

Proteomics‐Informed Prediction of Rosuvastatin Plasma Profiles in Patients with a Wide Range of Body Weight.
Abstract
Rosuvastatin is a frequently used probe to study transporter‐mediated hepatic uptake. Pharmacokinetic models have therefore been developed to predict transporter impact on rosuvastatin disposition in vivo. However, the interindividual differences in transporter concentrations were not considered in these models, and the predicted transporter impact was compared with historical in vivo data. In this study, we investigated the influence of interindividual transporter concentrations on the hepatic uptake clearance of rosuvastatin in 54 patients covering a wide range of body weight. The 54 patients were given an oral dose of rosuvastatin the day before undergoing gastric bypass or cholecystectomy, and pharmacokinetic (PK) parameters were established from each patient’s individual time‐concentration profiles. Liver biopsies were sampled from each patient and their individual hepatic transporter concentrations were quantified. We combined the transporter concentrations with in vitro uptake kinetics determined in HEK293‐transfected cells, and developed a semimechanistic model with a bottom‐up approach to predict the plasma concentration profiles of the single dose of rosuvastatin in each patient. The predicted PK parameters were evaluated against the measured in vivo plasma PKs from the same 54 patients. The developed model predicted the rosuvastatin PKs within two‐fold error for rosuvastatin area under the plasma concentration versus time curve (AUC; 78% of the patients; average fold error (AFE): 0.96), peak plasma concentration (Cmax; 76%; AFE: 1.05), and terminal half‐life (t1/2; 98%; AFE: 0.89), and captured differences in the rosuvastatin PKs in patients with the OATP1B1 521T<C polymorphism. This demonstrates that hepatic uptake clearance determined in transfected cell lines, together with proteomics scaling, provides a useful tool for prediction models, without the need for empirical scaling factors.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Rosuvastatin is a commonly used probe drug for pharmacokinetic (PK) investigations and has been used to establish prediction models of human hepatic uptake transport in average populations. These models have been validated with unrelated in vivo data from the literature.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can transfected cell lines and individual protein quantification be used in a bottom‐up mechanistic model to predict and capture interindividual differences of rosuvastatin plasma PKs in patients covering a wide body weight span?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ Interindividual differences in plasma concentration profiles of rosuvastatin are not directly associated with body weight and obesity. Transfected cell lines and protein quantification can be used to model rosuvastatin plasma profiles with an average fold error of 0.96 across patients covering a wide range of body weight. Interindividual differences in hepatic uptake transporters did not alone explain the large interindividual differences in rosuvastatin PKs.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study has identified in vitro kinetics and protein quantification as important in vitro parameters for predicting in vivo PKs without the need of arbitrary scaling factors. The successful example of rosuvastatin studied here should be investigated for other drugs and other clearance routes.
Transporter‐mediated uptake and efflux are important for the distribution and elimination of many drugs. 1 An example of such drugs are statins, or 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors, that are commonly used cholesterol lowering agents. Rosuvastatin is a frequently used probe for studying different transporter profiles and transporter‐mediated drug‐drug interactions both in vitro and in vivo, 2 , 3 , 4 , 5 as it has low metabolic clearance 6 and is a substrate of several uptake and efflux transporters. In humans, hepatic elimination of rosuvastatin accounts for ~ 70% of the total clearance, 7 where the major clearance mechanism is biliary excretion of the unchanged form. 6 The hepatic uptake transporters OATP1B1, OATP1B3, OATP2B1, 8 , 9 and NTCP, 10 which are located in the basolateral membrane (facing the blood) of the hepatocytes, play a major role in transporting rosuvastatin into the hepatocytes for hepatic elimination. Subsequently, BCRP 6 , 11 and possibly MDR1 and MRP2 6 eliminate rosuvastatin by efflux into the bile.
To study hepatic uptake of compounds, such as rosuvastatin, primary human hepatocytes is the gold standard in vitro system. Hepatocyte uptake is used in prediction models of hepatic disposition as it is easily scaled to in vivo uptake clearance by accounting for the number of hepatocytes per gram liver (hepatocellularity). 12 Hepatocytes can also be used to simulate the influence of hepatic disposition and metabolism on pharmacokinetic (PK) profiles. 13 , 14 , 15 A disadvantage of using hepatocytes is that the influence of individual transporters cannot easily be deconvoluted. Individual transporter data may be required for understanding, for example, drug‐drug interactions, the impact of genetic variants with altered function, or physiologically regulated and disease‐regulated transporter expression. Alternatively, the transport parameters can be established separately in cell lines transfected with individual human transporters. The uptake activities from cell lines expressing single transporters have been added up, combined with transporter protein concentrations in the cell lines and human tissues, and scaled to the in vivo situation. 8 , 9 , 16 , 17 , 18 , 19 , 20 , 21 Rosuvastatin has also been used in PK investigations and modeling of transporter‐mediated drug‐drug interactions, 22 , 23 and to establish prediction models of human hepatic uptake transport. 8 , 9 , 19 However, these studies did not consider the contribution of interindividual transporter concentrations and the predicted values were generally compared with unrelated in vivo data from the literature.
In contrast, we used hepatic transporter concentrations and in vivo PK data obtained from the same patients. This allowed us to investigate the influence of interindividual transporter concentrations on the hepatic uptake clearance of rosuvastatin in 54 patients undergoing gastric bypass surgery or cholecystectomy who were given an oral dose of rosuvastatin. For this purpose, we measured plasma rosuvastatin concentration profiles the day before surgery, and quantified the individual hepatic transporter concentrations in liver biopsies sampled at the time of surgery in each of the 54 patients. We then combined in vitro uptake kinetics from HEK293‐transfected cells with protein quantification in the cell lines and liver tissues to assess the contribution of OATP1B1, OATP1B3, OATP2B1, and NTCP to the rosuvastatin hepatic uptake clearance in the 54 patients. These parameters were included in a semimechanistic model developed by a bottom‐up approach to predict the rosuvastatin plasma concentration profiles, which were compared with the plasma PKs obtained from the same patients.
METHODS
Human liver tissue
Liver biopsies were obtained from 54 patients who provided informed consent as part of the COCKTAIL study, 24 and in agreement with the approval by the Regional Committee for Medical and Health Research Ethics. Biopsies were collected from 36 patients undergoing Roux‐en‐Y gastric bypass surgery and 18 patients undergoing cholecystectomy. Biopsies were snap frozen in liquid nitrogen directly upon sampling and stored at −80°C until analysis. See “Patient inclusion and data exclusion” in Supplementary Methods.
Plasma concentration‐time profile of rosuvastatin in vivo
A 20 mg rosuvastatin tablet was swallowed by the patients in a fasting state the day before surgery. Food was allowed after 2 hours. Blood samples were collected at time points from 0 to 24 hours, and were immediately placed on ice before 10 minutes centrifugation at 4°C at 1,800 g. Plasma was mixed with a matching volume of 0.1 M sodium acetate buffer and frozen at −70°C within 1 hour. Plasma concentrations of rosuvastatin were measured by Covance Laboratories, using a validated liquid chromatography tandem mass spectrometry method. 25
Protein quantification
Proteins were extracted from the liver biopsies by homogenization in a sodium dodecyl sulfate‐containing lysis buffer. Samples were processed using the MED‐Filter Aided Sample Preparation protocol, with LysC and trypsin. 26 Total protein and peptide amounts were determined based on tryptophan fluorescence. 27 Proteomics analysis was performed with Q Exactive HF and Q Exactive HF‐X mass spectrometers. Mass spectrometry (MS) data was processed with MaxQuant (version 1.6.0.16) 28 where proteins were identified by searching MS and MS/MS data of peptides against a decoy version of the human UniProtKB (UP000005640). Spectral raw intensities were normalized with variance stabilization, 29 and were subsequently used to calculate protein concentrations using the Total Protein Approach. 30 Batch effects were removed by geometric mean centering of proteins from samples analyzed at different time points.
In HEK293 cells, only the individual overexpressed transporters (OATP1B1, OATP1B3, OATP2B1, and NTCP) were of interest. Therefore, a targeted proteomics method, previously shown to be comparable with the label‐free method above, was used. 31 Proteins were extracted from the cells in an sodium dodecyl sulfate‐containing buffer. Samples were processed with Filter Aided Sample Preparation, 31 , 32 using trypsin. Targeted proteomics analysis was performed as previously described 31 using a QTRAP 6500 MS. Three transitions per surrogate peptide were monitored for quantification in the multiple reaction monitoring‐mode (Table S1 ). Data were processed using MultiQuant (version 3.0.5373.0). Protein concentrations were calculated by the peak area ratios of the internal standard peptide and sample peptide transitions.
Rosuvastatin uptake in vitro kinetics
Mock‐transfected HEK Flp‐In‐293 cells and cells stably expressing either OATP1B1, OATP1B3, OATP2B1, or NTCP 16 , 17 were incubated for 2 minutes at 37°C with varying concentrations of rosuvastatin (OATP1B1: 0.1–50 µM, OATP1B3: 0.1–60 µM, OATP2B1: 0.1–60 µM, and NTCP: 0.25–450 µM). Rosuvastatin accumulation was determined with a Waters Xevo TQ‐MS. Active transporter‐mediated uptake was calculated by subtracting the accumulation in mock‐transfected cells (passive diffusion) from the uptake in the transfected cells. The initial uptake rate was related to the substrate concentration and the uptake curve was fitted to the Michaelis‐Menten equation using GraphPad Prism (version 7.03):
| (1) |
where v is the uptake rate, Vmax is the maximal uptake rate, [S] is the substrate concentration, and Km is the substrate concentration at which the uptake rate is half of Vmax. Passive clearance (CLpassive) was determined by linear regression analysis in mock‐transfected cells. Uptake clearance of rosuvastatin by each transporter was further calculated by:
| (2) |
Hepatic uptake clearance prediction from proteomics and kinetic parameters from HEK293 cells
Hepatic uptake clearance was predicted from clearance and transporter protein concentrations in the HEK293‐cells and liver biopsies 16 , 17 , 33 :
| (3) |
where E liver and E cell are the transporter concentration (pmol/mg total protein) in the liver tissue and cell system, respectively, and HomPPGL is milligrams of homogenate protein per gram of liver tissue (88 mg protein/g liver 16 ).
Semimechanistic mathematical model for predicting rosuvastatin plasma concentration profiles
Rosuvastatin plasma concentration profiles were predicted using a semi‐mechanistic based PK compartment model developed by a bottom‐up approach. The three‐compartment model, including liver, gallbladder, and blood compartment, was built as described in Eqs. (4), (5), (6), (7). The oral dose was absorbed from the intestine into the liver, and subsequently distributed to the blood, assuming an instant mixing of blood from the portal and central vein. Enterohepatic recirculation was incorporated with the gallbladder compartment to capture the biphasic elimination of rosuvastatin:
| (4) |
| (5) |
| (6) |
| (7) |
where I, L, G, and B is the amount of rosuvastatin in the intestine, liver, gallbladder, and blood compartment, respectively. F is the intestinal fraction of drug that is (re)absorbed and reaches the liver, ka is the absorption constant, and kb is the biliary emptying rate. CLpassive is the passive clearance, CLefflux and CLuptake is the active efflux and uptake clearance of rosuvastatin between blood and liver. CLbile is the hepatic clearance of rosuvastatin to the gallbladder, CLmet is the metabolic clearance of rosuvastatin, and CLrenal is the renal rosuvastatin clearance. VL is the fractional liver volume of the total body weight, and VB is the volume of distribution. CLuptake and CLpassive are the experimental values described above. Other parameter values were obtained from the literature. VL and VB were scaled with individual total body weight (Table S2 ). Differential equations were solved using the deSolve package in R (version 3.4.4). The area under the plasma concentration versus time curve from zero to 24 hours (AUC) from the observed (AUCobs) and predicted (AUCpred) plasma concentration profiles were calculated with the MESS package in R using the linear interpolation (composite trapezoid rule) including all collected data points. Peak plasma concentration (Cmax) was determined based on the largest measured and predicted rosuvastatin plasma concentration. Terminal half‐life (t ½) was calculated from the log‐linear slope (k el) in the terminal elimination phase using at least three concentrations with t ½ = ln(2)/k el.
The predictive performance of the model was assessed using the predefined criteria of less than twofold difference in predicted/observed AUC, Cmax, and t ½ for the majority of the patients (> 70%). In addition, the approved interval of average fold errors of the PK parameters were set to 0.8 and 1.25 to ensure the model was not constantly underpredicting or overpredicting the plasma concentrations. Three models were primarily evaluated: model 1 – literature data for kb 34 (Table S2 ), model 2 – optimized kb based on the observed plasma concentration profile of rosuvastatin, and model 3 – as model 2, with correction for hepatic uptake activity reduction in carriers of the OATP1B1 521T>C polymorphism. To further evaluate models 1 and 2, we assessed the model performance after intravenous infusion in healthy male volunteers using data from Martin et al. 7 The patients undergoing cholecystectomy were assumed to be demographically more similar to the healthy volunteers as compared with the patients undergoing gastric bypass. Therefore, the mean demographic and proteomics data from the cholecystectomy patients was used to simulate the PK profile after a 4‐hour intravenous infusion of rosuvastatin, with the dose (8 mg) being added directly to the blood compartment in the model. The evaluation is presented in Figure S11 and Table S5 .
More detailed descriptions of the methods are provided in Supplementary Material.
RESULTS
Plasma concentration profiles of rosuvastatin
Rosuvastatin plasma concentration‐time profiles were determined in 54 patients following an oral dose of 20 mg rosuvastatin the day before gastric bypass or cholecystectomy surgery (Table 1 ). The profiles demonstrated a sharp initial peak in plasma concentration and a slower, biphasic elimination for most of the patients (Figure 1a ). Large interindividual differences of the measured rosuvastatin PK parameters were observed (Figure 1a,b ; Table 2 ). The AUCobs varied 13‐fold, from 18.3 to 237.1 ng/mL/h (median AUCobs 58.9 ng/mL/h), Cmax varied 30‐fold, from 1.9 (minimum value) to 56.6 (maximum value) ng/mL (median Cmax 7.9 ng/mL), and the t ½ varied 12‐fold, from 4.1 to 50.4 hours (median 7.7 hours) across all patients. The patients covered a large weight span with body weight ranging from 47 to 171 kg (Table 1 ). There were no correlations between the AUCobs, Cmax, or t ½ and body weight (Figure S2 a–c).
Table 1.
Patient information
| Total number of patients | 54 | |
| Median | Range a | |
| Age, years | 47 | 19–63 |
| Body weight, kg | 113 | 47.4–171.3 |
| BMI, kg/m2 | 39.6 | 18.3–64.5 |
| Number of patients b | ||
| Sex, M | 14 (26%) | |
| Sex, F | 40 (74%) | |
| Type 2 diabetes | 11 (20%) | |
| White | 53 (98%) | |
| Latin American | 1 (2%) |
BMI, body mass index.
Minimum and maximum values across all patients.
Number in parenthesis denotes percentages of the patients included in the study.
Figure 1.
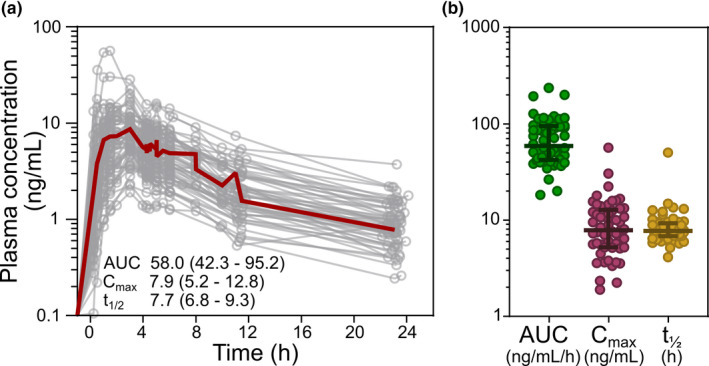
Plasma concentration profiles of rosuvastatin. (a) Plasma concentration‐time profiles of rosuvastatin in the 54 patients the day before gastric bypass or cholecystectomy surgery. Median values, with 25th and 75th percentiles in parenthesis, for area under the plasma concentration versus time curve (AUC; ng/mL/h), peak plasma concentration (Cmax; ng/mL), and terminal half‐life (t ½; hours) is given. Red thick line represents the mean plasma concentration profile of all patients. (b) Distribution of plasma concentration pharmacokinetic parameters in the 54 patients. Median values, with 25th and 75th percentiles are marked with lines. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 2.
Observed and predicted pharmacokinetic parameters for the 54 patients
| In vivo | Model 1 | Model 2 | Model 3 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Median (range) a | Median (range) a | AFE | Patients predicted within 2‐fold | Median (range) a | AFE | Patients predicted within 2‐fold | Median (range) a | AFE | Patients predicted within 2‐fold | |
| AUC, ng/mL/h | 58.9 (18.3–237.1) | 61.8 (36.5–121.5) | 0.95 | 85% | 51.0 (29.7–102.2) | 0.79 | 76% | 55.7 (29.7–115.0) | 0.96 | 78% |
| Cmax, ng/mL | 7.9 (1.9–56.6) | 7.4 (4.5–14.4) | 0.91 | 74% | 7.3 (4.4–14.1) | 0.90 | 74% | 8.3 (4.4–14.3) | 1.05 | 76% |
| t1/2, hours | 7.7 (4.1–50.4) | 5.4 (5.1–5.7) | 0.66 | 87% | 6.9 (6.5–9.3) | 0.91 | 98% | 6.9 (6.3–9.3) | 0.89 | 98% |
AFE, average fold error predicted/observed; AUC, area under the plasma concentration versus time curve; Cmax, peak plasma concentration.
Minimum and maximum values across all patients.
Protein quantification of uptake transporters in human liver
We also observed interindividual differences in protein concentrations of the hepatic uptake transporters OATP1B1, OATP1B3, OATP2B1, and NTCP across the 54 patients. OATP1B1 protein concentrations ranged 0.9–5.5 (6‐fold), OATP1B3 ranged 0.3–2.6 (9‐fold), OATP2B1 ranged 0.4–1.2 (3‐fold), and NTCP ranged 0.04–2.5 (57‐fold) pmol/mg protein (Figure 2a–e ) across the patients. Of the four transporters, only OATP1B3 concentrations were negatively correlated with body weight (rs = –0.35, P = 0.0085, followed by Benjamini–Hochberg correction for multiple comparisons; Figure S3 a–d).
Figure 2.
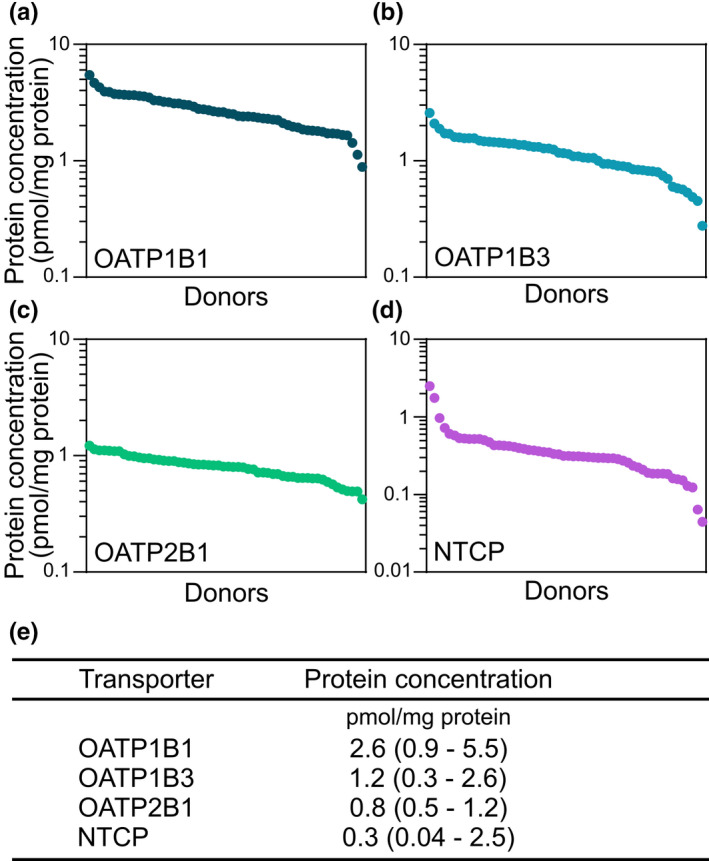
Protein quantification of uptake transproters in human liver. (a) Protein concentration of OATP1B1, (b) OATP1B3, (c) OATP2B1, and (d) NTCP in liver biopsies obtained from the 54 patients undergoing gastric bypass or cholecystectomy surgery. (e) Median protein concentration, with range in parenthesis, of OATP1B1, OATP1B3, OATP2B1, and NTCP in the patients. [Colour figure can be viewed at wileyonlinelibrary.com]
In vitro rosuvastatin transport in HEK293 cells
The in vitro kinetic parameters required for the prediction of rosuvastatin plasma concentration profiles were obtained using OATP1B1‐expressing, OATP1B3‐expressing, OATP2B1‐expressing, or NTCP‐expressing HEK293 cells. The transporter‐mediated uptake of rosuvastatin followed Michaelis–Menten kinetics for all four transporters (Figure 3a–e ). The obtained Km values were in line with, or slightly higher than, those previously described (Table S3 ). The rosuvastatin uptake clearance (CLtransporter) by the different transporters were calculated to 2.6 (OATP1B1), 1.3 (OATP1B3), 0.46 (OATP2B1), and 1.6 (NTCP) µL/min/mg protein. The concentration of each transporter in the respective cell systems ranged between 0.9 and 6.2 pmol/mg protein (Figure 3e ).
Figure 3.
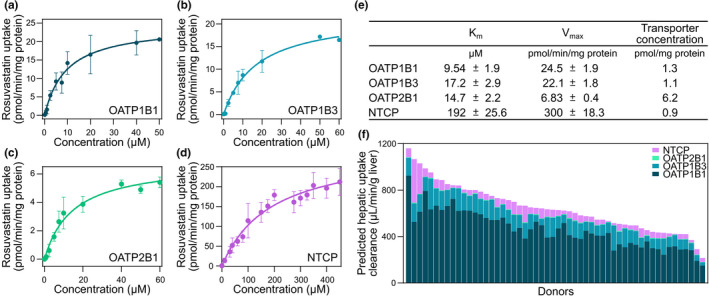
In vitro rosuvastatin transport in HEK293 cells. (a) Concentration dependent uptake of rosuvastatin in HEK293 cells overexpressing OATP1B1, (b) OATP1B3, (c) OATP2B1 (d), and NTCP, respectively. (e) Kinetic parameters of rosuvastatin uptake and transporter protein concentrations in HEK293‐transfected cells. (f) Predicted hepatic uptake clearance of rosuvastatin, obtained from clearance estimates in HEK293 cells and transporter concentrations in cells and liver biopsies in the 54 patients undergoing gastric bypass or cholecystectomy surgery. Km, Michaelis‐Menten constant; Vmax, maximal rate of transport. [Colour figure can be viewed at wileyonlinelibrary.com]
Prediction of intrinsic hepatic rosuvastatin uptake clearance
Hepatic uptake clearance (CLuptake) of rosuvastatin was predicted by scaling the CLtransporter to that in vivo using the transporter concentrations in the cell systems and liver biopsies (Eq. 3). In agreement with the interindividual differences in transporter concentrations, large differences of predicted hepatic transporter clearances were observed across the patients, ranging between 215 and 1,161 (median 630) µl/min/g liver (Figure 3f ). Similar to that observed for the in vivo plasma PK parameters, no correlation was found between the predicted hepatic uptake rosuvastatin clearance and body weight (Figure S4 a; rs = –0.15, P = 0.27). Nevertheless, a negative correlation was observed between AUCobs and predicted hepatic uptake clearance (rs = –0.30, P = 0.03) in the patients, showing that hepatic uptake clearance decreases the plasma concentration of rosuvastatin (Figure S4 b). This agreed well with the negative correlation between AUCobs and hepatic OATP1B1 protein concentration (Figure S4 c; rs = −0.31, P = 0.02), because OATP1B1 was found to be the major uptake transporter of rosuvastatin in most of the patients, with contributions of 49–86% of the total active uptake (Figure 3f ). This is in line with OATP1B1 being the major contributor to active uptake of rosuvastatin and other statins in human hepatocytes. 6 , 9 , 16 , 20 In general, OATP1B3 was the second largest contributor to the rosuvastatin uptake, with 9–41% of the total active uptake. However, in 5 of the 54 patients, NTCP was the second most important transporter, contributing with 13–35%. OATP2B1 played a minor role in the rosuvastatin uptake with contributions of 0.4–2% of the total active uptake, which agrees with previous findings. 6
Prediction of rosuvastatin plasma concentration profiles
Prediction of rosuvastatin plasma concentration profiles was performed using our established in vitro uptake kinetic parameters and individual protein concentrations from the patients (Eqs. (4), (5), (6), (7)), in the three‐compartment semimechanistic model (Figure 4a ). The gallbladder compartment represents enterohepatic recirculation described as a continuous process by the biliary emptying rate (kb). With model 1, when using a previously established emptying rate from the gallbladder to the intestine 34 (Table S2 ), AUC, Cmax, and t ½ were predicted within 2‐fold from the observed data for the majority of the patients (85%, 74%, and 87%, respectively; Table 2 ). Nevertheless, the model systematically underpredicted the t ½ (average fold error (AFE): 0.66, Figure S5 a), and did not fully capture the biphasic elimination behavior. The gallbladder emptying rate can decrease with body size and gallbladder volume, 34 and when lowering kb in model 2, the biphasic elimination was better captured (Figure 4b , Figure S5 b, Table 2 ). The t 1/2 was predicted within 2‐fold error for 98% of the patients, without bias toward underprediction or overprediction (AFE: 0.91; Figure S5 b). The AUC and Cmax were not markedly changed by the lowered biliary emptying rate. For AUC, 76% of the predictions were within 2‐fold of that observed of which 50% were within 1.5‐fold, without bias toward underprediction or overprediction (AFE: 0.79, respectively; Figure 4c , Figures S5b–S7 , Table 2 ). Further, sensitivity analysis showed that the hepatic uptake clearance of rosuvastatin influenced both AUC and Cmax, but not t ½ (Figure S8 ).
Figure 4.
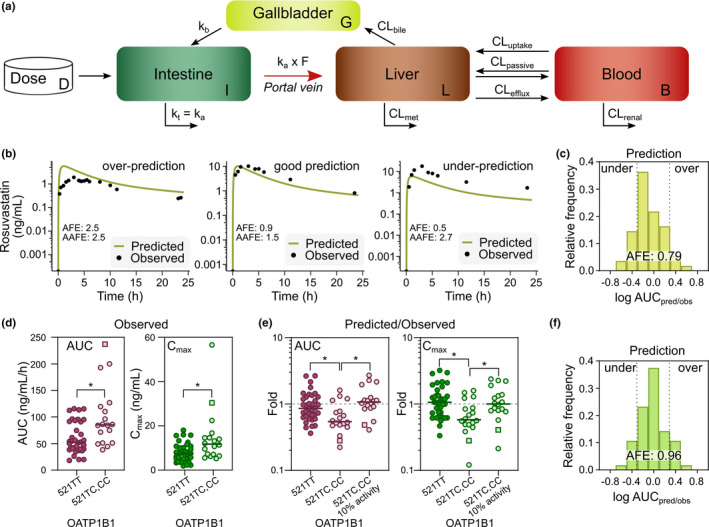
Prediction of intrinsic hepatic rosuvastatin uptake clearance. (a) Schematics of the three‐compartment semimechanistic model 2. (b) Representative examples of underpredictions, good, and overpredictions of rosuvastatin plasma concentration profiles, using model 2 with gallbladder‐emptying rate optimized from 34 . (c) Frequency distribution of rosuvastatin area under the plasma concentration versus time curve (AUC) predicted/observed fold errors across the 54 patients (model 2). Dotted lines represent twofold underprediction and overprediction. (d) Observed AUC and peak plasma concentration (Cmax) values from rosuvastatin plasma concentration profiles, separated by patients carrying the OATP1B1 521TT and OATP1B1 521TC,CC variants (square symbolizes the carrier of the 521CC allele). (e) Fold values from predicted and observed AUC and Cmax from the semimechanistic model, separated by patients with OATP1B1 521TT and OATP1B1 521TC,CC variants before (model 2) and after correcting for the 521C 10% reduced activity (model 3). 10 (f) Frequency distribution of rosuvastatin AUC predicted/observed fold errors across the 54 patients after adjusting for the 521C 10% reduced activity, where dotted lines represent twofold underprediction and overprediction (model 3). *P < 0.05, Kruskal–Wallis test followed by Dunn’s multiple comparisons test. AUC (ng/mL/h); Cmax (ng/mL); terminal half‐life (t 1/2) (h). CL, clearance. [Colour figure can be viewed at wileyonlinelibrary.com]
Influence of genetic variants on rosuvastatin PKs
One third of the patients (18 of 54) were carriers of the OATP1B1 521T>C polymorphism (rs 4149956; Val174Ala; included in the OATP1B1*5 variant), which is slightly higher than the previously estimated prevalence of 8–20% in a white population. 35 The 521T>C is associated with reduced hepatic uptake activity of, for example, rosuvastatin, leading to higher plasma concentrations. 36 This agreed well with our results where carriers of the 521C polymorphism showed higher rosuvastatin exposure, with 63% higher AUCobs and 59% higher Cmax than homozygotes for the 521T variant (P = 0.001 and P = 0.004, Mann–Whitney U test; Figure 4d ). 35 , 37
We also observed a significant underprediction of AUC and Cmax values for carriers of the OATP1B1 521C variant (P = 0.038 and P = 0.006, Kruskal–Wallis test followed by Dunn’s multiple comparisons test; Figure 4e ) in model 2. The median fold values of predicted/observed in 521C carriers were 0.54 and 0.59 for AUC and Cmax, respectively, compared with values of 0.86 and 1.1 for AUC and Cmax, respectively, for patients homozygous for the 521T variant. In HeLa cells, rosuvastatin uptake of the OATP1B1 521C variant (OATP1B1*5) is reduced to 10% of that from the reference allele (521T; OATP1B1*1a). 10 When adjusting for this reduced activity in model 3, the predictions of both AUC and Cmax for the 521C carriers were improved (fold AUC = 1.0, fold Cmax = 1.1) and became comparable to those observed in homozygotes for the 521T variant (Figure 4e , Figure S9 ). This adjustment improved the overall predictions of AUC and Cmax giving AFEs of 0.96 and 1.05 (Figure 4f , Table 2 ), where 78% of the predictions of AUC were within 2‐fold from that observed, and of which 50% were within 1.4‐fold.
DISCUSSION
We combined in vitro uptake kinetics from transporter transfected cells with human liver protein quantification to assess the contribution of four hepatic uptake transporters to the rosuvastatin plasma clearance in 54 patients enrolled in the COCKTAIL study. 24 Transporter concentrations were determined in liver biopsies obtained from each patient and we used a bottom‐up approach to build a semimechanistic model to predict the rosuvastatin plasma PKs in all 54 patients. The model predictions were assessed by comparing the predicted vs. observed in vivo rosuvastatin PK parameters and plasma concentration profiles from a single, oral dose of rosuvastatin for each patient. The model successfully predicted the rosuvastatin PKs for the majority of the patients with less than twofold difference in rosuvastatin AUC (78% of the patients), Cmax (76%), and t1/2 (98%), without the need for arbitrary scaling factors.
The observed 13‐fold interindividual differences in AUC across the 54 patients were in line with that previously observed (Figure S1 , Table S4 ). The 54 patients covered a large body weight span (47–171 kg), which gave us the opportunity to investigate the relationship between rosuvastatin disposition and body weight. Metabolic activity and protein concentrations of certain CYP enzymes have previously been shown to decrease with increasing body weight. 38 , 39 We observed a negative correlation between protein concentrations of OATP1B3 and body weight. However, this effect was not large enough to significantly influence the rosuvastatin disposition in vivo, as no correlation was observed among AUCobs, Cmax, or t ½, and body weight (Figure S2 a–f). This is most likely due to that OATP1B1 contributed to the majority of the hepatic uptake clearance (49–86%; Figure 1f ), which is in line with that observed by Kitamura et al. (66–84%), 6 but higher than that described by Bosgra et al. (23%). 8 However, the authors of the latter study acknowledge that they may have underestimated and/or overestimated the contribution of OATP1B1 and OATP2B1, respectively. Further, although the data were obtained from 54 patients with or without obesity, no significant differences in rosuvastatin PKs or modeling results were observed between the two groups (Figures S6, S7 ). However, other factors, including genetic variants with altered function and physiological variation affecting renal and hepatic clearance, may also have influenced the observed large interindividual differences in rosuvastatin PKs. 40
The observed higher in vivo exposure and underprediction of rosuvastatin plasma concentrations in patients with the OATP1B1 521C variant was likely caused by a reduced activity of the transporter. 10 It has been suggested that this activity reduction for the 521C allele is caused by a defect in transferring the transporter to the plasma membrane. 36 As the protein quantification method used here does not discriminate for plasma membrane proteins but measures all transporters in the cell, the active protein concentrations at the surface could be overestimated and consequently lead to underprediction of the rosuvastatin disposition. It could be reasoned that the plasma membrane could be isolated in order to capture the reduced plasma membrane concentration of the OATP1B1 521C variant. However, due to poor enrichment and protein loss during fractionation, it has proven difficult to impossible to successfully quantify proteins from isolated plasma membrane fractions. 31 , 41 , 42 , 43 Instead, we accounted for the reduced OATP1B1 521C activity (10%) in our model, and noted that the predictions for AUC and Cmax became comparable to those from patients homozygous for the 521T variant. Notably, a patient homozygote for the 521C variant were one of the patients most affected by the decreased activity (square symbol in Figure 4d,e ). Inclusion of other genetic variants could further improve the prediction models. 44
The rosuvastatin plasma concentration profiles displayed a biphasic elimination behavior, which could have several explanations. First, rosuvastatin could be distributed to low perfused peripheral tissues (e.g., adipose tissue), from which rosuvastatin would be slowly released to the blood and subsequently eliminated. However, as rosuvastatin is mainly present in vivo in its acid and hydrophilic form, 45 it is unlikely that the compound would distribute to low‐perfused, lipid rich, organs, such as the adipose tissue. 8 In agreement with this, no biphasic elimination was observed when testing the rosuvastatin distribution in a whole body physiologically‐based pharmacokinetic model, 46 using predicted tissue to plasma ratio and an experimental value of volume of distribution (VB) combined with lipophilicity. 47 Therefore, we instead used a V B value (0.227 L/kg) predicted from physicochemical parameters (logP, fu, and ion class) of rosuvastatin, which indicated that rosuvastatin is primarily distributed to the extracellular fluid. 48 This V B value is in agreement with those previously calculated (0.335 and 0.366 L/kg) for rosuvastatin. 8 Second, the biphasic behavior could be an effect of enterohepatic recirculation of rosuvastatin. 7 , 8 This is in line with that the gallbladder emptying rate was the only parameter in our model that affected the terminal elimination (Figure S5 ). The gallbladder emptying rate and delay is strongly influenced by meal intake, but accurate modeling of the latter is complex. 49 Therefore, complete and accurate modeling of the gallbladder emptying would require exact time points of food intake. In this study, the patients had several, unmonitored meals during the 24‐hour course of sample collection, and the time points required for the complex modeling of the gallbladder were not recorded. Thus, we assumed a continuous release model 49 without any delay, which could be the reason for our need to optimize the emptying rate.
Further, the observed interindividual difference in hepatic transporter concentrations (Figure 2a–e ) gave a large variability in the predicted rosuvastatin uptake clearance (Figure 3f ). However, when modeling how these interindividual differences affected the plasma concentrations—by comparing input values of mean concentrations for each of the four uptake transporters with input values based on the individual concentrations—only small, but insignificant changes in the predicted Cmax was obtained (AFE decreasing from 1.68 for mean protein concentrations to 1.63 with individual protein concentrations). This suggests that protein quantification is an appropriate, physiological scaling factor for in vitro‐in vivo scaling, albeit the interindividual differences in uptake transporter concentrations could not completely describe the observed interindividual variability of rosuvastatin PKs (Figure S10 ). Other factors that likely affect the rosuvastatin PKs after oral dosing are intestinal absorption, bile efflux, and renal elimination, all of which were kept constant in our model. For instance, the hydrophilic nature of rosuvastatin requires active uptake transport into the intestinal lumen and is determined by the apically located uptake transporter OATP2B1 and efflux transporter BCRP. 22 , 50 Interindividual variability of these intestinal proteins could affect the rosuvastatin PKs, and models incorporating intestinal absorption and distribution could shed light on these issues. 22 , 23 Furthermore, the effect of interindividual differences in hepatic biliary efflux of rosuvastatin, such as transport by BCRP (that was kept constant in this model) could also be of importance. Additionally, both intestinal and hepatic blood flow should also be considered for an improved in vivo‐relevant model, as it influences the drug transit. 8 This could be especially important for drug disposition predictions in patients with obesity, where increased blood volume is common. 51 Finally, as renal clearance accounts for ~ 28% of the total rosuvastatin clearance, 7 individual differences in kidney function and active transport (OAT3) could also impact the rosuvastatin plasma distribution. For further investigations of population variability, these parameters should be incorporated in more complex, whole body physiologically based pharmacokinetic models. However, as the scope of this study was to investigate if in vitro hepatic uptake kinetics combined with protein quantification could be used in a simple bottom‐up prediction model, we kept these parameters constant. It could be argued that the model could give better prediction of rosuvastatin PK after intravenous dosing, as compared with oral dosing as used in this study, because intravenous dosing would eliminate factors, such as limited absorption, active intestinal efflux, and poor solubility, that could confound the expression—activity correlation. To investigate how our model could recapitulate intravenous data, we compared previously published intravenous data from healthy male volunteers 7 with our model on a population level. Overall, intravenous data could be recapitulated acceptably, considering that the populations were different (Figure S11 , Table S5 ). We noted that the healthy male volunteers in Martin et al. 7 had a slightly higher clearance as compared with the patients evaluated in our study. Additionally, we noted that model 1, using the literature value of bile emptying rate 34 better described the PK of the healthy volunteers, as compared with the estimated value (model 2) used in our patient group. We refrained from any further interpretations of these findings, because this would require detailed demographic and proteomics data for the healthy male volunteers included in the study by Martin et al. 7
In summary, we used a bottom‐up approach to build a semimechanistic model for the prediction of rosuvastatin plasma concentration profiles, by combining uptake kinetics determined in transporter transfected cell lines with individual protein quantification. The model predictions were assessed against PK parameters and plasma concentration profiles obtained from the same patients, from which the liver biopsies were sampled, thus using each of the patients as their own control. With this approach, we predicted the PKs of rosuvastatin for the majority of the patients with less than twofold difference in AUC (78% of the patients, AFE: 0.96), Cmax (76%; AFE: 1.05), and t 1/2 (98%; AFE: 0.89). These results were obtained without arbitrary scaling factors, with a model that showed sensitivity to altered protein activity due to transporter genetic variation.
Funding
This study was supported by the Swedish Research Council, approval numbers 5715 and 01951.
Conflict of Interest
The authors declared no competing interests for this work.
Author Contributions
C.W. and P.A. wrote the manuscript. C.W., P.A., T.B.A., J.H., and A.Å. designed the research. C.W., I.R., and J.R.W. performed the research. C.W., I.R., L.P.G., S.K., and R.J.L. analyzed data.
Supporting information
Supplementary Data S1
Figures S1‐S11
Table S1‐S3
Acknowledgments
The authors are grateful to Maria Karlgren, Department of Pharmacy, Uppsala University for providing the HEK293 transfected cell lines and to Katharina Zettl, Biochemical Proteomics Group, Max Planck Institute of Biochemistry, for brilliant technical assistance. We also thank Rune Sandbu, Lars Thomas Seeberg, and colleagues at Department of Gastrointestinal Surgery, Vestfold Hospital Trust for providing liver biopsies. Finally, we are grateful to the patients at the Morbid Obesity Center, Vestfold Hospital Trust, for participating in the COCKTAIL study and accepting tissue biopsies to be taken. Protein concentrations and allometrics can be found in Supplementary Data S1 .
References
- 1. Giacomini, K.M. et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 9, 215 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shah, Y. et al. Effect of omeprazole on the pharmacokinetics of rosuvastatin in healthy male volunteers. Am. J. Ther. 23, e1514–e1523 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Stopfer, P. et al. Optimization of a drug transporter probe cocktail: potential screening tool for transporter‐mediated drug–drug interactions. Br. J. Clin. Pharmacol. 84, 1941–1949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emami Riedmaier, A. et al. More power to OATP1B1: an evaluation of sample size in pharmacogenetic studies using a rosuvastatin PBPK model for intestinal, hepatic, and renal transporter‐mediated clearances. J. Clin. Pharmacol. 56, S132–S142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wu, H.‐F. et al. Rosuvastatin pharmacokinetics in Asian and white subjects wild type for both OATP1B1 and BCRP under control and inhibited conditions. J. Pharm. Sci. 106, 2751–2757 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kitamura, S. et al. Involvement of multiple transporters in the hepatobiliary transport of rosuvastatin. Drug Metab. Dispos. 36, 2014 (2008). [DOI] [PubMed] [Google Scholar]
- 7. Martin, P.D. et al. Absolute oral bioavailability of rosuvastatin in healthy white adult male volunteers. Clin. Ther. 25, 2553–2563 (2003). [DOI] [PubMed] [Google Scholar]
- 8. Bosgra, S. et al. Predicting carrier‐mediated hepatic disposition of rosuvastatin in man by scaling from individual transfected cell‐lines in vitro using absolute transporter protein quantification and PBPK modeling. Eur. J. Pharm. Sci. 65, 156–166 (2014). [DOI] [PubMed] [Google Scholar]
- 9. Kunze, A. et al. Prediction of organic anion‐transporting polypeptide 1B1‐ and 1B3‐mediated hepatic uptake of statins based on transporter protein expression and activity data. Drug Metab. Dispos. 42, 1514 (2014). [DOI] [PubMed] [Google Scholar]
- 10. Ho, R.H. et al. Drug and bile acid transporters in rosuvastatin hepatic uptake: function, expression, and pharmacogenetics. Gastroenterology 130, 1793–1806 (2006). [DOI] [PubMed] [Google Scholar]
- 11. Huang, L. , Wang, Y. & Grimm, S. ATP‐dependent transport of rosuvastatin in membrane vesicles expressing breast cancer resistance protein. Drug Metab. Dispos. 34, 738 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Riccardi, K.A. et al. A novel unified approach to predict human hepatic clearance for both enzyme‐ and transporter‐mediated mechanisms using suspended human hepatocytes. Drug Metab. Dispos. 47, 484 (2019). [DOI] [PubMed] [Google Scholar]
- 13. Jones, H.M. et al. Mechanistic pharmacokinetic modeling for the prediction of transporter‐mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab. Dispos. 40, 1007 (2012). [DOI] [PubMed] [Google Scholar]
- 14. Sato, M. et al. Physiologically based pharmacokinetic modeling of Bosentan identifies the saturable hepatic uptake as a major contributor to its nonlinear pharmacokinetics. Drug Metab. Dispos. 46, 740 (2018). [DOI] [PubMed] [Google Scholar]
- 15. Varma, M.V.S. et al. Physiologically based modeling of pravastatin transporter‐mediated hepatobiliary disposition and drug‐drug interactions. Pharm. Res. 29, 2860–2873 (2012). [DOI] [PubMed] [Google Scholar]
- 16. Vildhede, A. et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug‐drug interactions. Drug Metab. Dispos. 42, 1210 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Karlgren, M. et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug‐drug interactions. J. Med. Chem. 55, 4740–4763 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Akazawa, T. et al. Quantitative targeted absolute proteomics of transporters and pharmacoproteomics‐based reconstruction of P‐glycoprotein function in mouse small intestine. Mol. Pharm. 13, 2443–2456 (2016). [DOI] [PubMed] [Google Scholar]
- 19. Rose, R.H. et al. Application of a physiologically based pharmacokinetic model to predict OATP1B1‐related variability in pharmacodynamics of rosuvastatin. CPT Pharmacometrics Syst. Pharmacol. 3, 124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hirano, M. et al. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J. Pharmacol. Exp. Ther. 311, 139 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Vildhede, A. et al. Mechanistic modeling of pitavastatin disposition in sandwich‐cultured human hepatocytes: a proteomics‐informed bottom‐up approach. Drug Metab. Dispos. 44, 505 (2016). [DOI] [PubMed] [Google Scholar]
- 22. Wang, Q. , Zheng, M. & Leil, T. Investigating transporter‐mediated drug‐drug interactions using a physiologically based pharmacokinetic model of rosuvastatin. CPT Pharmacometrics Syst. Pharmacol. 6, 228–238 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jamei, M. et al. A mechanistic framework for in vitro–in vivo extrapolation of liver membrane transporters: prediction of drug–drug interaction between rosuvastatin and cyclosporine. Clin. Pharmacokinet. 53, 73–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hjelmesæth, J. et al. Impact of body weight, low energy diet and gastric bypass on drug bioavailability, cardiovascular risk factors and metabolic biomarkers: protocol for an open, non‐randomised, three‐armed single centre study (COCKTAIL). BMJ Open 8, e021878 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Braamskamp, M.J.A.M. et al. Efficacy and safety of rosuvastatin therapy in children and adolescents with familial hypercholesterolemia: Results from the CHARON study. J. Clin. Lipidol. 9, 741–750 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Wiśniewski, J.R. & Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 84, 2631–2637 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Wiśniewski, J.R. & Gaugaz, F.Z. Fast and sensitive total protein and peptide assays for proteomic analysis. Anal. Chem. 87, 4110–4116 (2015). [DOI] [PubMed] [Google Scholar]
- 28. Tyanova, S. , Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat. Protoc. 11, 2301 (2016). [DOI] [PubMed] [Google Scholar]
- 29. Huber, W. et al. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18(suppl. 1), S96–S104 (2002). [DOI] [PubMed] [Google Scholar]
- 30. Wiśniewski, J.R. & Rakus, D. Multi‐enzyme digestion FASP and the ‘Total Protein Approach’‐based absolute quantification of the Escherichia coli proteome. J. Proteomics 109, 322–331 (2014). [DOI] [PubMed] [Google Scholar]
- 31. Wegler, C. et al. Variability in mass spectrometry‐based quantification of clinically relevant drug transporters and drug metabolizing enzymes. Mol. Pharm. 14, 3142–3151 (2017). [DOI] [PubMed] [Google Scholar]
- 32. Wiśniewski, J.R. et al. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Ishida, K. et al. Successful prediction of in vivo hepatobiliary clearances and hepatic concentrations of rosuvastatin using sandwich‐cultured rat hepatocytes, transporter‐expressing cell lines, and quantitative proteomics. Drug Metab. Dispos. 46, 66 (2018). [DOI] [PubMed] [Google Scholar]
- 34. Vezina, W.C. et al. Increased volume and decreased emptying of the gallbladder in large (morbidly obese, tall normal, and muscular normal) people. Gastroenterology 98, 1000–1007 (1990). [DOI] [PubMed] [Google Scholar]
- 35. Lee, H.H. & Ho, R.H. Interindividual and interethnic variability in drug disposition: polymorphisms in organic anion transporting polypeptide 1B1 (OATP1B1; SLCO1B1). Br. J. Clin. Pharmacol. 83, 1176–1184 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kameyama, Y. et al. Functional characterization of SLCO1B1 (OATP‐C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet. Genomics 15, 513–522 (2005). [DOI] [PubMed] [Google Scholar]
- 37. Pasanen, M.K. et al. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 82, 726–733 (2007). [DOI] [PubMed] [Google Scholar]
- 38. Ulvestad, M. et al. Impact of OATP1B1, MDR1, and CYP3A4 expression in liver and intestine on interpatient pharmacokinetic variability of atorvastatin in obese subjects. Clin. Pharmacol. Ther. 93, 275–282 (2013). [DOI] [PubMed] [Google Scholar]
- 39. Krogstad, V. et al. A comparative analysis of cytochrome P450 activities in paired liver and small intestinal samples from patients with obesity. Drug Metab. Dispos. 48, 8–17 (2019). [DOI] [PubMed] [Google Scholar]
- 40. Turner, R.M. , Park, B.K. & Pirmohamed, M. Parsing interindividual drug variability: an emerging role for systems pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 7, 221–241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumar, V. et al. Quantitative transporter proteomics by liquid chromatography with tandem mass spectrometry: addressing methodologic issues of plasma membrane isolation and expression‐activity relationship. Drug Metab. Dispos. 43, 284 (2015). [DOI] [PubMed] [Google Scholar]
- 42. Harwood, M.D. et al. Lost in centrifugation: accounting for transporter protein losses in quantitative targeted absolute proteomics. Drug Metab. Dispos. 42, 1766 (2014). [DOI] [PubMed] [Google Scholar]
- 43. Wiśniewski, J.R. , Wegler, C. & Artursson, P. Subcellular fractionation of human liver reveals limits in global proteomic quantification from isolated fractions. Anal. Biochem. 509, 82–88 (2016). [DOI] [PubMed] [Google Scholar]
- 44. Nies, A.T. et al. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 5, 1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam. Clin. Pharmacol. 19, 117–125 (2005). [DOI] [PubMed] [Google Scholar]
- 46. Langdon, G. et al. Linking preclinical and clinical whole‐body physiologically based pharmacokinetic models with prior distributions in NONMEM. Eur. J. Clin. Pharmacol. 63, 485–498 (2007). [DOI] [PubMed] [Google Scholar]
- 47. Jansson, R. , Bredberg, U. & Ashton, M. Prediction of drug tissue to plasma concentration ratios using a measured volume of distribution in combination with lipophilicity. J. Pharm. Sci. 97, 2324–2339 (2008). [DOI] [PubMed] [Google Scholar]
- 48. Davies, B. & Morris, T. Physiological parameters in laboratory animals and humans. Pharm. Res. 10, 1093–1095 (1993). [DOI] [PubMed] [Google Scholar]
- 49. Colburn, W.A. Pharmacokinetic and biopharmaceutic parameters during enterohepatic circulation of drugs. J. Pharm. Sci. 71, 131–133 (1982). [DOI] [PubMed] [Google Scholar]
- 50. Johnson, M. et al. Inhibition of intestinal OATP2B1 by the calcium receptor antagonist ronacaleret results in a significant drug‐drug interaction by causing a 2‐fold decrease in exposure of rosuvastatin. Drug Metab. Dispos. 45, 27 (2017). [DOI] [PubMed] [Google Scholar]
- 51. Knibbe, C.A.J. et al. Drug disposition in obesity: toward evidence‐based dosing. Annu. Rev. Pharmacol. Toxicol. 55, 149–167 (2015). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data S1
Figures S1‐S11
Table S1‐S3