

Abstract
One of the fundamental questions in bone biology is where osteoblasts originate and how osteoblast differentiation is regulated. The mechanism underlying which factors regulate chondrocyte to osteoblast lineage commitment remains unknown. Our data showed that Runt-related transcription factor 1 (Runx1) is expressed at different stages of both chondrocyte and osteoblast differentiation. Runx1 chondrocyte-specific knockout (Runx1f/fCol2α1-cre) mice exhibited impaired cartilage formation, decreased bone density, and an osteoporotic phenotype. The expressions of chondrocyte differentiation regulation genes, including Sox9, Ihh, CyclinD1, PTH1R, and hypertrophic chondrocyte marker genes including Col2α1, Runx2, MMP13, Col10α1 in the growth plate were significantly decreased in Runx1f/fCol2α1-cre mice chondrocytes. Importantly, the expression of osteoblast differentiation regulation genes including Osx, Runx2, ATF4, and osteoblast marker genes including osteocalcin (OCN) and osteopontin (OPN) were significantly decreased in the osteoblasts of Runx1f/fCol2α1-cre mice. Notably, our data showed that osteoblast differentiation regulation genes and marker genes are also expressed in chondrocytes and the expressions of these marker genes were significantly decreased in the chondrocytes of Runx1f/fCol2α1-cre mice. Our data showed that Chromatin immunoprecipitation (ChIP) and promoter mapping analysis revealed that Runx1 directly binds to the Indian hedgehog homolog (Ihh) promoter to regulate its expression, indicating that Runx1 directly regulates the transcriptional expression of chondrocyte genes. Collectively, we revealed that Runx1 signals chondrocyte to osteoblast lineage commitment and promotes endochondral bone formation through enhancing both chondrogenesis and osteogenesis genes expressions, indicating Runx1 may be a therapeutic target to enhance endochondral bone formation and prevent osteoporosis fractures.
Keywords: Runx1, chondrocyte, endochondral bone formation, osteoblast, Ihh
INTRODUCTION
One of the fundamental questions in bone biology is where osteoblasts originate and how osteoblast differentiation is regulated. Ono et al. reported that a subset of chondrogenic cells provides early mesenchymal progenitors in growing bones [1], indicating an important role of chondrogenic cells in endochondral bone formation. However, the mechanism underlying which factors regulate chondrocyte to osteoblast lineage commitment and the function of the mesenchymal progenitors in endochondral bone formation remain unknown. Our data showed that Runt-related transcription factor 1 (Runx1) is expressed at different stages of both chondrocyte and osteoblast differentiation, which led us hypothesize that Runx1 may be the factor that controls chondrocyte to osteoblast lineage commitment and the function of the mesenchymal progenitors in endochondral bone formation.
Proper skeletal formation and growth are strictly dependent on the orchestration of all the processes participating in endochondral bone formation at the growth plate. Endochondral ossification begins with mesenchymal cells condensations, and cells undergo differentiation into cartilage anlagen, followed by the steps of chondrocytes proliferation, differentiation, and apoptosis [2]. These processes are strictly regulated by different growth factors and transcription factors such as Sox9, Runx2, Ihh, PTH1R, and Bmps [2, 3].
Runx1, also known as AML1 and cbfa2, belongs to the Runt-Related Transcription Factor family, which is comprised of Runx1, Runx2, and Runx3. All three members share a highly homology DNA binding sequence Runt, and are expressed in a variety of tissues. They play different roles in the development process [4–7]. Runx2 is required for osteogenesis and is associated with cleidocranial dysplasia [8]. Runx3, an important factor in nerve cell differentiation, cooperates with Runx2 to regulate chondrocyte proliferation and hypertrophy [9–11]. Runx1 has been widely studied as the regulator in the hematopoietic development, and genetic ablation of Runx1 leads to lethality of newborn homozygous mice. In recent years, Runx1 has been reported to be linked with the lineage direction of the undifferentiated cells in the calvarial sutures, periosteum, and postnatal perichondrium, and may contribute to the earliest stages of skeletogenesis [12]. In vitro experiments also confirmed that Runx1 is important in the early stages of chondrocyte differentiation [12, 13]. Studies using stage specific Runx1 deficient mice showed Runx1 cooperatively with Runx2 regulates sternal morphogenesis and the commitment of mesenchymal cells to chondrocytes [14, 15]. However, the role of Runx1 in postnatal chondrocyte and osteoblast differentiation and endochondral bone formation remains unclear.
To test our hypothesis of the role of Runx1 in regulating chondrocyte to osteoblast lineage commitment and the function of the mesenchymal progenitors in endochondral bone formation, we generated chondrocyte-specific Runx1-deficient mice (Runx1f/fCol2α1-cre mice) by crossing Col2α1-cre mice with Runx1f/f mice. These mutant mice survived to adulthood and displayed compromised endochondral bone formation including delayed development of the vertebra and long bones, inhibited chondrocyte and osteoblast differentiation, and a decrease in the length of the hypertrophic zone, indicating that Runx1 deletion in chondrocytes and osteoblasts impaired growth plate development. Chromatin immunoprecipitation (ChIP) and promoter analysis revealed that Runx1 directly binds to the promoter regions of Ihh to regulate its expression, indicating that Runx1 can directly regulate the transcriptional expression of chondrocyte marker genes and osteoblast marker genes. We revealed that Runx1 governs chondrocyte to osteoblast lineage commitment and promotes endochondral bone formation through enhancing both chondrogenesis and osteogenesis genes expressions, indicating Runx1 may be a therapeutic target for the treatment of bone diseases such as osteoporosis.
MATERIALS AND METHODS
Generation of Runx1f/fCol2α1-cre mice
Runx1f/f mice on a C57/B6L background were purchased from Jackson Laboratory, and the Col2α1-Cre mouse line on a C57/B6L background was kindly provided by Dr. Rosa Serra from The University of Alabama at Birmingham (UAB) (Birmingham, AL, USA). Runx1f/f mice were crossed with Col2α1-cre mice to get Runx1f/+, Col2α1-cre, which were intercrossed to get homozygous Runx1f/fCol2α1-cre mice. All mice were maintained under a 12-hour light–dark cycle with ad libitum access to food and water at the UAB Animal Facility. The study was approved by the UAB Institutional Animal Care and Use Committee and conformed to NIH guidelines, and followed all recommendations of ARRIVE (Animal Research: Reporting in Vivo Experiments) guidelines. Animals were euthanized by Carbon dioxide inhalation anesthesia.
Radiographic Procedures
For X-ray analysis, radiography was performed using the Faxitron Model MX-20 at 26 kV by the University of Alabama at Birmingham (UAB) Small Animal Bone Phenotyping Core associated with the Center for Metabolic Bone Disease. The microcomputed tomography analysis was performed to determine the bone mass of fixed femurs by the UAB Small Animal Bone Phenotyping Core associated with the Center for Metabolic Bone Disease.
uCT Analysis
Excised mouse humerus and femurs were scanned using the Scanco CT40 desktop cone-beam micro–CT (mCT) scanner (Scanco Medical AG, Bruttisellen, Switzerland). The trabecular bone scanning was performed from the growth plate (310 slices at 12mm per slice) analyzed using the CT Evaluation Program (v5.0A; Scanco Medical). The scanning and analysis of the cortical bone were performed at the midshaft of the femur and consisted of 25 slices (12 mm per slice).
Skeletal staining
For skeletal preparations, mice were skinned, eviscerated, fixed in 95% (vol/vol) ethanol, cleared in acetone, stained with Alizarin red and/or Alcian blue stains, and sequentially cleared in 1% KOH. Cartilage and mineralized bone were characterized by different colors (blue and red, respectively) after the stain, according to standard protocols [16].
Bone tissue preparation
Femurs and tibiae of mice were harvested, skinned, and fixed in 4% (wt/vol) paraformaldehyde overnight. Samples were then dehydrated in ethanol solution and decalcified in 10% (wt/vol) EDTA for 1–4 week(s). For paraffin sections, samples were dehydrated in ethanol, cleared in xylene, embedded in paraffin, and sectioned at 6 μm with Leica microtome and mounted on Superfrost Plus slides (Fisher). For frozen sections, samples were infiltrated in 30% (wt/vol) sucrose, embedded in optimal cutting temperature compound, sectioned at 8 μm with a freezing microtome, and affixed to Superfrost Plus Gold slides (Fisher). Histological analysis was performed including staining with Von Kossa, Picro Sirius Red (ab150681), Safranin O, ALP and hematoxylin/eosin (H&E) stains using paraffin sections.
Immunohistochemistry (IHC)
6-μm paraffin sections were deparaffined and antigens retrieval was achieved by heat treatment with a commercial reagent (Abcam AB970). Immunohistochemistry staining was performed using a commercial kit (vector laboratories, Mouse on Mouse Basic kit CAT#BMK-2202). Primary antibodies: rabbit-anti-Col X (Abcam, ab58632), mice-anti-col2 (Santa cruz, sc-52658), rabbit-anti-mmp13 (Abcam, ab39012), rabbit-anti-Opn (osteopontin) (Abcam, ab8448), anti-Cbfβ (Santa Cruz, sc-56751), rabbit-anti-Runx1 (Abcam, ab23980), rabbit-anti-Runx2 (Abcam, ab23981), rabbit-anti-Osx (Osterix) (Abcam, ab22552). The procedure was following the manufacturer’s instructions. Slides were counterstained by hematoxylin.
Immunofluorescence analysis
Osteoblast and chondrocyte genes were analyzed by immunofluorescence using the following primary antibodies: mice-anti-PTHrP-R (Santa Cruz, sc-12722), rabbit-anti-Ihh (Abcam, ab39634), Rabbit-anti-cyclinD1 (Santa cruz, sc-8396) and these secondary antibodies: FITC-goat-anti-mouse IgG(H+L) and TR-goat-anti-rabbit IgG (H+L). Imaging was taken by Leica Confocal Microscope and Zeiss fluorescent microscope.
Quantitative Real-time PCR analysis
mRNA was extracted from cultured by using TRIzol (Invitrogen) and then reverse transcribed into cDNA according to the manufacturer’s manual (qScript cDNA Synthesis Kit, Quanta Biosciences Inc.). Genes were analyzed by quantitative real-time PCR (qRT-PCR) using the StepOne Real-Time PCR System (Life Technologies). Gapdh and Hprt were used as internal controls. The primer sequences are shown in Table S1.
Western blots assay
Proteins samples were loaded on SDS-PAGE and electric transferred to nitrocellulose membranes. Runx1, Ihh, PTH1R, Col2α1, Col10α1, Opn, Ocn, Runx2, and CBFβ protein levels were analyzed using GAPDH as a loading control, Primary antibodies are the antibodies used in the IHC and IF stain. Secondary blotting was performed using horseradish peroxidase-linked anti-rabbit IgG (7074) and horseradish peroxidase-linked anti-mouse IgG (7076) (Cell signaling).
Primary cell culture and ATDC5 cell transfection
Chondrocytes from the mice knee joint were harvested as described [1, 17] and micromass cultures- Plate cells as 10 μL micromass drop in a 24-mm tissue culture well and incubate at 5% CO2, 37°C in a humidified tissue culture incubator for 1.5 or 2 h to allow cell attachment- were applied for 7-day and 14-day [18]. Alcian blue staining was used to detect the chondrogenesis. Calvarial cells were isolated from newborn mice and seeded in cell culture dishes at a density of 3×103 cells/cm2, as previously described [19], and ALP and oil-red staining was conducted in cultured dishes as previously descried [20]. Von Kossa staining was operated in cultured wells as previously descried. For retrovirus production, pMXs vectors was transfected into 293GPG cells and for lentivirus production using a calcium phosphate co-precipitation method and retrovirus supernatant were harvested between 48–96 h [21]. And retrovirus titers were determined by transfecting HEK293T cells with serial dilutions of virus supernatant. ATDC5 cells were transduced with virus supernatant in the presence of 8 μg ml−1 polybrene (Sigma) for 24 h before induced with chondrogenesis induction medium.
Adipogenesis Assays
Confluent cultures of primary calvarial cells were subjected to adipogenic medium containing 0.1 μM dexamethasone, 50 μM indomethacin, and 5 μg/ml insulin for 14 days. The progression of adipogenesis was monitored under light microscope. At the end of culture period, cells were stained for lipid droplets using Oil Red-O stain as described [20].
Chromatin Immunoprecipitation
Chondrocytes were induced from ATDC5 cell line for 7 days and Chromatin immunoprecipitation (ChIP) was performed as previously described [22]. After immunoprecipitation using monoclonal anti-Runx1 antibody (ab23980; Abcam) and DNA extraction, quantitative PCR was performed using the primers in the promoter region of the mouse gene Ihh. Primer sequences are presented in the Supplementary Table S2.
Promoter Analyses.
Ihh promoter sequences were analyzed for putative Runx binding sites with PROMO3.0 (http://alggen.lsi.upc.es/) using version 8.3 of the TRANSFAC database. The promoter region (−) and (+) of the mouse Ihh gene was amplified by PCR from a murine Ihh BAC clone (CH29–567C21; CHORI). Primer sequences are available in Table S3. Then the promoter regions were inserted into the pGL3-basic vector to construct the pGL3-Ihh promoter fragments. ATDC5 cells (Sigma) were cultured in 24-well plates, were transfected with the DNA mixture containing different pGL3-Ihh construct (0.3μg) and β-GAL-expressing plasmids (0.06 μg), with or without Runx1 expressing vector (psport6-CMV-Runx1, 0.3μg) using a calcium phosphate co-precipitation method. Luciferase was detected using Glo Luciferase Assay System (Promega) as described [20, 22]. The β-GAL activity of the cell lysates was analyzed using β-Galactosidase Enzyme Assay System (E2000; Promega). The level of luciferase activity was normalized to the level of β-GAL activity.
Statistical analysis
Data were presented as mean ± SD (n>3). Student’s t-test was used to assess the statistical significance, values were considered statistically significant at P < 0.05. Results are representative of at least four individual experiments. Figures are representative of the data.
RESULTS
Chondrocyte-specific deficiency of Runx1 caused dwarfism, decreased body weight, and decreased bone density in mutant mice.
In order to study the function of Runx1 in chondrocyte differentiation and endochondral bone formation, we crossed Runx1f/f mice with Col2α1-cre transgenic mice to generate the Runx1f/fCol2α1-cre mutant mice. Genotyping was confirmed by PCR (Fig. 1A). Runx1f/fCol2-cre mice had shorter stature and lower body weight compared to Runx1f/f and Col2α1-cre controls (Fig. 1B–E). The growth curves of murine weight revealed that the weights of the newborn mutant mice were only slightly lower than the littermate controls, however, after 2 months, the mutant mice body weight remained significantly lower than that of the control groups (Fig. 1E). Furthermore, the body length of mutant mice was significantly shorter than that of WT mice from the same litter beginning from 8 weeks (Fig. 1D). X-ray examination the long bones of 4-week, 8-week, and 6-month old mice showed Runx1f/fCol2α1-cre mice had a significant reduction in ossification, bone density, and trabecular thickness (Fig. 1F). To determine the function of Runx1 in trabecular bone formation, we performed Micro computed tomography (μCT) analysis of 4-week-old murine femurs to analyze the differences of bone structural parameters, which demonstrated that in Runx1f/fCol2α1-cre mice, the ratio of bone volume/tissue volume (BV/TV) was decreased by 42%, the trabecular bone numbers (Tb. N) were decreased by 30%, trabecular bone thickness (Tb. Tn) was reduced by 42%, and connectivity density (Conn. D) was decreased by 13% compared to control mice (Fig. 1G, H). Taken together, these results demonstrated that Runx1 deficiency in chondrocytes and osteoblasts results in dwarfism, weight and body length reduction, and a significant decrease in bone density during postnatal skeletal development.
Figure 1. Deficiency of Runx1 in chondrocytes and osteoblasts caused the dwarfism, decreased body weight, and decreased bone density in mutant mice.
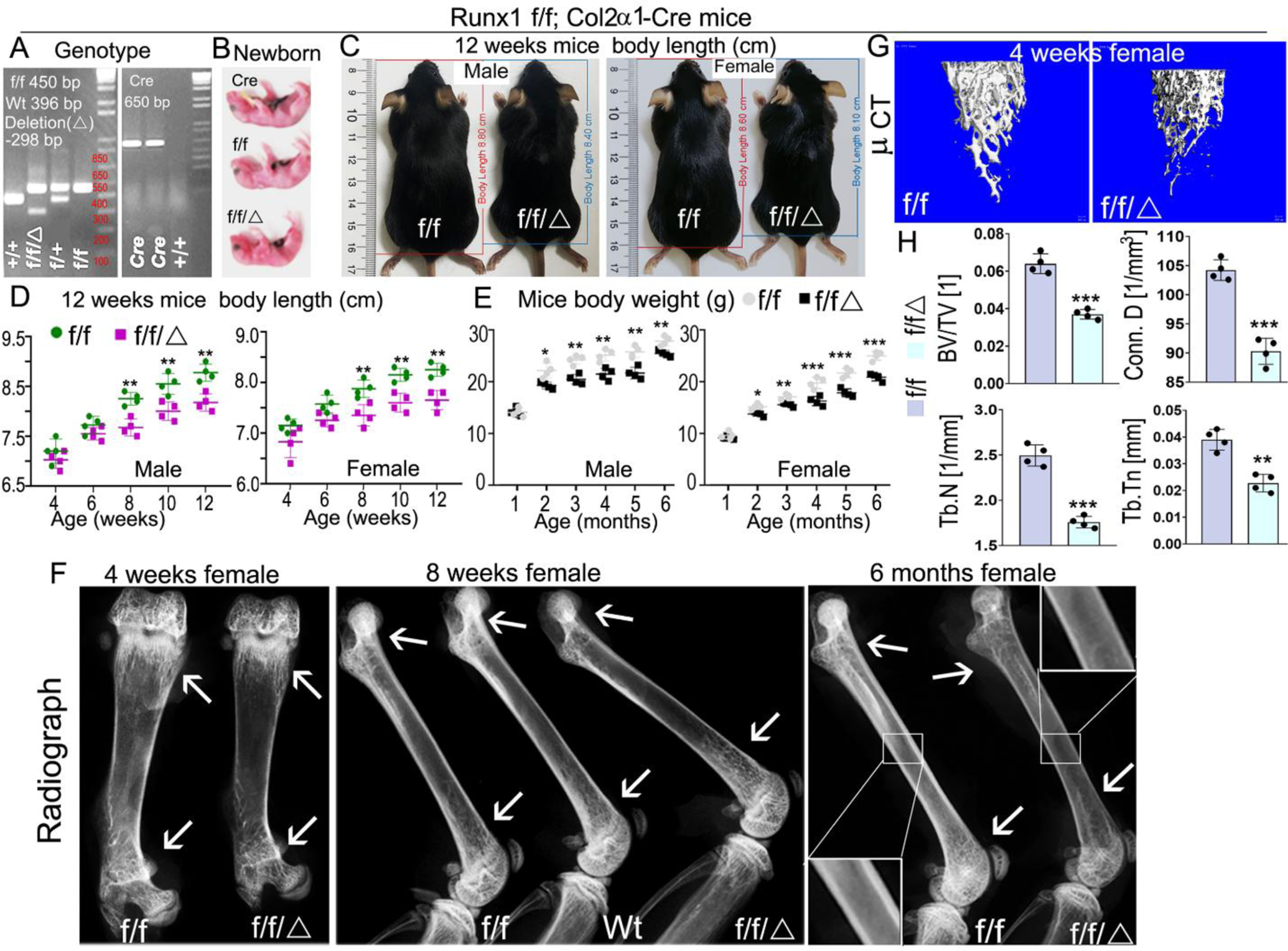
(A) PCR genotyping images of Runx1 f/f and Runx1f/fCol2α1-cre mice. f, Runx1 allele carrying loxP sites; +, WT Runx1 allele; Δ, Runx1 allele with loxP floxed sequence deleted. (B) Gross appearances of newborn Runx1f/fCol2α1-cre mice and littermates. (C) Measurement of mice body lengths from 12-week-old Runx1f/fCol2α1-cre and Runx1f/f mice. (D) Quantification of mouse body lengths from 4-weeks-old to 12-weeks-old. (E) Quantification of mouse body weights from 1-month-old to 6-months-old. (F) X-ray analysis long bones from 4-week, 8-week, and 6-month-old Runx1f/fCol2α1-cre mice and littermate controls (f/f), white arrows referred to cortical bone and trabecular bone density differences between wt and mutant mice. (G) Micro computed tomography (μCT) examination was performed to determine the bone formation in 4-week murine femurs. (H) Quantification data of bone volume/tissue volume (BV/TV), the trabecular bone numbers (Tb. N), trabecular bone thickness (Tb. Tn), and connectivity density (Conn. D) in G. All data are presented as mean ± SD, n=4, NS denotes not significant, *p<0.05, **p<0.01, ***p<0.001. f/f denotes Runx1f/f; f/f/Δ denotes Runx1f/fCol2α1-cre.
Postnatal Runx1f/fCol2α1-cre mice display impaired trabecular bone formation and deformed growth plates.
Alizarin red & Alcian blue staining of newborn, 1-week-old, and 2-week-old mice skeletons revealed that multiple parts of the Runx1f/fCol2α1-cre mice skeletons displayed delayed endochondral bone formation (Fig. S1A). The body length of mutant mice was shorter than the control groups (Fig. S1A, left panel), the vertebra in the mutant mice were less developed (Fig. S1A, middle panel), as shown by the red and yellow arrows, while the long bones of mutant mice were shorter and smaller than those in the control group (Fig. S1A, B). These data indicate that deficiency of Runx1 may result in delayed development of murine cartilage and long bones seen in Runx1f/fCol2α1-cre mice. In order to determine the whether the delayed bone formation in the mutant mice was the result of decreased trabecular bone formation, hematoxylin and eosin (H&E) staining was performed on the newborn, 3-week old, and 6-month-old Runx1f/fCol2α1-cre and Runx1f/f murine femurs. The results showed that trabecular bone formation in the mutant bone marrow was significantly decreased as compared with that in the control groups. Unexpectedly, adipocytes were greatly accumulated in the mutant bone marrow as show in both the 3-week-old and 6-month-old murine femur sections (Fig. 2B; Fig. S2F (blue arrows)). This result was confirmed by Von Kossa staining of the newborn and 3-week-old femurs and vertebra, which demonstrated that the mineral deposition was decreased by 50% in the mutant bones as compared with that in the control groups (Fig. 2C, E; Fig. S2C). Picro Sirius red staining of newborn femurs and 1-month-old mice tibias showed that collagen I expression level was dramatically reduced in the mutant bones compared to the control groups (Fig. S2A, B), which further demonstrates that the bone formation was compromised in mutant mice. Alkaline phosphatase (ALP) staining was also performed on 3-week-old and 6-month-old murine long bones to observe the differences in osteoblast differentiation between the two groups, which showed that the osteoblast activity in mutant long bones was decreased by 77% as compared with that in the control group (Fig. 2D, E; Fig. S2D). Immunohistochemistry (IHC) staining was performed on the newborn cartilage to examine Runx1 expression in murine growth plates. The results demonstrated that Runx1 is expressed in the proliferation zone (PZ) and hypertrophic zone (HZ); Runx1 deletion in chondrocytes and osteoblasts lead to significantly decreased expression of Runx1 in the proliferation zone, hypertrophic zone, and trabecular bone (Fig. 2F, H). Safranin O staining was performed on the newborn, postnatal day 9, 3-week-old, and 6-month-old murine tibias and the results showed the mutant mice had a significant reduction in the width of the epiphyseal plate (Fig. S2E, yellow outlined region), with shorter hypertrophic zones compared to littermate controls (Fig. 2G, I), which resulted in the malformed growth plates in the mutant mice long bones. Moreover, the hypertrophic chondrocytes in the mutant mice growth plates showed irregular arrangements and failed to organize into well-formed columns compared to those in the control groups (Fig. 2G). Collectively, our results demonstrated that Runx1 deficiency in chondrocytes led to the decreased postnatal cartilage and long bone formation, which may result from the impaired trabecular bone formation and deformed growth plate in the mutant mice.
Figure 2. Postnatal Runx1f/fCol2α1-cre mice display impaired trabecular bone formation and deformed growth plates.
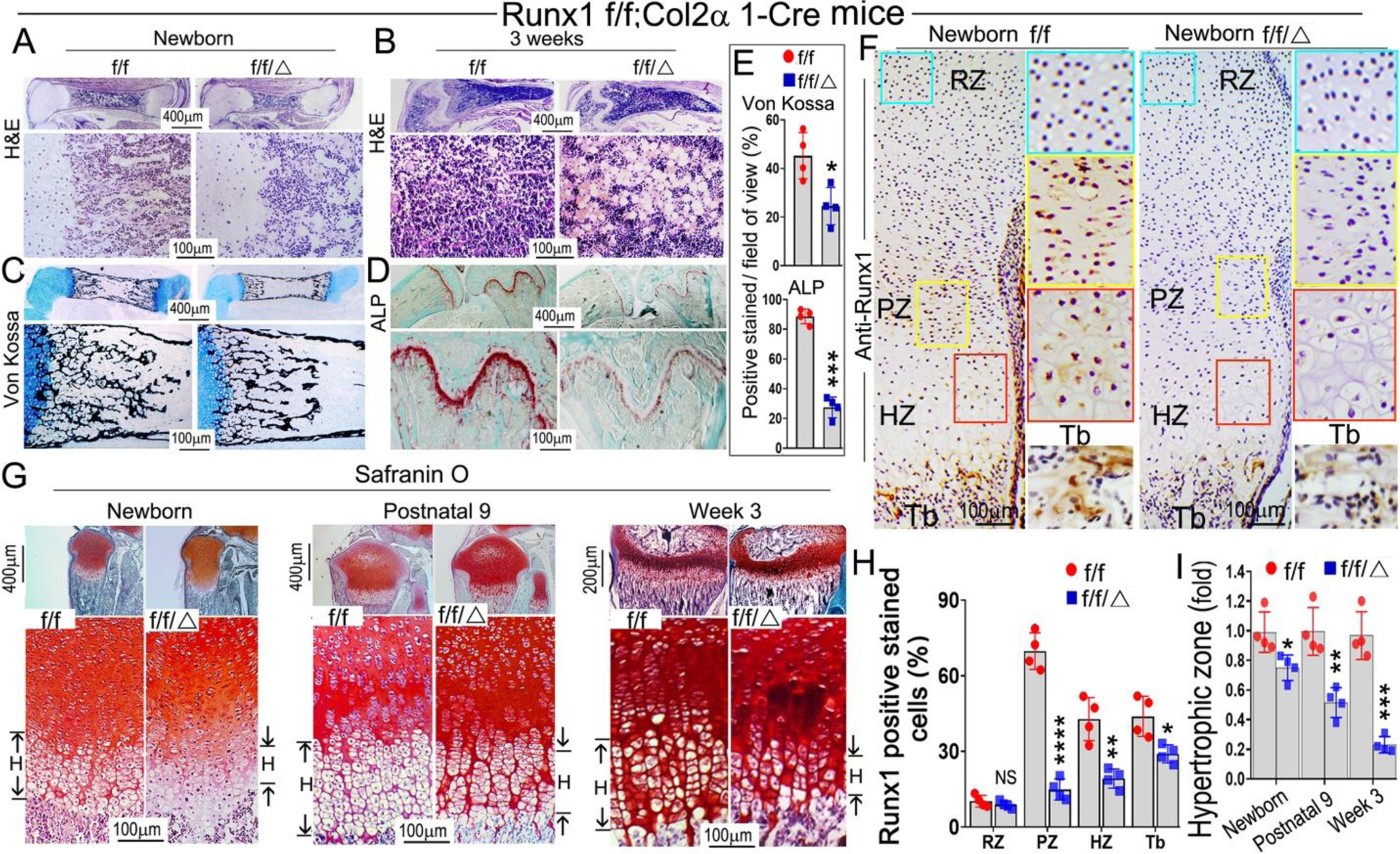
(A, B) Hematoxylin and eosin (H&E) staining of (A) newborn and (B) 3-week-old murine Runx1f/fCol2α1-cre and Runx1f/f femurs. (C) Von Kossa staining of the newborn murine femurs. (D) ALP staining of 3-week-old murine femurs and tibias. (E) Quantification of (C) Von Kossa staining and (D) ALP staining. (F) Immunohistochemistry (IHC) staining of newborn wild type and Runx1f/fCol2α1-cre mice femur sections by using anti-Runx1 antibodies to detect the expression of Runx1 in the growth plates. RZ, resting zone; PZ, proliferation zone; HZ, hypertrophic zone; Tb, trabecular bone. (G) Safranin O staining of the tibias from newborn, postnatal 9, and 3-week old mice. H denotes hypertrophic zone. (H) Quantification of Runx1 positive staining cells in (F). (I) Measurement the length of the hypertrophic zone in (G). All data are presented as mean ± SD, n=6, NS denotes not significant, *p<0.05, **p<0.01, ***p<0.001.
Runx1 deficiency in chondrocytes leads to the decreased expression of chondrocyte hypertrophy genes.
The results above showed Runx1f/fCol2α1-cre mice had shorter hypertrophic zones in the growth plate. To investigate the effects of Runx1 deletion on the chondrocytes in murine cartilage, we performed PCNA staining on 3-week old mice femurs (Fig. 3C). The results showed PCNA positive cells were decreased by 76% in the mutant growth plates compared with control (Fig. 3C, G). In order to identify the role of Runx1 in chondrocyte differentiation, we performed IHC staining by using anti-Runx2, anti-collagen II (Col2α1), anti-collagen X (Col10α1), and anti-MMP13 on the growth plates of 3-week old mice femurs (Fig. 3B, D–F). We confirmed conditional deletion of Runx1 by 80% in 3-week-old mutant mice (Fig. 3A, G). Col2α1, which is mostly expressed in the pre-hypertrophic chondrocytes of the growth plate was reduced by 57% (Fig. 3D, G). The expression of Col10α1 and MMP13, which are markers for hypertrophic and terminal stages of hypertrophic chondrocytes, were decreased in the 3-week old mutant mice by 73% and 62% respectively (Fig. 3E–G). Notably, Runx2 expression was decreased by 61% in the mutant mice (Fig. 3B, G). Collectively, these results suggest that the reduced length of the hypertrophic zones in mutant growth plates could be the result of the impaired differentiation of chondrocytes from the significantly decreased expression levels of key chondrocyte genes.
Figure 3. Runx1 deficiency in chondrocytes and osteoblasts leads to a decrease in the expression of chondrocyte hypertrophic related genes.
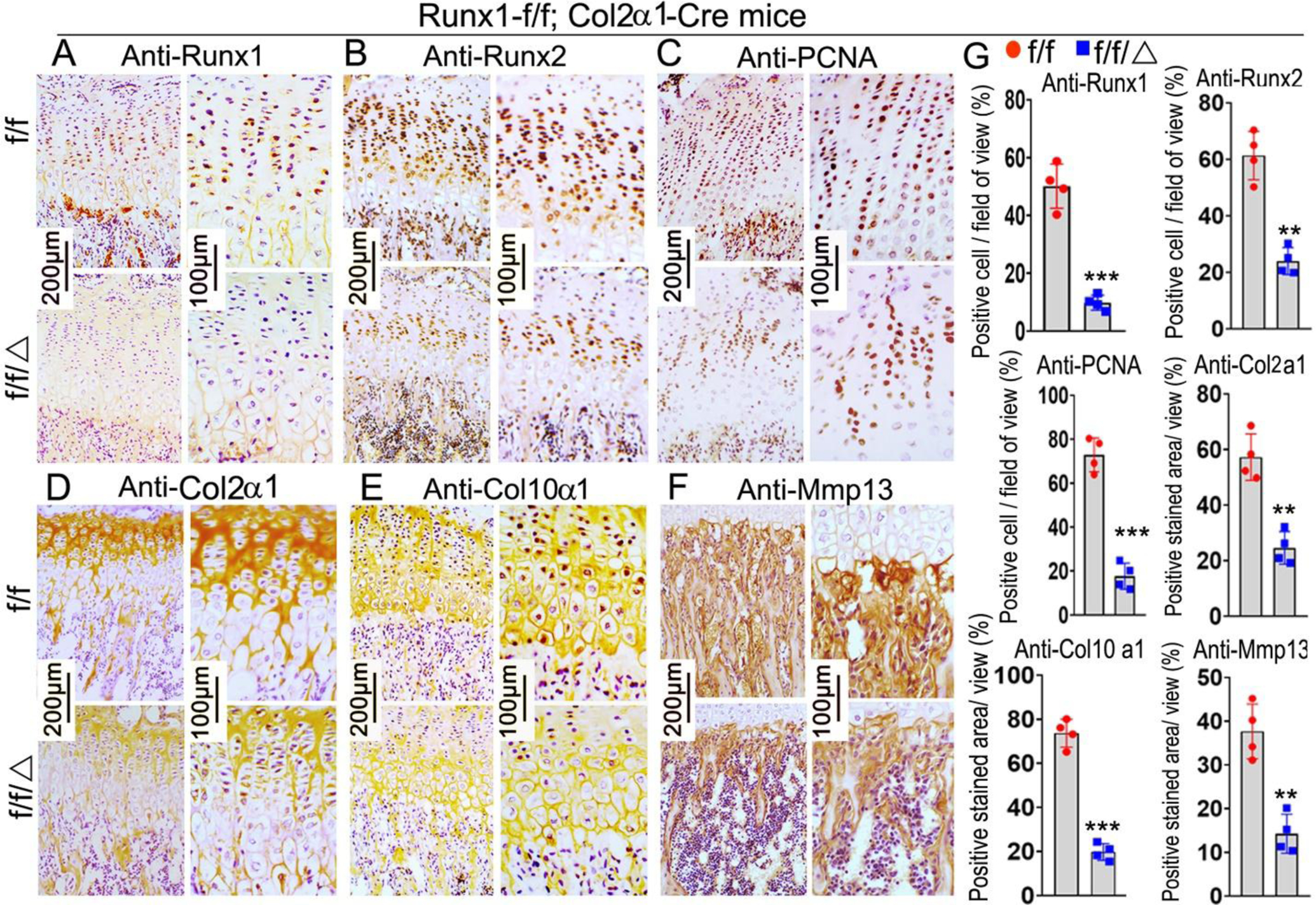
Immunohistochemistry (IHC) staining of chondrocyte differentiation marker genes in the femurs of 3-week old Runx1f/fCol2α1-cre and Runx1f/f mice, including anti-Runx1 (A), anti-Runx2 (B), anti-PCNA (C), anti-Col2α1 (D), anti-Col10α1 (E), and anti-MMP13 (F). (G) Quantification of positive staining cells and area in (A)-(F). All data are presented as mean ± SD, n=6, NS denotes not significant, **p<0.01, ***p<0.001.
Runx1 deficiency in chondrocytes leads to impaired osteoblast differentiation and down-regulation of Ihh and CyclinD1 in murine femurs.
To investigate the effect of chondrocyte derived Runx1 on osteoblast differentiation, we performed immunofluorescence (IF) staining on the postnatal day 9 murine tibias. The expression of the pre-osteoblast marker OSX was significantly decreased in the trabecular bone of the mutant mice (Fig. 4A). Moreover, a significant reduction in Osteopontin (OPN) and osteocalcin (OCN) was observed in the mutant trabecular bone (Fig. 4B, C). These results reveal that Runx1 expression in chondrocytes and osteoblasts is required for normal osteoblast differentiation. The rate of chondrocyte proliferation is regulated by cyclins, which form complexes with cyclin-dependent kinases. Recent data also suggest that IHH and PTH1R regulate cell cycle progression by the activation of cyclins [23]. Since our results showed decreased chondrocyte differentiation in mutant mouse chondrocytes, we examined the expression of cyclinD1, Indian hedgehog homolog (Ihh), and parathyroid hormone-related peptide receptor (PTH1R) by performing IF staining on the postnatal day 9 murine femoral slides (Fig. 4D–F). The results showed that the expression of cyclinD1 decreased by 60% in the growth plates of mutant mice femurs compared with that in the control group (Fig. 4E, G), which is consistent with decreased proliferation of chondrocytes observed in the mutant mice growth plates (Fig. 2G). Moreover, Ihh, which is the main regulator of CyclinD1, was decreased by 60% in the mutant mice (Fig. 4D, G). Consistently, PTH1R was decreased in the mutant growth plates by 57% (Fig. 4F, G). Since Ihh is the main regulator of chondrocyte and osteoblast differentiation during endochondral bone formation, the decreased expression of Ihh may contribute to the trabecular bone formation and deformed growth plate in the mutant mice. Collectively, our results demonstrated that Col2α1-cre mediated Runx1 deletion affects chondrocytes differentiation by interacting with Ihh, CyclinD1, and PTH1R and then leads to decreased osteoblasts differentiation.
Figure 4. Runx1 deficiency in chondrocytes and osteoblasts leads to the impaired osteoblast differentiation and down-regulation of Ihh, CyclinD1 in murine femurs.
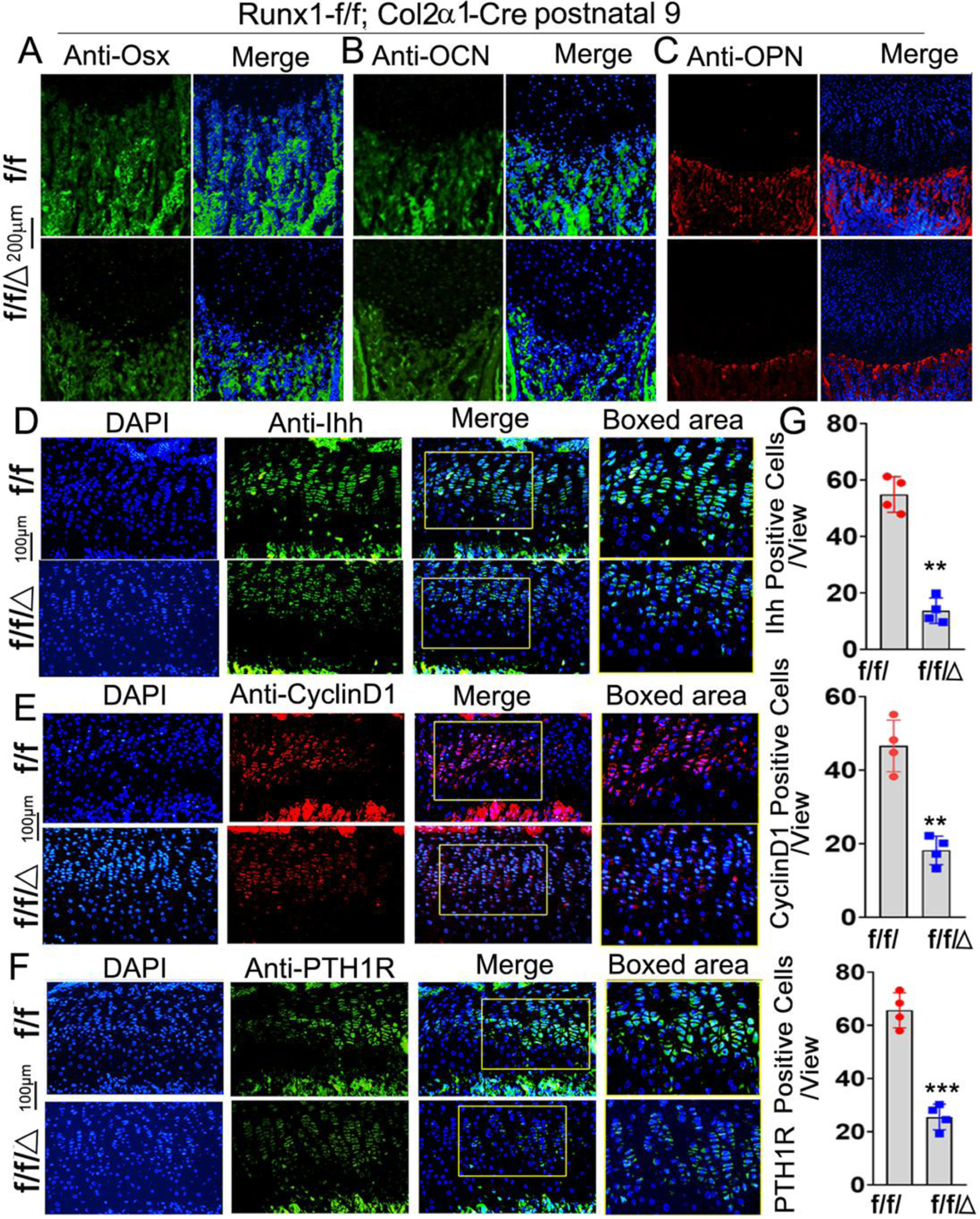
Immunofluorescence (IF) staining of osteoblast related genes in newborn murine femurs with anti-Osx (A), anti-OCN (B), and anti-OPN (C). (D) IF staining by using anti-Ihh, (E) anti-CylinD1, and (F) anti-PTH1R in postnatal 9 murine growth plates. (G) Quantification of IF staining positive cells and area in (D)-(F). All data are presented as mean ± SD, n=6, NS denotes not significant, **p<0.01, ***p<0.001.
BMSCs from Runx1f/fCol2α1-cre mice have inhibited osteogenic potency.
To further determine the role of Runx1 in osteoblast differentiation, we used bone marrow stromal cells (BMSCs) derived from bone marrow of 6-week-old murine long bones to examine the difference of osteoblast differentiation between mutant mice and control groups. After 14 days of culture in the osteogenic medium, ALP staining was performed on these cells, and the results revealed that osteoblast formation in mutant cells was significantly reduced compared to control cells (Fig. 5A). Deletion of Runx1 expression in BMSCs was confirmed by western blot (Fig. 5B) and qPCR (Fig. 5D). Analysis of protein levels of osteoblast-related genes demonstrated that protein levels of Runx2 were significantly decreased in Runx1f/fCol2α1-cre BMSCs at day 7 but not at day 14 compared to its control (Fig. 5B, C). We also found that protein levels of Runx1 binding partner Cbfβ were not significantly changed in Runx1f/fCol2α1-cre BMSCs (Fig. 5B, C). Notably, protein levels of osteoblast genes Opn, Osx, and Atf4 were decreased by 60%, 50%, and 80% respectively in day 14 Runx1f/fCol2α1-cre BMSCs (Fig. 5B, C). Interestingly, while the protein levels of Ihh were decreased by 70% in day 7 mutant BMSCs, there was no significant different in the Ihh protein levels between WT and mutant BMSCs at day 14, while the levels of Ihh were significantly higher in day 14 BMSCs compared to day 7 (Fig. 5B, C). Consistent with the western blot data, qPCR demonstrated significant decreases in the expression levels of Runx2, Opn, Osx, Ocn, and Atf4 (Fig. 5D). We suspect that Runx2 expression is directly dependent on Runx1, although Runx2 expression may also be regulated indirectly through Ihh signaling. However, further study is needed. Notably, there was no significant change in the expression levels of Cbfβ and Runx3 (Fig. 5D). Taken together, these results indicate that expression of Runx1 in chondrocytes is required for normal osteoblast differentiation.
Figure 5. BMSCs from Runx1f/fCol2α1-cre mice have inhibited osteogenesis.
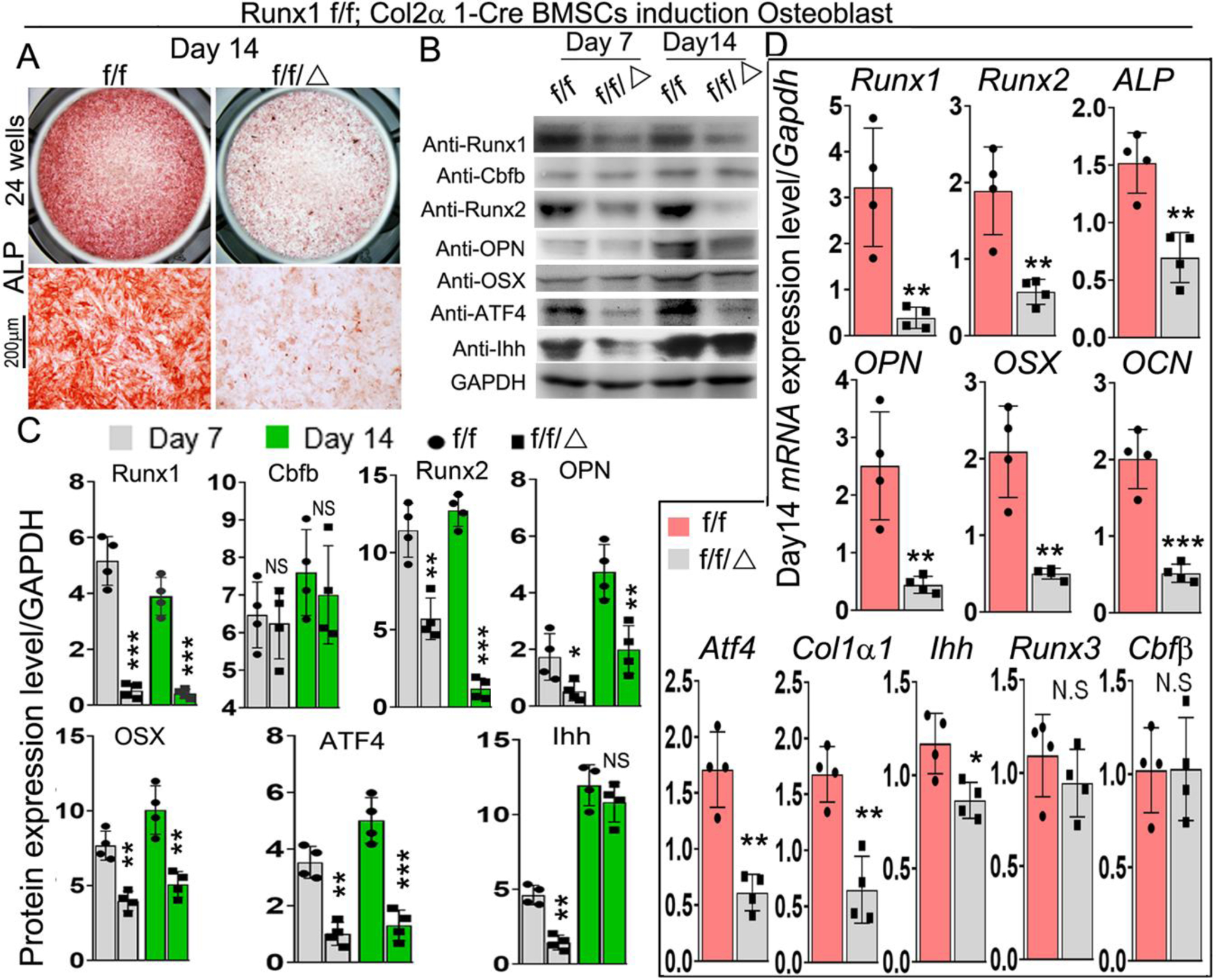
(A) ALP staining of day 14 bone marrow stem cells (BMSCs) from 6-week-old murine bone marrow cultured in osteogenic medium. (B) Western blot was performed to analyze the protein expression level of osteoblast differentiation related genes in day 7 and day 14 BMSCs. (C) Quantification of the results of Western blot in (B). (D) qPCR was used to analyze the expression levels of osteoblast and chondrocyte genes in day 14 BMSCs. All data are presented as mean ± SD, n=6, NS denotes not significant, *p<0.05, **p<0.01, ***p<0.001.
Runx1 may interfere with Ihh, CyclinD1, and PTH1R to control chondrocyte proliferation and differentiation in vitro.
In order to investigate the molecular mechanism for the delayed endochondral bone formation of Runx1f/fCol2α1-cre mice, in vitro chondrocyte differentiation assays were performed in a micromass culture pattern. Alcian blue staining of primary chondrocytes prepared from P0 Runx1f/fCol2α1-cre mice growth plates showed significantly reduced matrix deposition in mutant chondrocytes, which is reflected by weaker Alcian blue staining of the cells on days 7 and 14 (Fig. 6A), while ALP staining demonstrated a significant decrease in osteogenic capability during primary cartilage chondrogenic differentiation (Fig. 6B). Interestingly, we found adipocytes accumulation in Runx1f/fCol2α1-cre cell cultures (Fig. 6A (blue boxed region), B (red boxed region). mRNA and protein were harvested from the Day 7 and 14 cultured cells in Fig 6A. Consistent with the previous results, PCNA and chondrocyte differentiation markers were significantly decreased at the mRNA and protein levels (Fig. 6C, D, E). Consistently, the protein levels of Ihh, CyclinD1, and PTH1R were significantly downregulated in the mutant chondrocytes both in mRNA and protein levels (Fig. 6C, D, E). According to previous reports, Runx1 may function with Runx2 to regulate sternal development and chondrocyte commitment [14]. We examined the expression of Runx2 in chondrocytes from Runx1f/fCol2α1-cre mice through Western blot. Consistent with the results of IHC staining (Fig. 3B), the results revealed that the expression of Runx2 at protein level was decreased by 82% at day 7 (Fig. 6C, D) and 61% at day 14 at mRNA level (Fig. 6E). Notably, the levels of Sox9 were significantly decreased at both the mRNA and protein level (Fig. 6C, D, E), while the expression levels of Opn, Osx were similarly decreased (Fig. 6C, D, E).
Figure 6. Runx1 may interfere with Ihh signaling to regulate chondrocyte proliferation and differentiation in vitro.
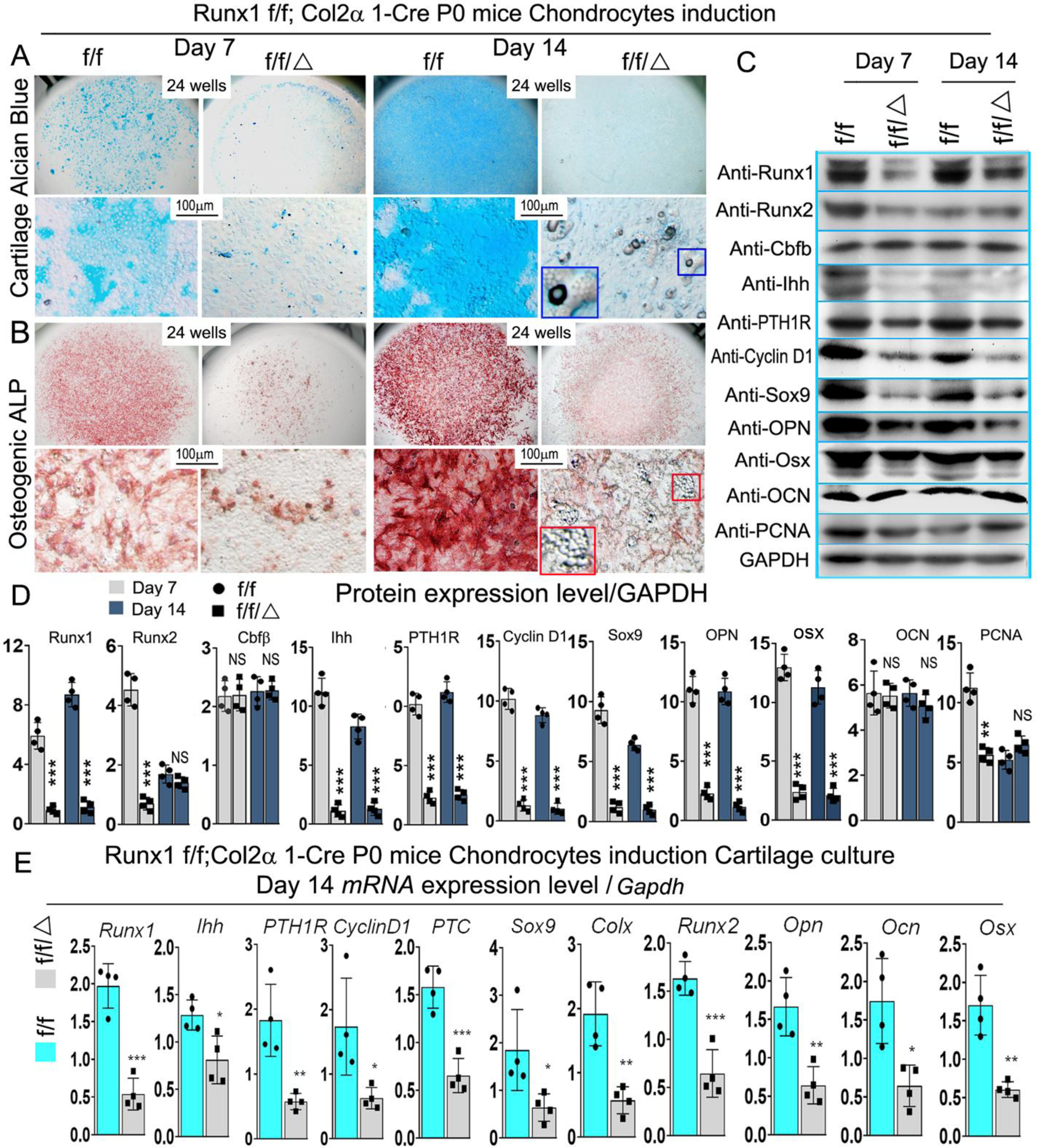
(A, B) Micromass culture of chondrocytes derived from Runx1f/fCol2α1-cre and Runx1f/f newborn mice growth plates. The blue and red boxed and zoomed area reffered to adipocytes. (A) Alcian blue and (B) ALP staining was performed at day 7 and day 14. (C) Western blot was performed on cultured chondrocytes of day 7 and day 14 from A to evaluate the protein levels of Runx1, Runx2, Cbfβ, Ihh, PTH1R, Cyclin D1, Sox9, Opn, Osx, Ocn, and PCNA. (D) Quantification data of (C). (E) qPCR was used to analysis the expression level of Ihh signaling related genes in day 14 chondrocytes. All data are presented as mean ± SD, n=4, NS denotes not significant, *p<0.05, **p<0.01, ***p<0.001.
Runx1 deficiency in chondrocytes leads to adipocyte differentiation after osteogenesis or adipogenesis induction for 14 and 21 days.
To further explore Runx1 regulation of lineage cell differentiation, we performed Oil-red staining of Runx1f/fCol2α1-cre and WT calvarial cells (Fig 7A, B; Fig. S3C). Our results demonstrate that after osteogenesis induction for 14 and 21 days, Oil-red staining was significantly increased in the mutant group compared with the WT group (Fig 7A, B; Fig. S3C). In addition, we cultured the calvarial cells with adipogenesis medium for 14 days followed by Oil-red staining, which was dramatically increased in the mutant group (Fig. S3D, E), which further demonstrated that adipocyte differentiation was promoted after Runx1 was deficient in chondrocytes. We also found significant reductions in osteogenesis and mineralization in newborn Runx1f/fCol2α1-cre calvarial cells after osteogenesis induction (Fig. S3A, B). Notably, we found that the protein levels of osteogenesis-related markers Runx2 and OCN were significantly decreased in the mutant mice compared with the control mice (Fig 7C, D). However, the protein levels of adipogenic marker C/ebpa were increased by over 4-fold in the mutant mice compared with control mice (Fig 7C, D). Similarly, we found that expression of osteoblast markers Runx2, OCN, and Osx were decreased by 95%, 90%, and 90%, respectively, while the expression of adipogenesis genes C/ebpα, Pparγ, and Fabp4 were increased by 85%, 92%, and 71%, respectively (Fig. 7E). These results suggest that Runx1 can cell non-autonomously regulate adipocytes differentiation.
Figure 7. Runx1 cell-autonomously regulated osteoblasts and adipocytes differentiation.
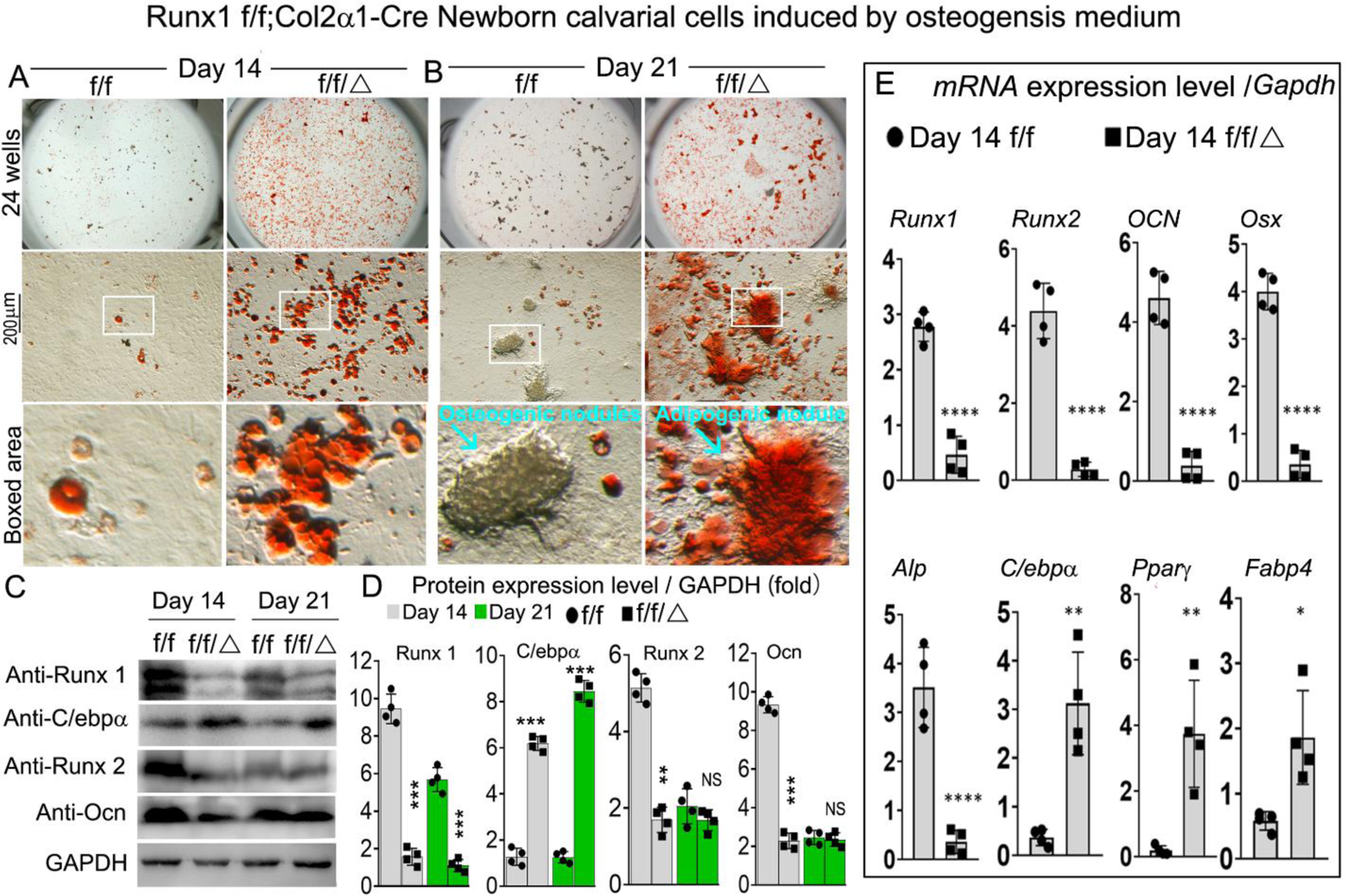
(A, B) Oil-Red staining of Runx1f/f and Runx1f/fCol2α1-cre newborn mice calvarial cells induced by osteogenesis medium for (A) 14 and (B) 21 days. (C) Western blot was performed to examine the protein levels of osteoblasts makers and adipocytes markers in Runx1f/f and Runx1f/fCol2α1-cre newborn mice calvarial cells induced by osteogenesis medium for 14 days and 21 days. (D) Quantification of (C). (E) qPCR for osteoblast markers and adipocytes markers expression in Runx1f/f and Runx1f/fCol2α1-cre newborn mice calvarial cells induced by osteogenesis medium for 14 days. All data are presented as mean ± SD, n=4; *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001.
Overexpression of Runx1 dramatically increases chondrocyte differentiation in ATDC5 cells.
Using a pMX-puro-GFP control vector, we generated a retrovirus encoding the GFP cDNA to infect ATDC5 cells, and we showed that GFP was highly expressed post-infection (Fig. 8A), confirming that this retroviral system can sustain high gene expression for our overexpression studies. Retrovirus-mediated overexpression of Runx1 in ATDC5 cells significantly increased chondrocyte differentiation (Fig. 8B), as well as increased the protein levels of Runx1, Sox9, PTC, and PTH1R by 90%, 80%, 85%, and 90%, respectively (Fig. 8C, D). Collectively, the results indicated that Runx1 may cooperate with Runx2 to regulate chondrocyte differentiation by interacting with Ihh signaling.
Figure 8. Overexpression of Runx1 dramatically increases chondrocyte differentiation in ATDC5 cells.
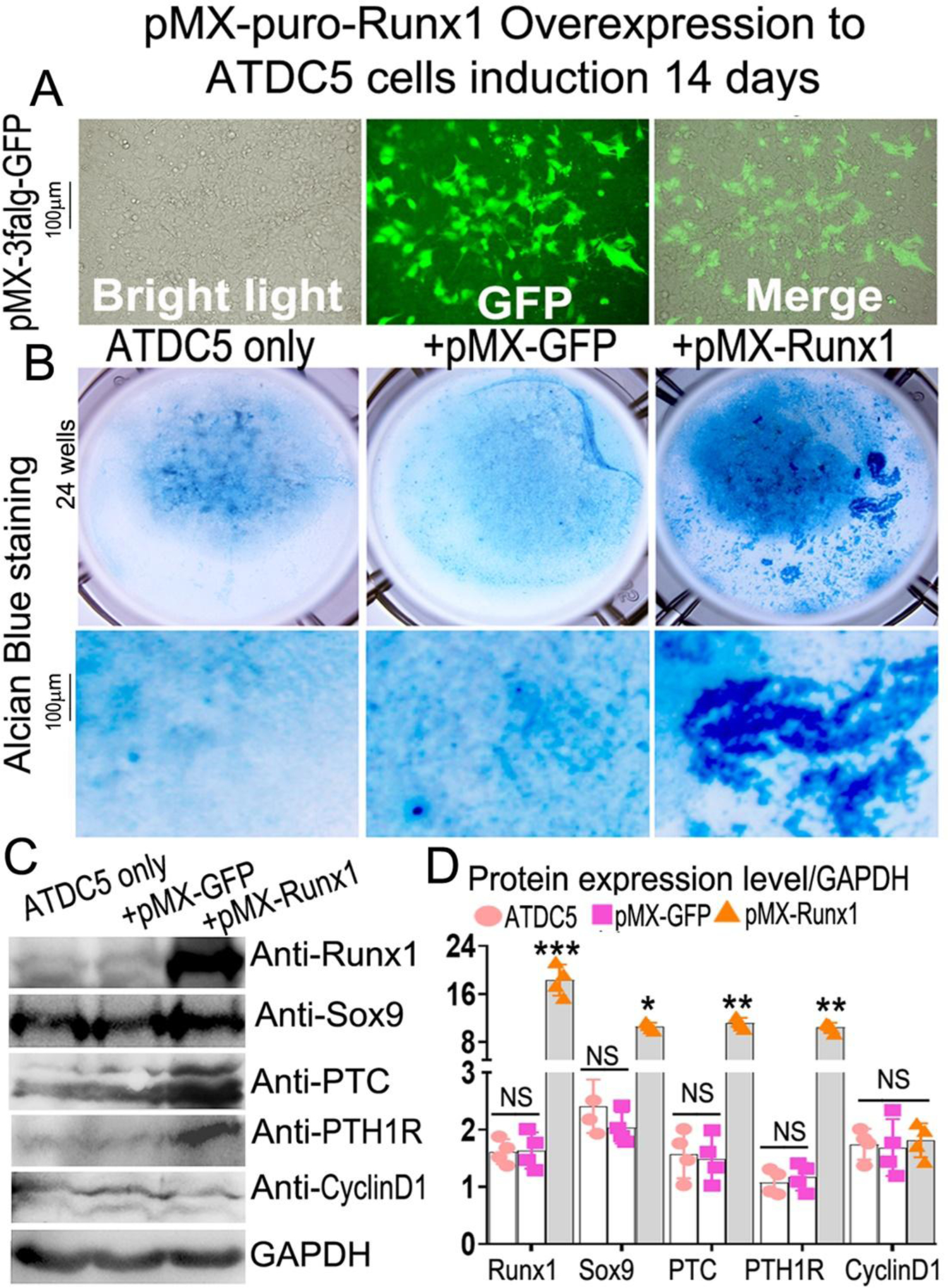
(A) overexpression of GFP in 14 days induced ATDC5 cells by retrovirus. (B) retrovirus-mediated overexpression of Runx1 in ATDC5 cells. (C) Western blot was performed on day 14 cultured chondrocytes to evaluate the protein levels of osteoblast genes and Ihh signaling-related genes. (D) Quantification of protein levels in C. All data are presented as mean ± SD, n=4, NS denotes not significant, *p<0.05, **p<0.01, ***p<0.001.
Runx1 regulates Ihh expression by directly binding to its promoter.
Chromatin immunoprecipitation (ChIP) assay was performed to determine whether Runx1 binds to the Ihh promoter to control osteoblast and chondrocyte differentiation. We analyzed the Ihh promoter regions and designed primers accordingly by using an online transcription factor binding site predictive tool (alggen.Lsi.Upc.es). We identified 5 binding sites in the Ihh promoter region (−3919 to +270) (Fig. 9A). ChIP assay was performed by using an anti-Runx1 antibody, and DNA was pulled down, amplified, and analyzed by using the primers. The ChIP input value represents the binding efficiency of the adjacent region around the location of the primer pair. The results showed that Ihh primer 4 resulted in the highest binding value compared with other primers, indicating Runx1 potentially binds to the Ihh promoter on the binding sites around site 4 (Fig. 9B, C). To investigate the functional role for Runx1 in binding the Ihh promoter we performed a luciferase assay by cloning the relevant region into pGL3. The promoter luciferase analysis showed that luciferase activity was highest when driven by the longest Ihh promoter fragment (−1280/+80) with co-expression of Runx1 and significantly lower when driven by the other Ihh promoter fragments or without co-expression of Runx1 (Fig. 9D). Taken together, these results demonstrate that Runx1 deletion impacts chondrocyte and osteoblast differentiation by affecting the expression of genes critical to osteoblast and choncrocyte differentiation, and that Runx1 regulates Ihh expression at the transcriptional level by directly binding its promoter.
Figure 9. Runx1 regulated Ihh expression by directly binding to its promoter.
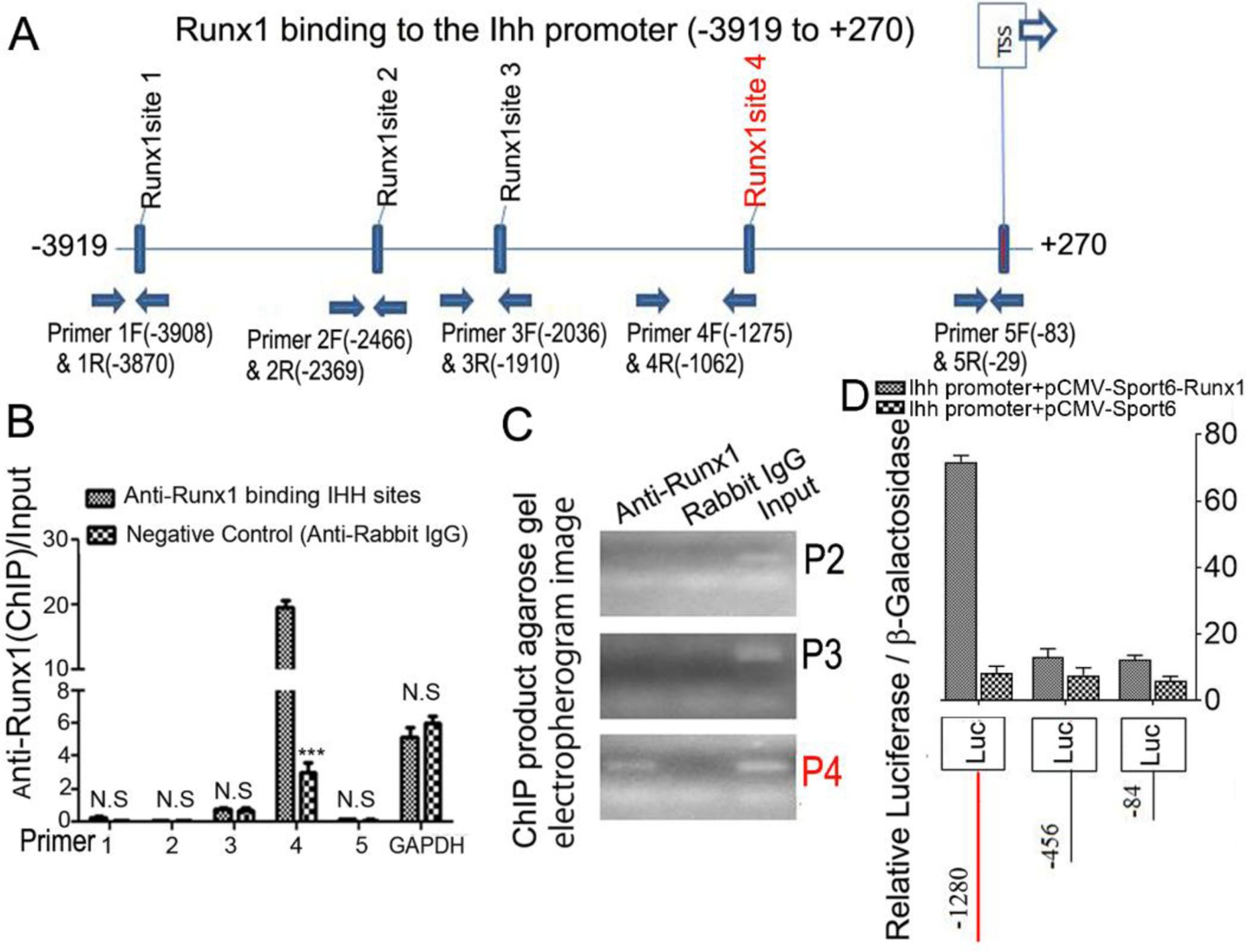
(A) Schematic display of Ihh promoter region (−3919 to +270): TSS, predicted Runx1-binding sites, and ChIP primers positions. (B) ChIP analysis of Runx1 binding to the Ihh promoter in ATDC5 cell line induced 7 days using primers as indicated on the x-axis. Results are presented as ChIP/Input. (C). Agarose gel image using ChIP qPCR products in (B). (D) Ihh promoter fragments were inserted into pGL3-basic vector. ATDC5 cells were co-transfected with pGL3-Ihh −84bp, −456bp, −1280bp and Runx1. Luciferase was detected at 48 hours post transfection and normalized to β-gal activity. All data are presented as mean ± SD, n=4, NS denotes not significant, ***p<0.001.
DISCUSSION
In this study, we found that Runx1 is expressed at different stages of both chondrocyte and osteoblast differentiation, and that Runx1 ablation in chondrocytes resulted in compromised endochondral bone formation including the delayed development of the vertebra and trabecular bones, inhibited chondrocyte and osteoblast differentiation, and decreased the length of the hypertrophic zone, indicating that Runx1 deletion in chondrocytes impaired growth plate development and hypertrophic chondrocytes. Further, Runx1 deficiency in chondrocytes resulted in decreased expression levels of Ihh, CyclinD1, and PTH1R. Chromatin immunoprecipitation (ChIP) and promoter analysis revealed that Runx1 directly binds to the Ihh promoter to regulate its expression, indicating that Runx1 directly regulates the transcriptional expression of chondrocyte marker genes and osteoblast marker genes. Collectively, we revealed that Runx1 governs chondrocyte to osteoblast lineage commitment and promotes endochondral bone formation through enhancing both chondrogenesis and osteogenesis genes expressions, indicating Runx1 may be a therapeutic target to enhance endochondral bone formation and prevent osteoporosis fractures.
Runx1 has been shown to be expressed in mesenchymal condensation sites, prechondrocytic tissues, resting and proliferative chondrocytes [12]. Due to its expression pattern, many studies have focused on the function of Runx1 in the early development stage of skeletal development, yet its role in chondrocyte differentiation and endochondral bone formation, and the mechanism underlying which factors regulate chondrocyte to osteoblast lineage commitment and the function of chondrocyte to osteoblast commitment remain unknown. Studies showed that Runx1 induces mesenchymal stem cell commitment to early stages of chondrogenesis [13]. Both Runx1 and sox9 are intensely expressed in skeletal elements that later give rise to bone and cartilage [9]. It has been demonstrated that during the early stage of skeletal mesenchymal cells differentiation into chondrocytes and osteoblasts, Runx1 and Runx2 are co-expressed [12, 13]. In addition, Runx1 overexpression in skeletal mesenchymal cells causes to chondrocyte differentiation, but Runx1 knockdown causes to Runx2 decline as well as chondrocyte and osteoblast differentiation defect [13]. Runx1 has also been shown to cooperate with Runx2 to regulate the commitment of mesenchymal cells to commit to chondrocytes [14]. However, the role of Runx1 in chondrocyte differentiation and endochondral ossification still remains largely unknown. Thus, in the current study we crossed Col2α1-Cre mice with Runx1f/f mice to delete Runx1 specifically in chondrocytes to study the function of chondrocyte derived Runx1 in endochondral bone formation. Our results have demonstrated the pivotal role of Runx1 in maintaining normal postnatal growth plate structures and trabecular bones.
Ono et al. reported that a subset of chondrogenic cells provides early mesenchymal progenitors in growing bones [1], indicating an important role of chondrogenic cells in endochondral bone formation. Yet the mechanism underlying which factors regulate chondrocyte to osteoblast lineage commitment remain unknown. Our data showed that Runx1 is expressed at different stages of both chondrocyte and osteoblast differentiation (Fig. 2F). Interestingly, our in vitro chondrocyte culture data showed that both chondrocyte and osteoblast marker genes are expressed in chondrocytes in vitro, and the expressions of these marker genes were significantly decreased in Runx1f/fCol2α1-cre chondrocytes (Fig. 6). This indicates that Runx1 may play a key role in regulating chondrocyte to osteoblast lineage commitment by regulating the expression of key chondrocyte and osteoblast genes.
In previous studies, Runx1 was selectively deleted in limb mesenchyme by using Prx1-cre, and only a slight and transient inhibition of sternal mineralization was observed, while no phenotypes were observed in the long bones [14]. Further, their studies also showed no abnormal skeletal morphogenesis in the Runx1f/fPrx1-cre mice [14, 24]. Prx1+ cells do not exist in the metaphysis mesenchymal progenitors which later differentiate into trabecular bone, and part of them residing in the periosteum of cortical bone, which leads to the growth of cortical bone [25]. In fact, Col2α1+ cells not only marked chondrocytes and perichondrial cells, but also osteoblasts and stromal cells progressively in the metaphysis [1] which later leads to the growth of trabecular bone. Thus, the cell clusters that Prx1-cre and Col2α1-cre marked have several key differences, which led to the fact that the Runx1f/fPrx1-cre mice model phenotypes were not obvious while our Runx1f/f Col2α1-cre mice model was apparent. In addition, the previous research was limited to P0 to 3-week-old mice using Prx1 Cre but our research was from P0 to 24 weeks old using Col2α1-cre. Liakhovitskaia et al. found that Runx1 plays an essential role in the development of the sternum and some skull elements, but is not essential for major skeletal development [26]. Their research only focused on the embryos of Runx1 reversible knockout mice. In our study, we examined mice from P0 to 24 weeks old to determine the role of Runx1 in endochondral ossification. Interestingly, we observed the delayed bone formation in vertebra (Fig. S2C) and trabecular bones (Fig. 1F, G) of Runx1f/fCol2α1-cre mice at different ages. These results were confirmed by our histological examinations of vertebra and trabecular bones, which showed decreased length of the hypertrophic zone, resulting in the shorted growth plates in the mutant cartilage (Fig. 2G). Furthermore, the expression of chondrocyte differentiation markers also decreased accompanied by the deformed chondrocytes and disorganized hypertrophic chondrocytes in the mutant growth plates (Fig.3). Runx1 is involved in chondrocyte proliferation and lineage determination [7], and whereas Runx1 is predominately expressed in the proliferative and pre-hypertrophic chondrocytes, Runx2 is expressed primarily by hypertrophic chondrocytes of the growth plates [27]. Thus, following conditional deletion of Runx1, resting and proliferative chondrocytes cannot differentiate into hypertrophic chondrocytes due to the loss of Runx2 regulation, leading to the significant reduction in trabecular bone observed in Runx1f/fCol2α1-cre mice. Moreover, the expression of PCNA decreased in Runx1f/fCol2α1-cre mice. Thus, the shorter stature of the mice may be due to the inhibition of the chondrocyte differentiation and terminal maturation. Given the reduced numbers of hypertrophic chondrocytes, osteoblasts, and reduced trabecular bone in Runx1f/fCol2α1-cre mice, Runx1 may maintain trabecular bone formation through its regulation of Runx2, Ihh, CyclinD1, and PTH1R in hypertrophic chondrocytes.
The deficiency of Runx1 can lead to the delayed endochondral bone formation, which may partly result from the impaired chondrocyte and osteoblast differentiation during postnatal skeletal development. Our findings demonstrate that Runx1 is required for chondrocyte development and maintaining a normal growth plate. Runx1 has been reported to exhibit a unique spatial and temporal expression pattern during bone formation, which is different from the expression patterns of other Runx proteins [12]. Previous studies mostly focus on the embryonic development of Runx1 deficiency in the chondrocytes [14], while our observation points were from newborn to 6-month old mice. Furthermore, due to the mutation’s phenotypic penetrance and expressivity, not all the mice that carry the mutation of Runx1 will also show the related phenotypes [28]. Studies also revealed that haploinsufficiency of Runx1 may not be sufficient to impair chondrogenesis [5, 24]. Taken together, our results provided the compelling evidence that Runx1 plays an important role in maintaining normal chondrocyte and osteoblast development.
Cell fate-mapping studies revealed that cells expressing cre-recombinases driven by the Col2α1 promoter contribute not only to chondrocytes and perichondrial cells, but also to osteoblasts and mesenchymal progenitor cells at later stages [1]. In our studies, the mutant mice displayed a reduced bone density and trabecular bone formation. Furthermore, the bone defects of mutant mice occurred predominately in the trabecular bone formation. These defects may be due to impaired osteoblast development. Furthermore, our IF staining results showed that the protein levels of Osx, Ocn, and Opn were significantly decreased in the mutant trabecular bone compared with control groups (Fig. 4A, B, C). In vitro data also confirmed that Runx1 deficiency in chondrocytes and osteoblasts resulted in decreased expression of osteoblast related genes at both the mRNA and protein levels. Interestingly, bone marrow adipocytes began to accumulate in the mutant limbs as early as 3-week-old (Fig. 2B). Since osteoblasts and adipocytes are derived from the same undifferentiated mesenchymal cells, these results suggest that Runx1 may also have a function in osteoblast cell fate determination, which warrants further study to elucidate the underlying mechanisms. Notably, the protein levels of adipogenesis genes were significantly increased in the mutant mice compared with control mice, while the expression of osteoblast markers were significantly decreased (Fig. 7). Our previous research shows that the Cbfβ/Runx heterodimer regulates osteoblast–adipocyte lineage commitment both cell nonautonomously through enhancing β-catenin signaling and cell autonomously through suppressing adipogenesis gene expression to maintain osteoblast lineage commitment and increase chondrocyte gene expression[20]. Thus, this could be the reason why chondrocyte-specific deletion of Runx1 can lead to increased adipogenesis. These results suggest that Runx1 may be required to suppress adipocyte lineage commitment.
Growth plate development is regulated by many signaling pathways such as BMPs, FGFs, and Wnts [2]. Ihh-PTH1R signaling is a key signaling regulator of chondrocyte proliferation and maturation [29]. Expression of Ihh by prehypertrophic chondrocytes leads to proliferation of growth plate chondrocytes, and up-regulation of PTH1R inhibits chondrocyte differentiation and down-regulates Ihh, thus completing the negative feedback loop [30]. The Runx family undergoes crosstalk with key signaling pathways and transcriptional programs that specify cell fate, especially including the osteogenesis, such as Wnt, TGFβ, and BMP signaling [31]. Runx proteins are highly regulated by several signaling networks to control musculoskeletal development and bone maintenance [32, 33]. Runx2 has been shown to regulate chondrocyte proliferation and differentiation by interacting with Ihh-PTH1R signaling [34], as Runx1 and Runx2 share a high homology DNA binding sequence, it has been speculated that Runx1 may also function with Ihh to regulate endochondral bone formation. By using ChIP and promoter assays, we demonstrated that Runx1 up-regulates Ihh expression by directly binding to its promoter to control chondrocyte differentiation. In the current study, Runx1 deficiency in chondrocytes lead to decreased expression of Ihh, CyclinD1, and PTH1R (Fig. 4D, E, F) in the murine growth plates, leading to the decreased expression of PCNA, Col2α1, Col10α1 and MMP13 in the mutant growth plates (Fig. 3C, E, E, F). These results suggest that Runx1 may regulate chondrocyte proliferation and hypertrophy to coordinate the timing and rate of endochondral bone formation through Ihh signaling.
In summary, our data demonstrate that Runx1 upregulates chondrocyte to osteoblast commitment and promotes endochondral bone formation through enhancing both chondrogenesis and osteogenesis. Moreover, by regulating osteoblast and chondrocyte differentiation, Runx1 plays a fundamental role in maintaining postnatal bone homeostasis. Our studies provided a new insight into the role of Runx1 during postnatal skeletal development and may open a therapeutic avenue to treat bone diseases, such as pectus excavatum and osteoporosis.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Guochun Zhu and Jun Tang for their excellent technical assistance, and Mrs. Diep Edwards for her excellent assistance with the manuscript. We are also grateful for the assistance from the Small Animal Phenotyping Core and Metabolism Core Laboratory at the University of Alabama at Birmingham.
Funding: This work was supported by National Institutes of Health [AR-070135 and AG-056438 to W.C.; DE-028264 and DE-023813 to Y.-P.L.]. C.-Y.T was sponsored by the China Scholarship Council [201706370159].
Abbreviations
- ALP
Alkaline phosphatase
- BMSCs
bone marrow stromal cells
- ChIP
chromatin immunoprecipitation
- HZ
hypertrophic zone
- IF
immunofluorescence
- IHC
immunohistochemistry
- Ihh
Indian hedgehog homolog
- OCN
osteocalcin
- OPN
osteopontin
- PZ
proliferation zone
Footnotes
Competing interests: The authors declare that there are no competing interests associated with the manuscript.
References
- 1.Ono N, Ono W, Nagasawa T and Kronenberg HM (2014) A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol 16, 1157–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kronenberg HM (2003) Developmental regulation of the growth plate. Nature. 423, 332–336 [DOI] [PubMed] [Google Scholar]
- 3.Weng T, Xie Y, Huang J, Luo F, Yi L, He Q, Chen D and Chen L (2014) Inactivation of Vhl in Osteochondral Progenitor Cells Causes High Bone Mass Phenotype and Protects Against Age-Related Bone Loss in Adult Mice. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 29, 820–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato S, Kimura A, Ozdemir J, Asou Y, Miyazaki M, Jinno T, Ae K, Liu X, Osaki M, Takeuchi Y, Fukumoto S, Kawaguchi H, Haro H, Shinomiya K, Karsenty G and Takeda S (2008) The distinct role of the Runx proteins in chondrocyte differentiation and intervertebral disc degeneration: findings in murine models and in human disease. Arthritis and rheumatism. 58, 2764–2775 [DOI] [PubMed] [Google Scholar]
- 5.Yoshida CA and Komori T (2005) Role of Runx proteins in chondrogenesis. Critical reviews in eukaryotic gene expression. 15, 243–254 [DOI] [PubMed] [Google Scholar]
- 6.Komori T (2005) Regulation of skeletal development by the Runx family of transcription factors. Journal of cellular biochemistry. 95, 445–453 [DOI] [PubMed] [Google Scholar]
- 7.Johnson K, Zhu S, Tremblay MS, Payette JN, Wang J, Bouchez LC, Meeusen S, Althage A, Cho CY, Wu X and Schultz PG (2012) A stem cell-based approach to cartilage repair. Science. 336, 717–721 [DOI] [PubMed] [Google Scholar]
- 8.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB and Owen MJ (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 89, 765–771 [DOI] [PubMed] [Google Scholar]
- 9.Yamashiro T, Wang XP, Li Z, Oya S, Aberg T, Fukunaga T, Kamioka H, Speck NA, Takano-Yamamoto T and Thesleff I (2004) Possible roles of Runx1 and Sox9 in incipient intramembranous ossification. J Bone Miner Res. 19, 1671–1677 [DOI] [PubMed] [Google Scholar]
- 10.Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, Yamana K, Zanma A, Takada K, Ito Y and Komori T (2004) Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev. 18, 952–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soung do Y, Dong Y, Wang Y, Zuscik MJ, Schwarz EM, O’Keefe RJ and Drissi H (2007) Runx3/AML2/Cbfa3 regulates early and late chondrocyte differentiation. J. Bone Miner. Res 22, 1260–1270 [DOI] [PubMed] [Google Scholar]
- 12.Lian JB, Balint E, Javed A, Drissi H, Vitti R, Quinlan EJ, Zhang L, Van Wijnen AJ, Stein JL, Speck N and Stein GS (2003) Runx1/AML1 hematopoietic transcription factor contributes to skeletal development in vivo. Journal of cellular physiology. 196, 301–311 [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Belflower RM, Dong YF, Schwarz EM, O’Keefe RJ and Drissi H (2005) Runx1/AML1/Cbfa2 mediates onset of mesenchymal cell differentiation toward chondrogenesis. J. Bone Miner. Res 20, 1624–1636 [DOI] [PubMed] [Google Scholar]
- 14.Kimura A, Inose H, Yano F, Fujita K, Ikeda T, Sato S, Iwasaki M, Jinno T, Ae K, Fukumoto S, Takeuchi Y, Itoh H, Imamura T, Kawaguchi H, Chung UI, Martin JF, Iseki S, Shinomiya K and Takeda S (2010) Runx1 and Runx2 cooperate during sternal morphogenesis. Development. 137, 1159–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith N, Dong Y, Lian JB, Pratap J, Kingsley PD, van Wijnen AJ, Stein JL, Schwarz EM, O’Keefe RJ, Stein GS and Drissi MH (2005) Overlapping expression of Runx1(Cbfa2) and Runx2(Cbfa1) transcription factors supports cooperative induction of skeletal development. Journal of cellular physiology. 203, 133–143 [DOI] [PubMed] [Google Scholar]
- 16.McLeod MJ (1980) Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 22, 299–301 [DOI] [PubMed] [Google Scholar]
- 17.Chen Q, Shou P, Zheng C, Jiang M, Cao G, Yang Q, Cao J, Xie N, Velletri T, Zhang X, Xu C, Zhang L, Yang H, Hou J, Wang Y and Shi Y (2016) Fate decision of mesenchymal stem cells: adipocytes or osteoblasts? Cell Death Differ. 23, 1128–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeLise AM, Stringa E, Woodward WA, Mello MA and Tuan RS (2000) Embryonic limb mesenchyme micromass culture as an in vitro model for chondrogenesis and cartilage maturation. Methods Mol. Biol 137, 359–375 [DOI] [PubMed] [Google Scholar]
- 19.Tian F, Wu M, Deng L, Zhu G, Ma J, Gao B, Wang L, Li YP and Chen W (2014) Core binding factor beta (Cbfbeta) controls the balance of chondrocyte proliferation and differentiation by upregulating Indian hedgehog (Ihh) expression and inhibiting parathyroid hormone-related protein receptor (PPR) expression in postnatal cartilage and bone formation. J. Bone Miner. Res 29, 1564–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu M, Wang Y, Shao JZ, Wang J, Chen W and Li YP (2017) Cbfbeta governs osteoblast-adipocyte lineage commitment through enhancing beta-catenin signaling and suppressing adipogenesis gene expression. Proc. Natl. Acad. Sci. U. S. A 114, 10119–10124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ory DS, Neugeboren BA and Mulligan RC (1996) A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U. S. A 93, 11400–11406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen W, Ma J, Zhu G, Jules J, Wu M, McConnell M, Tian F, Paulson C, Zhou X, Wang L and Li YP (2014) Cbfbeta deletion in mice recapitulates cleidocranial dysplasia and reveals multiple functions of Cbfbeta required for skeletal development. Proc. Natl. Acad. Sci. U. S. A 111, 8482–8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wuelling M and Vortkamp A (2010) Transcriptional networks controlling chondrocyte proliferation and differentiation during endochondral ossification. Pediatric Nephrology. 25, 625–631 [DOI] [PubMed] [Google Scholar]
- 24.Soung do Y, Talebian L, Matheny CJ, Guzzo R, Speck ME, Lieberman JR, Speck NA and Drissi H (2012) Runx1 dose-dependently regulates endochondral ossification during skeletal development and fracture healing. J. Bone Miner. Res 27, 1585–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han Y, Feng H, Sun J, Liang X, Wang Z, Xing W, Dai Q, Yang Y, Han A, Wei Z, Bi Q, Ji H, Kang T and Zou W (2019) Lkb1 deletion in periosteal mesenchymal progenitors induces osteogenic tumors through mTORC1 activation. J Clin Invest. 129, 1895–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liakhovitskaia A, Lana-Elola E, Stamateris E, Rice DP, van ‘t Hof RJ and Medvinsky A (2010) The essential requirement for Runx1 in the development of the sternum. Developmental biology. 340, 539–546 [DOI] [PubMed] [Google Scholar]
- 27.Wu M, Li C, Zhu G, Wang Y, Jules J, Lu Y, McConnell M, Wang YJ, Shao JZ, Li YP and Chen W (2014) Deletion of core-binding factor beta (Cbfbeta) in mesenchymal progenitor cells provides new insights into Cbfbeta/Runxs complex function in cartilage and bone development. Bone. 65, 49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Brugge JS and Janes KA (2011) Intersection of FOXO- and RUNX1-mediated gene expression programs in single breast epithelial cells during morphogenesis and tumor progression. Proceedings of the National Academy of Sciences of the United States of America. 108, E803–E812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minina E, Kreschel C, Naski MC, Ornitz DM and Vortkamp A (2002) Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Developmental cell. 3, 439–449 [DOI] [PubMed] [Google Scholar]
- 30.Minina E, Wenzel HM, Kreschel C, Karp S, Gaffield W, McMahon AP and Vortkamp A (2001) BMP and Ihh/PTHrP signaling interact to coordinate chondrocyte proliferation and differentiation. Development. 128, 4523–4534 [DOI] [PubMed] [Google Scholar]
- 31.Blyth K, Cameron ER and Neil JC (2005) The RUNX genes: gain or loss of function in cancer. Nat. Rev. Cancer 5, 376–387 [DOI] [PubMed] [Google Scholar]
- 32.Komori T (2011) Signaling networks in RUNX2-dependent bone development. Journal of cellular biochemistry. 112, 750–755 [DOI] [PubMed] [Google Scholar]
- 33.Levanon D and Groner Y (2004) Structure and regulated expression of mammalian RUNX genes. Oncogene. 23, 4211–4219 [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Ghori-Javed FY, Rashid H, Adhami MD, Serra R, Gutierrez SE and Javed A (2014) Runx2 regulates endochondral ossification through control of chondrocyte proliferation and differentiation. J Bone Miner Res. 29, 2653–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.