

Abstract
The advent of next generation DNA sequencing (NGS) has revolutionized clinical medicine by enabling wide‐spread testing for genomic anomalies and polymorphisms. With that explosion in testing, however, come several informatics challenges including managing large amounts of data, interpreting the results and providing clinical decision support. We present Flype, a web‐based bioinformatics platform built by a small group of bioinformaticians working in a community hospital setting, to address these challenges by allowing us to: (a) securely accept data from a variety of sources, (b) send orders to a variety of destinations, (c) perform secondary analysis and annotation of NGS data, (d) provide a central repository for all genomic variants, (e) assist with tertiary analysis and clinical interpretation, (f) send signed out data to our EHR as both PDF and discrete data elements, (g) allow population frequency analysis and (h) update variant annotation when literature knowledge evolves. We discuss the multiple use cases Flype supports such as (a) in‐house NGS tests, (b) in‐house pharmacogenomics (PGX) tests, (c) dramatic scale‐up of genomic testing using an external lab, (d) consumer genomics using two external partners, and (e) a variety of reporting tools. The source code for Flype is available upon request to the authors.
Keywords: bioinformatics, clinical decision support, EMR integration, personalized medicine, pharmacogenomics
1. INTRODUCTION
Personalized medicine (PMed) aims to stratify patients finely enough to predict response to various clinical interventions. While it has been a goal of medical practice for a long time, significant strides in this endeavor have been made since the publication of the human genome at the turn of the century and the more recent development of next generation DNA sequencing (NGS) technologies (Feero, Guttmacher, & Collins, 2010). However, in order to initiate an advanced PMed program in a community hospital setting, the biotechnology of NGS is necessary but not sufficient. What is also needed is the computational technology to incorporate the data resulting from NGS into the hospital's workflows in a tactical manner; specifically, to make it usable by clinicians without deluging them with unmanageable amounts of data. Both NGS and this computational technology could be deployed in a hospital through a combination of in‐house development and outsourcing to a vendor.
Outsourcing NGS usually takes the form of contracting with an external lab to sequence hospital specimens. Given the rapid pace of technology development in the NGS space, external labs contracted to do this work are constantly changing making it a challenge to fully integrate with clinical care for reasons explained below. Primarily for this difficulty of integration, external lab reports are frequently faxed back to the hospital and then scanned into the electronic health records (EHR) as static images rather than as usable discrete data elements. Even when not fax‐based, it is still very far away from full integration with the EHR and providing appropriate clinical decision support. When in‐house NGS development is thrown into the mix, as is often the case, a data management challenge ensues due to the increased variety of results that must now be incorporated back into the EHR. To solve this integration challenge the informatics platform must, at a minimum, satisfy the eight requirements summarized in Table 1.
TABLE 1.
Requirements of the computational technology
| Item | Description | Use cases that need item | ||
|---|---|---|---|---|
| NGS | PGX | DNA10K | ||
| 1 | Be able to securely accept data from a variety of sources, internal and external | ✓ | ✓ | ✓ |
| 2 | Be able to send order information to a variety of destinations, internal and external | ✓ | ✓ | ✓ |
| 3 | Perform secondary analysis and annotation of NGS data generated in‐house | ✓ | ✓ | |
| 4 | Act as a central repository for all genomic variants identified in patient specimens | ✓ | ✓ | ✓ |
| 5 | Assist with tertiary analysis and clinical interpretation of NGS data (potentially including sign out of in‐house tests) | ✓ | ✓ | |
| 6 | Send signed out data to EHR not only as a human readable PDF report but also as discrete data elements | ✓ | ✓ | ✓ |
| 7 | Allow population frequency analysis of variants among patients | ✓ | ✓ | ✓ |
| 8 | Update variant annotation when literature knowledge concerning those variants evolves | ✓ | ✓ | ✓ a |
Updates currently provided by Color, but could be transitioned to Flype if necessary in the future.
Implementing any rapidly evolving technology like genomics into clinical practice has long been a challenge due to financial and organizational concerns. Organizations must also solve financial challenges related to reimbursement for these novel tests. Reimbursement for these tests will become common in the future when these tests become routine; until then, it is useful to have patient advocates and other dedicated personnel to work with payers to get reimbursement. The costs for bioinformatics and additional IT infrastructure are fixed and cannot be assigned to any single test; they are usually amortized over a large number of tests in multiple NGS assays. Healthcare organizations usually prefer to have long‐term stable relationships with vendors but implementing genomics necessitates flexibility, which specifically means pursuing short‐term contracts. Implementing new genomic testing also requires a robust education environment for practitioners and nurses to ensure they use and interpret the tests appropriately. Finally, patient data safety and integrity are of primary concern for IT in every hospital and those concerns must be addressed even as large amounts of genomic data are analyzed and processed. Given the volume of data, there is an active debate in the community (Zandesh, Ghazisaeedi, Devarakonda, & Haghighi, 2019) regarding the pros and cons of storing and analyzing this data in the Cloud as opposed to on‐premises. These issues must be dealt with at an institutional level, and what is appropriate for each organization depends on the current status of their technology.
When our personalized medicine program began, the priority needs were variant annotation and repository (items 3 and 4, Table 1). However, as we attempted to fulfill these needs with a commercial software, it quickly became apparent that the other needs mentioned in Table 1 are equally important for personalized medicine to truly advance at our institution. For example, the commercial product we deployed adequately identified variants, but it was unable to transmit that information to the EHR. It also failed to retain interpretation information on variants from one sample to the next (when the same variant might be seen again). Further, it had limited ability to connect to ever‐changing external knowledge bases like OncoKB (Chakravarty et al., 2017); such connections entailed convincing the vendor to build the specific software modules. Finally, when we introduced an additional assay, namely pharmacogenomics (PGX) testing, basic interpretation of the data required combining genotypes at multiple loci in a gene into a single “star allele” diplotype for that gene. Even this basic requirement was entirely outside the purview of our installed commercial software and we had to look for other solutions including building a solution ourselves.
Given that developing new assays and partnering with external labs with novel assays was likely to be routine, what was required was an agile solution that does not need new software purchases at every new decision point. Failing to find commercial software that can satisfy all eight needs in Table 1 and still remain agile, we developed Flype: an informatics platform that allowed us to address all of the above requirements. Over time, Flype has grown as our hospital expanded our NGS services to include new gene panels and partnered with multiple commercial vendors.
Flype has been in operation at NorthShore for over 5 years. It is now used to support clinical reporting of interpreted NGS and PGX results to the EHR, with manual intervention required only in steps where human expertise is necessary. It is also used to transmit orders to and retrieve results from several commercial NGS labs.
In this article we describe the overall Flype architecture, which enables it stay agile while satisfying these diverse requirements. We describe the interactions between our custom software with other standard open source modules and how it is built so that a small team is able to build and maintain it. Flype's modular architecture means that it is configurable for use by other hospital systems which are interested in initiating PMed programs or those that have established PMed programs but are encountering some of the challenges mentioned above when attempting to ramp up their offering.
2. MATERIAL AND METHODS
2.1. Flype components
In the past 5 years at NorthShore, Flype has supported PGX testing on two different internal platforms, receiving data from several commercial laboratories, enabled rapid scale up of partnership programs with vendor laboratories as well as down‐scaling of other partnerships, incorporated tens of thousands of outside genetic tests including full EHR integration and has enabled the testing and launching of several in‐house lab‐developed NGS assays with custom software to support their data. The modules to accomplish all this can be divided into a user facing web portal and three distinct components on the backend: a relational database for storing data, a custom code base of bioinformatics pipelines and Concourse, a robust framework for connecting to a large number of external services.
2.1.1. Web portal
The most visible (user‐facing) component of Flype (Figure 1) is a web portal built using the Django framework (Django Project, n.d.) that uses an Apache web server to serve dynamic content. Flype authenticates users using either open standards such as lightweight directory access protocol (LDAP) for clinical use or Google Authentication for nonclinical use. This relieves Flype of the necessity to maintain a database of credentials and relieves our users of the need to remember yet another username and password. Once logged in, Flype's web interface allows authenticated users access to powerful bioinformatics workflows which they use to upload, import, analyze and interpret NGS or PGX data, sign‐out reports and perform audits of test performance. All this functionality is exemplified in greater depth among the descriptions of use cases below and satisfies requirements 3, 4, 7, and 8 listed in Table 1.
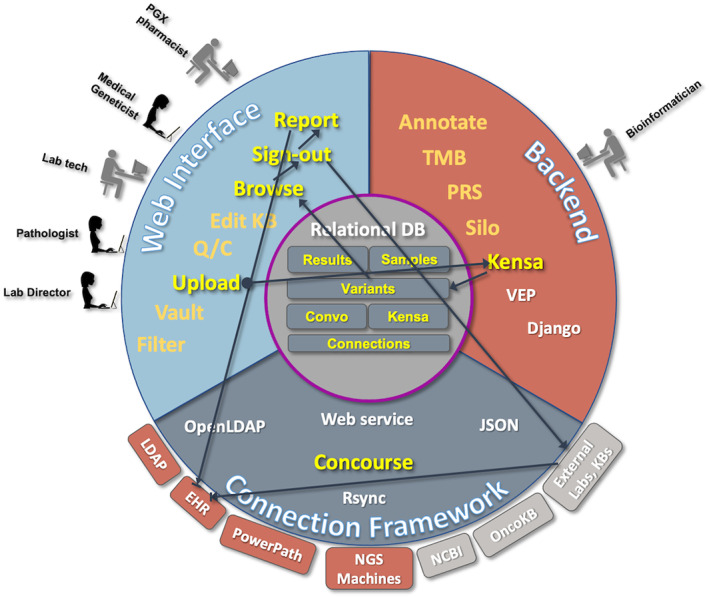
FIGURE 1.
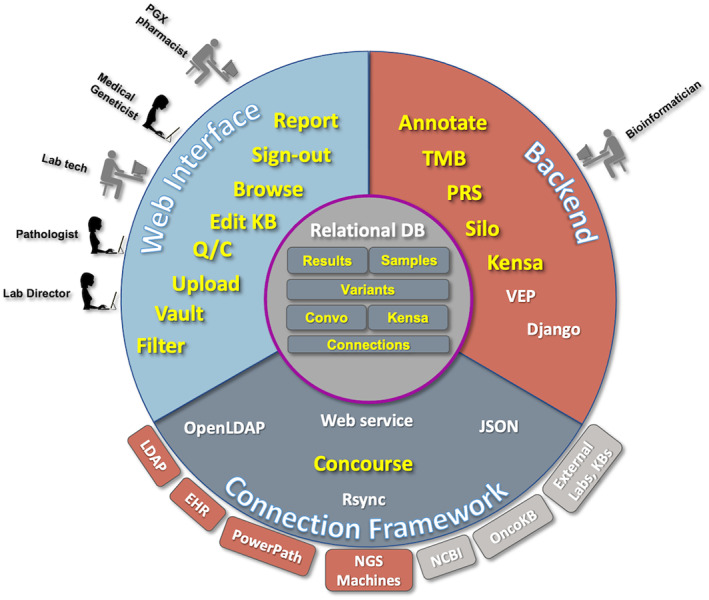
This figure illustrates the different components of the software and server environment which come together in Flype. The users see the Web Interface (top left), and use that to upload, analyze, and interpret NGS and PGX sample results. Expert pathologists and pharmacogenomicists also use the web interface to maintain the NGS Knowledge Base (Convo) and the PGX Knowledge Base (Kensa). On the Backend (top right), bioinformaticians manage code which computes tumor mutational burden (TMB), polygenic risk score (PRS), manages the Job Shop, provides a listing of files for the lab to manage (Vault), calculates copy number changes on certain sample types (Silo) and computes PGX diplotypes from genotype data uploaded by the lab (Kensa). New samples imported through the Vault are annotated using VEP, and results returned to the central Relational Database. The code to manage the bioinformatic scripts, VEP annotation and provide the web interface is served by a Django web framework hosted on a local server. The third component is Concourse, the Connection Framework (bottom) manages the connections and data retrieval from internal servers (rust) and external servers (gray) using web services, database calls, file transfers and other server functions. Data on each new sample, each new variant, and our knowledge bases for interpreting NGS and PGX assays are maintained within the central Relational Database. Modules developed for Flype are highlighted in yellow; open source applications and software packages are represented in white
2.1.2. Relational database
Flype uses a relational database (RDB) in the backend to keep track of patient samples and genomic variants identified in them. Flype uses the pyODBC module to connect to the RDB and currently supports the open‐source PostgreSQL RDB as well as Microsoft's SQL Server RDB. Other database platforms could be added relatively easily since most of Flype's interactions with the RDB use ANSI‐SQL compliant code. At NorthShore, compatibility with MS SQL Server allowed us to communicate with our Epic (Verona, WI) EHR and with SunQuest's PowerPath system (Sunquest Information System, Tucson, AZ), which our pathology lab uses for sample tracking. This illustrates how our strategy of leveraging open source standards has enabled Flype to satisfy requirements 1, 2, and 6 (Table 1).
The RDB comprises Flype's core concept of a variant repository (requirement 4, Table 1). Results from SNP‐based assays, such as some PGX genotyping arrays, are also converted into genomic coordinates so that similar tools can be used to annotate and interpret the clinical consequences of all variants. The RDB also stores knowledge bases (KB) that are internally developed (requirement 5, Table 1) including Convo (NGS KB) and Kensa (PGX KB). It also stores information about patient specimens and data about communications to external services.
2.1.3. Bioinformatics pipelines
Flype has the ability to annotate uploaded variants using widely available open source modules (requirement 3, Table 1) and comes with the ability to switch between them relatively easily. For example, we currently use the Ensembl variant effect predictor (VEP) (McLaren et al., 2016), but in the past have used snpEff (Cingolani et al., 2012). We migrated to VEP because it allows for better control of Human Genome Variation Society (HGVS) syntax, has better documentation about RefSeq release and provides overlapping dbSNP (Sherry et al., 2001) or COSMIC (Tate et al., 2019) records.
We maintain a local copy of VEP to avoid sending patient information to a remote server, guard against network disruptions and to better control the version of VEP used in annotation. We also maintain local copies of reference databases from other important sources like ClinVar (Landrum et al., 2020) and gnomAD (Karczewski et al., 2020) population frequencies so that variants in these databases are matched to variants in Flype using our own software. This separation allowed us to keep the core gene and RefSeq annotation stable while updating these other sources more frequently. For example, this allows Flype administrators to update ClinVar entries more frequently to highlight potentially actionable changes like a variant of uncertain significance (VUS) changing to Pathogenic. At NorthShore, we generate a report upon update summarizing the changes in variant classification and send that report to our center for medical genetics.
Other bioinformatics pipelines that are included in Flype solve one or more of the requirements enumerated earlier. These pipelines are: (a) Filtering: filters the variants output by the vendor‐provided Torrent Suite software (requirement 3, Table 1) to correct for typical artifacts we have observed. (b) Results dashboard: uses information like population frequency and ClinVar status to filter benign germline polymorphisms out when displaying somatic variants. This reduces the number of variants (requirement 5, Table 1) the Pathologists need to manually interpret before signing out a report. (c) Kensa: to use curated translation tables from PharmVar (Gaedigk, et al., 2019) and PharmGKB (Whirl‐Carrillo et al., 2012) for converting genotype results from our PGX panel to star allele diplotypes of genes (requirement 3, Table 1). Kensa also maintains a local KB of clinical interpretations of those diplotypes based on recommendations from CPIC (Caudle et al., 2017), from FDA labeling and from other guidelines. (d) SILO: to analyze changes in read depths for detecting gene amplifications in oncology samples (requirement 3, Table 1). (e) Convo: a KB to allow our pathologists to build and maintain a local library of clinical interpretations of different genetic variants in various tumors (requirement 5, Table 1). Since Convo is locally maintained within our RDB, it allows patient care to be systematically informed by NCCN guidelines and allows other data—which may not yet rise to the level of a guideline—to be used in suggesting the best‐informed clinical management for a patient. (f) Population frequencies: a pipeline to display the occurrence of a given variant in our patient population (requirement 4, Table 1). This allows us to identify patients that may be impacted by a new development in the KB and also to monitor for quality control whether any particular genotype is showing unexpectedly high or low levels of occurrence.
2.1.4. Concourse
This is Flype's powerful built‐in connection framework which enables it to connect to almost arbitrarily defined outside systems. Using open standards like web service calls and Javascript Online Notation (JSON) formatted data Flype connects to a wide array of external and internal systems (requirements 1, 2, and 6, Table 1) including: (a) NorthShore systems like our Epic EHR system (Verona, WI), SoftLab sample tracking system (SCC Soft Computer, Clearwater, FL), PowerPath (Sunquest Information System, Tucson, AZ) specimen tracking system, enterprise data warehouse system for accessing other clinical outcomes data and to our AD/LDAP server to authenticate user login credentials, (b) to external NGS labs, as described below, (c) to an external network of genetic counselors at Genome Medical Inc. (South San Francisco, CA), (d) external proprietary KBs like those at ActX (Seattle, WA) and PierianDx (Creve Coeur, MO) and (5) to external public domain KBs like OncoKB (Chakravarty et al., 2017), the IARC TP53 database (Bouaoun et al., 2016) and BRCAExchange (Cline et al., 2018).
Other connections with other potential partners are constantly being considered in this rapidly changing genomics world and Flype provides our organization with the unique ability to quickly launch tests and abandon those that prove unsatisfactory. The versatile connectivity of Concourse allowed Flype to help our institution streamline workflows in a surprising way during the recent COVID‐19 pandemic (Manuscript in preparation).
3. RESULTS
3.1. Flype use cases
Flype satisfies a number of uses, some of which are described below. Its open and modular architecture has enabled NorthShore to grow our PMed program by helping launch PGX testing (including switching between PGX platforms), enabling rapid scale up to incorporate over 10,000 genetic tests from external labs and helping launch several in‐house lab‐developed assays for somatic variants. Flype can also be used in a research environment, without connecting to an EHR, to help with sample analysis.
3.1.1. Supporting In‐house NGS tests
This workflow, illustrated in Figure 2, shows that all the requirements one through eight are important for a complete solution. NGS analysis begins when our Ion Torrent sequencing machines transfer the NGS data onto the scratch space of the Flype server after each run is completed. The data includes detailed wells files with raw base calls, BAM files with genome alignment of called bases and VCF files with variant calls. Additional calls such as those for FLT3 internal tandem duplications from a myeloid panel may also be included.
FIGURE 2.
The NGS Workflow is highlighted, illustrating sample flow from the internal NGS servers using rsync to allow new runs to be visible through Flype's Vault application. Extraction leads to entries in the variant database table, starting the annotation process, TMB calculation, and Silo copy number calculation, as appropriate. Once the bioinformatic calculations are complete, new samples are available on the web interface for browsing, analysis and sign‐out to EHR
The next step in the Flype NGS workflow happens when lab technologists import NGS runs using Flype's vault application. This application (Figure 3) allows specifying some ancillary data about a sequencing run, identifying patient samples, controls, and validation samples, then begin the import process. Flype's vault then extracts the associated BAMs, VCFs, and sample meta‐data to folders designated for permanent storage. Notably, massive amounts of data in the so‐called wells files are not transferred and would be purged as needed since it was determined that these files are not required to be stored permanently.
FIGURE 3.
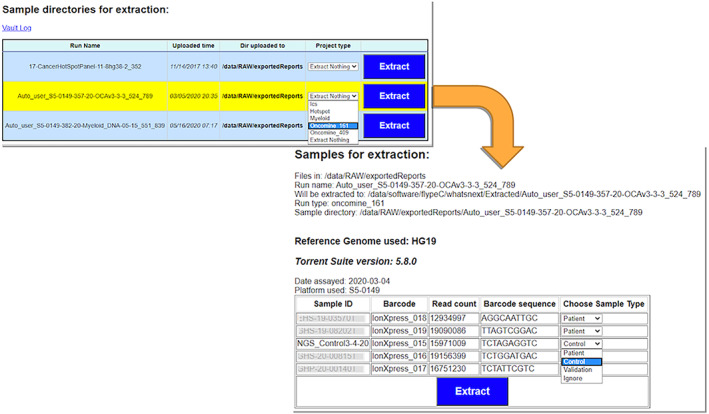
The Flype Vault sample flow illustrates importing a new run, choosing the type of run, and then identifying patient samples, controls, and validation before extracting the samples into Flype
Then vault analyzes the called variants in the VCF file (a) to filter out those which are likely to be sequencing artifacts (b) to normalize their representation to be the shortest variant moved left‐most on the genome and (c) to lift‐over (Zhao et al., 2014) to a genome version so that every variant in Flype is represented in both GRCh37 and GRCh38 versions of the human genome. All called variants are entered into our variant repository and annotated with VEP. Where appropriate, vault also applies our SILO algorithm (manuscript in preparation) which does a read‐depth analysis to identify gene amplifications. Finally, it does additional bioinformatic processing like identifying poorly covered regions and calculating Tumor Mutation Burden.
Sample import, moving of files and variant annotation through the vault can consume considerable server resources. To manage resources, Flype has a queueing system called Job Shop, to allow multiple users to independently start sample import without conflict. Job Shop has a configurable priority queueing system. For example, we set it so that smaller jobs, like pharmacogenomic tests or small gene panels, have higher priority and finish quickly. New NGS samples imported through the vault are displayed on Flype web pages, awaiting sample review and interpretation by the lab director or sign‐out pathologist (SP).
Flype currently supports the import and sign‐out of a 50 gene Cancer HotSpot panel, a 40 gene Myeloid panel and an expanded gene panel based on the NCI‐MATCH Trial Assay (Oncomine 161 and 409 panels). Additional NGS assays to detect cfDNA and an inherited cancer syndrome panel are also supported.
Flype provides the SP a dashboard (Figure 4) which lists relevant results of each sample along with custom links containing the gene and variant to external knowledge bases like OncoKB (Chakravarty et al., 2017), BRCAExchange (Cline et al., 2018), IARC TP53 database (Bouaoun et al., 2016) and Flype's Convo module. Variants are filtered to remove known germline polymorphisms based on population databases and also any variants in a lab‐maintained list of “frequently seen artifacts.” The listing for each variant also includes a link to view it in the genomic context using IGV.js, the web version of the Integrative Genome Viewer (IGV) (Robinson, Thorvaldsdóttir, Wenger, Zehir, & Mesirov, 2017). There is also a link to drill down to a full variant detail page with protein domain information (Jay & Brouwer, 2016) as well as additional information from external KBs like COSMIC (Tate et al., 2019), OncoKB (Chakravarty et al., 2017), AACR's GENIE project (AACR Project GENIE Consortium, 2017), RefSeq, gnomAD (Karczewski et al., 2020), ESP6500 and 1,000 Genomes. This variant detail page also links to Convo KB and to other NorthShore patients with this variant. Members of the pathology team without SP privileges can review variants for quality and add comments to them, mark a sample “Q/C complete” or enter variant interpretations, but only designated pathologists can sign out the final report to the EHR.
FIGURE 4.
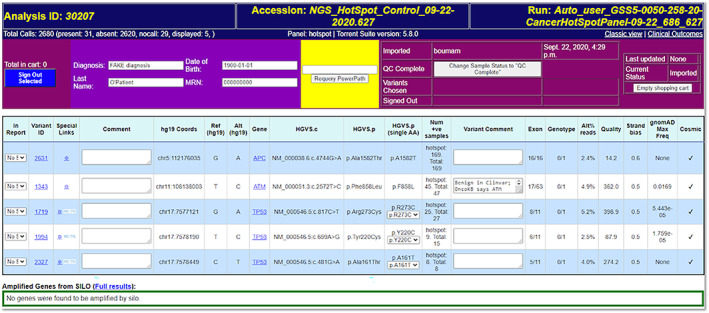
The Flype dashboard view of a 50‐gene cancer Hotspot sample is shown, with synonymous, benign, and common polymorphisms filtered to reduce the number of variants to evaluate in this example from 31 to 5. Laboratory technologists enter comments regarding sample Q/C, Pathologists enter comments regarding their interpretation of the variant and select from the drop down next to each variant whether it should be on the front page of the final report, in the VUS section or not be included in the final report. Dynamic links allow Pathologists to lookup that gene/variant combination at OncoKB (Chakravarty et al., 2017) or specialty databases. In addition, Pathologists can choose to use an aggregated variant description (such as “KRAS activating” or “TP53 inactivating”) instead of the individual variant where appropriate. These values are passed to the sign‐out page, so that aggregated interpretations in Convo can be brought into the report when appropriate
It is important to note that the results dashboard connects to an internal NorthShore system: PowerPath (Sunquest Information System, Tucson, AZ), the sample tracking system used by our Pathology department. Through this connection, Flype gathers patient demographics and some preliminary diagnosis information which may be edited by the SP. Using this diagnosis information, Flype identifies any existing relevant Convo interpretations and brings them into the report to be edited and included in the report. When the SP writes a brand‐new interpretation, it gets added to Convo and is available for the next sample in which the same variant with the same diagnosis may be encountered.
Using all this information, the SP uses a drop‐down next to each variant on the dashboard to assign it to be (a) included on the front page of the report, (b) included in the report as a VUS, or (c) not included in the report. The SP can also manually add findings from additional assays like findings from the ArcherDX (Boulder, CO) gene fusion assay.
Finally, Flype displays a PDF report using the SP's detailed interpretations of all front‐page variants and list of identified VUS as well as boiler plate information about the assay. Upon sign‐out, this PDF report is sent to the EHR. Discrete data elements from the report are also available but, at present, we do not transmit them to the EHR for tumor genomic testing. Once a report has been signed out, the PDF can be reviewed but cannot be changed; however, users with SP privileges can “unlock” a signed‐out sample to prepare an addendum or amendment.
3.1.2. Supporting In‐house Pharmacogenomics Tests
Pharmacogenomics is the application of PMed that promises to optimize drug therapy through knowledge of a patient's genome (Dunnenberger et al., 2016). The PGX workflow in Flype (Figure 5) follows a similar path to NGS and illustrates all eight of the requirements enumerated in Table 1. (a) The workflow starts when permissioned lab personnel use a page on Flype to upload the PGX data. (b) Next, Flype's Kensa (manuscript in preparation) module analyzes the PGX results using translation tables to determine the diplotype for each gene, (c) permissioned PGX experts in our pharmacy review the results and use the Kensa KB to match computed diplotypes against prescribing recommendations for each gene and finally, (d) they review the results and sign‐out, at which time Flype generates a PDF report of their recommendations.
FIGURE 5.
The workflow for in‐house PGX testing is highlighted: sample flow starting with a browser upload of genotype data, which is processed by the Kensa algorithm, inserting diplotypes and genotypes into the variant database for each sample, which generates new entries in the Flype sample view available for the pharmacogenomicist to browse. After review, the pharmacogenomicist signs out the sample, which generates a PDF report as well as a set of discrete data elements, both of which are sent to the EHR. The sign‐out button also generates an encrypted VCF which is sent to an external knowledge base partner
Illustrating the power of Concourse, Flype's connection framework, sign‐out causes several out‐going messages: (a) the PDF report is transmitted to the EHR, (b) discrete data about the patient's genotypes and gene diplotypes are sent to the EHR, (c) an email is sent with the PDF report to a specified distribution list to enable some downstream aspects of the workflow and finally, (d) a VCF formatted version of the variants is created, encrypted and sent via secure file transfer to ActX (Seattle, WA), our knowledge base partner. When a physician writes a prescription through our EHR, this triggers a communication with the ActX rules engine to check whether there is any concern about the script based on the patient's PGX results; some of those concerns trigger an interruptive warning (Wake, Ilbawi, Dunnenberger, & Hulick, 2019) to complete the clinical decision support loop.
Only authorized individuals can submit a patient report to the EHR, but any user can leverage the Kensa KB to create an anonymous report of prescribing recommendations by specifying diplotypes of various pharmacogenes on Flype. The Kensa KB is organized by genes, diplotypes and drugs metabolized by those genes. It is based upon standards developed by CPIC (Caudle et al., 2017) and PharmVar (Gaedigk, et al., 2019) and is maintained by permissioned members of the PGX team through Flype's Kensa page.
3.1.3. Supporting dramatic scale‐up: DNA10K
Flype played a central role (Figure 6) in our recent DNA10K initiative (NorthShore and Color complete delivery of clinical genomics, 2020), where 10,000 of our patients were offered a free test from Color Genomics Inc. (Burlingame, CA) which included (a) PGX, (b) screening for hereditary cancer risk and (c) screening for hereditary cardiovascular risk. This workflow requires items 1, 2, 4, 6, 7, and 8 in Table 1.
FIGURE 6.
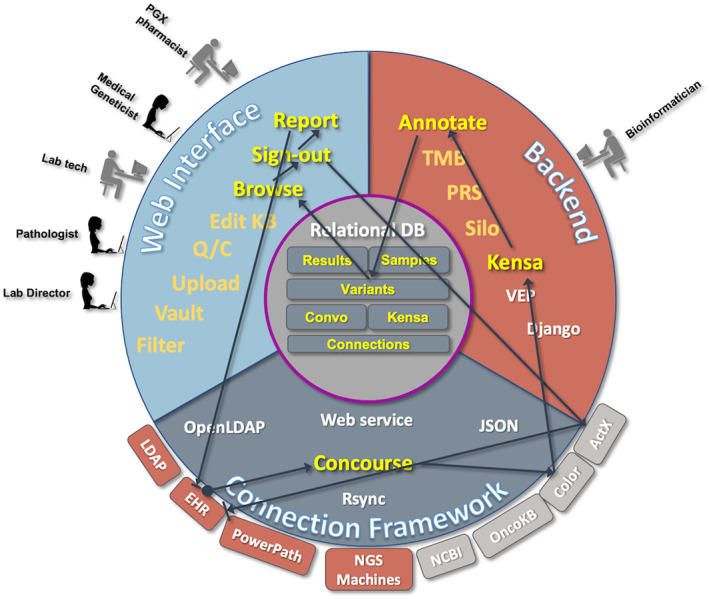
Workflow for NorthShore's DNA10K project involving partnership with Color Genomics Inc. (Burlingame, CA)
The workflow starts when an order for DNA10K is submitted by a clinician through the EHR. Using Concourse, Flype receives this order and transmits it out to Color. Next, a blood specimen is shipped from the clinic to Color and when results are signed out at Color, Flype retrieves the results through a web services call. Color provides the results as discrete data as well as PDF reports; both of these are downloaded by Flype and transmitted to the EHR. Flype also repackages the PGX data into a VCF and sends that file to ActX (Seattle, WA), so that Color testing has the same level of clinical decision support as in‐house PGX testing.
Of 10,691 patients tested through this partnership, 802 (7%) were positive for one or more pathogenic variants in the Hereditary Cancer panel and 107 (1%) for one or more pathogenic variant in the Hereditary Heart Health test (Manuscript submitted).
Color also provides continuous updates to patient results including reclassification of variants for example, from VUS to Likely pathogenic. Flype retrieves these updates from Color and transmits them to the EHR (requirement 8, Table 1).
The DNA10K PGX test combined with our in‐house PGX test now drive PGX prescribing interactions through our EHR for over 12,500 patients, representing a dramatic scale‐up of our PGX program. An assessment of the changes to prescribing patterns, based upon these interruptive alerts and other observations from the implementation of PGX at NorthShore, will be reported in a separate communication.
3.1.4. Supporting consumer genomics: Polygenic risk scores
Using a recently developed algorithm for calculating the genetic risk of developing prostate cancer during a man's lifetime (Chen et al., 2016) NorthShore offers the Prostate Cancer Genetic Risk Score (GRS), a risk score test for men in most of the United States. To offer this nationwide (NorthShore is a community hospital whose network is restricted to Northern Illinois), we partnered with Helix (San Mateo, CA), a direct to consumer genomic testing company and with Genome Medical (South San Francisco, CA), which has licensed physicians in each state to provide return of results and genetic counseling where needed.
The process begins when a consumer anywhere in the country purchases a Prostate Cancer GRS test through the Helix web site and completes a simple questionnaire about family history. Flype polls the Helix servers through web services and identifies the purchase and transmits the order to NorthShore's customer relationship management server, which contacts the customer via encrypted e‐mail to obtain their consent for testing and then transmits this consent back to Helix. When the customer sends their specimen to Helix and analysis is complete, Flype retrieves the data from Helix through their API, calculates the customer's lifetime risk of prostate cancer. Next, our Medical Geneticist logs into Flype to review the results and produce, and sign out a PDF report. Flype then transmits the discrete data as well as the PDF to Genome Medical who return results to the customer.
With the use of an internal bioinformatic pipeline for calculating the prostate cancer GRS based on genetic results, with external connections to two external partners in Helix and Genome Medical and connection to NorthShore's internal marketing server, this complicated pipeline illustrates the extraordinary range of Concourse, Flype's connectivity framework.
3.1.5. Supporting reporting tools
Statistics of aggregated patient results in the variant repository, including PGX results (Figure 7), can be quickly visualized for laboratory quality review as well as for trend analysis and usage reporting. Pie charts and tables summarize the results for each gene or SNP for PGX data and they also provide details of patients with specific genetic polymorphism.
FIGURE 7.
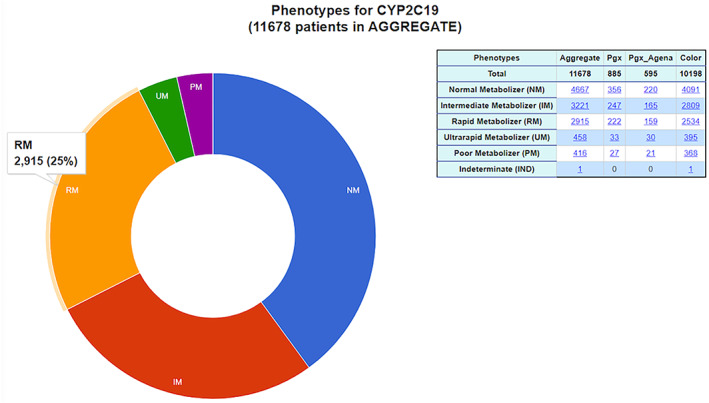
One of Flype's reporting tools are illustrated in this diagram, which shows representative Phenotype data for CYP2C19. The reporting tools also generate genotype‐ and diplotype‐level views of PGX results and can illustrate performance of individual SNPs or alternate assays such as copy number which are useful for evaluating assay performance. Data are also segregated by assay type
Examples of other reporting tools include comparing multiple NGS samples side‐by‐side (useful for laboratory quality review of replicates) and trend lines for Silo over time (to monitor for any changes related to new reagent batches). Flype also helps provide performance statistics and other metrics used during our Molecular Pathology laboratory's periodic audits by accrediting organizations.
3.1.6. Flype for pharmacogenomics collaborations
We also host an instance of Flype outside our firewall for sharing the Kensa KB with other collaborating institutions. Those interested in such collaboration can contact the authors.
4. DISCUSSION
4.1. Future directions
Flype is continuously being extended to support several new features. We are building a clinical outcomes module that will allow us to link genetic changes recorded in Flype with outcomes data pulled from the EHR. Using functionality of this module, we have done a retrospective analysis of outcomes in patients of metastatic castrate‐resistant prostate cancer (mCRPC; manuscript in preparation). We expect this module to help with clinical research use cases like cohort generation and quality control use cases like retrospective utility analysis. It should also be helpful for management of individual patients by locating previous patients that match a profile and showing their previous outcomes.
We are also adding a module to track sample status in the NGS sequencing lab. This module will take over the NGS‐specific elements of sample tracking away from a Laboratory Information Management System (LIMS), which is usually too generic and not efficiently usable by NGS. Other additions to Flype include integrating newer bioinformatics algorithms like those for detecting micro‐satellite instability. We are exploring ways to use APIs to external organizations like CPIC (Caudle et al., 2017) and RxNorm (Nelson, Zeng, Kilbourne, Powell, & Moore, 2011) for updates to our internal KBs.
While pulling down data from external KBs is indispensable for signing out patients, we are constantly reminded that these KBs and published literature are but a minute part of the experience of the medical community. It would be very desirable to expand and learn from the clinical outcomes of the many patients that are not participating in any research protocol. We hope that in the future, issues related to data governance, patient privacy and data consistency can be addressed to allow clinical outcomes from many different organizations can be shared so that real time evaluation of the performance of various interventions can be done. The immediate goal of Flype's clinical outcomes module is to accomplish this within our organization but by sharing Flype we hope to contribute to this sharing, collaborative future. Our mCRPC study mentioned above has taught us that this can be especially useful in managing oncology patients.
When we initiated Flype almost 5 years ago, the field appeared to be in a flux and we expected that by about now, it would have settled. We expected that the clinical use cases and workflows would be well understood and streamlined. Instead, what we find is that the use cases today are even more diverse than they were then. Even so, some general trends are strongly apparent.
First, the dichotomy between clinically actionable findings and all other genomic findings is getting weaker with time. It is a common occurrence for patients to want to discuss with their Medical Geneticist their report from a consumer genomics company even though most of the data in those reports is not clinically actionable today. However, it is very clear that over time the actionable portion will grow. Therefore, for the foreseeable future, it is important to have a clearing house for holding genomic data, that is, a non‐EHR destination which is capable for holding genomic findings considered non‐actionable at the moment.
Second, the $1,000 genome is still far away for clinical purposes. Even though it will be achieved in purely NGS technology terms, it is very unclear how a whole genome would be even represented, let alone utilized, in clinical care. Interfacing with multiple KBs, providing clinical decision support in the EHR and periodic update of variant status for all patients would be valuable. All three of these capabilities are bundled in with Flype and will be put to the test in the future as more and more genomic data are marked up as clinically relevant.
Third, given the economics of genome sequencing technology, it appears likely that commercial NGS labs will remain a strong presence for the foreseeable future. With newer techniques like cell free DNA analysis (so‐called liquid biopsies) making their appearance routinely, the need to interface with multiple labs will remain important. At present, most labs only report clinically actionable findings. While this is understandable for a signed‐out report which can only contain a reasonably small number of variants, we expect that labs will start reporting out (perhaps through an API layer) all variants that are reliably detected, whether or not those variants are clinically actionable at this moment. Making this a routine practice should allow appropriately equipped organizations to track the status of these variants and provide a more robust integration of genomic results into clinical practice.
Writing modular, extensible code as a part of an ecosystem like Flype will allow organizations to respond to changing data standard formats (JSON, HL7, FIHR) and treat them as just another third‐party connection to snap on to the existing framework. The plug‐and‐play aspect of Flype's Concourse module allows us to contemplate this and add or remove functionality in an agile paradigm.
5. SUMMARY
We present Flype, a web‐based bioinformatics platform for use on an organization's intranet. Flype allows molecular pathologists, laboratory technologists, pharmacists, oncologists and medical geneticists to review, interpret and sign out NGS tests, PGX tests and polygenic risk score tests. It supports extraordinary connectivity to other servers inside and outside the organization including, most prominently, getting orders from, and sending results to the EHR. Flype is written with open source software and can easily be maintained and customized by a small group. The source code for Flype is available upon request to the authors.
DECLARATION OF INTERESTS
All authors are employees of NorthShore University HealthSystem.
ACKNOWLEDGMENTS
The development of Flype has benefited from the continuous feedback from Drs. Mir B. Alikhan, Kathy A. Mangold, and Nora E. Joseph and from Mike Akroush, MS. We also acknowledge technical support from Charlie Cron, Eric Cron and the rest of the Unix team in our Health Information Technology department. The authors gratefully acknowledge the support from multiple anonymous Foundations.
Helseth DL Jr, Gulukota K, Miller N, et al. Flype: Software for enabling personalized medicine. Am J Med Genet Part C. 2021;187C:37–47. 10.1002/ajmg.c.31867
Donald L. Helseth, Jr and Kamalakar Gulukota contributed equally to this work
Contributor Information
Donald L. Helseth, Jr, Email: lhelseth@northshore.org.
Kamalakar Gulukota, Email: kgulukota@northshore.org.
DATA AVAILABILITY STATEMENT
The source code is available upon request to the authors.
REFERENCES
- AACR Project GENIE Consortium . (2017). AACR project GENIE: Powering precision medicine through an international consortium. Cancer Discovery, 7(8), 818–831. 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun, L. , Sonkin, D. , Ardin, M. , Hollstein, M. , Byrnes, G. , Zavadil, J. , & Olivier, M. (2016). TP53 variations in human cancers: New lessons from the IARC TP53 database and genomics data. Human Mutation, 37(9), 865–876. 10.1002/humu.23035 [DOI] [PubMed] [Google Scholar]
- Caudle, K. E. , Dunnenberger, H. M. , Freimuth, R. R. , Peterson, J. F. , Burlison, J. D. , Whirl‐Carrillo, M. , … Hoffman, J. M. (2017). Standardizing terms for clinical pharmacogenetic test results: Consensus terms from the clinical pharmacogenetics implementation consortium (CPIC). Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19(2), 215–223. 10.1038/gim.2016.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty, D. , Gao, J. , Phillips, S. M. , Kundra, R. , Zhang, H. , Wang, J. , … Schultz, N. (2017). OncoKB: A precision oncology Knowledge Base. JCO Precision Oncology. 10.1200/PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, H. , Liu, X. , Brendler, C. B. , Ankerst, D. P. , Leach, R. J. , Goodman, P. J. , … Xu, J. (2016). Adding genetic risk score to family history identifies twice as many high‐risk men for prostate cancer: Results from the prostate cancer prevention trial. The Prostate, 76(12), 1120–1129. 10.1002/pros.23200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, l. , Coon, M. , Nguyen, T. , Wang, L. , … Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso‐2; iso‐3. Fly, 6(2), 80–92. 10.4161/fly.19695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline, M. S. , Liao, R. G. , Parsons, M. T. , Paten, B. , Alquaddoomi, F. , Antoniou, A. , … Spurdle, A. B. (2018). BRCA challenge: BRCA exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genetics, 14(12), e1007752. 10.1371/journal.pgen.1007752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Django project . (n.d.). Django Project. Retrieved from https://www.djangoproject.com/
- Dunnenberger, H. M. , Biszewski, M. , Bell, G. C. , Sereika, A. , May, H. , Johnson, S. G. , … Khandekar, J. (2016). Implementation of a multidisciplinary pharmacogenomics clinic in a community health system. American Journal of Health‐System Pharmacy: AJHP: Official Journal of the American Society of Health‐System Pharmacists, 73(23), 1956–1966. 10.2146/ajhp160072 [DOI] [PubMed] [Google Scholar]
- Feero, W. G. , Guttmacher, A. E. , & Collins, F. S. (2010). Genomic medicine‐ ‐an updated primer. The New England Journal of Medicine, 362(21), 2001–2011. 10.1056/NEJMra0907175 [DOI] [PubMed] [Google Scholar]
- Gaedigk, A. , Sangkuhl, K. , Whirl‐Carrillo, M. , Twist, G. P. , Klein, T. E. , Miller, N. A. , & PharmVar Steering Committee . (2019). The evolution of PharmVar. Clinical Pharmacology and Therapeutics, 105(1), 29–32. 10.1002/cpt.1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay, J. J. , & Brouwer, C. (2016). Lollipops in the clinic: Information dense mutation plots for precision medicine. PLoS One, 11(8), e0160519. 10.1371/journal.pone.0160519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum, M. J. , Chitipiralla, S. , Brown, G. R. , Chen, C. , Gu, B. , Hart, J. , … Kattman, B. L. (2020). ClinVar: Improvements to accessing data. Nucleic Acids Research, 48(D1), D835–D844. 10.1093/nar/gkz972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren, W. , Gil, L. , Hunt, S. E. , Riat, H. S. , Ritchie, G. R. , Thormann, A. , … Cunningham, F. (2016). The Ensembl variant effect predictor. Genome Biology, 17(1), 122. 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, S. J. , Zeng, K. , Kilbourne, J. , Powell, T. , & Moore, R. (2011). Normalized names for clinical drugs: RxNorm at 6 years. Journal of the American Medical Informatics Association: JAMIA, 18(4), 441–448. 10.1136/amiajnl-2011-000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NorthShore and Color complete delivery of clinical genomics in routine care to 10,000 patients in the largest U.S. program to date, with plans to expand in 2020. (2020). PR Newswire .https://www.prnewswire.com/news-releases/northshore-and-color-complete-delivery-of-clinical-genomics-in-routine-care-to-10-000-patients-in-the-largest-us-program-to-date-with-plans-to-expand-in-2020-300985704.html
- Robinson, J. T. , Thorvaldsdóttir, H. , Wenger, A. M. , Zehir, A. , & Mesirov, J. P. (2017). Variant review with the integrative genomics viewer. Cancer Research, 77(21), e31–e34. 10.1158/0008-5472.CAN-17-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry, S. T. , Ward, M. H. , Kholodov, M. , Baker, J. , Phan, L. , Smigielski, E. M. , & Sirotkin, K. (2001). dbSNP: The NCBI database of genetic variation. Nucleic Acids Research, 29(1), 308–311. 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate, J. G. , Bamford, S. , Jubb, H. C. , Sondka, Z. , Beare, D. M. , Bindal, N. , … Forbes, S. A. (2019). COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Research, 47(D1), D941–D947. 10.1093/nar/gky1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake, D. T. , Ilbawi, N. , Dunnenberger, H. M. , & Hulick, P. J. (2019). Pharmacogenomics: Prescribing precisely. The Medical Clinics of North America, 103(6), 977–990. 10.1016/j.mcna.2019.07.002 [DOI] [PubMed] [Google Scholar]
- Whirl‐Carrillo, M. , McDonagh, E. M. , Hebert, J. M. , Gong, L. , Sangkuhl, K. , Thorn, C. F. , … Klein, T. E. (2012). Pharmacogenomics knowledge for personalized medicine. Clinical Pharmacology and Therapeutics, 92(4), 414–417. 10.1038/clpt.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandesh, Z. , Ghazisaeedi, M. , Devarakonda, M. V. , & Haghighi, M. S. (2019). Legal framework for health cloud: A systematic review. International Journal of Medical Informatics, 132, 103953. 10.1016/j.ijmedinf.2019.103953 [DOI] [PubMed] [Google Scholar]
- Zhao, H. , Sun, Z. , Wang, J. , Huang, H. , Kocher, J. P. , & Wang, L. (2014). CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinformatics (Oxford, England), 30(7), 1006–1007. 10.1093/bioinformatics/btt730 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The source code is available upon request to the authors.