

Metabotropic glutamate receptor 7 (mGlu7) is an inhibitory heterotrimeric G-protein-coupled receptor that modulates neurotransmitter release and synaptic plasticity at presynaptic terminals in the mammalian central nervous system. Recent studies have shown that rare mutations in glutamate receptors and synaptic scaffold proteins are associated with neurodevelopmental disorders (NDDs). However, the role of presynaptic mGlu7 in the pathogenesis of NDDs remains largely unknown.
Keywords: degradation, GRM7, neurodevelopmental disorder, point mutation
Abstract
Metabotropic glutamate receptor 7 (mGlu7) is an inhibitory heterotrimeric G-protein-coupled receptor that modulates neurotransmitter release and synaptic plasticity at presynaptic terminals in the mammalian central nervous system. Recent studies have shown that rare mutations in glutamate receptors and synaptic scaffold proteins are associated with neurodevelopmental disorders (NDDs). However, the role of presynaptic mGlu7 in the pathogenesis of NDDs remains largely unknown. Recent whole-exome sequencing (WES) studies in families with NDDs have revealed that several missense mutations (c.1865G>A:p.R622Q; c.461T>C:p.I154T; c.1972C>T:p.R658W and c.2024C>A:p.T675K) or a nonsense mutation (c.1757G>A:p.W586X) in the GRM7 gene may be linked to NDDs. In the present study, we investigated the mechanistic links between GRM7 point mutations and NDD pathology. We find that the pathogenic GRM7 I154T and R658W/T675K mutations lead to the degradation of the mGlu7 protein. In particular, the GRM7 R658W/T675K mutation results in a lack of surface mGlu7 expression in heterologous cells and cultured neurons isolated from male and female rat embryos. We demonstrate that the expression of mGlu7 variants or exposure to mGlu7 antagonists impairs axon outgrowth through the mitogen-activated protein kinase (MAPK)-cAMP-protein kinase A (PKA) signaling pathway during early neuronal development, which subsequently leads to a decrease in the number of presynaptic terminals in mature neurons. Treatment with an mGlu7 agonist restores the pathologic phenotypes caused by mGlu7 I154T but not by mGlu7 R658W/T675K because of its lack of neuronal surface expression. These findings provide evidence that stable neuronal surface expression of mGlu7 is essential for neural development and that mGlu7 is a promising therapeutic target for NDDs.
SIGNIFICANCE STATEMENT Neurodevelopmental disorders (NDDs) affect brain development and function by multiple etiologies. Metabotropic glutamate receptor 7 (mGlu7) is a receptor that controls excitatory neurotransmission and synaptic plasticity. Since accumulating evidence indicates that the GRM7 gene locus is associated with NDD risk, we analyzed the functional effects of human GRM7 variants identified in patients with NDDs. We demonstrate that stable neuronal surface expression of mGlu7 is essential for axon outgrowth and presynaptic terminal development in neurons. We found that mitogen-activated protein kinase (MAPK)-cAMP-protein kinase A (PKA) signaling and subsequent cytoskeletal dynamics are defective because of the degradation of mGlu7 variants. Finally, we show that the defects caused by mGlu7 I154T can be reversed by agonists, providing the rationale for proposing mGlu7 as a potential therapeutic target for NDDs.
Introduction
Neurodevelopmental disorders (NDDs) are a group of disorders that manifest a wide range of neurologic and psychiatric symptoms beginning in the development period. According to the Diagnostic and Statistical Manual of Mental Disorders (DSM-V), NDDs include autism spectrum disorders (ASDs), intellectual disabilities (IDs), attention-deficit hyperactivity disorders (ADHDs), specific learning disorders, communication disorders, and motor disorders (American Psychiatric Association, 2013).
Metabotropic glutamate (mGlu) receptors are G-protein-coupled receptors that modulate neurotransmission and synaptic plasticity throughout the central nervous system. Of the eight mGlu receptors, mGlu receptor 7 (mGlu7) is coupled with inhibitory Gαi/o protein and localized primarily in the presynaptic active zone (Okamoto et al., 1994; Shigemoto et al., 1996; Schoepp, 2001; Dalezios et al., 2002). It has been shown that stable surface expression of mGlu7, which is regulated by posttranslational modifications and receptor-scaffold protein interactions, is critical for receptor function and synaptic plasticity (Pelkey et al., 2005; Suh et al., 2008, 2013; Choi et al., 2016; Lee et al., 2019). Recently, it was found that mGlu7 mRNA levels decreased in the P8 cerebral cortex of X-linked methyl-CpG binding protein 2 (Mecp2)-knock-out mice, a model of human Rett syndrome (RTT), that exhibit developmental regression (Bedogni et al., 2016). In particular, expression levels of mGlu7 protein were also reduced in the postmortem motor cortex of RTT patients (Gogliotti et al., 2017), suggesting a key role for the stable expression of mGlu7 in the pathogenesis of NDDs (Palazzo et al., 2016; Fisher et al., 2018a).
Several genetic association studies have identified human GRM7 as a potential NDD risk locus (Elia et al., 2011; Gai et al., 2012; Park et al., 2013; Yang and Pan, 2013; Liu et al., 2015; Noroozi et al., 2016). These studies have revealed inherited or de novo point mutations or deletions in GRM7 introns and/or exons in cohorts of ASD or ADHD patients. In addition, a de novo missense mutation (c.1865G>A:p.R622Q) was reported to be associated with ASD on the basis of large-scale whole-exome sequencing (WES) studies in families with ASD (Sanders et al., 2012; Iossifov et al., 2014). A recent WES study on consanguineous families identified GRM7 as the candidate gene for the highest risk of NDDs, including DD/ID and brain malformations (Charng et al., 2016). This study identified a homozygous missense mutation (c.461T>C:p.I154T) from two affected siblings with DDs/IDs, seizures, hypotonia, and brain atrophy. The compound heterozygous missense mutations (c.1972C>T:p.R658W and c.2024C>A:p.T675K) was also identified in another sibling with similar clinical features (Charng et al., 2016). In a different set of study, a homozygous nonsense mutation (c.1757G>A:p.W586X, in which X designates a translation termination codon) was identified in families with NDDs (Reuter et al., 2017).
In this study, we investigated the mechanism by which human GRM7 variants carrying mutations in protein-coding sequences lead to the pathologic phenotypes observed in NDD patients. Specifically, we characterized the function of the GRM7 variants identified from the existing WES literature for NDD patients (Table 1; Sanders et al., 2012; Iossifov et al., 2014; Charng et al., 2016; Reuter et al., 2017). When we expressed human GRM7 variants in heterologous cells and rat primary cultured neurons, we found a profound reduction in the protein expression of mGlu7 variants. The instability of mGlu7 variant proteins is caused by protein degradation through the proteasomal or autophagosomal-lysosomal degradation pathway. We show that the GRM7 variants cause a severe impairment in axon outgrowth during early neuronal development, which subsequently leads to a deficit in the number of presynaptic terminals in mature neurons. We discovered that the mitogen-activated protein kinase (MAPK)-cAMP-protein kinase A (PKA) pathway is perturbed by the GRM7 variants. Of particular importance, we found that the deficits in axon outgrowth and presynaptic terminal development induced by mGlu7 I154T were restored by treatment with an mGlu7 agonist during early development. Thus, our study provides mechanistic insight into the development of NDDs by the GRM7 variants and suggests mGlu7 as a potential therapeutic target for NDD treatment.
Table 1.
Pathogenic human GRM7 variants and clinical features
| Nucleotide change | Amino acid substitution | Clinical features of patients | References |
|---|---|---|---|
| c.1865G>A | p.Arg622Gln | ASD | Sanders et al. (2012), Iossifov et al. (2014) |
| c.461T>C | p.Ile154Thr | DD, hypotonia, seizures, brain malformation | Charng et al. (2016) |
| c.1972C>T; c.2024C>A | p.Arg658Trp; p.Thr675Lys | DD, ID, hypotonia, seizures, hypomyelination, brain atrophy | Charng et al. (2016) |
| c.1757G>A | p.Trp586X | ID, seizures, limb hypertonia, microcephaly, cerebral atrophy, leukodystrophy | Reuter et al. (2017) |
ASD, autism spectrum disorder; DD, developmental delay; ID, intellectual disability.
Materials and Methods
Plasmid DNA constructs and cloning
We generated a human wild-type (WT) GRM7 cDNA plasmid from rat WT Grm7 cDNA (GenBank accession No. NM_000844 and NM_031040) by substituting four amino acid mismatches in the coding sequence using site-directed mutagenesis. The following oligonucleotide primers were used: H454N-forward (F), 5′-gtatatccgcAatgttaacttcaatgg-3′ and H454N-reverse (R), 5′-ccattgaagttaacatTgcggatatac-3′; T488S-F, 5′-caacaaacacaaGcaaccctggtta-3′ and T488S-R, 5′-taaccagggttgCttgtgtttgttg-3′; S520A-F, 5′-gagagatcccaGcctctgtgtgtac-3′ and S520A-R, 5′-gtacacacagaggCtgggatctctc-3′; N578D-F, 5′-ctggctgtcagGaTatcccaatcatc-3′ and N578D-R, 5′-gatgattgggatAtCctgacagccag-3′ (the mutated nucleotides are indicated by capital letters). Using human WT GRM7 cDNA as a template, we generated pathogenic mutants by site-directed mutagenesis using the following oligonucleotide primers: I154T-F, 5′-tagttggagtgaCtggggcttcggg-3′ and I154T-R, 5′-cccgaagccccaGtcactccaacta-3′; W586X-F, 5′-tcaaactggagtAgcactccccctg-3′ and W586X-R, 5′-cagggggagtgcTactccagtttga-3′; R622Q-F, 5′-cacccattgtcCAggcatctgggcg-3′ and R622Q-R, 5′-cgcccagatgccTGgacaatgggtg-3′; R658W-F, 5′-tgtgttctttcTgGcgtgtcttctt-3′ and R658W-R, 5′-aagaagacacgCcAgaaagaacaca-3′; T675K-F, 5′-ctgcccttttaaAGaagaccaatcg-3′ and T675K-R, 5′-cgattggtcttCTttaaaagggcag-3′. The PCRs were performed using Phusion DNA polymerase (catalog #M0530, New England Biolabs) according to the manufacturer's instructions.
Antibodies and reagents
The antibodies used in this study were obtained from the following commercial sources: anti-mGluR7a (RRID:AB_310459), anti-VGLUT1 (RRID:AB_2301751; Millipore), anti-VGAT (RRID:AB_887873, Synaptic Systems), anti-α-tubulin (RRID:AB_477583, Sigma-Aldrich), anti-LC3B (RRID:AB_881433), anti-Na+/K+ ATPase α1 (RRID:AB_306023; Abcam), horseradish peroxidase (HRP)-conjugated anti-rabbit IgG (RRID:AB_2313567, Jackson ImmunoResearch), anti-SMI312 (RRID:AB_2566782), anti-β-III tubulin (TUJ1, RRID:AB_2313773; BioLegend), anti-Ub (RRID:AB_628423, Santa Cruz Biotechnology), anti-GFP (RRID:AB_221569), anti-transferrin receptor (TfR; RRID:AB_86623), HRP-conjugated anti-mouse IgG (RRID:AB_2536527), Alexa Fluor 488-conjugated anti-mouse or rabbit IgG (RRID:AB_2534088 or RRID:AB_2576217), Alexa Fluor 568-conjugated anti-mouse or rabbit IgG (RRID:AB_144696 or RRID:AB_10563566), Alexa Fluor 488 phalloidin (RRID:AB_2315147; Thermo Fisher Scientific). Anti-c-myc (9E10) antibody was generated from a 9E10 clone (RRID:CVCL_G671, ATCC) in our laboratory. The following reagents were purchased from commercial sources: endoglycosidase H (Endo Hf, catalog #P0703S), peptide-N-glycosidase F (PNGase F, catalog #P0704S; New England Biolabs), MG132 (catalog #1748), chloroquine (catalog #4109), L-AP4 (catalog #0103), AMN082 (catalog #2385), MSOP (catalog #66515-29-5), MMPIP (catalog #2963), PD98059 (catalog #1213), PP2 (catalog #1407; Tocris Bioscience), bafilomycin-A1 (catalog #B1793), U73122 (catalog #U6756), nocodazole (catalog #M1404), paclitaxel (catalog #T7402), latrunculin A (catalog #L5163), forskolin (catalog #F3917), PMA (catalog #P1585), rolipram (catalog #R6520), FK506 (catalog #F4679), pertussis toxin (catalog #P2980; Sigma-Aldrich), H89 (catalog #371963), Go6976 (catalog #365250), dibutyryl-cAMP (db-cAMP, catalog #28745), KN93 (catalog #422708; Calbiochem), LY294002 (catalog #9901, Cell Signaling Technology), acridine orange (AO)/propidium iodide (PI) staining solution (catalog #CS2-0106, Nexcelom Bioscience).
Primary cortical or hippocampal neuron culture
All animal experiments were conducted in accordance with the guidelines of the Seoul National University Institutional Animal Care and Use Committee (protocol no. SNU-161222-2-4). Pregnant female Sprague Dawley rats on gestational day 18 were purchased from a commercial source (ORIENT BIO). Day 18 embryos of either sex were collected by cesarian section and decapitated. The hippocampi or cortices of the embryos were isolated in dissecting solution [HBSS (catalog #14170-161, Thermo Fisher Scientific) with 10 mm HEPES (catalog #15630-080, Thermo Fisher Scientific) and penicillin-streptomycin (catalog #15070-063, Thermo Fisher Scientific)]. The dissected tissues were incubated for 12 min in dissecting solution with 0.05% trypsin (catalog #T1005, Sigma-Aldrich) and 0.157 mg/ml deoxyribonuclease I (catalog #D5025, Sigma-Aldrich) at 37°C. The trypsin-treated tissues were dissociated by trituration using a fire-polished Pasteur pipette. The triturated cells were plated on poly-D-lysine-coated culture plates in serum-free Neurobasal media (catalog #21103-049, Thermo Fisher Scientific) with B-27 supplement (catalog #17504-044, Thermo Fisher Scientific) and 1% L-glutamine (catalog #G7513, Sigma-Aldrich), in which they were maintained before use. The number of days in vitro (DIV) varied depending on the assay as detailed below.
Western blotting
Primary cultured neurons or transfected HEK 293T cells were lysed in TNE lysis buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, and 0.1% SDS) with protease and phosphatase inhibitor cocktails (catalog #11873580001, Roche; catalog #P3200-005, GenDEPOT). After the lysates were centrifuged at 20,000 × g for 15 min at 4°C, the insoluble materials were removed. The supernatants were then mixed with 6× Laemmli SDS sample buffer and denatured by incubating at 37°C for 20 min. The samples were resolved by SDS-PAGE and transferred onto PVDF membranes (catalog #IPVH00010, Millipore). The membrane was blocked in Tris-buffered saline (TBS) with 5% nonfat skim milk and 0.1% Tween 20 (catalog #T1003, Anatrace) at room temperature (RT) for 1 h. After blocking, the membrane was incubated overnight with primary antibodies at 4°C. After several washes with TBS containing 0.1% Tween 20, the membrane was incubated with HRP-conjugated secondary antibodies. The washed membrane was briefly incubated with chemiluminescent substrate (catalog #34580, Thermo Fisher Scientific) and exposed on x-ray film (catalog #CP-BU, AGFA).
Surface biotinylation assay
For the surface biotinylation assay, surface proteins were labeled using membrane-impermeable biotin. Primary cortical neurons were incubated with 0.5 mg/ml EZ-Link sulfo-NHS-SS-biotin (catalog #21331, Thermo Fisher Scientific) in 1× PBS with 1 mm MgCl2 and 0.1 mm CaCl2 for 20 min at 4°C. Surface biotinylated neurons were lysed in TNE lysis buffer and pulled down with streptavidin-agarose beads (catalog #20347, Thermo Fisher Scientific) for 3 h at 4°C with gentle rotation. After washing the beads four times with TNE lysis buffer, the bound proteins were analyzed by Western blotting.
Glycosidase assay
The lysates from primary cultured neurons were immunoprecipitated using anti-myc antibody bound to protein G Sepharose beads (catalog #17-0618-01, GE Healthcare) for 4 h at 4°C. After washing, the proteins bound to beads were incubated overnight with 10× GlycoBuffer and Endo Hf (1500 units) or PNGase F (500 units) at 37°C. The deglycosylated samples were mixed with 6× Laemmli buffer, incubated for 5 min at 100°C, and analyzed by Western blotting.
Virus production
To knock down endogenous mGlu7, small hairpin RNA (shRNA) with a short hairpin was cloned under the H1 promoter (target shRNA sequence for rat mGlu7: 5′-gct tac ttc aca tcc cgg aca-3′) in the FUGW lentiviral vector (Lichnerova et al., 2015). For the mGlu7 rescue study, endogenous mGlu7 was knocked down, and mGlu7 WT or mutant cDNAs harboring silent mutations (5′-gct tac ttT acG AGc cgg aca-3′, capital letters indicate mutated nucleotides that resist targeted shRNA) with IRES-EGFP were simultaneously expressed under the Ub promoter. Lentivirus particles were produced in HEK 293T cells by cotransfection with Δ 8.9 and VSVG vectors (Lichnerova et al., 2015). Supernatants containing the viral particles were collected 60 h after transfection.
Real-time RT-PCR
Total RNA was extracted from DIV3 primary cortical neurons using TRIzol reagent (catalog #9109, Takara Bio). Two micrograms of total RNA were converted to cDNA using AMV reverse transcriptase (catalog #M0277L, New England Biolabs) according to the manufacturer's instructions. Real-time RT-PCR was performed using a CFX Connect real-time system (Bio-Rad) and SYBR Green reagent (catalog #RR820A, Takara Bio). The oligonucleotides used to detect the human GRM7 gene were myc-hmGlu7-F, 5′-agaagctgatcagcgaggc-3′ and myc-hmGlu7-R, 5′-atcgctgttgatctggtcca-3′; rat-GAPDH-F, 5′-actctacccacggcaagttc-3′ and rat-GAPDH-R, 5′-tactcagcaccagcatca-3′.
Translating ribosome affinity purification (TRAP) assay
EGFP-Rpl10a was cotransfected with the GRM7 variants in HEK 293T cells. Thirty-six hours after transfection, the cells were washed three times on ice with cold 1× diethyl pyrocarbonate (DEPC)-PBS. The cells were then lysed by lysis buffer [20 mm HEPES KOH (pH 7.4), 150 mm KCl, 10 mm MgCl2 and 1% IGEPAL CA-630 (catalog #I8896, Sigma-Aldrich) in DEPC water] with protease inhibitor cocktails, 0.5 mm DTT, and RNase inhibitor (catalog #10777019, Thermo Fisher Scientific). The cell lysates were further homogenized with a glass-glass homogenizer. The homogenates were centrifuged at 2000 × g for 10 min at 4°C and the supernatant was harvested. IGEPAL CA-630 was added to the supernatant at a final concentration of 2%. The supernatant was incubated on ice for 5 min, centrifuged at 18,000 × g for 10 min at 4°C, and the final supernatant was harvested. 10% of the final supernatant was saved and used for extraction of total RNA. The final supernatant was immunoprecipitated using anti-GFP antibody bound to protein A Sepharose beads (catalog #P3391, Sigma-Aldrich) for 3 h at 4°C. After the beads were washed four times with high-salt lysis buffer [20 mm HEPES KOH (pH 7.4), 350 mm KCl, 10 mm MgCl2, and 1% IGEPAL CA-630 in DEPC water] with 0.5 mm DTT, TRIzol reagent was immediately added to the beads. Rpl10a-bound GRM7 mRNA was isolated and analyzed by real-time RT-PCR as described above.
Immunofluorescence microscopy
Primary cortical neurons (DIV1) were plated on coverslips and transfected with mGlu7 and EGFP. Twenty-four hours after transfection, the neurons were treated with the indicated reagents for 24 h. At DIV3, the neurons were rinsed with PBS three times and fixed with 4% paraformaldehyde/4% sucrose for 15 min. After washing with PBS, the neurons were mounted on slides using ProLong antifade reagent (catalog #P7481, Thermo Fisher Scientific). For antibody staining, the fixed neurons were permeabilized with 0.25% Triton X-100 for 5 min and blocked in 10% normal goat serum (catalog #S-1000, Vector Laboratories) for 1 h at RT. The neurons were incubated overnight with primary antibodies at 4°C, washed with PBS three times, and incubated with Alexa Fluor secondary antibodies for 1 h at RT. Z-stacked maximum projection images were obtained using a Zeiss LSM 800 confocal microscope (RRID:SCR_015963). The length of the longest axon was analyzed by NeuronJ software (NIH).
For the synaptic development assay, primary cultured hippocampal neurons were infected with lentivirus particles expressing human GRM7 WT or variants in the rescue construct at DIV1. After treatment with AMN082, the neurons were maintained for 16 d. At DIV17, neurons were fixed, permeabilized, and immunostained. Images of randomly selected neurons were acquired using a Zeiss LSM 800 confocal microscope with a 63× objective lens. All image settings remained constant during scanning. Z-stacked images were converted to maximal projections and used to analyze the density, size, and intensity of presynaptic puncta from the axons identified by SMI312 staining. Quantification was performed in a blind manner using Imaris software (Bitplane). Specifically, z-stack images were analyzed with the FilamentTracer tool in auto depth mode. VGLUT1-positive or VGAT-positive puncta within 50-μm axons were masked using the Surface tool. The surface algorithm was used to calculate the number, area (size), and intensity of each puncta.
Lactate dehydrogenase (LDH) cytotoxicity assay
The cell viability of primary cultured hippocampal neurons (DIV3 or DIV17) infected with virus was measured using a LDH-Cytotoxicity Colorimetric Assay Kit II (catalog #K313-500, BioVision) according to the manufacturer's recommendations. The cytosolic LDH enzyme is released from damaged cells into the extracellular medium. Briefly, culture supernatants were harvested by centrifugation at 600 × g for 10 min. Ten microliters of the collected supernatant was mixed with 100-μl LDH reaction mixture and incubated for 30 min at RT. The reaction was stopped by the addition of 10-μl stop solution. The absorbance was measured at 450 nm using a microplate reader (AT/Sunrise, TECAN). The cytotoxicity (%) was calculated according to the manufacturer's guidelines.
Neuron counting and viability
The plated neurons (DIV3) were detached from the culture plate using 0.05% trypsin at 37°C for 10 min. Equal volumes of suspended neurons were mixed with staining solution containing 1 μg/ml AO and 20 μg/ml PI (Nexcelom Bioscience). AO Stains All nucleated cells, whereas PI only stains dead nucleated cells with compromised membranes. Twenty microliters of stained neurons were loaded into a disposable Cellometer counting chamber and placed in a Cellometer K2 instrument (Nexcelom Bioscience) for automated counting and viability measurements.
Statistical analysis
The data show the mean and the SEM based on at least three independent experiments and are presented as a proportion of the control values. The significance of the differences between the mean values of the datasets was compared by Student's paired t test or one-way ANOVA followed by Tukey's post hoc test or Bonferroni's post hoc test using GraphPad Prism (RRID:SCR_002798) software; p < 0.05 was considered statistically significant.
Results
Pathogenic mGlu7 variant proteins are expressed at low levels in heterologous cells and neurons
To investigate the functional effect of the GRM7 missense and nonsense variants, we first generated human GRM7 I154T, W586X, R622Q, and R658W/T675K (dual mutations) cDNA plasmids using site-directed mutagenesis. All mutant constructs were validated by Sanger sequencing. The amino acid Ile154 is located at the Venus flytrap domain (VFD) within the orthosteric ligand binding site. Trp586 is located at the end of the extracellular domain of mGlu7. Arg622 is located at the first intracellular loop. Arg658 is located in the first extracellular loop between the second and third transmembrane domains (TMs), and Thr675 is in the third TM (Fig. 1A). When we expressed extracellular c-myc-tagged human GRM7 variants in HEK 293T cells, we found that the protein expression levels of mGlu7 I154T and R658W/T675K were markedly reduced to 11 ± 1% or 5 ± 0% of the WT level, respectively, whereas the R622Q level was not changed compared with the WT level (*p < 0.0001; n.s., p > 0.9999; n = 3, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 1B). Although W586X, a truncated nonsense mutant, does not contain any TMs, it was expressed at the expected size (∼70 kDa; Fig. 1B). Compared with the reduced expression of mGlu7 I154T in the high molecular weight (HMW) dimer, the expression level of the low molecular weight (LMW) monomer of mGlu7 I154T was comparable to that of the WT protein (Fig. 1B).
Figure 1.
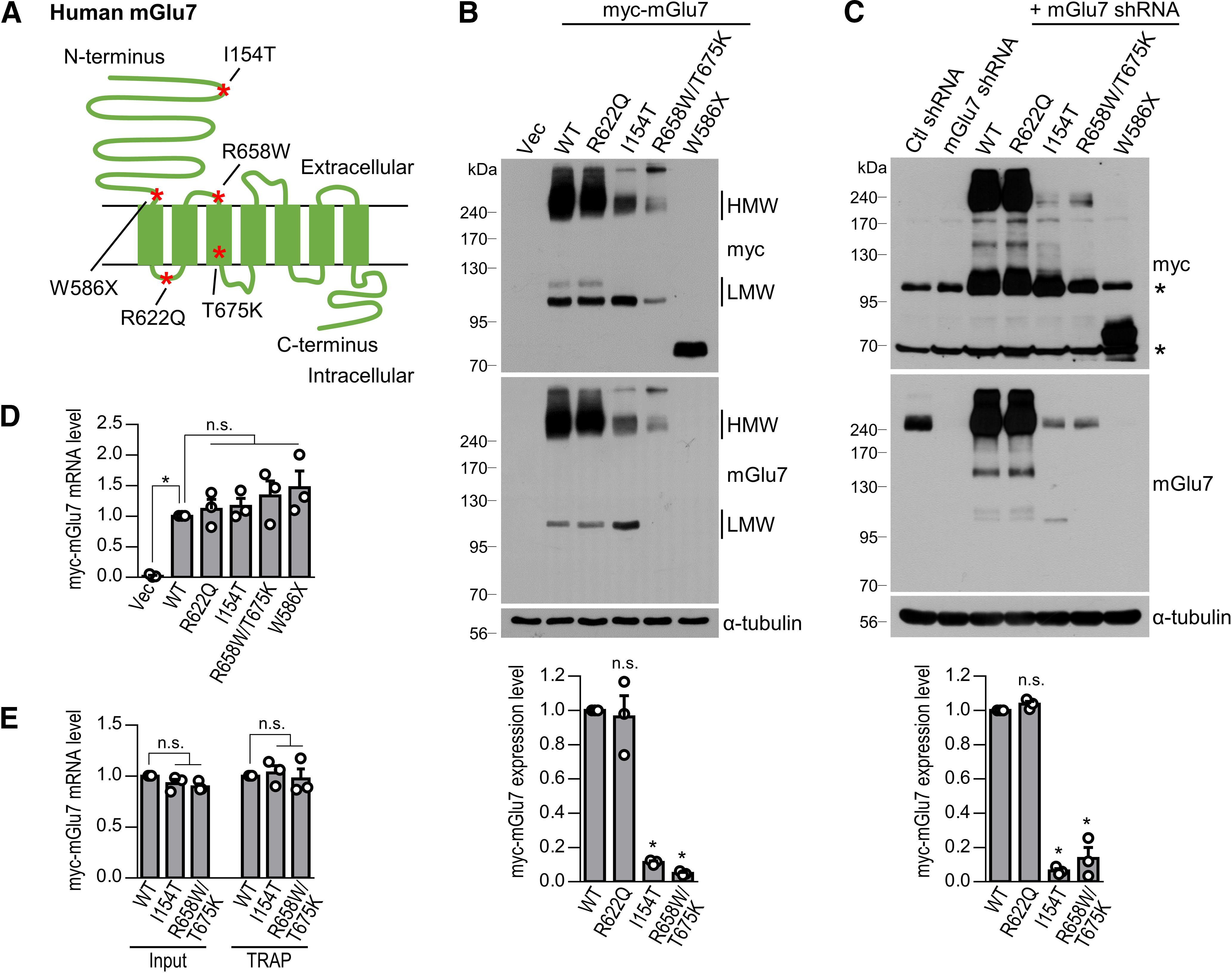
Expression of human GRM7 variants identified in NDD patients. A, Schematic diagram shows the location of the GRM7 pathologic variants identified in NDD patients. B, HEK 293T cells were transfected with myc-tagged human mGlu7 WT, R622Q, I154T, R658W/T675K, or W586X. Western blotting was performed with anti-myc or anti-mGlu7 antibodies. Note that the epitope of the anti-mGlu7 antibody is located at the extreme C terminus of mGlu7a; therefore, the anti-mGlu7 antibody did not recognize W586X, a truncated mutant of mGlu7. The HMW (>200 kDa) and LMW (∼100 kDa) forms of mGlu7 are labeled. Vec, vector control. Bar graph with scatter plots represent the mean ± SEM of the expression levels of HMW myc-mGlu7; n.s., not significant, *p < 0.0001. C, Lentivirus-mediated rescue expression of human myc-mGlu7 WT and variants. Endogenous rat mGlu7 was knocked down by shRNA, and human myc-mGlu7 WT, R622Q, I154T, R658W/T675K, or W586X were expressed with the same vector in DIV10 cortical neurons. At DIV17, Western blotting was performed with the indicated antibodies. Asterisks denote nonspecific bands. Bar graph with scatter plots represent the mean ± SEM of the expression levels of HMW myc-mGlu7; n.s., not significant, *p < 0.0001. D, After lentivirus-mediated infection, the mRNA expression levels of GRM7 WT or mutants in DIV3 cortical neurons were analyzed by real-time RT-PCR. Relative mRNA expression was calculated by the 2-ΔΔCT method using GAPDH as an internal control. Bar graph with scatter plot represents mean ± SEM, n.s., not significant, p = 0.0086. E, The efficiency of GRM7 mRNA translation was analyzed by TRAP assay. EGFP-tagged Rpl10a was cotransfected with the GRM7 variants into HEK 293T cells. The cell lysates were immunoprecipitated with anti-GFP antibody, and the bound mRNA was isolated. The amount of GRM7 mRNA bound to ribosomes was determined by a real-time RT-PCR assay. Input indicates the amount of total GRM7 mRNA. Bar graph with a scatter plot represents the mean ± SEM, n.s., not significant.
To evaluate the expression levels of the human GRM7 variants in neurons, we generated FHUW lentiviral constructs with endogenous mGlu7 expression knocked down by shRNA under the H1 promoter and expressed human GRM7 variants in the same construct downstream of the ubiquitin-C promoter (hereafter called the “rescue” construct; Lois et al., 2002). The rescue constructs were used to express the GRM7 WT or mutants throughout this study. Consistent with the results from the HEK 293T cells, we found that the protein expression levels of GRM7 I154T and R658W/T675K were markedly reduced to 6 ± 1% or 14 ± 2% of the WT level in cortical neurons at DIV17, whereas the GRM7 R622Q expression level was not changed compared with the WT level (*p < 0.0001; n.s., p > 0.9999; n = 3, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 1C). To determine whether the low protein expression of the GRM7 variants was caused by mRNA instability or transcription defects, a real-time RT-PCR assay was performed to determine the amount of exogenous myc-tagged GRM7-specific mRNA. We found that mRNA levels of the GRM7 variants were not significantly altered compared with that of the WT (*p = 0.0086; n.s., p > 0.9999, p > 0.9999, p = 0.9868, and p = 0.3959, respectively; n = 3, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 1D). Next, we examined the rate of GRM7 mRNA translation into protein using the TRAP method (Doyle et al., 2008; Heiman et al., 2008, 2014). EGFP-tagged Rpl10a, which encodes ribosomal protein L10a, was cotransfected with the GRM7 variants into HEK 293T cells. Ribosomes bound to mRNA transcripts were selectively isolated by immunoprecipitation using anti-GFP antibody. After mRNA was isolated from ribosomes, the amount of exogenous myc-GRM7-specific transcripts was determined by real-time RT-PCR. We found that ribosome-bound GRM7 mRNA levels were not significantly different in the WT and variants (input, n.s., p = 0.2487 and p = 0.0902, respectively; TRAP, n.s., p > 0.9999; n = 3, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 1E), suggesting that the efficiency of the mRNA translation was not changed in the GRM7 variants. These results indicate that the reduction in mGlu7 variant proteins was primarily determined at the protein level, not at the mRNA level.
mGlu7 variant proteins are degraded via the proteasomal and/or autophagosomal-lysosomal degradation pathway
To investigate the degradation pathway of mGlu7 variant proteins, we treated neurons expressing the variants with specific chemical inhibitors for 15 h: MG132 (5 μm, a proteasome inhibitor), chloroquine (100 μm, a lysosome inhibitor), and bafilomycin-A1 (100 nm, an autophagosome-lysosome fusion inhibitor). We found that treatment with MG132 significantly increased the expression levels of mGlu7 I154T and R658W/T675K (Fig. 2A). Treatment with chloroquine or bafilomycin-A1 also increased the expression level of I154T but not R658W/T675K, suggesting that mGlu7 I154T is degraded via proteasomes and autophagolysosomes, whereas mGlu7 R658W/T675K is degraded via proteasomes (*p = 0.0012, **p < 0.0001, ***p = 0.0010; n = 5, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 2A). The constitutive expression of WT mGlu7 was stable with a long half-life; therefore, treatment with degradation inhibitors had little effect on the expression level of mGlu7 WT.
Figure 2.

Degradation pathways and surface expression of mGlu7 pathologic variants. A, Primary cortical neurons were infected with rescue lentivirus expressing human myc-mGlu7 WT, I154T, or R658W/T675K. At DIV17, cortical neurons were incubated with 5 μm MG132, 100 μm chloroquine (CQ), or 100 nm bafilomycin-A1 (Baf-A1) for 15 h. Expression of exogenous mGlu7 was analyzed by Western blotting. LC3B and ubiquitin (Ub) blotting was performed to confirm the integrity of the assay. Bar graph with scatter plot represents the mean ± SEM normalized to the vehicle band intensity. *p = 0.0012, **p < 0.0001, ***p = 0.0010. B, Neuronal surface expression of mGlu7 variants was examined by a cell surface biotinylation assay. Cortical neurons were infected with rescue lentiviruses expressing myc-mGlu7 WT, R622Q, I154T, or R658W/T675K. At DIV17, the neurons were biotinylated using membrane-impermeable sulfo-NHS-SS-biotin and pulled down using streptavidin-agarose resin. Western blotting was conducted using the indicated antibodies. The short-exposed blot to x-ray film is presented for the myc-mGlu7 WT and R622Q. Na+/K+ ATPase α1 blot indicates a loading control for total and surface-expressed receptors, and α-tubulin blot shows the integrity of the assay. Bar graph with scatter plot represents a ratio of surface-to-total expression levels and is displayed as the mean ± SEM normalized to the WT level; n.s., not significant, *p = 0.0005, **p < 0.0001. C, EGFP-DNM1-K44A was cotransfected with myc-mGlu7 WT, I154T, or R658W/T675K in HEK 293T cells. Thirty-six hours after transfection, the surface-expressed receptors were analyzed by a cell surface biotinylation assay. TfR, transferrin receptor. D, Gel migration properties of the Endo Hf-treated or PNGase F-treated mGlu7 variants. Cortical neurons were infected by rescue lentivirus expressing myc-mGlu7 WT, R622Q, I154T, or R658W/T675K. At DIV17, neuronal lysates were immunoprecipitated with anti-myc antibody. The immunoprecipitates were incubated overnight with Endo Hf or PNGase F at 37°C and subjected to Western blotting using anti-myc antibody. Arrow, Endo Hf-resistant mature mGlu7; arrowhead, Endo Hf-sensitive immature mGlu7.
Since stable surface expression of mGlu7 has been shown to be important for bidirectional synaptic plasticity at mossy fiber (MF)–CA3 stratum lucidum interneuron (SLIN) synapses (Pelkey et al., 2005; Suh et al., 2008), we examined the protein maturation and surface expression of mGlu7 variants in neurons. After expression of mGlu7 WT or variants in primary cortical neurons using rescue lentivirus, surface-expressed receptors were labeled with membrane-impermeable sulfo-NHS-SS-biotin and pulled down using streptavidin-agarose beads. The relative surface expression level of mGlu7 I154T, the ratio of surface band intensity to total band intensity, was reduced by 53 ± 10% compared with that of WT and R622Q (Fig. 2B). In contrast, mGlu7 R658W/T675K was not expressed on the neuronal plasma membrane (*p = 0.0005, **p < 0.0001; n.s., p = 0.6595; n = 3, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 2B). To determine whether the lack of surface expression of mGlu7 R658W/T675K is caused by rapid endocytosis, we used DNM1-K44A, a dominant negative mutant of dynamin that inhibits clathrin-mediated endocytosis (Lee and De Camilli, 2002). Coexpression of DNM1-K44A increased the surface and total expression of mGlu7 I154T but did not restore the lack of surface expression of mGlu7 R658W/T675K in HEK 293T cells (Fig. 2C). The surface expression of TfR, which is endocytosed by the clathrin-mediated pathway, was increased by the coexpression of DNM1-K44A (Fig. 2C). Next, we evaluated the maturation status of mGlu7 N-glycans using N-glycosidase enzymes. Glycosidase-sensitive proteins exhibit rapid electrophoretic gel mobility during SDS-PAGE (Lichnerova et al., 2015; Skrenkova et al., 2018; Park et al., 2020). Endo Hf cleaves the “immature” type of N-glycans present in the ER or early Golgi apparatus but does not cleave the “mature” complex types of N-glycans present in the late Golgi or on the plasma membrane. PNGase F removes both the immature and mature forms of N-glycans. When we treated the lysates from cortical neurons expressing mGlu7 WT or variants with Endo Hf, mGlu7 WT and R622Q were resistant to Endo Hf, as indicated by identical gel mobility for the untreated samples in both dimer and monomer form (Fig. 2D). The mGlu7 I154T dimer was partially Endo Hf-resistant (arrow), while the mGlu7 I154T monomer was an Endo Hf-sensitive immature form (Fig. 2D, arrowhead). In contrast, mGlu7 R658W/T675K in both the dimer and monomer form was immature and sensitive to Endo Hf (Fig. 2D). These results indicate that mGlu7 WT, R622Q, and dimeric I154T contain mature N-glycans and are expressed on the neuronal surface plasma membrane, whereas neither mGlu7 R658W/T675K nor monomeric I154T are delivered to the neuronal surface and may exist in intracellular compartments such as the ER.
mGlu7 variants regulate axon outgrowth during early neuronal development
A recent study demonstrated that mGlu7 regulates the proliferation and differentiation of neural progenitor cells (NPCs) during early cortical development (Xia et al., 2015). Specifically, Grm7 knock-down increased NPC proliferation and decreased neuronal differentiation and neurite outgrowth by inhibiting NPC exit from the cell cycle, leading to defective neuronal development in the cortex of mouse embryos (Xia et al., 2015). Consistent with this report, we observed that mGlu7 knock-down reduced axon outgrowth of DIV3 primary cultured neurons (Fig. 3A). Immunostaining with anti-neurofilament antibody (SMI312), which specifically labels the axonal process, shows that the longest neurites were axonal processes (*p < 0.0001; n = 3, total neurons > 38; Student's unpaired t test; Fig. 3A).
Figure 3.

The expression of mGlu7 shRNA or pathologic variants inhibits axon outgrowth during early neuronal development. A, Cortical neurons (DIV1) were transfected with lentiviral vector harboring control (Ctl) shRNA or mGlu7 shRNA. The longest neurite was confirmed to be an axonal branch by immunostaining with SMI312, a specific marker for axons. The length of the longest axons was measured using NeuronJ software. Scale bar: 50 μm. Scatter plots represent the mean ± SEM. *p < 0.0001. B, Hippocampal neurons (DIV1) were transfected with the rescue constructs in which mGlu7 shRNA and myc-mGlu7 WT-IRES-EGFP or myc-mGlu7 variant-IRES-EGFP were expressed in the same vector. Because the GFP signal in a bicistronic IRES-EGFP construct was weak, neurons (DIV3) were stained with anti-GFP and then Alexa Fluor 488 secondary antibodies. Representative images of these GFP signals are displayed in black. Scale bar: 50 μm. Scatter plots represent the mean ± SEM. *p = 0.0012, **p = 0.0302, ***p = 0.0034, #p = 0.0011, ##p = 0.0075.
This finding prompted us to examine the effect of the GRM7 variants on axon outgrowth in the primary cultured neurons. We expressed the GRM7 rescue variants in the DIV1 cultured hippocampal neurons. Forty-eight hours after transfection, the length of the longest neurite (axon branch) was measured based on the GFP signal. We found that the expression of mGlu7 WT increased the growth of axonal processes at DIV3 by ∼2-fold, whereas the expression of mGlu7 I154T, R658W/T675K, or W586X markedly reduced axon outgrowth compared with the effect of the control vector (∼0.5-fold) and WT mGlu7 (∼2.5-fold; Fig. 3B). The expression of mGlu7 R622Q did not significantly alter axon outgrowth compared with the effect of WT expression (*p = 0.0012, **p = 0.0302, ***p = 0.0034, #p = 0.0011, ##p = 0.0075; n = 3, total neurons > 31; one-way ANOVA followed by Tukey's post hoc test; Fig. 3B). These results suggest that the degradation of mGlu7 causes defects in axon outgrowth during early neuronal development.
mGlu7 activity regulates axon outgrowth and restores defects in axon outgrowth
Next, we investigated the effects of mGlu7-specific agonists and antagonists on axon outgrowth of primary cultured neurons. We treated cultured DIV2 neurons with L-AP4 (200 μm, a Group III mGlu receptor agonist), AMN082 [1 μm, an mGlu7-specific positive allosteric modulator (PAM)], MSOP (100 μm, a Group III mGlu receptor antagonist), or MMPIP (10 μm, an mGlu7-specific negative allosteric modulator) for 24 h. We found that L-AP4 and AMN082 increased axon outgrowth, whereas MSOP and MMPIP decreased axon outgrowth by ∼2-fold (*p < 0.0001, **p = 0.0004, ***p = 0.0348, #p = 0.0008; n = 3, total neurons > 25; one-way ANOVA followed by Tukey's post hoc test; Fig. 4A). Thus, along with the importance of mGlu7 protein expression levels, the activity of mGlu7 is essential for axon outgrowth during early neuronal development.
Figure 4.

The impaired axon outgrowth by mGlu7 variants is restored by treatment with mGlu7 agonists. A, After primary hippocampal neurons (DIV1) were transfected with EGFP, an mGlu7 agonist (200 μm L-AP4, 1 μm AMN082) or antagonist (100 μm MSOP, 10 μm MMPIP) was administered for 24 h. At DIV3, axonal outgrowth was visualized by confocal microscopy. Scale bar: 50 μm. Scatter plots represent the mean ± SEM. *p < 0.0001, **p = 0.0004, ***p = 0.0348, #p = 0.0008. B, Control vector (Vec), mGlu7 WT, I154T, or R658W/T675K rescue construct was cotransfected with EGFP into primary cortical neurons (DIV1). Twenty-four hours after transfection, the neurons were treated with AMN082 for 24 h. At DIV3, the neurons were fixed and visualized by confocal microscopy. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001.
Since we observed an increase in axon outgrowth on cell treatment with mGlu7 agonists, we investigated whether treatment with mGlu7 agonist can restore the impaired axon outgrowth by mGlu7 variants. We transfected mGlu7 WT, I154T, or R658W/T675K into cultured neurons and treated the neurons with 1 μm AMN082 for 24 h. AMN082 did not significantly alter the WT mGlu7-induced increase in axon outgrowth. However, the reduction in axon outgrowth by mGlu7 I154T was restored to the control level by treatment with AMN082, whereas the reduction by mGlu7 R658W/T675K was not restored (*p < 0.0001; WT, n.s., p = 0.7925, R658W/T675K, n.s., p = 0.9997; n = 3, total neurons > 29; one-way ANOVA followed by Tukey's post hoc test; Fig. 4B). This difference is probably because of the availability of mGlu7 on the neuronal membrane surface, as mGlu7 I154T is expressed on the neuronal surface, but mGlu7 R658W/T675K is not.
mGlu7-mediated axon outgrowth is regulated through cytoskeletal organization via MAPK-cAMP-PKA signaling
We examined whether mGlu7 is expressed in the growth cone, the tip of the axonal process of the primary cultured neurons. At DIV3, cortical neurons were immunostained with anti-mGlu7 antibody (red) and TUJ1 (pseudo-blue), an antibody that selectively labels the shaft of neurites and the central domain of the growth cone. The neurons were also labeled with phalloidin (green) to visualize filamentous actin (F-actin) present at the peripheral domain of the growth cone (Turlova et al., 2016). Confocal microscopy imaging revealed that mGlu7 is present in the central and peripheral domains of the growth cone and in the somata and axonal shaft (Fig. 5A). We transfected myc-mGlu7 into cortical neurons, which was immunostained with anti-myc antibody. The overexpression of myc-mGlu7 followed the same pattern as the distribution of endogenous mGlu7 (Fig. 5B). The distribution pattern of mGlu7 suggested that mGlu7 may play a role in axon outgrowth through microtubule dynamics and actin polymerization.
Figure 5.
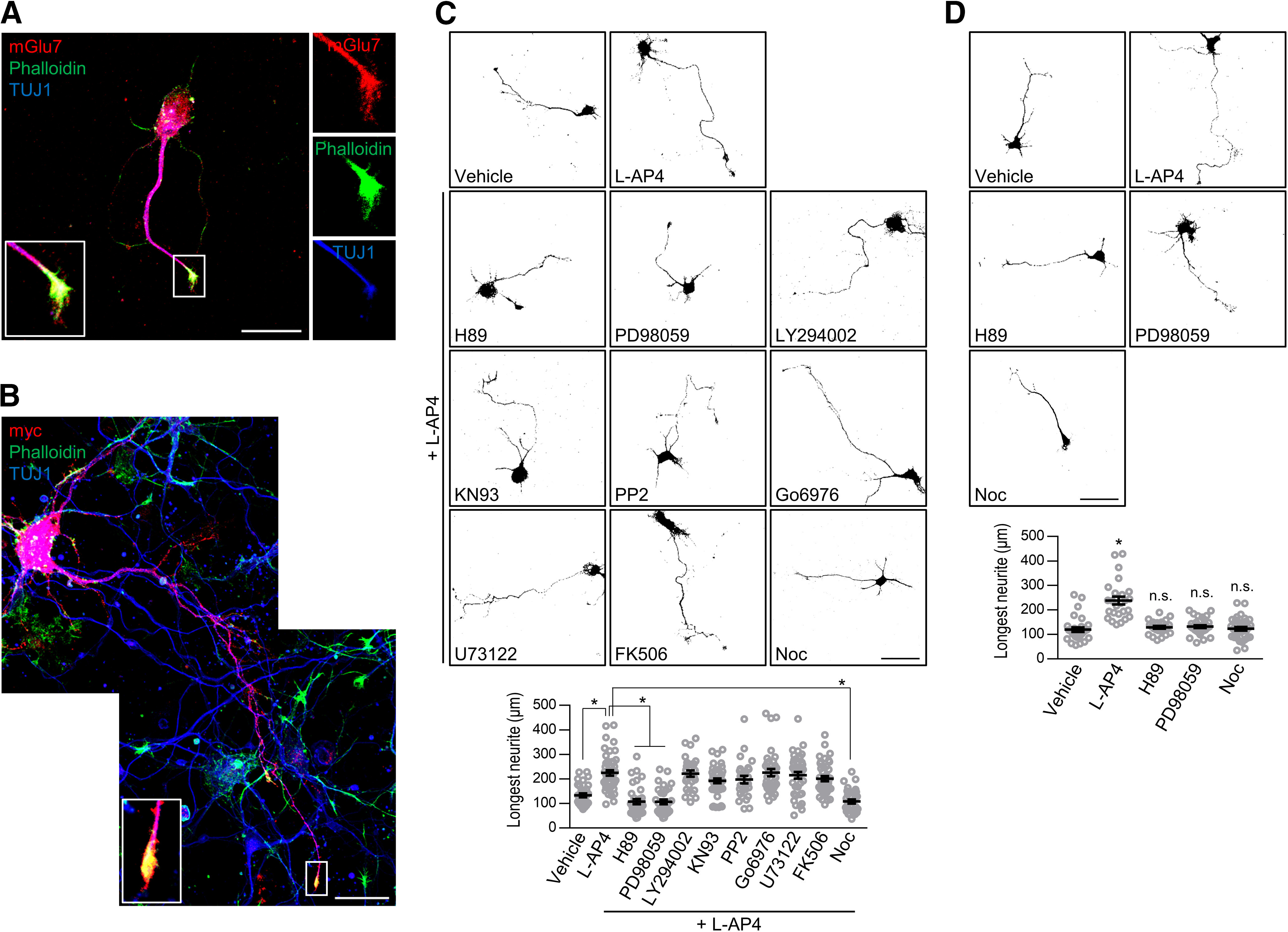
mGlu7-dependent axon outgrowth is regulated by the MAPK-cAMP-PKA pathway and microtubule dynamics. A, Localization of endogenous mGlu7 in early developing cortical axons. Cortical neurons (DIV3) were permeabilized and coimmunostained with rabbit anti-mGlu7 and mouse TUJ1 antibodies. After washing, the neurons were stained with anti-rabbit Alexa Fluor 568 or anti-mouse Alexa Fluor 647 secondary antibodies. F-actin was visualized with Alexa Fluor 488 phalloidin. The enlarged image reveals endogenous mGlu7 on the axonal shaft and tip of the growth cone. Scale bar: 20 μm. B, Human myc-mGlu7 cDNA was transfected into DIV1 cortical neurons. At DIV3, the distribution of mGlu7 was evaluated, as shown in panel A, using anti-myc and TUJ1 antibodies. Scale bar: 20 μm. C, The mGlu7 agonist-induced increase in axon outgrowth was abolished by treatment with a PKA inhibitor, MEK inhibitor, or microtubule polymerization inhibitor. After hippocampal neurons (DIV1) were transfected with EGFP, the neurons were treated with L-AP4 for 24 h in the presence of the following inhibitors: H89 (10 μm), PD98059 (50 μm), LY294002 (50 μm), KN93 (10 μm), PP2 (25 μm), Go6976 (200 nm), U73122 (2 μm), FK506 (1 μm), or nocodazole (Noc, 100 nm). At DIV3, axon outgrowth was visualized by confocal microscopy, and axon length was measured by NeuronJ software. Scale bar: 50 μm. Scatter plots represent the mean ± SEM. *p < 0.0001. D, The effect on axon outgrowth by the reagents involved in the mGlu7 signaling pathway. mGlu7 agonist L-AP4 increased axonal outgrowth at DIV3, whereas treatment with the indicated reagents alone did not affect axon outgrowth. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001.
To identify the molecular pathways involved in mGlu7-mediated axon outgrowth, we aimed to examine several pathways known to be involved in the function of Group III mGlu receptors including mGlu7. It was shown that Group III mGlu receptors are coupled with the phosphatidylinositol-3 kinase (PI3K) and MAPK pathways of agonist-induced neuroprotection in cerebellar granule cells (Iacovelli et al., 2002; Wang et al., 2012). The MAPK kinase (MEK)-extracellular signal-regulated kinase (ERK) signaling pathway is thought to be downstream of mGlu7 associated with β-arrestins (Jiang et al., 2006; Gu et al., 2012; Iacovelli et al., 2014; Lee et al., 2019). Src is involved in the Group III mGlu receptor-mediated ERK activation in midbrain neurons and heterologous cells (Iacovelli et al., 2004; Jiang et al., 2006). Protein kinase C (PKC), which is associated with protein interacting with C-kinase 1 (PICK1), plays essential roles in the stabilization of mGlu7 on the neuronal surface (Dev et al., 2000; Pelkey et al., 2005; Suh et al., 2008, 2018). mGlu7 regulates P/Q-type calcium channels and glutamate release via a phospholipase C (PLC)-dependent pathway in cerebellar granule cells and hippocampus (Perroy et al., 2000, 2002; Martín et al., 2007, 2010). Thus, we screened a series of chemical reagents at the following concentrations: H89 (10 μm, a PKA inhibitor), PD98059 (50 μm, an MEK inhibitor), LY294002 (50 μm, a PI3K inhibitor), KN93 (10 μm, a CaMKII inhibitor), PP2 (25 μm, a Src family kinase inhibitor), Go6976 (200 nm, a PKC inhibitor), U73122 (2 μm, a PLC inhibitor), FK506 (1 μm, a calcineurin inhibitor), and nocodazole (100 nm, a microtubule polymerization inhibitor) in the presence of the mGlu7 agonist L-AP4. We found that H89, PD98059, and nocodazole inhibited the agonist-induced increase in axonal outgrowth in the cultured neurons, while the other reagents had no effect (*p < 0.0001; n = 3, total neurons > 27; one-way ANOVA followed by Tukey's post hoc test; Fig. 5C). Treatment with these reagents alone had no effect on axon outgrowth (*p < 0.0001; H89, n.s., p = 0.9572, PD98059, n.s., p = 0.8670, Noc, n.s., p = 0.9987; n = 3, total neurons > 25; one-way ANOVA followed by Tukey's post hoc test; Fig. 5D). We further screened the following chemical reagents in the presence of the mGlu7 antagonist MMPIP: paclitaxel (3 nm, a microtubule disassembly inhibitor), latrunculin A (100 nm, an actin polymerization inhibitor), db-cAMP (500 μm, a cAMP analog), forskolin (3 μm, an adenylyl cyclase activator), and PMA (3 μm, a PKC activator). Treatment with paclitaxel, latrunculin A, db-cAMP, or forskolin restored the antagonist-induced decrease in axon outgrowth, whereas PMA did not restore it (*p < 0.0001, **p = 0.0003; n.s., p > 0.9999; n = 3, total neurons > 28; one-way ANOVA followed by Tukey's post hoc test; Fig. 6A). Although we were unable to observe the effect of these reagents alone on axon outgrowth under our experimental conditions (Fig. 6B), it has often been reported that these reagents can affect axon outgrowth (Qiu et al., 2002; Yamada et al., 2005; Aglah et al., 2008; Shelly et al., 2010; Batty et al., 2017). Thus, we tested the effect of these reagents on axon outgrowth at higher concentrations. We observed that axon outgrowth increased only when these reagents were treated at more than twice the concentration we had originally used, as shown in Figure 6A (6 nm paclitaxel, 200 nm latrunculin A, or 1 mm db-cAMP; *p = 0.0121, **p = 0.0233, ***p < 0.0001; paclitaxel, n.s., p > 0.9999, LacA, n.s., p = 0.7163, db-cAMP, n.s., p = 0.8515; n = 3, total neurons > 30; one-way ANOVA followed by Tukey's post hoc test; Fig. 6B). Taken together, microtubule polymerization and actin depolymerization through MAPK-cAMP-PKA signaling were required for mGlu7-mediated axon outgrowth.
Figure 6.

Impaired axon outgrowth induced by mGlu7 antagonist is regulated by cytoskeletal dynamics and cAMP signaling. A, mGlu7 antagonist-induced decrease in axon outgrowth was reversed by treatment with a microtubule disassembly inhibitor, actin polymerization inhibitor, cAMP analog, or adenylyl cyclase activator. The neurons were treated with MMPIP for 24 h in the presence of the following reagents: paclitaxel (3 nm), latrunculin A (LacA, 100 nm), db-cAMP (500 μm), forskolin (3 μm), or PMA (3 μm). Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001, **p = 0.0003. B, Dose-dependent effect of the reagents on axon outgrowth. Cortical neurons (DIV1) were transfected with EGFP. Twenty-four hours after transfection, the neurons were treated with paclitaxel, LacA, or db-cAMP at the indicated concentration for 24 h. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p = 0.0121, **p = 0.0233, ***p < 0.0001.
Impaired axon outgrowth by mGlu7 variants is restored by cAMP-elevating agents or cytoskeletal drugs
Next, we tested whether specific signaling can restore defective axon outgrowth by mGlu7 variants. Treatment with paclitaxel, latrunculin A, or db-cAMP reversed the reduced axonal outgrowth induced by mGlu7 I154T or R658W/T675K (*p < 0.0001; paclitaxel, n.s., p = 0.8526, LacA, n.s., p = 0.9979, db-cAMP, n.s., p = 0.9616; n = 3, total neurons > 30; one-way ANOVA followed by Tukey's post hoc test; Fig. 7A). In addition, treatment with these reagents also reversed the reduced axon outgrowth induced by mGlu7 knock-down, but PMA did not restore it (*p < 0.0001, **p = 0.0002; n.s., p = 0.9892; n = 3, total neurons > 23; one-way ANOVA followed by Tukey's post hoc test; Fig. 7B). To confirm that cAMP influences the extent of axon outgrowth by mGlu7 variants, we treated the cultured neurons with rolipram, a selective phosphodiesterase 4 (PDE4) inhibitor that suppresses cAMP breakdown. We found that rolipram had little effect on axon outgrowth in the vector control but reversed the reduced axonal outgrowth induced by mGlu7 I154T or R658W/T675K (*p < 0.0001; n.s., p = 0.5591; n = 3, total neurons > 30; one-way ANOVA followed by Tukey's post hoc test; Fig. 8A). Because mGlu7 is coupled with heterotrimeric Gαi/o protein, we tested whether Gαi/o protein is an upstream signaling mediator critical for axon outgrowth. We treated cortical neurons with pertussis toxin (PTX), which catalyzes the ADP-ribosylation of Gαi/o and prevents its interaction with GPCRs. PTX treatment had no effects on agonist-induced axon outgrowth (*p < 0.0001, **p = 0.0041, ***p = 0.0085; n = 3, total neurons > 37; one-way ANOVA followed by Tukey's post hoc test; Fig. 8B), suggesting that the Gαi/o-independent pathway is involved in mGlu7-mediated axon outgrowth. In addition, when we treated neurons with PD98059 in the presence of rolipram, PD98059 no longer inhibited agonist-induced axon outgrowth (*p < 0.0001; n.s., p = 0.7828; n = 3, total neurons > 33; one-way ANOVA followed by Tukey's post hoc test; Fig. 8C). Taken together, MAPK, but not the Gαi/o protein, is an upstream signaling mediator of the cAMP in regulating mGlu7-mediated axon outgrowth (Gao et al., 2003; Dumaz and Marais, 2005).
Figure 7.

The impaired axon outgrowth by mGlu7 variants or knock-down is restored by adjusting dysregulated cytoskeletal dynamics or cAMP signaling. A, Cortical neurons (DIV1) were cotransfected with EGFP and control vector (Vec), mGlu7 I154T, or R658W/T675K rescue construct. Twenty-four hours after transfection, the neurons were treated with the indicated reagents for 24 h. At DIV3, axonal outgrowth was visualized by confocal microscopy and measured by NeuronJ software. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001. B, Hippocampal neurons (DIV1) were transfected with lentiviral vector harboring control (Ctl) shRNA or mGlu7 shRNA. Twenty-four hours after transfection, the neurons were treated with the indicated reagents for 24 h. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001, **p = 0.0002.
Figure 8.

mGlu7-mediated axon outgrowth is regulated by the MAPK-cAMP pathway, but is independent of the Gαi pathway. A, Cortical neurons (DIV1) were cotransfected with EGFP and the control vector (Vec), mGlu7 I154T or R658W/T675K rescue constructs in the absence or presence of rolipram (500 nm). At DIV3, axonal outgrowth was visualized by confocal microscopy and measured by NeuronJ software. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001. B, mGlu7-mediated axon outgrowth is not related to the Gαi signaling pathway. After cortical neurons (DIV1) were transfected with EGFP, the neurons were treated with AMN082 in the presence or absence of 2, 4, or 10 μg/ml PTX, an inhibitor of Gα subunits of the heterotrimeric G-protein. At DIV3, axonal outgrowth was visualized by confocal microscopy. Scale bar: 50 μm. Scatter plots represent the mean ± SEM. *p < 0.0001, **p = 0.0041, ***p = 0.0085. C, MAPK is an upstream mediator of cAMP in mGlu7-mediated axon outgrowth. After transfection with EGFP, the cortical neurons (DIV1) were treated with MEK inhibitor (PD98059) and/or 500 nm rolipram, a PDE inhibitor that suppresses cAMP degradation, in the absence or presence of the mGlu7 agonist AMN082. At DIV3, axonal outgrowth was visualized by confocal microscopy. Scale bar: 50 μm. Scatter plots represent the mean ± SEM, n.s., not significant, *p < 0.0001.
The number of mature synapses is reduced by the early expression of the GRM7 variants
We questioned whether defects in early neuronal development could lead to impairment of synapse development in mature neurons. Primary cultured hippocampal neurons (DIV1) were infected with rescue lentivirus expressing the mGlu7 WT or variants and maintained for two weeks. At DIV17, excitatory or inhibitory synapses were visualized by immunostaining VGLUT1 or VGAT, which are excitatory or inhibitory presynaptic terminal markers, respectively. We analyzed the number, size, and intensity of VGLUT1-positive or VGAT-positive puncta distributed along the SMI312-positive axon branch. The number of both VGLUT1-positive and VGAT-positive synapses was markedly reduced by ∼2-fold for the neurons expressing mGlu7 I154T or mGlu7 R658W/T675K compared with those expressing WT mGlu7 (Fig. 9A,B). The size and intensity of the VGLUT1-positive or VGAT-positive puncta did not change significantly (Fig. 9A,B). In particular, consistent with the effect of the agonists on axon outgrowth, treatment with AMN082 at DIV2 restored the number of VGLUT1-positive and VGAT-positive puncta in the mature neurons (DIV17) expressing mGlu7 I154T (Fig. 9A,B). However, treatment with AMN082 failed to restore the number of synapses that had been reduced by mGlu7 R658W/T675K, which is not expressed on the neuronal surface membrane [number, *p < 0.0001; WT, n.s., p = 0.8688, R658W/T675K, n.s., p = 0.9378; size, n.s., p = 0.2109, p = 0.7541, p = 0.9984, p = 0.9678, and p = 0.8407, respectively; intensity, n.s., p = 0.9971, p > 0.9999, p = 0.0653, p = 0.0684, and p = 0.0855, respectively (Fig. 9A); number, *p < 0.0001; WT, n.s., p = 0.4210, R658W/T675K, n.s., p = 0.9907; size, n.s., p = 0.8363, p = 0.8780, p = 0.0587, p = 0.0759, and p = 0.2405, respectively; intensity, n.s., p = 0.9989, p > 0.9999, p = 0.1494, p = 0.8129, and p = 0.5573, respectively; n = 3; one-way ANOVA followed by Tukey's post hoc test (Fig. 9B)]. To determine whether changes in the number of mature synapses were because of neuronal cell death, we performed an LDH-cytotoxicity colorimetric assay. We confirmed that neither mGlu7 knock-down nor the expression of mGlu7 WT or mutants caused changes in cell viability (DIV3, n.s., p > 0.9999, DIV17, n.s., p > 0.9999; n = 3, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 9C). In addition, we counted the number of virus-infected neurons (DIV3) using the fluorescent automated cell counter Cellometer K2. The neurons were trypsinized, and thereafter stained with AO and PI to label all nucleated neurons and dead nucleated neurons, respectively. We found that ∼5% of neurons were dead among both uninfected and virus-infected neurons, but there was no significant difference in the number of live or dead neurons following infection with mGlu7 knock-down or mutant virus (live neurons, n.s., p > 0.9999, dead neurons, n.s., p > 0.9999; n = 4, one-way ANOVA followed by Bonferroni's post hoc test; Fig. 9D). These results indicate that neuronal viability was not altered by infection with mGlu7 knock-down or mutant virus.
Figure 9.
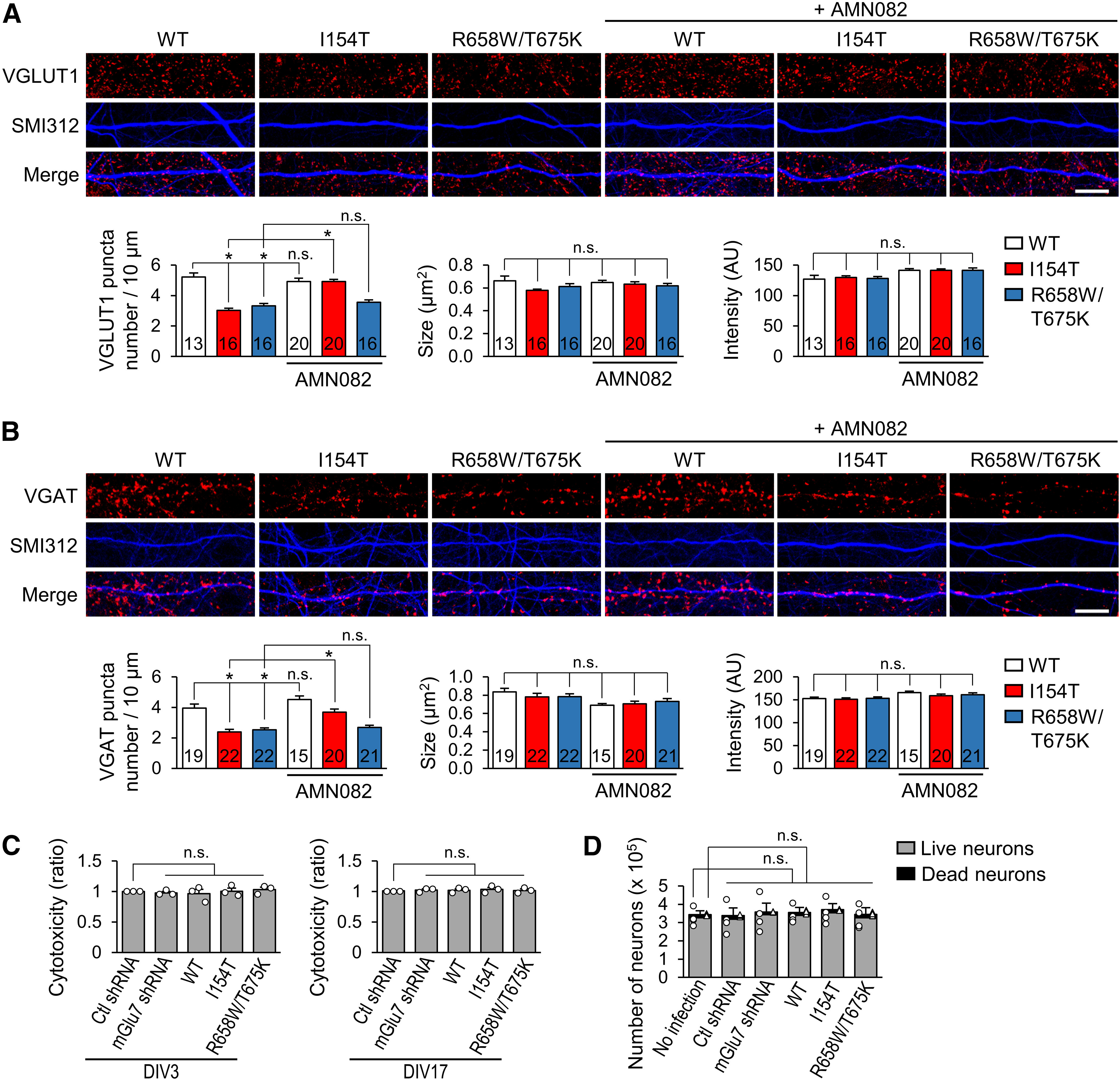
Presynaptic terminal development is deficient on expression of the mGlu7 rescue variants, and AMN082 restores the defect induced by mGlu7 I154T. A, Hippocampal neurons (DIV1) were infected with rescue lentivirus expressing mGlu7 WT, I154T, or R658W/T675K. Twenty-four hours after infection, the neurons were treated with AMN082. At DIV17, the neurons were permeabilized and immunostained with anti-VGLUT1 (red) and SMI312 (pseudo-blue) antibodies. Scale bar: 10 μm. Bar graphs show a summary of the effects of mGlu7 variants on the number of VGLUT1-positive puncta per 10-µm axon (left), size of the VGLUT1-positive puncta (middle), and intensity of the VGLUT1-positive puncta (right). The number in the bar indicates the total number of neuron analyzed. AU, arbitrary unit. Bar graphs represent mean ± SEM, n.s., not significant, *p < 0.0001. B, Hippocampal neurons were infected as shown in panel A and immunostained with anti-VGAT (red) and SMI312 (pseudo-blue) antibodies. Scale bar: 10 μm. Summary graphs show the effect of the GRM7 variants on the number of VGAT-positive puncta per 10-µm axon (left), size of the VGAT-positive puncta (middle), and intensity of the VGAT-positive puncta (right). The number in the bar indicates the total number of neurons analyzed. AU, arbitrary unit. Bar graphs represent mean ± SEM, n.s., not significant, *p < 0.0001. C, Cell viability was measured using an LDH cytotoxicity colorimetric assay in DIV3 (left panel) or DIV17 (right panel) hippocampal neurons infected with the indicated virus. Bar graph with scatter plots represent the mean ± SEM, n.s., not significant. D, After staining the neurons (DIV3) with AO/PI, the number of live neurons and the number of dead neurons in one well of a six-well culture plate were automatically counted using Cellometer K2. Bar graph represents the mean ± SEM. Scatter plots with open circle and triangle represent the live neuron number and dead neuron number, respectively; n.s., not significant.
Discussion
Neurite outgrowth and synapse formation during early brain development are fundamental processes for establishing functional neural circuits. Abnormal synapse assembly leads to circuit dysfunction, brain malformation, and NDDs. Early pharmacological interventions targeting glutamate receptors have successfully ameliorated synaptic and social deficits in animal models (Washbourne, 2015; Berry-Kravis et al., 2018). In the present study, we demonstrate that GRM7 is a candidate gene and a potential target for the therapeutic intervention of NDDs. Rare biallelic variants in GRM7 have been identified in six consanguineous families with NDDs, including three families recently characterized (Marafi et al., 2020). These patients commonly share clinical neurologic phenotypes such as DD/ID, early onset seizures, and microcephaly, suggesting GRM7 as a candidate gene for autosomal recessive NDDs (Charng et al., 2016; Reuter et al., 2017; Marafi et al., 2020). Using mutation information from these published studies, we demonstrate that mGlu7 is essential for axon outgrowth during early neuronal development. We show that the I154T and R658W/T675K missense mutations in GRM7 cause rapid degradation of the mGlu7 protein via proteasomes and/or autophagosomes-lysosomes. The degradation of mGlu7 variant proteins prevents axon outgrowth via the impairment of the MAPK-cAMP-PKA signaling pathway accompanied by dysregulation of cytoskeletal dynamics in primary cultured neurons. In addition, impaired axon outgrowth results in defect in presynaptic terminal development in mature excitatory and inhibitory synapses. Of particular interest, the defects caused by GRM7 I154T in axon outgrowth and presynaptic terminal development can be reversed by the activation of mGlu7 with an agonist, whereas the defects caused by GRM7 R658W/T675K are not because of its lack of surface expression (Fig. 10).
Figure 10.

A proposed model for NDD development induced by pathogenic GRM7 mutations and rescued by AMN082. WT mGlu7 is stably expressed on the neuronal surface and necessary for axon outgrowth and presynaptic terminal development. Rapid degradation of mGlu7 variant proteins prevents axon outgrowth and presynaptic terminal development via the defective MAPK-cAMP-PKA signaling pathway and subsequent dysregulation of cytoskeletal dynamics. As some mGlu7 I154T is expressed on the neuronal surface, an mGlu7 agonist can restore impaired axon outgrowth and presynaptic terminal development (blue arrows). We propose that this scenario is the molecular basis for the development of mGlu7-related DDs/IDs and suggest mGlu7 as a potential therapeutic target for NDDs.
The mGlu7 I154T and R658W/T675K proteins show the same pathologic phenotype, but their degradation pathways are different. The R658W/T675K protein was preferentially degraded via proteasomes, whereas the I154T protein was degraded via both proteasomes and autophagosomes-lysosomes. Although there was a marked reduction in the protein expression levels of both mGlu7 I154T and R658W/T675K, mGlu7 I154T was delivered to the surface of the plasma membrane, but R658W/T675K was not. Recently, we reported that ubiquitinated mGlu7 on the neuronal surface is degraded by both proteasomes and lysosomes (Lee et al., 2019). Because mGlu7 I154T was detected on the neuronal surface and its protein level was increased by blocking clathrin-mediated endocytosis, we propose that surface-expressed mGlu7 I154T is degraded via both proteasomes and lysosomes. However, neither mGlu7 R658W/T675K nor monomeric mGlu7 I154T was expressed on the neuronal surface, so misfolded mGlu7 R658W/T675K or I154T might be retained in the ER and trigger ER-associated protein degradation (ERAD) by cytosolic proteasomes.
We demonstrated that the mGlu7 protein expression level affects axon outgrowth in early neuronal development. It has been shown that the genetic reduction of GRM7 is associated with the development of RTT, a type of NDD caused by loss-of-function mutations in the MECP2 gene (Bedogni et al., 2016; Gogliotti et al., 2017; Fisher et al., 2018a,b). These reports showed that the mGlu7 protein expression level was reduced in the brains of RTT patients and Mecp2-null mice, which was attributed to the loss of mGlu7 mRNA transcription as regulated by MeCP2. In utero electroporation experiments revealed that embryonic Mecp2 knock-down impaired the migration of cortical neurons, which is a morphologic change similar to that observed in a study of mGlu7-knock-down neurons (Xia et al., 2015; Bedogni et al., 2016). Because we observed a profound reduction in the protein expression in the GRM7 variants, the pathologic phenotype of these GRM7 variants is likely attributable to the loss-of-function of mGlu7. However, since mGlu7 variants was partially expressed compared with the endogenous mGlu7 (Fig. 1C), we cannot exclude the possibility that mGlu7 variants may deliver a dominant-negative signal that is detrimental to early neuronal development. In addition to the protein expression level of mGlu7, using agonists or antagonists, we showed that the degree of mGlu7 activation is closely correlated with axonal outgrowth. The surface expression levels of mGlu7 I154T or R658W/T675K were significantly reduced or deficient, respectively (Fig. 2B). These findings indicate that proper surface expression of mGlu7 during the early developmental stage is essential for neuronal development and synapse formation.
We were not able to observe any phenotypic change by GRM7 R622Q, a heterozygous point mutation found in ASD patients. In addition, heterozygous deletions (exon 2 or exons 3–7) in coding sequences of GRM7 have been identified in ASD patients (Gai et al., 2012; Sanders et al., 2012; Iossifov et al., 2014; Liu et al., 2015; Fisher et al., 2018a). Deletion of exon 2 or exons from 3 to 7 will cause a frameshift in the GRM7 coding sequence, resulting in a truncated mGlu7 protein of 181 or 252 amino acids, respectively. Since we found defective axon outgrowth by GRM7 W586X, a homozygous deletion found in two patients with DD/ID and brain malformation (Reuter et al., 2017), these ASD-related deletions may lead to the same phenotype as acquired by GRM7 W586X in vitro. Thus, the GRM7 gene dosage may manifest the different phenotype in vivo as either ASD or DD/ID. Homozygous loss-of-function mutations in GRM7 can cause DD/ID with brain malformation, while heterozygous loss-of-function mutations with a dominant-negative effect on neuronal development may lead to ASD in vivo. The phenotypes unique to ASD may not be detected by our analysis because the molecular pathways in ASD differ from those in DD/ID and brain anomalies.
Our study suggests that aberrant GRM7 function may be one of the possible molecular pathways for NDD development (Fig. 10). Using a series of signaling inhibitors, we report that the MAPK-cAMP-PKA signaling and cytoskeletal dynamics are defective in the GRM7 variants. In particular, mGlu7-dependent MAPK signaling, but not G-protein signaling, appears to be a candidate pathway for the development of NDDs. Our previous study showed that β-arrestin 2 complexed with Nedd4 regulates mGlu7-mediated ERK signaling in primary cultured neurons (Lee et al., 2019). It has also been reported that β-arrestin 2 modulates Raf-MEK-ERK signaling of mGlu5, independent of Gαq-mediated signaling (Stoppel et al., 2017). Consistent with our discovery that MAPK is upstream of cAMP-PKA, activated ERK was shown to increase cAMP signaling by inhibiting PDE4 (Gao et al., 2003). Furthermore, accumulating evidence indicates that cAMP can regulate axon outgrowth via cytoskeleton remodeling (Qiu et al., 2002; Yamada et al., 2005; Aglah et al., 2008; Shelly et al., 2010; Batty et al., 2017). We show that defective axon outgrowth by mGlu7 variants can be reversed by modifying cytoskeleton dynamics; therefore, we propose a model in which the dysregulated MAPK-cAMP-PKA-cytoskeleton dynamics is a central signaling pathway for the impairment of axon outgrowth and synapse development by the GRM7 variants (Fig. 10).
Several studies on mGlu7-knock-out mice have shown that mGlu7 is involved in cognitive function, social behavior, motor coordination, epilepsy, depression, and anxiety. These behavioral phenotypes overlap with the phenotypes characteristic of human NDDs (Palazzo et al., 2016; Fisher et al., 2020). Specifically, mGlu7-knock-out mice exhibited diminished short-term plasticity in the hippocampus and impairment in short-term working memory and spatial learning (Bushell et al., 2002; Hölscher et al., 2004; Goddyn et al., 2015). mGlu7-knock-out mice also showed anxiolytic and antidepressant behavior, delayed extinction of a conditioned fear response, deficits in motor coordination and strength, and increased seizure susceptibility (Masugi et al., 1999; Sansig et al., 2001; Cryan et al., 2003; Callaerts-Vegh et al., 2006; Fisher et al., 2020). Although our data and another study support morphologic changes by acute mGlu7 knock-down in primary cultured neurons and mouse embryos (Xia et al., 2015), the mGlu7-knock-out mice do not display any brain malformation, such as microcephaly or cortical atrophy, which is observed in patients with the GRM7 variants. Although the scenario remains unclear because human and mouse mGlu7 proteins are highly homologous (99.5% identical), but we postulate the following explanations for this discrepancy. First, the mGlu7-mutant proteins, in addition to the effect of loss-of-function, may transmit detrimental signals during early neuronal development. Second, mGlu7 may be less critical for early neuronal development in mice than it is in humans. Third, mGlu7-knock-out neurons are more likely to compensate for mGlu7 loss than acute knock-down neurons.
It was shown that mGlu7 PAMs restore mGlu7-mediated altered synaptic transmission, deficits of long-term potentiation at Schaffer collateral (SC)–CA1 synapses, and attenuated social and cognitive behaviors in the RTT mouse model (Jalan-Sakrikar et al., 2014; Gogliotti et al., 2017; Fisher et al., 2018a). We show that mGlu7 activation by AMN082, the first selective PAM for mGlu7, restores the defects of axon outgrowth and synapse development if mGlu7 is expressed on the neuronal surface plasma membrane. Therefore, modulation of mGlu7 activity and stabilization of mGlu7 on the neuronal surface are promising therapeutic targets for controlling DD/ID phenotypes in NDDs, including RTT (Fisher et al., 2018a). Further cell type-specific genetic analysis on the role of the GRM7 variants will be required to understand the pathophysiology of the GRM7-related NDDs.
Footnotes
This work was supported by National Research Foundation of Korea Grants NRF-2020R1A5A1019023, NRF-2018R1A2B6004759, and NRF-2017M3C7A1029611 and by the Korea Health Technology R&D Project Grant HI18C0789 through the Korea Health Industry Development Institute.
The authors declare no competing financial interests.
References
- Aglah C, Gordon T, Posse de Chaves EI (2008) cAMP promotes neurite outgrowth and extension through protein kinase A but independently of Erk activation in cultured rat motoneurons. Neuropharmacology 55:8–17. 10.1016/j.neuropharm.2008.04.005 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders, Ed 5. Arlington: American Psychiatric Publishing. [Google Scholar]
- Batty NJ, Fenrich KK, Fouad K (2017) The role of cAMP and its downstream targets in neurite growth in the adult nervous system. Neurosci Lett 652:56–63. 10.1016/j.neulet.2016.12.033 [DOI] [PubMed] [Google Scholar]
- Bedogni F, Cobolli Gigli C, Pozzi D, Rossi RL, Scaramuzza L, Rossetti G, Pagani M, Kilstrup-Nielsen C, Matteoli M, Landsberger N (2016) Defects during Mecp2 null embryonic cortex development precede the onset of overt neurological symptoms. Cereb Cortex 26:2517–2529. 10.1093/cercor/bhv078 [DOI] [PubMed] [Google Scholar]
- Berry-Kravis EM, Lindemann L, Jønch AE, Apostol G, Bear MF, Carpenter RL, Crawley JN, Curie A, Des Portes V, Hossain F, Gasparini F, Gomez-Mancilla B, Hessl D, Loth E, Scharf SH, Wang PP, Von Raison F, Hagerman R, Spooren W, Jacquemont S (2018) Drug development for neurodevelopmental disorders: lessons learned from fragile X syndrome. Nat Rev Drug Discov 17:280–299. 10.1038/nrd.2017.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell TJ, Sansig G, Collett VJ, van der Putten H, Collingridge GL (2002) Altered short-term synaptic plasticity in mice lacking the metabotropic glutamate receptor mGlu7. ScientificWorldJournal 2:730–737. 10.1100/tsw.2002.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaerts-Vegh Z, Beckers T, Ball SM, Baeyens F, Callaerts PF,F, Cryan J, Molnar E, Hooge R (2006) Concomitant deficits in working memory and fear extinction are functionally dissociated from reduced anxiety in metabotropic glutamate receptor 7-deficient mice. J Neurosci 26:6573–6582. 10.1523/JNEUROSCI.1497-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charng WL, Karaca E, Coban Akdemir Z, Gambin T, Atik MM, Gu S, Posey JE, Jhangiani SN, Muzny DM, Doddapaneni H, Hu J, Boerwinkle E, Gibbs RA, Rosenfeld JA, Cui H, Xia F, Manickam K, Yang Y, Faqeih EA, Al Asmari A, et al. (2016) Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med Genomics 9:42. 10.1186/s12920-016-0208-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Park JY, Park SP, Lee H, Han S, Park KH, Suh YH (2016) Regulation of mGluR7 trafficking by SUMOylation in neurons. Neuropharmacology 102:229–235. 10.1016/j.neuropharm.2015.11.021 [DOI] [PubMed] [Google Scholar]
- Cryan JF, Kelly PH, Neijt HC, Sansig G, Flor PJ, Van Der Putten H (2003) Antidepressant and anxiolytic-like effects in mice lacking the group III metabotropic glutamate receptor mGluR7. Eur J Neurosci 17:2409–2417. 10.1046/j.1460-9568.2003.02667.x [DOI] [PubMed] [Google Scholar]
- Dalezios Y, Luján R, Shigemoto R, Roberts JD, Somogyi P (2002) Enrichment of mGluR7a in the presynaptic active zones of GABAergic and non-GABAergic terminals on interneurons in the rat somatosensory cortex. Cereb Cortex 12:961–974. 10.1093/cercor/12.9.961 [DOI] [PubMed] [Google Scholar]
- Dev KK, Nakajima Y, Kitano J, Braithwaite SP, Henley JM, Nakanishi S (2000) PICK1 interacts with and regulates PKC phosphorylation of mGLUR7. J Neurosci 20:7252–7257. 10.1523/JNEUROSCI.20-19-07252.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N (2008) Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135:749–762. 10.1016/j.cell.2008.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumaz N, Marais R (2005) Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft für Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J 272:3491–3504. 10.1111/j.1742-4658.2005.04763.x [DOI] [PubMed] [Google Scholar]
- Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D, Sleiman PMA, Zhang H, Kim CE, Robison R, Lyon GJ, Flory JH, Bradfield JP, Imielinski M, Hou C, Frackelton EC, Chiavacci RM, Sakurai T, Rabin C, Middleton FA, et al. (2011) Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet 44:78–84. 10.1038/ng.1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher NM, Seto M, Lindsley CW, Niswender CM (2018a) Metabotropic glutamate receptor 7: a new therapeutic target in neurodevelopmental disorders. Front Mol Neurosci 11:387. 10.3389/fnmol.2018.00444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher NM, Gogliotti RG, Vermudez SAD, Stansley BJ, Conn PJ, Niswender CM (2018b) Genetic reduction or negative modulation of mGlu7 does not impact anxiety and fear learning phenotypes in a mouse model of MECP2 duplication syndrome. ACS Chem Neurosci 9:2210–2217. 10.1021/acschemneuro.7b00414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher NM, Gould RW, Gogliotti RG, McDonald AJ, Badivuku H, Chennareddy S, Buch AB, Moore AM, Jenkins MT, Robb WH, Lindsley CW, Jones CK, Conn PJ, Niswender CM (2020) Phenotypic profiling of mGlu(7) knockout mice reveals new implications for neurodevelopmental disorders. Genes Brain Behav 19:e12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gai X, Xie HM, Perin JC, Takahashi N, Murphy K, Wenocur AS, D'Arcy M, O'Hara RJ, Goldmuntz E, Grice DE, Shaikh TH, Hakonarson H, Buxbaum JD, Elia J, White PS (2012) Rare structural variation of synapse and neurotransmission genes in autism. Mol Psychiatry 17:402–411. 10.1038/mp.2011.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nikulina E, Mellado W, Filbin MT (2003) Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci 23:11770–11777. 10.1523/JNEUROSCI.23-37-11770.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddyn H, Callaerts-Vegh Z, D'Hooge R (2015) Functional dissociation of group III metabotropic glutamate receptors revealed by direct comparison between the behavioral profiles of knockout mouse lines. Int J Neuropsychopharmacol 18:pyv053. 10.1093/ijnp/pyv053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogliotti RG, Senter RK, Fisher NM, Adams J, Zamorano R, Walker AG, Blobaum AL, Engers DW, Hopkins CR, Daniels JS, Jones CK, Lindsley CW, Xiang Z, Conn PJ, Niswender CM (2017) mGlu7 potentiation rescues cognitive, social, and respiratory phenotypes in a mouse model of Rett syndrome. Sci Transl Med 9:eaai7459. 10.1126/scitranslmed.aai7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Liu W, Wei J, Yan Z (2012) Regulation of N-methyl-D-aspartic acid (NMDA) receptors by metabotropic glutamate receptor 7. J Biol Chem 287:10265–10275. 10.1074/jbc.M111.325175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N (2008) A translational profiling approach for the molecular characterization of CNS cell types. Cell 135:738–748. 10.1016/j.cell.2008.10.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Kulicke R, Fenster RJ, Greengard P, Heintz N (2014) Cell type-specific mRNA purification by translating ribosome affinity purification (TRAP). Nat Protoc 9:1282–1291. 10.1038/nprot.2014.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher C, Schmid S, Pilz PKD, Sansig G, van der Putten H, Plappert CF (2004) Lack of the metabotropic glutamate receptor subtype 7 selectively impairs short-term working memory but not long-term memory. Behav Brain Res 154:473–481. 10.1016/j.bbr.2004.03.015 [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Bruno V, Salvatore L, Melchiorri D, Gradini R, Caricasole A, Barletta E, De Blasi A, Nicoletti F (2002) Native group-III metabotropic glutamate receptors are coupled to the mitogen-activated protein kinase/phosphatidylinositol-3-kinase pathways. J Neurochem 82:216–223. 10.1046/j.1471-4159.2002.00929.x [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Capobianco L, Iula M, Di Giorgi Gerevini V, Picascia A, Blahos J, Melchiorri D, Nicoletti F, De Blasi A (2004) Regulation of mGlu4 metabotropic glutamate receptor signaling by type-2 G-protein coupled receptor kinase (GRK2). Mol Pharmacol 65:1103–1110. 10.1124/mol.65.5.1103 [DOI] [PubMed] [Google Scholar]
- Iacovelli L, Felicioni M, Nisticò R, Nicoletti F, De Blasi A (2014) Selective regulation of recombinantly expressed mGlu7 metabotropic glutamate receptors by G protein-coupled receptor kinases and arrestins. Neuropharmacology 77:303–312. 10.1016/j.neuropharm.2013.10.013 [DOI] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, Smith JD, Paeper B, Nickerson DA, Dea J, Dong S, Gonzalez LE, Mandell JD, Mane SM, Murtha MT, Sullivan CA, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan-Sakrikar N, Field JR, Klar R, Mattmann ME, Gregory KJ, Zamorano R, Engers DW, Bollinger SR, Weaver CD, Days EL, Lewis LM, Utley TJ, Hurtado M, Rigault D, Acher F, Walker AG, Melancon BJ, Wood MR, Lindsley CW, Conn PJ, et al. (2014) Identification of positive allosteric modulators VU0155094 (ML397) and VU0422288 (ML396) reveals new insights into the biology of metabotropic glutamate receptor 7. ACS Chem Neurosci 5:1221–1237. 10.1021/cn500153z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Yan Z, Feng J (2006) Activation of group III metabotropic glutamate receptors attenuates rotenone toxicity on dopaminergic neurons through a microtubule-dependent mechanism. J Neurosci 26:4318–4328. 10.1523/JNEUROSCI.0118-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, De Camilli P (2002) Dynamin at actin tails. Proc Natl Acad Sci USA 99:161–166. 10.1073/pnas.012607799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Park S, Lee H, Han S, Song JM, Han D, Suh YH (2019) Nedd4 E3 ligase and beta-arrestins regulate ubiquitination, trafficking, and stability of the mGlu7 receptor. Elife 8:e44502. 10.7554/eLife.44502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichnerova K, Kaniakova M, Park SP, Skrenkova K, Wang YX, Petralia RS, Suh YH, Horak M (2015) Two N-glycosylation sites in the GluN1 subunit are essential for releasing N-methyl-d-aspartate (NMDA) receptors from the endoplasmic reticulum. J Biol Chem 290:18379–18390. 10.1074/jbc.M115.656546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang Y, Zhao D, Dong R, Yang X, Tammimies K, Uddin M, Scherer SW, Gai Z (2015) Rare de novo deletion of metabotropic glutamate receptor 7 (GRM7) gene in a patient with autism spectrum disorder. Am J Med Genet B Neuropsychiatr Genet 168b:258–264. 10.1002/ajmg.b.32306 [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295:868–872. 10.1126/science.1067081 [DOI] [PubMed] [Google Scholar]
- Marafi D, Mitani T, Isikay S, Hertecant J, Almannai M, Manickam K, Abou Jamra R, El-Hattab AW, Rajah J, Fatih JM, Du H, Karaca E, Bayram Y, Punetha J, Rosenfeld JA, Jhangiani SN, Boerwinkle E, Akdemir ZC, Erdin S, Hunter JV, et al. (2020) Biallelic GRM7 variants cause epilepsy, microcephaly, and cerebral atrophy. Ann Clin Transl Neurol 7:610–627. 10.1002/acn3.51003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín R, Torres M, Sánchez-Prieto J (2007) mGluR7 inhibits glutamate release through a PKC-independent decrease in the activity of P/Q-type Ca2+ channels and by diminishing cAMP in hippocampal nerve terminals. Eur J Neurosci 26:312–322. 10.1111/j.1460-9568.2007.05660.x [DOI] [PubMed] [Google Scholar]
- Martín R, Durroux T, Ciruela F, Torres M, Pin JP, Sánchez-Prieto J (2010) The metabotropic glutamate receptor mGlu7 activates phospholipase C, translocates munc-13-1 protein, and potentiates glutamate release at cerebrocortical nerve terminals. J Biol Chem 285:17907–17917. 10.1074/jbc.M109.080838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masugi M, Yokoi M, Shigemoto R, Muguruma K, Watanabe Y, Sansig G, van der Putten H, Nakanishi S (1999) Metabotropic glutamate receptor subtype 7 ablation causes deficit in fear response and conditioned taste aversion. J Neurosci 19:955–963. 10.1523/JNEUROSCI.19-03-00955.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noroozi R, Taheri M, Movafagh A, Mirfakhraie R, Solgi G, Sayad A, Mazdeh M, Darvish H (2016) Glutamate receptor, metabotropic 7 (GRM7) gene variations and susceptibility to autism: a case-control study. Autism Res 9:1161–1168. 10.1002/aur.1640 [DOI] [PubMed] [Google Scholar]
- Okamoto N, Hori S, Akazawa C, Hayashi Y, Shigemoto R, Mizuno N, Nakanishi S (1994) Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J Biol Chem 269:1231–1236. [PubMed] [Google Scholar]
- Palazzo E, Marabese I, de Novellis V, Rossi F, Maione S (2016) Metabotropic glutamate receptor 7: from synaptic function to therapeutic implications. Curr Neuropharmacol 14:504–513. 10.2174/1570159x13666150716165323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park DH, Park S, Song JM, Kang M, Lee S, Horak M, Suh YH (2020) N-linked glycosylation of the mGlu7 receptor regulates the forward trafficking and transsynaptic interaction with Elfn1. FASEB J 34:14977–14996. [DOI] [PubMed] [Google Scholar]
- Park S, Jung SW, Kim BN, Cho SC, Shin MS, Kim JW, Yoo HJ, Cho DY, Chung US, Son JW, Kim HW (2013) Association between the GRM7 rs3792452 polymorphism and attention deficit hyperacitiveity disorder in a Korean sample. Behav Brain Funct 9:1. 10.1186/1744-9081-9-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ (2005) mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron 46:89–102. 10.1016/j.neuron.2005.02.011 [DOI] [PubMed] [Google Scholar]
- Perroy J, Prezeau L, De Waard M, Shigemoto R, Bockaert J, Fagni L (2000) Selective blockade of P/Q-type calcium channels by the metabotropic glutamate receptor type 7 involves a phospholipase C pathway in neurons. J Neurosci 20:7896–7904. 10.1523/JNEUROSCI.20-21-07896.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perroy J, El Far O, Bertaso F, Pin JP, Betz H, Bockaert J, Fagni L (2002) PICK1 is required for the control of synaptic transmission by the metabotropic glutamate receptor 7. EMBO J 21:2990–2999. 10.1093/emboj/cdf313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT (2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34:895–903. 10.1016/s0896-6273(02)00730-4 [DOI] [PubMed] [Google Scholar]
- Reuter MS, Tawamie H, Buchert R, Hosny Gebril O, Froukh T, Thiel C, Uebe S, Ekici AB, Krumbiegel M, Zweier C, Hoyer J, Eberlein K, Bauer J, Scheller U, Strom TM, Hoffjan S, Abdelraouf ER, Meguid NA, Abboud A, Al Khateeb MA, et al. (2017) Diagnostic yield and novel candidate genes by exome sequencing in 152 consanguineous families with neurodevelopmental disorders. JAMA Psychiatry 74:293–299. 10.1001/jamapsychiatry.2016.3798 [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, et al. (2012) De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485:237–241. 10.1038/nature10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansig G, Bushell TJ, Clarke VR, Rozov A, Burnashev N, Portet C, Gasparini F, Schmutz M, Klebs K, Shigemoto R, Flor PJ, Kuhn R, Knoepfel T, Schroeder M, Hampson DR, Collett VJ, Zhang C, Duvoisin RM, Collingridge GL, van Der Putten H (2001) Increased seizure susceptibility in mice lacking metabotropic glutamate receptor 7. J Neurosci 21:8734–8745. 10.1523/JNEUROSCI.21-22-08734.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD (2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299:12–20. [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM (2010) Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science 327:547–552. 10.1126/science.1179735 [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Kulik A, Roberts JD, Ohishi H, Nusser Z, Kaneko T, Somogyi P (1996) Target-cell-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature 381:523–525. 10.1038/381523a0 [DOI] [PubMed] [Google Scholar]
- Skrenkova K, Lee S, Lichnerova K, Kaniakova M, Hansikova H, Zapotocky M, Suh YH, Horak M (2018) N-glycosylation regulates the trafficking and surface mobility of GluN3A-containing NMDA receptors. Front Mol Neurosci 11:188. 10.3389/fnmol.2018.00188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppel LJ, Auerbach BD, Senter RK, Preza AR, Lefkowitz RJ, Bear MF (2017) β-Arrestin2 couples metabotropic glutamate receptor 5 to neuronal protein synthesis and is a potential target to treat fragile X. Cell Rep 18:2807–2814. 10.1016/j.celrep.2017.02.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Pelkey KA, Lavezzari G, Roche PA, Huganir RL, McBain CJ, Roche KW (2008) Corequirement of PICK1 binding and PKC phosphorylation for stable surface expression of the metabotropic glutamate receptor mGluR7. Neuron 58:736–748. 10.1016/j.neuron.2008.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Park JY, Park S, Jou I, Roche PA, Roche KW (2013) Regulation of metabotropic glutamate receptor 7 (mGluR7) internalization and surface expression by Ser/Thr protein phosphatase 1. J Biol Chem 288:17544–17551. 10.1074/jbc.M112.439513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YH, Chang K, Roche KW (2018) Metabotropic glutamate receptor trafficking. Mol Cell Neurosci 91:10–24. 10.1016/j.mcn.2018.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turlova E, Bae CYJ, Deurloo M, Chen W, Barszczyk A, Horgen FD, Fleig A, Feng ZP, Sun HS (2016) TRPM7 regulates axonal outgrowth and maturation of primary hippocampal neurons. Mol Neurobiol 53:595–610. 10.1007/s12035-014-9032-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Wang H, Luo Y, Jia LJ, Zhao JN, Zhang HH, Ma ZW, Xue QS, Yu BW (2012) The effects of metabotropic glutamate receptor 7 allosteric agonist N,N'-dibenzhydrylethane-1,2-diamine dihydrochloride on developmental sevoflurane neurotoxicity: role of extracellular signal-regulated kinase 1 and 2 mitogen-activated protein kinase signaling pathway. Neuroscience 205:167–177. 10.1016/j.neuroscience.2011.12.039 [DOI] [PubMed] [Google Scholar]
- Washbourne P (2015) Synapse assembly and neurodevelopmental disorders. Neuropsychopharmacology 40:4–15. 10.1038/npp.2014.163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Liu Y, Jiao J (2015) GRM7 regulates embryonic neurogenesis via CREB and YAP. Stem cell reports 4:795–810. 10.1016/j.stemcr.2015.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada RX, Matsuki N, Ikegaya Y (2005) cAMP differentially regulates axonal and dendritic development of dentate granule cells. J Biol Chem 280:38020–38028. 10.1074/jbc.M503800200 [DOI] [PubMed] [Google Scholar]
- Yang Y, Pan C (2013) Role of metabotropic glutamate receptor 7 in autism spectrum disorders: a pilot study. Life Sci 92:149–153. 10.1016/j.lfs.2012.11.010 [DOI] [PubMed] [Google Scholar]