture. Fibrosis occurs when this balance is disrupted, favoring matrix production over degradation (McKleroy et al. 2013). The pathogenesis of pulmonary fibrosis is complex and the composition of collagen is different in fibrotic tissues. Previous studies have shown that in idiopathic chronic pulmonary fibrosis (IPF), in addition to an increase in total collagen, the relative content of type III collagen ranges from 12 to 24% in different patients, with the remainder being primarily type I collagen, indicating a shift of the ratio of collagen I/III from ~2:1 in normal lung to ~3–7:1 in IPF (Madri and Furthmayr 1980; Selman et al. 1982; Selman et al. 1986; Seyer et al. 1976; Snider 1981). This shift was also observed in the fibrotic lungs from patients with respiratory distress syndrome (RDS in infants or ARDS in adults), in which the ratio of type I to type III collagen increased to 3–4:1 (Last et al. 1983; Shoemaker et al. 1984). However, in early lung fibrosis, an increase in the proportion of type III collagen and an increase in total type I and type III collagen were observed in a study examining the collagen composition of biopsy samples obtained from patients with a variety of fibrotic diseases (Bateman et al. 1981). In more established areas of scar formation, there was almost exclusively type I collagen (Bateman et al. 1981). Although we did not determine the ratio of type I to type III collagen in mouse lungs after Nano-Ni exposure, the above studies clearly indicate that there is an alteration in tissue collagen polymorphism in the fibrotic process in the diseased state, which may have possible pathogenetic implications.
The mechanism of pulmonary toxic effects of nanoparticles is very complex. Several factors, such as small size, shape, and chemical toxicity of the native material, may be involved in the toxic effects of nanoparticles. It is possible that the soluble components of Nano-Ni may mediate or contribute to the pulmonary toxicity and fibrosis or to other biological effects. However, cells do not efficiently uptake extracellular Ni ions and lungs are able to rapidly clear water-soluble Ni species (Benson et al. 1995; Gillespie et al. 2010). Only approximately 1–3% (wt%) of Ni was released into cell culture medium following 18–24 h incubation of 10–50 μg/mL of Nano-Ni suspension at 37°C (Latvala et al. 2016; Pietruska et al. 2011). Previous studies have shown that Ni and NiO nanoparticles cause toxicity, including DNA damage and mutations, in human lung epithelial cells, but soluble Ni ions/complexes from NiCl2 or aqueous extract of nickel nanoparticles could not (Akerlund et al. 2018; Cho et al. 2012). Cho et al. also found that NiO nanoparticles, but not the released Ni fraction, caused pulmonary inflammatory responses when instilled into the rat lungs (Cho et al. 2012). These results clearly suggest that Ni and NiO nanoparticles show more pronounced pulmonary effects than Ni ions/complexes, indicating a more serious health concern. In this study, both Nano-Ni and nickel ions released from Nano-Ni may be involved in Nano-Ni-induced pulmonary toxicity and fibrosis. We cannot distinguish which one plays a larger role in our current in vivo studies. Further studies are needed to compare the toxicity of nickel nanoparticles and ‘water-soluble’ Ni compounds such as NiCl2.
Taken together, our study herein demonstrates that Nano-Ni can cause lung inflammation, injury, and fibrosis in both WT and miR-21 KO mice; however, Nano-Ni-induced pulmonary toxicity is of much lesser extent in miR-21 KO mice. The results support that miR-21 may be involved in Nano-Ni-induced pulmonary toxicity. miR-21 may regulate proinflammatory cytokines to fulfill Nano-Ni-induced pulmonary inflammation and injury. Furthermore, upregulation of miR-21 reduces Smad7 expression that further up-regulates profibrotic mediators and eventually causes lung fibrosis. The results provide further understanding of pulmonary toxicity caused by Nano-Ni exposure. However, the precise mechanisms by which miR-21 influences lung inflammation and fibrosis need to be further investigated; other putative targets of miR-21 and associated signaling pathways which may be involved in Nano-Ni-induced lung inflammation and fibrosis cannot be ignored.
Acknowledgements
The results were presented in part at 2019 Society of Toxicology (SOT) Annual Conference; March 10-14, 2019; Baltimore, Maryland, USA.
Funding
This work was partly supported by NIH (ES023693, ES028911 and HL147856), KSEF-148-RED-502-16-381, and Kentucky Lung Cancer Research Program to Dr. Qunwei Zhang.
Abbreviations
- Nano-Ni
Nickel nanoparticles
- WT
Wild-type
- miR-21
microRNA-21
- BAL
Bronchoalveolar lavage
- BALF
Bronchoalveolar lavage fluid
- CXCL1/KC
Chemokine (C-X-C motif) ligand 1 / keratinocyte chemoattractant
- LDH
Lactate dehydrogenase
- MMP-2
Matrix metalloproteinase-2
- MMP-9
Matrix metalloproteinase-9
- IL-6
Interleukin 6
- TNFα
Tumor necrosis factor alpha
- TGF-β1
Transforming growth factor beta 1
- COL1A1
Collagen, type I, alpha 1
- COL3A1
Collagen, type III, alpha 1
- Smad2
SMAD Family Member 2
- Smad7
SMAD Family Member 7
- DAPI
4′,6-diamidino-2-phenylindole
- TTF-1
Thyroid transcription factor 1
- α-SMA
Alpha-smooth muscle actin
Footnotes
Ethics approval and consent to participate
The protocols and the use of animals were approved by and in accordance with the University of Louisville Animal Care and Use Committee.
Consent for publication
Not applicable.
Availability of data and materials
All data and materials are included in the manuscript.
Disclosure of interest
The authors report no conflict of interest.
References
- Aillon KL, Xie Y, El-Gendy N, Berkland CJ, and Forrest ML 2009. “Effects of nanomaterial physicochemical properties on in vivo toxicity.” Adv Drug Deliv Rev 61: 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerlund E, Cappellini F, Di Bucchianico S, Islam S, Skoglund S, Derr R, Odnevall Wallinder I, Hendriks G, and Karlsson HL 2018. “Genotoxic and mutagenic properties of Ni and NiO nanoparticles investigated by comet assay, gamma-H2AX staining, Hprt mutation assay and ToxTracker reporter cell lines.” Environ Mol Mutagen 59: 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon 1990. “Report of the International Committee on Nickel Carcinogenesis in Man.” Scand J Work Environ Health 16: 1–82. [DOI] [PubMed] [Google Scholar]
- Bajpai R, Roy S, kulshrestha N, Rafiee J, Koratkar N, and Misra DS 2012. “Graphene supported nickel nanoparticle as a viable replacement for platinum in dye sensitized solar cells.” Nanoscale 4: 926–930. [DOI] [PubMed] [Google Scholar]
- Barnett RE, Conklin DJ, Ryan L, Keskey RC, Ramjee V, Sepulveda EA, Srivastava S, Bhatnagar A, and Cheadle WG 2016. “Anti-inflammatory effects of miR-21 in the macrophage response to peritonitis.” J Leukoc Biol 99: 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman ED, Turner-Warwick M, and Adelmann-Grill BC 1981. “Immunohistochemical study of collagen types in human foetal lung and fibrotic lung disease.” Thorax 36: 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum J, and Duffy HS 2011. “Fibroblasts and myofibroblasts: what are we talking about?” J Cardiovasc Pharmacol 57: 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson JM, Chang IY, Cheng YS, Hahn FF, Kennedy CH, Barr EB, Maples KR, and Snipes MB 1995. “Particle clearance and histopathology in lungs of F344/N rats and B6C3F1 mice inhaling nickel oxide or nickel sulfate.” Fundam Appl Toxicol 28: 232–244. [DOI] [PubMed] [Google Scholar]
- Blazquez-Prieto J, Lopez-Alonso I, Amado-Rodriguez L, Huidobro C, Gonzalez-Lopez A, Kuebler WM, and Albaiceta GM 2018. “Impaired lung repair during neutropenia can be reverted by matrix metalloproteinase-9.” Thorax 73: 321–330. [DOI] [PubMed] [Google Scholar]
- Bollati V, Marinelli B, Apostoli P, Bonzini M, Nordio F, Hoxha M, Pegoraro V, Motta V, Tarantini L, Cantone L, Schwartz J, Bertazzi PA, and Baccarelli A 2010. “Exposure to metal-rich particulate matter modifies the expression of candidate microRNAs in peripheral blood leukocytes.” Environ Health Perspect 118: 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C, Chou J, and Werb Z 2014. “Remodelling the extracellular matrix in development and disease.” Nat Rev Mol Cell Biol 15: 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronnum H, Andersen DC, Schneider M, Sandberg MB, Eskildsen T, Nielsen SB, Kalluri R, and Sheikh SP 2013. “miR-21 promotes fibrogenic epithelial-to-mesenchymal transition of epicardial mesothelial cells involving Programmed Cell Death 4 and Sprouty-1.” PLoS One 8: e56280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Rahman M, and Nana-Sinkam SP 2014. “MicroRNAs in respiratory disease. A clinician’s overview.” Ann Am Thorac Soc 11: 1277–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capasso L, Camatini M, and Gualtieri M 2014. “Nickel oxide nanoparticles induce inflammation and genotoxic effect in lung epithelial cells.” Toxicol Lett 226: 28–34. [DOI] [PubMed] [Google Scholar]
- Chen S, Xue Y, Wu X, Le C, Bhutkar A, Bell EL, Zhang F, Langer R, and Sharp PA 2014. “Global microRNA depletion suppresses tumor angiogenesis.” Genes Dev 28: 1054–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WS, Duffin R, Poland CA, Duschl A, Oostingh GJ, Macnee W, Bradley M, Megson IL, and Donaldson K 2012. “Differential pro-inflammatory effects of metal oxide nanoparticles and their soluble ions in vitro and in vivo; zinc and copper nanoparticles, but not their ions, recruit eosinophils to the lungs.” Nanotoxicology 6: 22–35. [DOI] [PubMed] [Google Scholar]
- Colvin VL 2003. “The potential environmental impact of engineered nanomaterials.” Nat Biotechnol 21: 1166–1170. [DOI] [PubMed] [Google Scholar]
- Davis BN, Hilyard AC, Lagna G, and Hata A 2008. “SMAD proteins control DROSHA-mediated microRNA maturation.” Nature 454: 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang Y, and Feng XH 1998. “Smads: transcriptional activators of TGF-beta responses.” Cell 95: 737–740. [DOI] [PubMed] [Google Scholar]
- Dick CA, Brown DM, Donaldson K, and Stone V 2003. “The role of free radicals in the toxic and inflammatory effects of four different ultrafine particle types.” Inhal Toxicol 15: 39–52. [DOI] [PubMed] [Google Scholar]
- Driscoll KE, Costa DL, Hatch G, Henderson R, Oberdorster G, Salem H, and Schlesinger RB 2000. “Intratracheal instillation as an exposure technique for the evaluation of respiratory tract toxicity: uses and limitations.” Toxicol Sci 55: 24–35. [DOI] [PubMed] [Google Scholar]
- Feng L, Zhang Y, Jiang M, Mo Y, Wan R, Jia Z, Tollerud DJ, Zhang X, and Zhang Q 2015. “Up-regulation of Gadd45alpha after exposure to metal nanoparticles: the role of hypoxia inducible factor 1alpha.” Environ Toxicol 30: 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohlich E, Mercuri A, Wu S, and Salar-Behzadi S 2016. “Measurements of Deposition, Lung Surface Area and Lung Fluid for Simulation of Inhaled Compounds.” Front Pharmacol 7: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, and Krichevsky AM 2008. “MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators.” Mol Cell Biol 28: 5369–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie PA, Kang GS, Elder A, Gelein R, Chen L, Moreira AL, Koberstein J, Tchou-Wong KM, Gordon T, and Chen LC 2010. “Pulmonary response after exposure to inhaled nickel hydroxide nanoparticles: short and long-term studies in mice.” Nanotoxicology 4: 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glista-Baker EE, Taylor AJ, Sayers BC, Thompson EA, and Bonner JC 2014. “Nickel nanoparticles cause exaggerated lung and airway remodeling in mice lacking the T-box transcription factor, TBX21 (T-bet).” Part Fibre Toxicol 11: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee KJ, Werb Z, and Kheradmand F 2007. “Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted.” Physiol Rev 87: 69–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinea-Viniegra J, Jimenez M, Schonthaler HB, Navarro R, Delgado Y, Concha-Garzon MJ, Tschachler E, Obad S, Dauden E, and Wagner EF 2014. “Targeting miR-21 to treat psoriasis.” Sci Transl Med 6: 225re221. [DOI] [PubMed] [Google Scholar]
- Gushima Y, Ichikado K, Suga M, Okamoto T, Iyonaga K, Sato K, Miyakawa H, and Ando M 2001. “Expression of matrix metalloproteinases in pigs with hyperoxia-induced acute lung injury.” Eur Respir J 18: 827–837. [DOI] [PubMed] [Google Scholar]
- Ha TY 2011a. “MicroRNAs in Human Diseases: From Cancer to Cardiovascular Disease.” Immune Netw 11: 135–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha TY 2011b. “MicroRNAs in Human Diseases: From Lung, Liver and Kidney Diseases to Infectious Disease, Sickle Cell Disease and Endometrium Disease.” Immune Netw 11: 309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hance AJ, and Crystal RG 1975. “The connective tissue of lung.” Am Rev Respir Dis 112: 657–711. [DOI] [PubMed] [Google Scholar]
- Hannocks MJ, Zhang X, Gerwien H, Chashchina A, Burmeister M, Korpos E, Song J, and Sorokin L 2019. “The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes.” Matrix Biol 75–76: 102–113. [DOI] [PubMed] [Google Scholar]
- Howrylak JA, and Nakahira K 2017. “Inflammasomes: Key Mediators of Lung Immunity.” Annu Rev Physiol 79: 471–494. [DOI] [PubMed] [Google Scholar]
- Hsu AT, Barrett CD, DeBusk GM, Ellson CD, Gautam S, Talmor DS, Gallagher DC, and Yaffe MB 2015. “Kinetics and Role of Plasma Matrix Metalloproteinase-9 Expression in Acute Lung Injury and the Acute Respiratory Distress Syndrome.” Shock 44: 128–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC 1990. “Chromium, nickel and welding.” IARC Monogr Eval Carcinog Risks Hum 49: 1–648. [PMC free article] [PubMed] [Google Scholar]
- ISO 2015. “International Organization for Standardization (ISO). Nanotechnologies — Vocabulary — Part 2: Nano-objects.” ISO/TS 80004–2:2015. [Google Scholar]
- Jiang C, Guo Y, Yu H, Lu S, and Meng L 2019. “Pleiotropic microRNA-21 in pulmonary remodeling: novel insights for molecular mechanism and present advancements.” Allergy Asthma Clin Immunol 15: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Journeay WS, and Goldman RH 2014. “Occupational handling of nickel nanoparticles: a case report.” Am J Ind Med 57: 1073–1076. [DOI] [PubMed] [Google Scholar]
- Last JA, Siefkin AD, and Reiser KM 1983. “Type I collagen content is increased in lungs of patients with adult respiratory distress syndrome.” Thorax 38: 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latronico MV, and Condorelli G 2009. “MicroRNAs and cardiac pathology.” Nat Rev Cardiol 6: 419–429. [DOI] [PubMed] [Google Scholar]
- Latvala S, Hedberg J, Di Bucchianico S, Moller L, Odnevall Wallinder I, Elihn K, and Karlsson HL 2016. “Nickel Release, ROS Generation and Toxicity of Ni and NiO Micro- and Nanoparticles.” PLoS One 11: e0159684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY, Pedroza M, Zhou Y, Davies J, Philip K, Molina J, Luo F, George AT, Garcia-Morales LJ, Bunge RR, Bruckner BA, Loebe M, Seethamraju H, Agarwal SK, and Blackburn MR 2014. “Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis.” J Immunol 193: 3755–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei D, Lee DC, Magasinski A, Zhao E, Steingart D, and Yushin G 2016. “Performance Enhancement and Side Reactions in Rechargeable Nickel-Iron Batteries with Nanostructured Electrodes.” ACS Appl Mater Interfaces 8: 2088–2096. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, and Bartel DP 2005. “Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets.” Cell 120: 15–20. [DOI] [PubMed] [Google Scholar]
- Li Q, Zhang D, Wang Y, Sun P, Hou X, Larner J, Xiong W, and Mi J 2013. “MiR-21/Smad 7 signaling determines TGF-beta1-induced CAF formation.” Sci Rep 3: 2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Gan H, Zhang H, Tang W, Sun Y, Tang X, Kong D, Zhou J, Wang Y, and Zhu Y 2014. “MicroRNA21 inhibits SMAD7 expression through a target sequence in the 3’ untranslated region and inhibits proliferation of renal tubular epithelial cells.” Mol Med Rep 10: 707–712. [DOI] [PubMed] [Google Scholar]
- Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, Kaminski N, and Abraham E 2010. “miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis.” J Exp Med 207: 1589–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD 2001. “Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.” Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Loffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermuller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, and Horn F 2007. “Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer.” Blood 110: 1330–1333. [DOI] [PubMed] [Google Scholar]
- Long G, Mo Y, Zhang Q, and Jiang M 2019. “Analysis of Nanomaterial Toxicity by Western Blot.” Methods Mol Biol 1894: 161–169. [DOI] [PubMed] [Google Scholar]
- Lu TX, Munitz A, and Rothenberg ME 2009. “MicroRNA-21 is up-regulated in allergic airway inflammation and regulates IL-12p35 expression.” J Immunol 182: 4994–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Kumar M, Choudhury SN, Becker Buscaglia LE, Barker JR, Kanakamedala K, Liu MF, and Li Y 2011. “Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis.” Proc Natl Acad Sci U S A 108: 10144–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madri JA, and Furthmayr H 1980. “Collagen polymorphism in the lung. An immunochemical study of pulmonary fibrosis.” Hum Pathol 11: 353–366. [DOI] [PubMed] [Google Scholar]
- McKleroy W, Lee TH, and Atabai K 2013. “Always cleave up your mess: targeting collagen degradation to treat tissue fibrosis.” Am J Physiol Lung Cell Mol Physiol 304: L709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merline R, Moreth K, Beckmann J, Nastase MV, Zeng-Brouwers J, Tralhao JG, Lemarchand P, Pfeilschifter J, Schaefer RM, Iozzo RV, and Schaefer L 2011. “Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21.” Sci Signal 4: ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Deng JJ, Gowda PS, Rao MK, Lin CL, Chen CL, Huang T, and Sun LZ 2014. “Androgen receptor and microRNA-21 axis downregulates transforming growth factor beta receptor II (TGFBR2) expression in prostate cancer.” Oncogene 33: 4097–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Zhu X, Hu X, Tollerud DJ, and Zhang Q 2008. “Cytokine and NO release from peripheral blood neutrophils after exposure to metal nanoparticles: in vitro and ex vivo studies.” Nanotoxicology 2: 79–87. [Google Scholar]
- Mo Y, Wan R, Chien S, Tollerud DJ, and Zhang Q 2009a. “Activation of endothelial cells after exposure to ambient ultrafine particles: the role of NADPH oxidase.” Toxicol Appl Pharmacol 236: 183–193. [DOI] [PubMed] [Google Scholar]
- Mo Y, Wan R, Wang J, Chien S, Tollerud DJ, and Zhang Q 2009b. “Diabetes is associated with increased sensitivity of alveolar macrophages to urban particulate matter exposure.” Toxicology 262: 130–137. [DOI] [PubMed] [Google Scholar]
- Mo Y, Wan R, Feng L, Chien S, Tollerud DJ, and Zhang Q 2012. “Combination effects of cigarette smoke extract and ambient ultrafine particles on endothelial cells.” Toxicol In Vitro 26: 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Chen J, Schlueter CF, and Hoyle GW 2013. “Differential susceptibility of inbred mouse strains to chlorine-induced airway fibrosis.” Am J Physiol Lung Cell Mol Physiol 304: L92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Chen J, Humphrey DM Jr., Fodah RA, Warawa JM, and Hoyle GW 2015. “Abnormal epithelial structure and chronic lung inflammation after repair of chlorine-induced airway injury.” Am J Physiol Lung Cell Mol Physiol 308: L168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo Y, Jiang M, Zhang Y, Wan R, Li J, Zhong CJ, Li H, Tang S, and Zhang Q 2019. “Comparative mouse lung injury by nickel nanoparticles with differential surface modification.” J Nanobiotechnology 17: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan LG, and Usher V 1994. “Health problems associated with nickel refining and use.” Ann Occup Hyg 38: 189–198. [DOI] [PubMed] [Google Scholar]
- Murphy G, Willenbrock F, Crabbe T, O’Shea M, Ward R, Atkinson S, O’Connell J, and Docherty A 1994. “Regulation of matrix metalloproteinase activity.” Ann N Y Acad Sci 732: 31–41. [DOI] [PubMed] [Google Scholar]
- Niska K, Zielinska E, Radomski MW, and Inkielewicz-Stepniak I 2018. “Metal nanoparticles in dermatology and cosmetology: Interactions with human skin cells.” Chem Biol Interact 295: 38–51. [DOI] [PubMed] [Google Scholar]
- O’Connell RM, Rao DS, and Baltimore D 2012. “microRNA regulation of inflammatory responses.” Annu Rev Immunol 30: 295–312. [DOI] [PubMed] [Google Scholar]
- Oberdorster G, Oberdorster E, and Oberdorster J 2005. “Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles.” Environ Health Perspect 113: 823–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, Nishimura S, Imamura Y, Kitayama H, Alexander DB, Ide C, Horan TP, Arakawa T, Yoshida H, Nishikawa S, Itoh Y, Seiki M, Itohara S, Takahashi C, and Noda M 2001. “The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis.” Cell 107: 789–800. [DOI] [PubMed] [Google Scholar]
- Pace E, Di Vincenzo S, Di Salvo E, Genovese S, Dino P, Sangiorgi C, Ferraro M, and Gangemi S 2019. “MiR-21 upregulation increases IL-8 expression and tumorigenesis program in airway epithelial cells exposed to cigarette smoke.” J Cell Physiol 234: 22183–22194. [DOI] [PubMed] [Google Scholar]
- Parks WC, and Shapiro SD 2001. “Matrix metalloproteinases in lung biology.” Respir Res 2: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks WC 2003. “Matrix metalloproteinases in lung repair.” Eur Respir J Suppl 44: 36s–38s. [DOI] [PubMed] [Google Scholar]
- Phillips JI, Green FY, Davies JC, and Murray J 2010. “Pulmonary and systemic toxicity following exposure to nickel nanoparticles.” Am J Ind Med 53: 763–767. [DOI] [PubMed] [Google Scholar]
- Pietruska JR, Liu X, Smith A, McNeil K, Weston P, Zhitkovich A, Hurt R, and Kane AB 2011. “Bioavailability, intracellular mobilization of nickel, and HIF-1alpha activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles.” Toxicol Sci 124: 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port JD, and Sucharov C 2010. “Role of microRNAs in cardiovascular disease: therapeutic challenges and potentials.” J Cardiovasc Pharmacol 56: 444–453. [DOI] [PubMed] [Google Scholar]
- Qi W, Li H, Cai XH, Gu JQ, Meng J, Xie HQ, Zhang JL, Chen J, Jin XG, Tang Q, Hao Y, Gao Y, Wen AQ, Xue XY, Gao Smith F, and Jin SW 2015. “Lipoxin A4 activates alveolar epithelial sodium channel gamma via the microRNA-21/PTEN/AKT pathway in lipopolysaccharide-induced inflammatory lung injury.” Lab Invest 95: 1258–1268. [DOI] [PubMed] [Google Scholar]
- Reis ST, Pontes-Junior J, Antunes AA, Dall’Oglio MF, Dip N, Passerotti CC, Rossini GA, Morais DR, Nesrallah AJ, Piantino C, Srougi M, and Leite KR 2012. “miR-21 may acts as an oncomir by targeting RECK, a matrix metalloproteinase regulator, in prostate cancer.” BMC Urol 12: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roggli E, Britan A, Gattesco S, Lin-Marq N, Abderrahmani A, Meda P, and Regazzi R 2010. “Involvement of microRNAs in the cytotoxic effects exerted by proinflammatory cytokines on pancreatic beta-cells.” Diabetes 59: 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman M, Chapela R, Montano M, Soto H, and Diaz de Leon L 1982. “Changes of collagen content in fibrotic lung disease.” Arch Invest Med (Mex) 13: 93–100 passim. [PubMed] [Google Scholar]
- Selman M, Montano M, Ramos C, and Chapela R 1986. “Concentration, biosynthesis and degradation of collagen in idiopathic pulmonary fibrosis.” Thorax 41: 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S, Perez GA, Tewari K, Comas X, and Kim M 2018. “Catalytic activity of nickel nanoparticles stabilized by adsorbing polymers for enhanced carbon sequestration.” Sci Rep 8: 11786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa R, and Hata A 2013. “Role of microRNAs in lung development and pulmonary diseases.” Pulm Circ 3: 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyer JM, Hutcheson ET, and Kang AH 1976. “Collagen polymorphism in idiopathic chronic pulmonary fibrosis.” J Clin Invest 57: 1498–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Hickman J, Gazit N, Rabkin E, and Mishin Y 2018. “Nickel nanoparticles set a new record of strength.” Nat Commun 9: 4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ 2015. “Turning 21: Induction of miR-21 as a Key Switch in the Inflammatory Response.” Front Immunol 6: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Liang Y, Yang J, Xia Y, Chen H, Han H, Yang Y, Wu W, Gao R, and Qin H 2013. “MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury.” PLoS One 8: e66814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SW, Song IH, and Um SH 2015. “Role of Physicochemical Properties in Nanoparticle Toxicity.” Nanomaterials (Basel) 5: 1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CT, Reiser KM, Goetzman BW, and Last JA 1984. “Elevated ratios of type I/III collagen in the lungs of chronically ventilated neonates with respiratory distress.” Pediatr Res 18: 1176–1180. [DOI] [PubMed] [Google Scholar]
- Shukla MN, Rose JL, Ray R, Lathrop KL, Ray A, and Ray P 2009. “Hepatocyte growth factor inhibits epithelial to myofibroblast transition in lung cells via Smad7.” Am J Respir Cell Mol Biol 40: 643–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider GL 1981. “Collagen concentration and rates of synthesis in idiopathic pulmonary fibrosis.” Am Rev Respir Dis 124: 341–342. [DOI] [PubMed] [Google Scholar]
- Sobral LM, Montan PF, Zecchin KG, Martelli-Junior H, Vargas PA, Graner E, and Coletta RD 2011. “Smad7 blocks transforming growth factor-beta1-induced gingival fibroblast-myofibroblast transition via inhibitory regulation of Smad2 and connective tissue growth factor.” J Periodontol 82: 642–651. [DOI] [PubMed] [Google Scholar]
- Stankic S, Suman S, Haque F, and Vidic J 2016. “Pure and multi metal oxide nanoparticles: synthesis, antibacterial and cytotoxic properties.” J Nanobiotechnology 14: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun KH, Chang Y, Reed NI, and Sheppard D 2016. “alpha-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFbeta activation or collagen production across multiple models of organ fibrosis.” Am J Physiol Lung Cell Mol Physiol 310: L824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi C, Sheng Z, Horan TP, Kitayama H, Maki M, Hitomi K, Kitaura Y, Takai S, Sasahara RM, Horimoto A, Ikawa Y, Ratzkin BJ, Arakawa T, and Noda M 1998. “Regulation of matrix metalloproteinase-9 and inhibition of tumor invasion by the membrane-anchored glycoprotein RECK.” Proc Natl Acad Sci U S A 95: 13221–13226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker SZ, Standley EA, and Jamison TF 2014. “Recent advances in homogeneous nickel catalysis.” Nature 509: 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Roberts JR, Hubbs AF, Reasor MJ, and Antonini JM 2002. “Quantitative image analysis of drug-induced lung fibrosis using laser scanning confocal microscopy.” Toxicol Sci 67: 295–302. [DOI] [PubMed] [Google Scholar]
- Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, and Engelhardt S 2008. “MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts.” Nature 456: 980–984. [DOI] [PubMed] [Google Scholar]
- Tsezou A 2014. “Osteoarthritis year in review 2014: genetics and genomics.” Osteoarthritis Cartilage 22: 2017–2024. [DOI] [PubMed] [Google Scholar]
- Upagupta C, Shimbori C, Alsilmi R, and Kolb M 2018. “Matrix abnormalities in pulmonary fibrosis.” Eur Respir Rev 27: 180033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennegaard MT, Bonefeld CM, Hagedorn PH, Bangsgaard N, Lovendorf MB, Odum N, Woetmann A, Geisler C, and Skov L 2012. “Allergic contact dermatitis induces upregulation of identical microRNAs in humans and mice.” Contact Dermatitis 67: 298–305. [DOI] [PubMed] [Google Scholar]
- Wan R, Mo Y, Zhang X, Chien S, Tollerud DJ, and Zhang Q 2008. “Matrix metalloproteinase-2 and −9 are induced differently by metal nanoparticles in human monocytes: The role of oxidative stress and protein tyrosine kinase activation.” Toxicol Appl Pharmacol 233: 276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Mo Y, Chien S, Li Y, Li Y, Tollerud DJ, and Zhang Q 2011. “The role of hypoxia inducible factor-1alpha in the increased MMP-2 and MMP-9 production by human monocytes exposed to nickel nanoparticles.” Nanotoxicology 5: 568–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan R, Mo Y, Zhang Z, Jiang M, Tang S, and Zhang Q 2017. “Cobalt nanoparticles induce lung injury, DNA damage and mutations in mice.” Part Fibre Toxicol 14: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, McDaniel K, Zhou T, Ramos-Lorenzo S, Wu C, Huang L, Chen D, Annable T, Francis H, Glaser S, Alpini G, and Meng F 2018. “Knockout of microRNA-21 attenuates alcoholic hepatitis through the VHL/NF-kappaB signaling pathway in hepatic stellate cells.” Am J Physiol Gastrointest Liver Physiol 315: G385–G398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Liang J, Guo R, Liu N, Noble PW, and Jiang D 2011. “Comprehensive microRNA analysis in bleomycin-induced pulmonary fibrosis identifies multiple sites of molecular regulation.” Physiol Genomics 43: 479–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Liao H, Cheng M, Shi X, Lin X, Feng XH, and Chen YG 2016. “Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-beta (TGF-beta)/Smad Signaling.” J Biol Chem 291: 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Banerjee S, de Freitas A, Sanders YY, Ding Q, Matalon S, Thannickal VJ, Abraham E, and Liu G 2012. “Participation of miR-200 in pulmonary fibrosis.” Am J Pathol 180: 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Mo Y, Wan R, Chien S, Zhang X, and Zhang Q 2010. “Regulation of plasminogen activator inhibitor-1 expression in endothelial cells with exposure to metal nanoparticles.” Toxicol Lett 195: 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Kusaka Y, Sato K, Mo Y, Fukuda M, and Donaldson K 1998. “Toxicity of ultrafine nickel particles in lungs after intratracheal instillation.” J Occup Health 40: 171–176. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Kusaka Y, Zhu X, Sato K, Mo Y, Kluz T, and Donaldson K 2003. “Comparative toxicity of standard nickel and ultrafine nickel in lung after intratracheal instillation.” J Occup Health 45: 23–30. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Mo Y, Gu A, Wan R, Zhang Q, and Tollerud DJ 2016. “Effects of urban particulate matter with high glucose on human monocytes U937.” J Appl Toxicol 36: 586–595. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wan R, Zhang Q, and Mo Y 2019. “Application of Gelatin Zymography in Nanotoxicity Research.” Methods Mol Biol 1894: 133–143. [DOI] [PubMed] [Google Scholar]
- Zhong X, Chung AC, Chen HY, Dong Y, Meng XM, Li R, Yang W, Hou FF, and Lan HY 2013. “miR-21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes.” Diabetologia 56: 663–674. [DOI] [PubMed] [Google Scholar]
- Zhou F, Li S, Jia W, Lv G, Song C, Kang C, and Zhang Q 2015. “Effects of diesel exhaust particles on microRNA-21 in human bronchial epithelial cells and potential carcinogenic mechanisms.” Mol Med Rep 12: 2329–2335. [DOI] [PubMed] [Google Scholar]