

Abstract
We have previously reported that dual 5-HT1A and 5-HT7 receptor ligands might find utility as treatment options for various CNS related conditions including cognitive and anxiolytic impairments. We have also more recently reported that SYA16263 has antipsychotic-like properties with an absence of catalepsy in animal models ascribed to its ability to recruit β-arrestin to the D2 receptor. However, SYA16263 also binds with very high affinity to 5-HT1AR (Ki = 1.1 nM) and a moderate affinity at 5-HT7R (Ki = 90 nM). Thus, it was of interest to exploit its pharmacophore elements in designing new dual receptor ligands. Using SYA16263 as the lead molecule, we have conducted a limited structure-affinity relationship (SAFIR) study by modifying various structural elements in the arylalkyl moiety, resulting in the identification of a new dual 5-HT1AR and 5-HT7R ligand, 6-chloro-2-methyl-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (21), which unlike SYA16263, has a sub-nanomolar (5-HT1AR, Ki = 0.74 nM) and a low nanomolar (5-HT7R, Ki = 8.4 nM ) affinity for these receptors. Interestingly, 21 is a full agonist at 5-HT1AR and antagonist at the 5-HT7R, functional characteristics which point to its potential as an antidepressant agent.
Keywords: Dopamine and serotonin receptors, pyridinyl piperazine, indanones, 5-HT subtype receptors, Dual 5-HT1A and 5-HT7 receptors, SYA16263
Graphical Abstract

1. Introduction
The pharmacotherapy of major central nervous system (CNS) disorders relies on multi-CNS receptor targeting. In fact, atypical antipsychotic agents such as cariprazine, aripiprazole, clozapine, ziprasidone, and lurasidone owe their superior efficacy and tolerable side-effect profiles to their ability to modulate dopamine (DA), serotonin (5-HT), and even cholinergic (ACh) receptors simultaneously [1–3].
Serotonin receptors (5-HTRs) are found predominantly on neurons in the dorsal and median raphe nuclei of the brainstem that project into other brain regions such as the hypothalamus, thalamus, hippocampus, and cortex where they regulate mood, perception, reward, anger, aggression, appetite, memory, sexuality, and attention [4–6]. Not surprisingly, perturbation in serotonergic neurotransmission is implicated in several neuropsychiatric illnesses including major depressive disorder (MDD), bipolar disorder, anxiety, attention deficit and hyperactivity disorder (ADHD) and schizophrenia [7, 8]. Several lines of evidence have revealed that the 5-HT1A receptor subtype (5-HT1AR) is involved in the pathophysiology of the aforementioned mental disorders making it a tenable target for antidepressant, antianxiety and antipsychotic drug discovery. For instance, there is a growing body of evidence that supports the anti-negative symptom and pro-cognitive effects of ligands that activate 5-HT1AR in schizophrenia [9–11]. In addition, 5-HT1A knockout mice display increased anxiety and depressive behaviors, and have been used as animal models for these disorders, further highlighting the key roles that the 5-HT1AR plays in the pathophysiology of CNS disorders [12, 13]. In fact, antidepressants such as vilazodone that inhibit 5-HT reuptake and partially activate 5-HT1ARs are more efficacious, fast acting, and tolerable [14, 15].
Another closely related 5-HTR, the serotonin-7 receptor (5-HT7R), mediates key physiological functions that include sleep, mood, learning memory and cognition [16–19]. Indeed, the antidepressant effects of amisulpride are attributed to its antagonism at the 5-HT7 receptor [20]. A growing wealth of evidence suggests that 5-HT7 is co-localized with 5-HT1A, and cross talks resulting from heterodimers of these receptors have been implicated in CNS diseases such as depression [21]. Not only do these receptors share over 40% sequence homology, ligands that activate these receptors tend to exhibit cross activity [22, 23]. We posit that dual ligands that activate 5-HT1A and antagonize 5-HT7 receptors may serve as potential treatment options for depression, anxiety and other diseases characterized by cognitive deficits.
N-Aryl/heteroaryl piperazines are of interest due in part to their unique ability to impart desirable pharmacological properties to clinically useful CNS drugs including aripiprazole and cariprazine (Fig. 1) [24]. In fact, evidence from pharmacophore models suggests that the pyridinyl-piperazine moiety may provide key structural elements necessary for binding to both 5-HT1A and 5-HT7 [25]. In our previous publications [26, 27] we identified the deoxygenated azaperone SYA 16263 (2), (Fig. 1) as a ligand that displayed a desirable in-vitro binding affinity profile to clinically relevant CNS receptors. In animal studies, SYA 16263 possesses moderate antipsychotic-like activity with low propensity to cause motoric side effects [28]. We reported in a recent patent [29] that, as a possible mechanism of action, 2 displayed a biased β-arrestin functionality at the D2 receptor. However, compound 2 also displayed a very high affinity at the 5-HT1AR,sparking our interest in exploring its structure-affinity relationship (SAFIR) at relevant CNS receptors and further expand and probe the potential utility of its analogs in CNS diseases. To that end, we have designed, synthesized and evaluated several N-alkyl substituted pyridinyl-piperazines to identify new leads for further development.
Fig. 1.

Structures of some FDA approved CNS drugs containing N-Aryl/heteroaryl piperazine moieties along with Azaperone (1) and SYA16263 (2).
2. Chemistry
Azaperone (1) is a commercially available compound and we previously reported SYA16263 (2) [27]. In general, the final compounds evaluated in this manuscript were obtained by refluxing or carrying out a microwave-assisted synthesis (MWAS) reaction of the amine portion, 1-(pyridin-2-yl)piperazine, with various alkylating agents in DME, CH3CN or toluene in the presence of K2CO3 or NaHCO3 as a base and a catalytic amount of KI. Final target compounds, where applicable, were converted to the HCl or oxalate salts. Comprehensive descriptions of the synthetic methods and accompanying data are provided in the experimental section. Briefly, in order to produce the alkylating agent 1c and 1d, aniline (1a) was N-acetylated under basic conditions and the resulting acetanilide (1b) acylated with 4-chlorobutyryl chloride under Friedel-Crafts conditions to produce intermediate 1c (Scheme 1). The keto group in 1c was removed under a modified phenyl-keto reduction reaction described by West and colleagues [30] to afford the intermediate 1d (Scheme 1). Alkylating agents 1c and 1d were then separately reacted with 1-(pyridin-2-yl)piperazine to produce acetamides 3 and 4 respectively. Deacetylation of 3 and 4 under acidic conditions following a modified method described by Kilbourn et al. [31] afforded 5 and 6 respectively (Scheme 1).
Scheme 1.

Synthesis of compounds 3, 4, 5 and 6. Reagents and conditions: (a) acetyl chloride, DCM, Et3N; (b) 4-chlorobutyryl chloride, AlCl3, CS2, reflux; (c) triethylsilane (TES), TFA, 0 °C-rt, 15 mins; (d) 1-(pyridin-2-yl)piperazine, K2CO3/KI, DME, MWAS; (e) conc. HCl/AcOH, MWAS, 30 mins.
Compounds 7 and 8 were prepared by first acylating dihydrocarbostyril 2a ated under Friedel-Crafts acylation conditions as described in Scheme 1 to produce ketone 2b which was subsequently reduced, similar to 1c, to produce the alkyl halide 2c (Scheme 2). The alkyl halides 2b and 2c were separately reacted with 1-(pyridin-2-yl)piperazine leading to compounds 7 and 8 respectively (Scheme 2).
Scheme 2.

Synthesis of compounds 7 and 8. Reagents and conditions: (a) 4-chlorobutyryl chloride, AlCl3, CS2, reflux; (b) triethylsilane, TFA, 0 °C-rt, 15mins; (c) 1-(pyridin-2-yl)piperazine, K2CO3/KI, DME, MWAS.
Commercially available 6-hydroxy-dihydrocarbostyril 3a was O-alkylated with 1-bromo-3-chloropropane to give intermediate 3b which was then used to react separately with 1-(pyridin-2-yl)piperazine and 1-(pyridin-2-yl)-1,4-diazepane under the general alkylation procedure to produce compounds 9 and 10 respectively (Scheme 3).
Scheme 3.

Synthesis of compounds 9 and 10. Reagents and conditions: (a) 1-bromo-3-chloropropane, K2CO3, KI, DME, MWAS; (b) amine [(9, amine = 1-(pyridin-2-yl)piperazine; 10, amine = 1-(pyridin-2-yl)-1,4-diazepane)], K2CO3, KI, DME, MWAS.
To access the alkylating agent 4b, 5-hydroxy-2,3-dihydro-1H-inden-1-one 4a was O-alkylated under similar condition as 3a (Scheme 3). The resulting alkylating agent was subsequently reacted with 1-(pyridin-2-yl)piperazine to afford compound 11 (Scheme 4).
Scheme 4.

Synthesis of compound 11. Reagents and conditions: (a) 1-bromo-3-chloropropane, K2CO3, KI, toluene, MWAS; (b) 1-(pyridin-2-yl)piperazine, K2CO3, KI, toluene, MWAS.
To prepare compound 12, the indane 5a was similarly acylated as 1c and 2b (Schemes 1 and 2) to produce the ketone 5b which was then used to react with 1-(pyridin-2-yl)piperazine under the general alkylation conditions to produce compound 12 (Scheme 5).
Scheme 5.

Synthesis of compound 12. Reagents and conditions: (a) 4-chlorobutyryl chloride, AlCl3, DCM, rt; (b) 1-(pyridin-2-yl)piperazine, K2CO3/KI, DME, MWAS.
In order to obtain compound 13, the alkylating agent 5-chloro-2-(2-chloroethyl)-2,3-dihydro-1H-inden-1-one 6a previously reported by us [27, 32, 33], was reacted with 1-pyridin-2-yl-piperazine under general alkylation reaction conditions (Scheme 6).
Scheme 6.

Synthesis of compound 13. Reagents and conditions: (a) NaHCO3, KI, toluene, reflux.
Ketone 6a was reduced under Luche reduction [34] conditions to yield alcohol 7a that was subsequently used to N-alkylate 1-pyridin-2-yl-piperazine to produce compound 14 (Scheme 7).
Scheme 7.

Synthesis of compound 14. Reagents and conditions: (a) CeCl3, NaBH4, MeOH, rt, 1 h; (b) 1-pyridin-2-yl-piperazine, KI, K2CO3, DME, reflux.
Alkylating agent 8a, previously reported by us [27, 33], was reduced to produce indane 8b under Clemmensen reduction conditions. Under the general alkylation conditions, alkylating agents 8a and 8b were separately reacted with 1-pyridin-2-yl-piperazine to produce compounds 15 and 16 respectively (Scheme 8).
Scheme 8.

Synthesis of compounds 15 and 16. Reagents and conditions: (a) Zn amalgam, conc. HCl, toluene, reflux; (b) 1-pyridin-2-yl-piperazine, K2CO3, KI, DME, reflux.
To synthesize compounds 17 and 18, a five-step procedure (Scheme 9) similar to that reported by us [32] was followed. Briefly, commercially available chloroindanones 9a and 9b were separately refluxed with glyoxylic acid in an aqueous sulfuric acid under a cross-aldol condensation reaction to afford the α,β-unsaturated ketones 9c and 9d respectively. Next, the obtained ketones were reduced under the palladium-carbon catalyzed hydrogenation reaction to afford the γ-keto-carboxylic acids 9e and 9f respectively. Ethylene ketal protection of the keto groups in each intermediate and subsequent reduction of the carboxylic function to an alcohol using LiAlH4 produced the corresponding alcohols 9g and 9h. The alcohol functions were separately converted to the tosylate to form alkylating agents 9i and 9j respectively, and were used to react with 1-pyridin-2-yl-piperazine followed by deprotection to make the final compounds 17 and 18 (Scheme 9) respectively.
Scheme 9.

Synthesis of compounds 17 and 18. Reagents and conditions: (a) glyoxylic acid, H2SO4, H2O, reflux; (b) Pd/C(10%) H2, MeOH/Dioxane 48 h; (c) i) ethylene glycol, pTSA, reflux, ii) LiAlH4 dry THF, 0 °C; d) p-TsCl, Et3N, CH2Cl2; e) i) 1-pyridin-2-yl-piperazine, K2CO3, KI, DME, reflux, ii) TsOH, MeOH, rt, 12 h.
Synthesis of 5-chloro-1-(4-chlorophenyl)pentan-1-one 10a followed a similar procedure as reported by Komissarov et al [35] and the resulting alkyl halide was subjected to the same two-step reaction reported by us [27, 33] to prepare the indanone 10b. Following same general alkyaltion procedure compound 19 was prepared in good yield (Scheme 10).
Scheme 10.

Synthesis of compound 19. Reagents and conditions: (a) i) hexamethylenetetramine, Ac2O, 105 °C, 18 h, (ii) H2SO4, 60 °C [33]; (b) 1-pyridin-2-yl-piperazine NaHCO3, KI, toluene, reflux.
Claisen condensation of the 5-substitutted-indanone 11a with diethylcarbonate (DEC) under basic conditions afforded the key β-keto ester 11b. Subsequent alkylation of the β-keto ester with the 1-bromo-4-chlorobutane produced the C-disubstituted β-keto esters 11c. An acid catalyzed decarboxylation of 11c under microwave heating conditions produced the alkylating agent 11d in good to moderate yield (Scheme 11). This decarboxylation method was a variant of what was described by Ulrich, Jordis, and colleagues [36]. The obtained alkylating agent 11d was reacted with 1-pyridin-2-yl-piperazine under the general alkylation conditions to produce compound 20 (Scheme 11).
Scheme 11.

Synthesis of compound 20. Reagents and conditions: (a) NaH, DEC, 0 °C-rt, 12h; (b) NaH, 1-bromo-4-chlorobutane, DMF, rt, 18–24h; (c) Conc. HCl in AcOH (glacial), MWAS; (d) i)1-(pyridin-2-yl)piperazine, K2CO3/KI, toluene, MWAS, ii) ethereal HCl.
To prepare the α-methylated 5-chloroindanone 12b, the β-keto ester 11a obtained from Scheme 11 was deprotonated and methylated under basic conditions to produce intermediate 12a (Scheme 12) which was decarboxylated to yield 12b. The alkylation step was repeated using 1-bromo-3-chloropropane and 1-bromo-4-chlorobutane to produce alkylating agents 12c and 12d respectively which were reacted separately with 1-(pyridin-2-yl)piperazine to afford compounds 21 and 22 respectively.
Scheme 12.

Synthesis of compounds 21 and 22. Reagents and conditions: (a) NaH, DMF, MeI, rt, 24h; (b) Conc. HCl in AcOH (glacial), MWAS; (c) NaH, DMF, 1-bromo-3-chloropropane (for 12c) or 1-bromo-4-chlorobutane (for 12d), rt, 24h; (d) 1-(pyridin-2-yl)piperazine, K2CO3/KI, toluene, MWAS.
3. Results and discussion
Our previous campaign expanded the antipsychotic potential of butyrophenones, and we reported that 1-(4-(4-fluorophenyl)butyl)-4-(pyridin-2-yl)piperazine (SYA16263 (2)) displays desirable multi-receptor binding affinity for D2-like receptors (Ki : D2 = 124 nM, D3 = 86 nM, D4 = 3.5 nM), and 5-HTRs (Ki : 5-HT2A, = 50 nM, 5-HT7 = 90 nM), particularly the 5-HT1A receptor (Ki 5-HT1A = 1.1 nM), (Table 1). Previous functional and in vivo behavioral studies indicated that compound 2 is a β-arrestin biased ligand at D2R and has low propensity to cause catalepsy in rats, a key early test to screen antipsychotic agents with the potential to cause motoric side effects [29]. Inspired by these seminal results, we embarked on SAFIR studies to explore the effects of structural changes in compound 2 on binding affinity at key CNS receptors. Due to the metabolic side effects associated with ligands that interact with 5-HT2CR, all compounds in this study were evaluated for binding affinity at this receptor.
Table 1:
Binding affinity constantsa [mean Ki, nM & (pKi ±SEM)b,c] at relevant CNS receptors and SERT transporter.
| Compound | D2 | D3 | D4 | 5-HT1A | 5-HT2A | 5-HT2C | 5-HT7 | SERT |
|---|---|---|---|---|---|---|---|---|
|
1 Azaperone |
228 (6.64 ± 0.06) | 89.7 (7.12 ± 0.09) | 6.7 (8.2 ± 0.07) | 8.1 (8.12 ± 0.05) | 62.7 (7.34 ± 0.06) | 1,540.3 (5.87 ± 0.07) | 120.5 (6.94 ± 0.08) | MTb |
|
2 (SYA16263)d |
124 ± 10 | 86 ± 4 | 3.5 ± 0.2 | 1.1 ± 0.05 | 50 ± 3 | MT | 90 ± 4 | MT |

|
1092 (5.96 ± 0.07) | 218 (6.66 ± 0.05) | 2384 (5.62±0.04) | 93 (7.03 ± 0.07) | 108 (6.97 ± 0.04) | 244 (6.61 ± 0.07) | 420 6.38 ± 0.06) | MT |

|
1930 (5.71 ± 0.08) | 203 (6.69 ± 0.05) | >10000 (<5) | 14 (7.84 ± 0.06) | 384.5 (6.45 ± 0.05) | 242 (6.62 ± 0.07) | 134 (6.87±0.05) | MT |
|
1095 (5.96 ± 0.07) | 469 (6.33 ± 0.05) | 577 (6.24 ± 0.03) | 27 7.57 ± 0.07) | 176.5 (6.81 ± 0.05) | 35 (7.5 ± 0.1) | 273 (6.56 ± 0.05) | MT |

|
674 (6.17 ± 0.08) | 391 (6.41 ± 0.05) | 1872 (5.73 ± 0.03) | 13 (7.87 ± 0.06) | 626 (6.2 ± 0.04) | MT | 222 6.65 ± 0.06) | MT |

|
>10,000 (<5) | 382.5 (6.4 ± 0.07) | 471 (6.39 ± 0.05) | 47.3 (7.3 ± 0.08) | 3111 (5.5 ± 0.06) | MT | 4.8 (8.32 ± 0.05) | MT |

|
526.5 (6.3 ± 0.1) | 205.5 (6.69 ± 0.08) | 146.3 (6.87 ± 0.09) | 4.5 (8.39 ± 0.07) | 564.3 (6.27 ± 0.05) | 1,130 (5.95 ± 0.06) | 24.5 (7.64 ± 0.05) | 1,143 (5.96 ± 0.1) |

|
2,667 (5.57 ± 0.08) | 647.5 (6.23 ± 0.05) | 496 (6.3 ± 0.05) | 20.3 (7.72 ± 0.08) | 318.3 (6.54 ± 0.07) | 1,192 (5.92 ± 0.07) | 187.5 (6.73 ± 0.05) | 3,064.6 (5.58 ± 0.1) |

|
2,798 (5.55 ± 0.08) | 2,098 (5.68 ± 0.05) | MT | 411 (6.39 ± 0.06) | 1,132 (5.95±0.07) | 6,662 (5.18 ± 0.08) | 957 (6.02 ± 0.06) | 248 6.61±0.09) |

|
MT | 286 (6.54 ± 0.05) | 121 (6.92 ± 0.07) | 19 (7.72 ± 0.07) | 161 (6.79 ± 0.05) | 1005 (6 ± 0.06) | 1598 (5.85±0.1) | MT |

|
699 (6.16 ± 0.07) | 376 (6.43 ± 0.05) | 14 (7.86±0.05) | 5.2 (8.28±0.06) | 345 (6.46 ± 0.05) | 111 (6.95 ± 0.09) | 77 (7.12 ± 0.05) | MT |
To probe the effects of electronic features of the para position of the phenyl ring in compounds 1 and 2, we introduced acetanilide (-NHC(O)CH3) and amino (-NH2) groups to produce compounds 3-6 and the binding affinity constants are reported in Table 1. Generally, these compounds showed no improvement in affinity for the clinically relevant DA and 5-HT receptors, compared to SYA16263. In particular, these changes were not tolerated at the D4 receptor. However, these compounds maintained moderate binding affinity at 5-HT1A, 5-HT2A and 5-HT7 receptors.
Replacing the p-fluoro-phenyl group in azaperone 1 with dihydrocarbostyril moiety to form compound 7 led to a 25-fold increase in potency at the 5-HT7R (Ki = 4.8 nM), moderate binding affinity at 5-HT1AR (Ki = 47.3nM) and loss of activity at D2R. Deoxygenating the keto function in compound 7 to obtain 8 restored the high binding potency at 5-HT1AR (Ki = 4.5 nM) with moderate to low affinity at all other receptors as reported in Table 1. Replacing the benzylic carbon at position 6 of the dihydrocarbostyril moiety found in 8 with an oxygen atom to give compound 9 did not produce any significant improvement in binding affinity at all receptors evaluated. Similarly, expanding the piperazine ring in compound 9 to a homopiperazine in 10 resulted in diminished affinity for all receptors under consideration, especially at 5-HT1AR where there was a 20-fold decrease in potency compared to compound 9 (9: Ki (5-HT1A) = 20.3 nM vs 10: Ki (5-HT1AR) = 411 nM) (Table 1). This suggests that the six-membered piperazine ring is essential for maintaining activity at 5-HT1AR. Removing the anilino-nitrogen in the dihydrocarbostyril moiety in 9 gave 11, which displayed no apparent improvement in affinity across all evaluated receptors. Further modification by replacing p-fluoro-phenyl in azaperone with dihydro-indene to obtain compound 12 restored the high binding potency at 5-HT1AR (Ki = 5.2 nM) and D4R (Ki = 14 nM) while binding affinity for other receptors was either moderate or diminished.
To further explore the SAR of the lead compounds (1 and 2), we synthesized partially restrained butyl spacer analogs 13-18, and their binding affinity constants are reported in Table 2. Restraining the keto group in azaperone (1) into an indanone to produce 13 resulted in complete loss of binding affinity at D2R. However, we observed a 20-fold improvement in binding affinity at the 5-HT7R (1: Ki = 120.5 nM vs 13: Ki = 11.91 nM) for 13, with no significant change in activity at other 5-HTRs. Reduction of the keto group in 13 to produce alcohol 14 led to restoration of moderate binding affinity for the D2R (Ki = 510 nM) while maintaining a similar trend, but with reduced affinity, at other receptors in comparison to SYA16263 (2). Compounds 15 and 16, chloro-indanone analogs of 1 and 2 respectively, were synthesized to explore the effects of isosteric replacement of the fluoro atom with a chloro atom. Interestingly, 15 displayed significant selectivity for the D4R (Ki = 65 nM) as affinities at D2R and D3R were lost while maintaining moderate binding activity at other 5-HTRs (Table 2). Compound 16 displayed high binding affinity for 5-HT1AR (Ki = 6.1 nM) and moderate affinity for 5-HT7R (Ki = 45 nM). Moving the chloro atom (para to the keto group) in compound 15 to position 6 (meta to the keto group) of the indanone moiety produced 17, which resulted in little or no improvement in binding affinity at the 5-HTRs (except at 5-HT1A where it displayed high affinity (Ki = 5.1 nM)) with no apparent gain in binding affinity for the D2R. Further, we introduced an additional chloro group into 15 to obtain compound 18 which can be viewed as a merge of 15 and 17. Again, this resulted in no significant gain in affinity at either DA or 5-HT receptors (Table 2).
Table 2:
Binding affinity Constantsa [mean Ki, nM & (pKi ±SEM)b,c] at relevant CNS receptors and SERT transporter.
| Compound | D2 | D3 | D4 | 5-HT1A | 5-HT2A | 5-HT2C | 5-HT7 | SERT |
|---|---|---|---|---|---|---|---|---|

|
MTb | 1469.8 | 41.39 | 11.39 | 75.09 | MT | 11.91 | MT |

|
510 ± 37 | 347 ± 24 | 7.9 ± 0.5 | 8 ± 0.5 | 580 ± 37 | MT | 62 ± 3 | 392 ± 33 |

|
MT | MT | 65 (7.19 ± 0.08) | 32 (7.5±0.09) | 113 (6.95±0.04) | 1,980 (5.7±0.05) | 14 (7.84±0.06) | MT |

|
1,234 (5.91 ± 0.07) | 247 (6.61 ± 0.09) | 44 (7.36 ± 0.06) | 6.1 (8.21 ± 0.07) | 110 (6.96 ± 0.04) | 2,729 (5.56 ± 0.08) | 45 (7.34 ± 0.05) | MT |

|
4,513 (5.35 ± 0.06) | 407 (6.39 ± 0.04) | 10 (7.99 ± 0.05) | 5.1 (8.3 ± 0.04) | 122 (6.91 ± 0.09) | 1,680 (5.77 ± 0.06) | 63 (7.2 ± 0.06) | MT |

|
3,071 (5.51 ± 0.06) | 707 (6.15 ± 0.04) | 45.5 (7.35 ± 0.06) | 46 (7.33 ± 0.04) | 159 (6.8 ± 0.09) | 1,644 (5.78 ± 0.06) | 23 (7.64 ± 0.06) | MT |
Results from NIMH PDSP
MT = missed 50% threshold inhibition at 10 μM concentration
Data points without standard error (SE) have errors below 20% of the mean value.
Inspired by the interesting binding affinity profile of the chloro-indanones in Table 2, and using 15 as the new lead compound, we further explored the effect of chain length between the indanone moiety and the pyridinyl-piperazine group on binding affinity. To this end, we synthesized compounds 19-22 and their binding affinities are reported in Table 3. Increasing the chain length from two (ethylenes) in 15 to three (propylenes) in 19 produced the much desired dual 5-HT1A and 5-HT7 ligands with low nanomolar binding affinity constants (19: Ki (5-HT1AR) = 2.3 nM, Ki (5-HT7R) = 7.8 nM). Further elongation of the chain length to four in compound 20 resulted in a similar binding affinity profile as observed with 19, particularly at the 5-HT1AR and 5-HT7R targets. Next, we explored the effects of introducing a methyl (CH3) group at the alpha (α)-position of the indanone segment to replace the labile α-hydrogen in 19 and 20 to obtain compounds 21 and 22 respectively. In addition, this modification served as a perfect handle to mitigate keto-enol tautomerism that occurs in indanones, as reported by us [37]. Interestingly, 5-chloro-2-methyl-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (21) produced the highest affinity ligand at 5-HT1AR (Ki = 0.74 nM) and 5-HT7R (Ki = 8.4 nM) among the indanone-pyridinylpiperazine series. Taken together, the indanones identified in this study tend to have poor affinity for D2R, varying binding affinity for D3R, moderate for D4R (10 nM < Ki < 100 nM) and moderate to high affinity at 5-HT1AR, 5-HT2AR, and 5-HT7R. In fact, compound 21 has a higher affinity at the 5-HT1AR and 5-HT7R (Ki: = 0.74 and 8.4 nM respectively) compared to aripiprazole (Ki: 5-HT1AR and 5-HT7R = 5.6 and 10.3 nM respectively) [38].
Table 3:
| Compound | D2 | D3 | D4 | 5-HT1A | 5-HT2A | 5-HT2C | 5-HT7 | SERT |
|---|---|---|---|---|---|---|---|---|

|
MTb | MT | 65 (7.19 ± 0.08) | 32 (7.5 ± 0.09) | 113 (6.95 ± 0.04) | 1,980 (5.7 ± 0.05) | 14 (7.84 ± 0.06) | MT |

|
MT | 80 (7.1 ± 0.09) | 31 (7.51 ± 0.04) | 2.3 (8.63 ± 0.08) | 533 (6.27 ± 0.05) | 1875 (5.73 ± 0.04) | 7.8 (8.11 ± 0.06) | MT |

|
1,056 (5.98 ± 0.08) | 1,167.5 (6.01 ± 0.06) | 64 (7.19 ± 0.05) | 4.4 (8.39 ± 0.08) | 440.3 (6.4 ± 0.07) | 367 (6.44 ± 0.07) | 15 (7.83 ± 0.05) | 2184 (5.72 ± 0.1 |

|
1096 (5.96 ± 0.05) | >10,000 (<5) | 48 (7.32 ± 0.09) | 0.74 (9.13 ± 0.08) | 200 (6.7 ± 0.07) | 1448 (5.84 ± 0.07) | 8.4 (8.07 ± 0.08) | >10,000 (<5) |

|
3260 (5.49 ± 0.09) | >10,000 (<5) | 143 (6.8 ± 0.1) | 2.45 (8.62 ± 0.09) | 578 (6.24 ± 0.07) | 1100 (5.96 ± 0.07) | 15 (7.82 ± 0.08) | >10,000 (<5) |

|
3.3±0.1 | 9.7±5.4 | 510±93 | 5.6±0.8 | 8.7±2.0 | 180± 37 | 10.3±3.7 | 1080±180 |
Results from NIMH PDSP
MT = missed 50% threshold inhibition at 10 μM concentration.
Data points without standard error (SE) have errors below 20% of the mean value.
Aripiprazole data was obtained from reference 43.
The target compounds in this study were also evaluated at the 5-HT2CR and the serotonin transporter (SERT). Overall, compounds reported in this study displayed little if any affinity for SERT while no definitive trend was observed at the 5-HT2CR, with most of the target compounds showing poor to moderate binding affinities, particularly among the indanones.
The functional characteristics of compound 21, the highest dual affinity ligand identified in this study were assessed at the 5-HT1AR and 5-HT7AR (Tables 4 and 5, Fig 2 and 3). As an agonist, serotonin has an EC50 of 2.03 nM (%Max = 101.5), whereas compound 21 has an EC50 of 2.74 nM and is fully efficacious (%Max = 100.5) (Table 4 & Fig. 2). As expected, serotonin similarly showed agonist properties at the 5-HT7AR but compound 21 did not display any agonist activity (Table 4 & Fig. 3). Looking at compound 21 in the antagonist assay, it had an IC50 value of 100 nM and 101.5% inhibition of adenylate cyclase compared to lurasidone’s IC50 value of 15.72 nM and 100.0% adenylate cyclase inhibition (Table 5 & Fig. 3). Thus, compound 21 acts as a full agonist at the 5-HT1AR but acts as an antagonist at the 5-HT7AR. This functional characteristic is consistent with characteristics for treating cognitive impairments and for developing antidepressant drugs.
Table 4:
Serotonin receptors functional assays, agonist effects
| 5-HT1AR GTPγS binding | 5-HT7AR adenylate cyclase | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | EC50 | SEM | n | % max | SEM | EC50 | SEM | n | %5-HT | SEM |
| 21 | 2.74 | 0.91 | 4 | 100.5 | 3.6 | >10μM | 1 | 0 | ||
| Serotonin | 2.03 | 0.56 | 6 | 101.5 | 1.5 | 3.66 | 0.52 | 2 | 96.9 | 1.9 |
Table 5:
Serotonin receptors functional assays, antagonist effects
| 5-HT1AR GTPγS binding | 5-HT7AR adenylate cyclase | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | IC50 | SEM | n | %max inhibition | SEM | IC50 | SEM | n | %max inhibition | SEM |
| 21 | - | 100 | 14 | 2 | 101.5 | 0.1 | ||||
| Lurasidone | 15.72 | 0.26 | 2 | 100.0 | 0.0 | |||||
Fig. 2.

In vitro serotonin receptor, HEK-5-HT1A [35S]GTPγS binding functional assay results showing the agonistic activity for Compound 21 in comparison with serotonin.
Fig. 3.

In vitro serotonin receptor, 5-HT7A adenylate cyclase functional assay results for compound 21. Graphs showing the specific cAMP formation for agonist activity of compound 21 in comparison to Serotonin (top graph) and antagonist activity of Compound 21 in comparison to Lurasidone (bottom graph).
In conclusion, using SYA16263 (2), which displayed high selectivity (>80 fold) for 5-HT1AR compared to 5-HT7R as the lead molecule, we have now identified very high affinity dual acting ligands for 5-HT1A and 5-HT7 receptors. Notably, the chloro-indanone pyridinyl-piperazine compound, 21 showed sub-nanomolar and low nanomolar binding affinities at the 5-HT1AR and 5-HT7R and with full agonist and antagonist effects at the respective receptors. Compound 21 is thus a valuable addition to the rather scarce group of reported dual 5-HT1AR and 5-HT7R selective ligands. It is anticipated that 21 may serve as a chemical probe for further exploitation of the SAR of multi-target ligands for CNS drug discovery.
4. Experimental section
4.1. Chemistry
Melting points were determined on a Gallenkamp (UK) apparatus and are uncorrected. All NMR spectra were obtained on a Varian 300 MHz Mercury Spectrometer and the free induction decay (FID) data were processed using Mestrelab’s Mnova NMR software (version 8.1) to obtain the reported NMR data. Elemental analyses were carried out by Atlantic Microlab, Inc., Norcross, GA, and are within 0.4% of theory unless otherwise noted. Flash chromatography was performed using a Teledyne CombiFlash® with Davisil grade 634 silica gel. Starting materials were obtained from Sigma–Aldrich and were used without further purification. All microwave assisted syntheses (MWAS) were carried out using a Biotage Initiator®.
4.1.1. Acetanilide (1b)
Acetanilide 1b was prepared by acetylating aniline following a simple method described by Zhang and co-workers [39]. Briefly, to a mixture of aniline (3.7 mL, 40 mmol) and triethylamine (7.3 mL) in dry CH2Cl2 (50 mL) was added acetyl chloride (3.2 mL, 44 mmol) dropwise while stirring. After addition, the mixture was allowed to stir for 4 h at room temperature. The reaction was quenched by adding water (20 mL) and the organic layer washed further with water (2 × 20 mL), brine (50 mL), dried using Na2SO4 and concentrated in-vacuo to obtain acetanilide (5 g, 93%) as solid needle-like crystals. 1H NMR (300 MHz, CDCl3) δ 8.77 (1H, s), 7.59 – 7.48 (2H, m), 7.33 – 7.20 (2H, m), 7.13 – 7.01 (1H, m), 2.12 (3H, s). 13C NMR (75 MHz, CDCl3) δ 169.50, 138.22, 128.83, 124.25, 120.38, 24.29.
4.1.2. N-(4-(4-chlorobutanoyl)phenyl)acetamide (1c)
A modified acylation reaction described by Lackey et al. [40] was followed to access intermediate 1c. Briefly, to a dry 100 mL round-bottomed flask equipped with a stirrer was added AlCl3 (5g, 37.5 mmol) carbon disulfide (CS2) (30 mL) and 4-chlorobutyryl chloride (2.5 mL, 22.5 mmol) at 0 °C with stirring. To the mixture obtained was added 1b (15 mmol) portionwise over 20 minutes. After addition is complete, the reaction mixture was allowed to warm to room temperature and stirred overnight. The content was dumped into a beaker containing 100 g of ice with 5 mL conc. HCl and stirred thoroughly. The brick red precipitate obtained was dissolved in methanol and loaded onto silica column and subsequently separated by flash chromatography (gradient elution up to 50% EtOAc in hexanes) to afford N-(4-(4-chlorobutanoyl)phenyl)acetamide (1c). Yellowish solid. Yield: 45%. 1H NMR (300 MHz, DMSO-d6) δ 10.26 (s, 1H), 7.92 (2H, dd, J = 6.5, 2.0 Hz), 7.71 (2H, dd, J = 6.5, 2.4 Hz), 3.73 – 3.60 (2H, m), 3.10 (2H, t, J = 6.8 Hz), 2.12 – 1.96 (5H, m). 13C NMR (75 MHz, DMSO-d6) δ 197.85, 169.36, 144.13, 131.49, 129.58, 118.62, 45.34, 35.22, 27.39, 24.61.
4.1.3. N-(4-(4-chlorobutyl)phenyl)acetamide (1d)
A modified phenyl-keto reduction reaction described by West and colleagues [30] was followed to access alkylating agent 1d. In brief, to a stirred trifluoroacetic acid (1.5 mL) solution of N-(4-(4-chlorobutanoyl)phenyl)acetamide 1c (0.33 g, 1.3 mmol) was added triethylsilane (0.5 mL) gradually over 10 min keeping the temperature at room temperature. After addition, the mixture was allowed to stir for 15 mins. The reaction was then quenched by neutralizing the trifluoroacetic acid with saturated NaHCO3 solution. The mixture was extracted with CH2Cl2 (3×25 mL) and the organic layer pulled together, washed with brine, dried over Na2SO4, and concentrated in-vacuo to afford N-(4-(4-chlorobutyl)phenyl)acetamide 1d as a pale yellow solid. Yield: 77%. 1H NMR (300 MHz, DMSO-d6) δ 9.83 (1H, s), 7.45 (2H, dd, J = 8.8, 2.4 Hz), 7.08 (2H, dd, J = 8.8, 2.4 Hz), 3.62 (2H, t, J = 6.1 Hz), 2.52 (2H, t, J = 7.0 Hz), 2.00 (3H, s), 1.78 – 1.50 (4H, m). 13C NMR (75 MHz, DMSO-d6) 168.48, 137.61, 136.75, 128.85, 119.48, 45.66, 34.07, 32.01, 28.63, 24.38.
4.1.4. General alkylation procedure:
A mixture of alkylating agent (1.1 equiv), base (1.0 equiv) K2CO3 (1.1 equiv), and KI (catalytic) in DME (10 mL) was placed in a 20 mL microwave vial with a stirrer and tightly sealed. The mixture was subjected to microwave heating (Biotage, 120–140 °C, 200–400W, 15±5 bar) for 30–60 mins. The mixture was directly purified on silica by flash chromatography (Hexanes: EtOAc gradient up to 70% EtOAc) to afford the titled compound. The free base where necessary, was converted to the HCl or oxalate salt.
Following the general alkylation reaction described in 4.1.4, alkylating agents 1c and 1d were reacted separately with 1-(pyridin-2-yl)piperazine to afford compounds 3 and 4 respectively
4.1.4.1. N-(4-(4-(4-(pyridin-2-yl)piperazin-1-yl)butanoyl)phenyl)acetamide oxalate (3)
White amorphous powder. Yield: 48%. Mp: 140–142 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.32 (1H, s), 8.18 – 8.10 (1H, m), 7.91 (2H, d, J = 8.4 Hz), 7.71 (2H, d, J = 8.5 Hz), 7.63 – 7.54 (1H, m), 6.92 (1H, d, J = 8.6 Hz), 6.72 (1H, dd, J = 7.1, 4.9 Hz), 3.86 – 3.62 (4H, m), 3.34 – 3.19 (4H, m), 3.17 – 3.07 (4H, m), 2.11 – 1.95 (5H, m). 13C NMR (75 MHz, DMSO-d6) δ 197.59, 169.42, 163.37, 158.44, 148.02, 144.20, 138.36, 131.39, 129.62 (2C), 118.62, 114.55, 108.12, 55.69, 51.02 (2C), 42.42 (2C), 35.04, 24.62, 18.53. Anal. C21H26N4O2·2.4(COOH)2 (C, H, N).
4.1.4.2. N-(4-(4-(4-(pyridin-2-yl)piperazin-1-yl)butyl)phenyl)acetamide (4)
White crystals. Yield: 50%. Mp: 135–136 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.82 (1H, s), 8.13 – 8.03 (1H, m), 7.50 – 7.41 (2H, m), 7.08 (2H, d, J = 8.3 Hz), 6.76 (1H, d, J = 8.6 Hz), 6.64 – 6.54 (2H, m), 3.47 – 3.38 (4H, m), 2.51 (2H, t, J = 7.3 Hz), 2.42 – 2.33 (4H, m), 2.28 (2H, t, J = 7.2 Hz), 2.00 (3H, s), 1.61 – 1.36 (4H, m). 13C NMR (75 MHz, DMSO-d6) δ 168.44, 159.51, 147.96, 137.86, 137.49, 137.20, 128.82 (2C), 119.44 (2C), 113.34, 107.42, 58.17, 53.05 (2C), 45.09 (2C), 34.85, 29.33, 26.25, 24.38. Anal. C21H28N4O·0.2H2O (C, H, N).
4.1.5. Deacetylation of compounds 3 and 4
Under acidic conditions following a slightly modified method described by Kilbourn and co-workers [31] afforded 1-(4-aminophenyl)-4-(4-(pyridin-2-yl)piperazin-1-yl)butan-1-one, 5 and 4-(4-(4-(pyridin-2-yl)piperazin-1-yl)butyl)aniline 6 respectively. Briefly, to an acetic acid (glacial, 7 mL) solution of the acetamide 3 or 4 (1 mmol) in a 20-mL microwave vial was added conc. HCl (3 mL). The homogeneous solution obtained was sealed and subjected to microwave heating (Biotage, 145 °C, 200–400W, 10 bar) for 15–30 mins. The internal pressure in the vial was released prior to opening and the content neutralized with saturated NaHCO3 until slightly alkaline (pH = 8). The aqueous mixture was extracted with CH2Cl2 (3×50 mL), organic layers pulled together, washed with brine (50 mL), dried over Na2SO4, and the excess solvent removed in-vacuo to afford the corresponding compounds 5 and 6 as free bases.
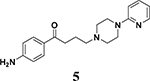
4.1.5.1. 1-(4-aminophenyl)-4-(4-(pyridin-2-yl)piperazin-1-yl)butan-1-one (5)
White crystals. Yield: 78%. Mp: 203–204 °C 1H NMR (300 MHz, DMSO-d6) δ 8.07 (1H, s), 7.67 (2H, dd, J = 5.1, 1.8 Hz), 7.52 – 7.45 (1H, m), 6.77 (1H,d, J = 8.8 Hz), 6.62 – 6.56 (1H, m), 6.54 (2H, dd, J = 5.0, 1.8 Hz), 5.98 (2H, s), 3.43 – 3.37 (4H, m), 2.81 (2H, t, J = 7.1 Hz), 2.40 (4H, t, J = 5.1 Hz), 2.31 (2H, t, J = 7.2 Hz), 1.81 – 1.70 (2H, m). 13C NMR (75 MHz, DMSO-d6) δ 200.34, 161.63, 155.90, 150.05, 140.29, 132.98 (2C), 127.27, 115.70, 115.14 (2C), 109.81, 59.92, 54.98 (2C), 47.11 (2C), 37.38, 24.18. Anal. C19H24N4O (C, H, N).
4.1.5.2. 4-(4-(4-(pyridin-2-yl)piperazin-1-yl)butyl)aniline (6)
White solid. Yield: 69%. Mp: 90–91 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.07 (1H, d, J = 4.9 Hz), 7.48 (1H, d, J = 13.6 Hz), 6.87 – 6.72 (3H, m), 6.60 (1H, dd, J = 7.0, 4.9 Hz), 6.51 – 6.42 (2H, m), 4.79 (2H, s), 3.41 (4H, t, J = 5.1 Hz), 2.44 – 2.34 (6H, m), 2.27 (2H, t, J = 7.0 Hz), 1.55 – 1.34 (4H, m). 13C NMR (75 MHz, DMSO-d6) δ 159.51, 147.96, 146.76, 137.87, 129.57 (2C), 129.04, 114.39 (2C), 113.34, 107.43, 58.26, 53.05 (2C), 45.08 (2C), 34.70, 29.71, 26.25. Anal. C19H26N4·0.3H2O (C, H, N).
4.1.6. 6-(4-chlorobutanoyl)-3,4-dihydroquinolin-2(1H)-one (2b)
Commercially available 3,4-dihydroquinolin-2(1H)-one 2a was acylated similarly as 1a to produce the intermediate 6-(4-chlorobutanoyl)-3,4-dihydroquinolin-2(1H)-one 2b as yellow crystals. Yield: 68%. 1H NMR (300 MHz, DMSO-d6) δ 10.40 (1H, s), 7.78 (1H, s), 7.75 (1H, d, J = 2.0 Hz), 6.91 (1H, d, J = 8.2 Hz), 3.67 (2H, t, J = 6.7 Hz), 3.07 (2H, t, J = 7.1 Hz), 2.92 (2H, t, J = 7.6 Hz), 2.47 (2H, t, J = 6.5, 8.6 Hz), 2.04 (2H, q, J = 6.9 Hz). 13C NMR (75 MHz, DMSO-d6) δ 197.81, 170.88, 143.17, 130.86, 128.32, 128.16, 123.90, 115.22, 45.37, 35.20, 30.53, 27.43, 24.95.
4.1.7. 6-(4-chlorobutyl)-3,4-dihydroquinolin-2(1H)-one (2c)
Deoxygenation of intermediate 2b following the TES-mediated reaction described above for 1d afforded the alkylating agent 2c in 50% yield. 1H NMR (300 MHz, DMSO-d6) δ 9.99 (1H, s), 6.96 – 6.86 (2H, m), 6.77 (1H, d, J = 7.2 Hz), 3.57 (2H, t, J = 6.2 Hz), 2.78 (2H, t, J = 7.0 Hz), 2.50 – 2.36 (4H, m), 1.72 – 1.57 (4H, m). 13C NMR (75 MHz, DMSO-d6) δ 170.52, 136.60, 135.71, 127.94, 127.18, 123.80, 115.38, 45.53, 34.12, 32.05, 30.93, 28.74, 25.33.
4.1.8. The synthesis of compounds 7 and 8 followed the same alkylation general conditions as described in section 4.1.4.
4.1.8.1. 6-(4-(4-(pyridin-2-yl)piperazin-1-yl)butanoyl)-3,4-dihydroquinolin-2(1H)-one (7)
White crystalline solid. Yield: 71%. Mp: 192–193 °C. 1H NMR (300 MHz, DMSO-d6) δ 10.39 (1H, s), 8.07 (1H, d, J = 4.4 Hz), 7.79 (2H, s), 7.48 (1H, ddd, J = 2.1, 7.1, 8.9 Hz), 6.91 (1H, d, J = 8.1 Hz), 6.76 (1H, d, J = 8.6 Hz), 6.59 (1H, dd, J = 4.8, 7.1 Hz), 3.43 – 3.33 (4H, m), 2.98 – 2.86 (4H, m), 2.47 (2H, t, J = 7.7 Hz), 2.43 – 2.36 (4H, m), 2.32 (2H, t, J = 7.1 Hz), 1.79 (2H, p, J = 7.1 Hz). 13C NMR (75 MHz, DMSO-d6) δ 198.81, 170.87, 159.50, 147.96, 142.92, 137.86, 131.33, 128.34, 128.18, 123.82, 115.18, 113.33, 107.42, 57.65, 52.93 (2C), 45.04 (2C), 35.76, 30.56, 24.99, 21.89. Anal. C22H26N4O2 (C, H, N).
4.1.8.2. 6-(4-(4-(pyridin-2-yl)piperazin-1-yl)butyl)-3,4-dihydroquinolin-2(1H)-one (8)
Solid white crystals. Yield: 62%. Mp: 144–145 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.98 (1H, s), 8.07 (1H, d, J = 4.8 Hz), 7.49 (1H, dd, J = 1.8, 6.8 Hz), 7.00 – 6.88 (2H, m), 6.81 – 6.70 (2H, m), 6.65 – 6.54 (1H, m), 3.45 – 3.38 (4H, m), 3.37 – 3.32 (3H, m), 2.84 – 2.75 (2H, m), 2.48 (2H, t, J = 7.2 Hz), 2.44 – 2.33 (3H, m), 2.28 (2H, t, J = 7.0 Hz), 1.59 – 1.47 (2H, m), 1.47 – 1.32 (2H, m). 13C NMR (75 MHz, DMSO-d6) δ 170.51, 159.51, 147.96, 137.87, 136.50, 136.18, 128.01, 127.22, 123.79, 115.30, 113.34, 107.43, 58.19, 53.06 (2C), 45.10, 34.85, 30.95, 29.44, 26.31, 25.31, 25.22. Anal. C22H28N4O· 0.8H20 (C, H, N).
4.1.9. 6-(3-chloropropoxy)-3,4-dihydroquinolin-2(1H)-one (3b)
A modified O-alkylation method reported by Rampa and colleagues [41] was followed to access intermediate 3b. Succinctly, 6-hydroxy-3,4-dihydroquinolin-2(1H)-one 3a (0.82 g, 5 mmol), 1-bromo-3-chloropropane (1 mL, 10 mmol), K2CO3, and KI (catalytic) were suspended in DME (15 mL) placed in a 20-mL Biotage microwave vial equipped with a vial. The vial was sealed and subjected to microwave heating (Biotage, 130°C, 200W, 5 bar) for 1h. After this period, the vial was removed from the microwave, allowed to cool, internal pressure released and opened. The excess alkylating agent was removed in-vacuo and the residue purified directly on silica using a Combiflash ® (gradient elution: up to 40% EtOAc in hexanes) to afford 6-(3-chloropropoxy)-3,4-dihydroquinolin-2(1H)-one, 3b (0.98 g, 82%) as white crystals. 1H NMR (300 MHz, DMSO-d6) δ 9.88 (1H, s), 6.82 – 6.71 (3H, m), 4.00 (2H t, J = 6.2 Hz,), 3.75 (2H, t, J = 6.8 Hz), 2.81 (2H, t, J = 7.7 Hz), 2.38 (2H, t, J = 7.7 Hz), 2.16 – 2.06 (2H, m). 13C NMR (75 MHz, DMSO-d6) δ 170.18, 153.94, 132.37, 125.32, 116.21, 114.53, 113.47, 64.92, 42.46, 32.20, 30.78, 25.52.
4.1.10. Synthesis of compounds 9 and 10 used the same alkylation conditions described in section 3.1.4.
4.1.10.1. 6-(3-(4-(pyridin-2-yl)piperazin-1-yl)propoxy)-3,4-dihydroquinolin-2(1H)-one (9)
Using alkylating agent 3b and 1-(pyridin-2-yl)piperazine, compound 9 was obtained as its free base as white solid. Yield: 54%. Mp: 154–155 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.88 (1H, s), 8.08 (1H, d, J = 5.0 Hz), 7.49 (1H, ddd, J = 8.9, 7.1, 2.0 Hz), 6.80 – 6.66 (4H, m), 6.60 (1H, dd, J = 7.0, 4.9 Hz), 3.93 (2H, t, J = 6.3 Hz), 3.44 (4H, t, J = 5.0 Hz), 2.80 (2H, t, J = 7.3 Hz), 2.47 – 2.41 (6H, m), 2.40 – 2.39 (1H, m), 2.38 – 2.35 (1H, m), 1.93 – 1.77 (2H, m).13C NMR (75 MHz, DMSO-d6) δ 170.17, 159.51, 154.25, 147.97, 137.87, 132.12, 125.26, 116.20, 114.46, 113.43, 113.36, 107.45, 66.51, 54.99, 53.07, 45.11, 30.81, 26.73, 25.55. Anal. C21H26N4O2 (C, H, N).
4.1.10.2. 6-(3-(4-(pyridin-2-yl)-1,4-diazepan-1-yl)propoxy)-3,4-dihydroquinolin-2(1H)-one oxalate (10)
Using alkylating agent 3b and 1-(pyridin-2-yl)-1,4-diazepane as the amine, compound 10 was obtained as an oxalate salt. Yield: 30%. Mp: 190–192 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.90 (1H, s), 8.08 (1H, d, J = 4.8 Hz), 7.62 – 7.46 (1H, m), 6.86 – 6.51 (5H, m), 4.11 – 3.78 (4H, m), 3.64 – 3.47 (2H, m), 3.41 – 3.06 (6H, m), 2.80 (2H, t, J = 7.4 Hz), 2.37 (2H, t, J = 7.4 Hz), 2.26 – 1.96 (4H, m). 13C NMR (75 MHz, DMSO-d6) δ 170.17, 163.94, 157.89, 153.79, 147.94, 138.13, 132.44, 125.30, 116.20, 114.55, 113.52, 112.66, 106.29, 65.67, 54.96, 54.05, 53.93, 45.61, 30.78, 25.52, 24.49, 24.19. Anal. C22H28N4O2·1.7(COOH)2 (C, H, N).
4.1.11. 5-(3-chloropropoxy)-2,3-dihydro-1H-inden-1-one (4b)
Using similar O-alkylation reaction method described for 3b above, intermediate 4b was obtained as white solid crystal by reacting commercially available 5-hydroxy-2,3-dihydro-1H-inden-1-one,4a with 1-bromo-3-chloropropane. Yield: 73%.1H NMR (300 MHz, CDCl3) δ 7.64 (1H, d, J = 8.3 Hz), 6.92 – 6.82 (2H, m), 4.16 (2H, t, J = 5.8 Hz), 3.72 (2H, t, J = 6.3 Hz), 3.04 (2H, t, J = 5.7 Hz), 2.67 – 2.58 (2H, m), 2.31 – 2.17 (2H, m). 13C NMR (75 MHz, CDCl3) δ 205.17, 164.29, 158.13, 130.51, 125.29, 115.58, 110.29, 64.62, 41.25, 36.39, 31.97, 25.84.
4.1.12. 5-(3-(4-(pyridin-2-yl)piperazin-1-yl)propoxy)-2,3-dihydro-1H-inden-1-one (11)
Following the general alkylation reaction 4.1.4, intermediate 4b was reacted with 1-(pyridin-2-yl)piperazine to produce compound 11 as a white solid crystal. Yield: 68% Mp: 103–104 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.08 (1H, s), 7.59 – 7.42 (2H, m), 7.04 (1H, s), 6.92 (1H, d, J = 8.0 Hz), 6.76 (1H, d, J = 8.6 Hz), 6.66 – 6.55 (1H, m), 4.10 (2H, t, J = 6.3 Hz), 3.52 – 3.38 (4H, m), 3.05 – 2.95 (2H, m), 2.59 – 2.50 (4H, m), 2.00 – 1.80 (2H, m), 1.63–1.22 (4H, m). 13C NMR (75 MHz, DMSO-d6) δ 204.58, 164.60, 159.49, 158.66, 147.98, 137.85, 130.21, 124.97, 116.02, 113.36, 110.99, 107.42, 66.86, 54.79, 53.05 (2C), 45.10 (2C), 36.44, 26.48, 25.90. Anal. C21H25N3O2.0.1H2O (C, H, N).
4.1.13. 1-(2,3-dihydro-1H-inden-5-yl)-4-(4-(pyridin-2-yl)piperazin-1-yl)butan-1-one (12)
The commercially available indane, 5a was acylated under Friedel-Crafts acylation conditions and the intermediate 5b obtained was subsequently reacted with 1-(pyridin-2-yl)piperazine under general N-alkylation conditions to afford the final compound 12 as white solid crystals. Yield: 59%, Mp: 74–75 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.07 (1H, s), 7.78 (1H, s), 7.72 (1H, d, J = 8.0 Hz), 7.52 – 7.42 (1H, m), 7.29 (1H, d, J = 7.8 Hz), 6.73 (1H, d, J = 8.6 Hz), 6.63 – 6.52 (1H, m), 3.44 – 3.30 (4H, m), 2.96 (2H, t, J = 7.0 Hz), 2.91 – 2.79 (4H, m), 2.44 – 2.26 (6H, m), 2.06 – 1.92 (2H, m), 1.87 – 1.69 (2H, m). 13C NMR (75 MHz, DMSO-d6) δ 199.95, 159.49, 149.82, 147.95, 144.70, 137.80, 135.89, 126.78, 124.69, 124.14, 113.29, 107.37, 57.63, 52.93 (2C), 45.03 (2C), 36.15, 32.85, 32.47, 25.42, 21.83. Anal. C22H27N3O (C, H, N).
4.1.14. 5-fluoro-2-(2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl)-2,3-dihydro-1H-inden-1-one (13)
A mixture of 2-(2-chloroethyl)-5-fluoro-2,3-dihydro-1H-inden-1-one, 6a (1.1 g, 5.2 mmol), 1-(pyridin-2-yl)piperazine (0.90 g, 5.5 mmol), KI (100 mg), NaHCO3 (1.0 g, 11.9 mmol) in toluene (10 mL) was heated to reflux under N2 for 12 h. After cooling to rt, the mixture was diluted with EtOAc (500 mL) and followed by washing with water (2×300 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo to dryness and followed by column chromatography on silica gel afforded 5-fluoro-2-(2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl)-2,3-dihydro-1H-inden-1-one, 13. The crystals were obtained by crystallization from EtOAc/Hexane in a yield of 26%. 1H NMR (300 MHz CDCl3): 8.16 (1H, m), 7.73 (1H, dd, J = 5.1, 8.1 Hz), 7.46 (1H, ddd, J = 1.8, 6.9, 8.7 Hz), 7.02 – 7.13 (2H, m), 6.58 – 6.63 (2H, m), 3.38 – 2.90 (4H, m), 3.32 (1H, dd, J = 7.5, 16.8 Hz), 2.86 (1H, dd, J = 4.2, 16.8 Hz), 2.75 – 2.81 (1H, m), 2.45 – 2.60 (6H, m), 2.12 – 2.21 (1H, m), 1.78 – 1.88 (1H, m). 13C NMR (300 MHz, CDCl3): 206.4, 167.0 (d, J = 254.2 Hz), 159.5, 156.2 (d, J = 10.4 Hz), 147.9, 137.4, 133.4, 126.0 (d, J = 10.4 Hz), 115.6 (d, J = 24.0 Hz), 113.2 (d, J = 3.4 Hz), 112.9, 107.0, 55.7, 53.0 (2C), 45.6, 45.0 (2C), 32.2, 28.3. Anal. C20H22FN3O (C, H, N).
4.1.15. 2-(2-Chloro-ethyl)-5-fluoro-indan-1-ol (7a)
To a solution of 2-(2-chloro-ethyl)-5-fluoro-indan-1-one (2 g, 9.4 mmol) and CeCl3 (160 mg, 0.65 mmol) in MeOH (10 mL) was added with stirring NaBH4 (0.7 g, 18.5 mmol) at rt. After stirring at rt for 1h, the mixture was diluted with EtOAc (200 mL) and washed with sat NaHCO3 (50 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo and followed by column chromatography on silica gel afforded 2-(2-Chloro-ethyl)-5-fluoro-indan-1-ol, 1.85 g, yield = 92%. 1H NMR (CDCl3): 7.33 (1H, m), 6.91 (2H, m), 4.82 (1H, brs), 3.73 (2H, m), 3.15 (1H, dd, J = 7.2, 15.3 Hz), 2.50 (1H, m), 2.42 (1H, m), 2.34 (1H, m), 2.02 (1H, m).
4.1.16. 5-fluoro-2-[2-(4-pyridin-2-yl-piperazin-1-yl)-ethyl]-indan-1-ol (14)
A mixture of 2-(2-Chloro-ethyl)-5-fluoro-indan-1-ol (0.60 g, 2.8 mmol) 7a, 1-pyridin-2-yl-piperazine dihydrochloride (0.67 g, 2.84 mmol), KI (0.48 g), K2CO3 (1.2 g, 8.7 mmol) in DME (10 mL) was heated to reflux under N2 for 12 h. Then the mixture was directly purified through column chromatography on silica gel afforded 5-fluoro-2-[2-(4-pyridin-2-yl-piperazin-1-yl)-ethyl]-indan-1-ol (14) which was converted to the hydroiodide salt. Crystallization from MeOH-Et2O gave a solid, 0.6 g, yield 46%. Mp: 174–175 °C. 1H NMR (DMSO-d6): 9.39 (1H, brs), 8.15 (1H, dd, J = 1.5, 4.5 Hz), 7.60 (1H, dt, J = 2.1, 8.7 Hz), 7.29 (1H, dd, J = 5.4, 7.5 Hz), 6.97 (3H, m), 6.74 (1H, dd, J = 4.8, 6.6 Hz), 5.52 (1H, brs), 4.68 (1H, brs), 4.38 (2H, brs), 3.59 (2H, brs), 3.04 (6H, m), 2.44 (1H, m), 2.08 (3H, m), 1.89 (1H, m). 13C NMR (150 MHz, CDCl3): 13C NMR (151 MHz, DMSO) δ 162.47 (d, J = 241.6 Hz, C-F), 158.33, 148.02, 143.64, 142.35, 138.45, 125.74, 114.75, 113.81, 111.74, 108.25, 79.02, 55.06, 48.01 (2C), 42.49 (2C), 40.57, 35.39, 27.38. Anal. C20H25FIN3O (C, H, N).
4.1.17. 5-chloro-2-(2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl)-2,3-dihydro-1H-inden-1-one (15)
Compound 15 was prepared similarly to 14 by reacting previously reported alkylating agent 5-chloro-2-(2-chloroethyl)-2,3-dihydro-1H-inden-1-one, 8a with 1-pyridin-2-yl-piperazine under same alkylation reaction conditions. The title compound was obtained as solid white crystal. Mp: 132–135 °C. 1H NMR (300 MHz, DMSO-d6’): 8.05 (1H, dd, J = 1.8, 4.8 Hz), 7.66 (1H, s), 7.59 (1H, d, J = 8.4 Hz), 7.48 (1H, dt, J = 1.8, 8.4 Hz), 7.42 (1H, dd, J = 1.8, 8.4 Hz), 6.75 (1H, d, J = 8.1 Hz), 6.59 (1H, dd, J = 5.4, 7.2 Hz), 3.18 – 3.28 (4H, m), 2.88 (1H, dd, J = 4.2, 18.0 Hz), 2.70 – 2.79 (1H, m), 2.34 – 2.45 (4H, m), 2.25 – 2.33 (3H, m), 1.88 – 1.97 (1H, m), 1.74 – 1.86 (1H, m). 13C NMR (150 MHz, CDCl3): 206.7, 159.4, 154.8, 147.9, 142.9, 137.4, 135.5, 128.1, 126.6, 124.9, 113.2, 107.0, 55.5, 52.9 (2C), 45.4, 45.0 (2C), 32.0, 28.2. Anal. C20H22ClN3O (C, H, N).
4.1.18. 5-chloro-2-(2-chloroethyl)-2,3-dihydro-1H-indene (8b)
Amalgamated zinc was prepared by stirring a mixture of zinc (1.2 g), HgCl2 (120 mg) in 5 ml water with conc HCl (0.1 mL) at room temperature. After stirring for 5 min, the mixture was decanted and followed by adding in order water (1 mL), conc HCl (1.75 mL), toluene (10 mL), and starting material 5-chloro-2-(2-chloroethyl)-2,3-dihydro-1H-inden-1-one, 8a (2 g, 8.73 mmol). The mixture was refluxed with stirring for 12 h. The solid was filtered off, aqueous layer was diluted with EtOAc (200 mL), washed with water, and sat. NaHCO3 (50 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo and used in the next step without further purification.
4.1.19. 1-(2-(5-chloro-2,3-dihydro-1H-inden-2-yl)ethyl)-4-(pyridin-2-yl)piperazine (16)
A mixture of 5-chloro-2-(2-chloroethyl)-2,3-dihydro-1H-indene, 8b (1 g, 4.6 mmol), 1-(pyridin-2-yl)piperazine ( 0.9 g, 5.6 mmol), KI (100 mg), K2CO3 (1.2 g, 9.2 mmol) in DME (10 mL) was heated to reflux under N2 for 12 h. Then the mixture was directly purified through column chromatography on silica gel to afforded the titled compound 1-(2-(5-chloro-2,3-dihydro-1H-inden-2-yl)ethyl)-4-(pyridin-2-yl)piperazine, 16 as an HCl salt after crystallization from MeOH-Et2O. Mp: 267–270 °C. 1H NMR (300 MHz, CD3OD) δ 8.16 (1H, ddd, J = 9.1, 7.1, 1.8 Hz), 8.12 – 8.04 (1H, m), 7.48 (1H, t, J = 10.4 Hz), 7.28 – 7.01 (4H, m), 3.79 (4H, s), 3.58 (1H, dd, J = 17.5, 7.8 Hz), 3.46 (1H, d, J = 6.0 Hz), 3.30 (4H, s), 3.17 – 3.01 (2H, m), 2.77 – 2.60 (2H, m), 2.52 (1H, td, J = 15.0, 7.5 Hz), 2.14 – 1.96 (2H, m). 13C NMR (151 MHz, CD3OD) δ 152.35, 145.01, 144.72, 141.15, 136.92, 131.66, 126.11, 125.29, 124.14, 114.48, 113.12, 55.73, 50.42 (2C), 43.35 (2C), 38.26, 37.78, 37.63, 29.03. Anal. C20H24ClN3.2HCl (C, H, N).
4.1.20. (E)-2-(6-chloro-1-oxo-1,3-dihydro-2H-inden-2-ylidene)acetic acid (9c)
A mixture of 6-chloro-2,3-dihydro-1H-inden-1-one (5 g, 30 mmol), glyoxylic acid (50% aqueous solution, 6.6 g, 72 mmol), and conc. H2SO4 (1 mL) in dioxane (5 mL) were stirred at refluxing temperature for 12 h. The mixture was cooled, the product filtered off, washed with water and dried to give the acid (E)-2-(6-chloro-1-oxo-1,3-dihydro-2H-inden-2-ylidene)acetic acid, 9c as a white solid. 1H NMR (600 MHz, DMSO-d6): δ 13.8 (1H, s), 7.86 – 7.58 (3H, m), 6.54 (1H, t, J = 2.38 Hz) 4.05 (2H, s).
4.1.21. (E)- 2-(5,6-dichloro-1-oxo-1,3-dihydro-2H-inden-2-ylidene)acetic acid (9d)
Preparation of intermediate 9d followed similar procedure as 9c to afford (E)- 2-(5,6-dichloro-1-oxo-1,3-dihydro-2H-inden-2-ylidene)acetic acid (86%) as a white solid. 1H NMR (300 MHz, CDCl3): δ 7.97 (1H, s), 7.92 (1H, s), 6.50 (1H, s), 4.00 (2H, s).
4.1.22. 2-(6-chloro-1-oxo-2,3-dihydro-1H-inden-2-yl)acetic acid (9e)
(E)- 2-(6-Chloro-1-oxo-1,3-dihydro-2H-inden-2-ylidene)acetic acid, 9c (3 g, 13.4 mmol) in MeOH (30 mL) and dioxane (120 mL) with Pd/C(10%, 1 g) was stirred under H2 (40 psi) for 48 h. The mixture was filtered through celite and the solvent evaporated to give 2-(6-chloro-1-oxo-2,3-dihydro-1H-inden-2-yl)acetic acid as an off-white solid. 1H NMR (600 MHz, DMSO-d6): δ 12.32 (1H, s), 7.7.75 – 7.51 (3H, m), 3.37–3.25 (1H, m), 2.94– 2.83 (2H, m), 2.76 – 2.62 (2H, m).
4.1.23. 2-(5,6-dichloro-1-oxo-2,3-dihydro-1H-inden-2-yl)acetic acid (9f)
Synthesis of 9f followed same procedure as 9e to give 2-(5,6-dichloro-1-oxo-2,3-dihydro-1H-inden-2-yl)acetic acid as an off-white solid. 1H NMR (300 MHz, CDCl3): δ 12.32 (s, 1H), 7.91 (s, 1H), 7.82 (s, 1H), 3.36–3.28 (m, 1H), 2.97–2.79 (m, 2H), 2.72–2.68 (m, 2H).
4.1.24. 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethan-1-ol (9g)
A solution of 2-(6-chloro-1-oxo-2,3-dihydro-1H-inden-2-yl)acetic acid (1.1 g, 4.8 mmol), ehtylene glycol (2 mL), TsOH (300 mg) in toluene (10 mL) was refluxed under N2 for 48 hrs and H2O was removed by azeotropic distillation. The reaction was monitored by 1H NMR in a maximum conversion of 80% after which the reaction was quenched by addition of Et3N (1 mL), diluted with EtOAc (250 mL), washed with sat NaHCO3, (25 mL) and H2O (25 mL). The organic layer was dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo to dryness and was used as such for the next reaction.
A solution of 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)acetic acid (1.3 g, 4.8 mmol) in dry THF (20 mL) was added dropwise to a suspension of LiAlH4 (0.37 g, 9.7 mmol) in dry THF (15 mL) at 0°C and the resulting mixture was stirred at refluxing temperature for 12 h. EtOAc was added to quench excess LiAlH4 and then aqueous HCl solution (10%, 50 mL) was added and the organic fraction separated. The aqueous solution was extracted with EtOAc (3×50 mL), and the combined organic fraction dried and the solvent evaporated to give alcohol 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethan-1-ol as a yellow oil which was used for the next step without further purification. 1H NMR (300 MHz, CDCl3): δ 7.26–7.22 (2H, m), 7.14 (1H, s),4.29 – 4.13 (4H, m), 3.77 – 3.73 (2H, m), 3.05 – 2.95 (1H, m), 2.69 – 2.63 (2H, m), 1.99 – 1.91 (1H, m), 1.77 – 1.63 (1H, m).
4.1.25. 2-(5,6-dichloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethan-1-ol (9h)
Preparation of 9h followed the same procedure as 9g to give the alcohol 2-(5,6-dichloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethan-1-ol as a yellow oil which was used as such for the next step. 1H NMR (300 MHz, CDCl3): δ 7.36 (1H, s), 7.30 (1H, s), 4.26 – 4.22 (2H, m), 4.16 – 4.12 (4H, m), 3.05 – 2.95 (1H, m), 2.69 – 2.60 (2H, m), 1.99 – 1.91 (1H, m), 1.77 – 1.63 (1H, m).
4.1.26. 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethyl 4-methylbenzenesulfonate (9i)
To a solution of 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethan-1-ol (0.55 g, 2.1 mmol), Et3N (0.66 mL, 6.3 mmol) in CH2Cl2 (10 mL) was added at room temperature p-TsCl (0.62 mg mL, 3.1 mmol). The mixture was stirred at room temperature for 12 h, and then followed by directly purification through column chromatography on silica gel, and provided 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethyl 4-methylbenzenesulfonate, yield 95%. 1H NMR (300 MHz, CDCl3): δ 7.81 (2H, d, J = 31.3 Hz), 7.37 −7.32 (2H, d, J = 31.3 Hz), 7.26–7.23 (2H, m), 7.10–7.08 (1H, m), 4.24 – 3.99 (6H, m), 2.93 – 2.83 (1H, m), 2.59 – 2.51 (2H, m), 2.46 (3H, s), 1.99 – 1.91 (1H, m), 1.77–1.63 (1H, m).
4.1.27. 2-(5,6-dichloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethyl 4-methylbenzene sulfonate (9j)
Synthesis of alkylating agent 9j followed the same procedure as 9i. White solid. Yield 95%. 1H NMR (300 MHz, CDCl3): δ 7.81 (1H, d, J = 31.3 Hz), 7.37 −7.32 (3H, m), 7.26 (1H, m), 4.23 – 3.97 (6H, m), 2.93 – 2.83 (1H, m), 2.59 – 2.51 (2H, m), 2.46 (3H, s), 1.99 – 1.91 (1H, m), 1.77–1.63 (1H, m).
4.1.28. 6-chloro-2-(2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl)-2,3-dihydro-1H-inden-1-one (17)
A mixture of 2-(6-chloro-2,3-dihydrospiro[indene-1,2’-[1,3]dioxolan]-2-yl)ethyl 4-methylbenzene sulfonate (263 mg, 0.64 mmol), 1-(pyridin-2-yl)piperazine (104 mg, 0.64 mmol), KI (50 mg), K2CO3 (266 mg, 1.9 mmol) in DME (10 mL) was heated 90 °C N2 for 12 h. The reaction was diluted with EtOAc (25 mL), washed with sat NaHCO3, (10 mL), water (10 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo to dry and proceeded for the further reaction. The crude product was dissolved in wet MeOH, TsOH (25 mg) was added with stirring at rt. After stirring at rt for 12 h, the solution was diluted with EtOAc (50 mL), and followed by washing with sat NaHCO3 (10 mL). The organic layer was dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo to dry and followed by column chromatography on silica gel afforded 6-chloro-2-(2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl)-2,3-dihydro-1H-inden-1-one. The product was converted to a dihydrochloride salt, further crystallization from MeOH-Et2O afforded the target product. Mp: 161–164 °C, 1H NMR (600 MHz, CD3OD) δ 8.23 – 8.06 (2H, m), 7.69 (2H, s), 7.59 (1H, d, J = 7.9 Hz), 7.48 (1H, d, J = 8.6 Hz), 7.15 (1H, s), 3.64 (4H, d, J = 31.1 Hz), 3.54 – 3.37 (3H, m), 3.32 (4H, s), 2.94 (2H, d, J = 14.9 Hz), 2.39 (1H, s), 2.10 (1H, s). 13C NMR (151 MHz, CD3OD) δ 206.64, 152.90, 152.08, 144.49, 137.85, 137.40, 135.03, 133.66, 128.16, 122.91, 114.59, 112.73, 55.13, 50.67, 45.18 (2C), 43.32 (2C), 32.00, 25.03. Anal. C20H22ClN3O•2HCl•1.2H2O (C, H, N).
4.1.29. 5,6-dichloro-2-(2-(4-(pyridin-2-yl)piperazin-1-yl)ethyl)-2,3-dihydro-1H-inden-1-one (18)
Following similar procedure as compound 17, the target compound 5,6-dichloro-2-(2-(3,4-dihydroisoquinolin-2(1H)-yl)ethyl)-2,3-dihydro-1H-inden-1-one, 18 was prepared in good yield, converted to hydrochloride salt and further crystallization from MeOH-Et2O to afford a white solid. Mp: 246–248 °C. 1H NMR (600 MHz, DMSO-d6) δ 11.63 (1H, s), 8.13 (1H, dd, J = 5.6, 1.2 Hz), 7.97 (1H, s), 7.92 (1H, t, J = 7.7 Hz), 7.86 (1H, s), 7.28 (1H, d, J = 8.8 Hz), 6.95 (1H, t, J = 6.3 Hz), 4.48 (3H, d, J = 12.7 Hz), 3.45 – 3.29 (4H, m), 3.27 – 3.10 (4H, m), 3.01 – 2.89 (1H, m), 2.87 (1H, dd, J = 17.4, 4.2 Hz), 2.35 – 2.24 (1H, m), 1.97 (1H, td, J = 13.3, 4.4 Hz). 13 C NMR (151 MHz, DMSO-d6) δ 205.22, 153.74, 142.32, 138.16, 136.49, 131.42, 129.60 (2C), 125.16 (2C), 114.43, 111.42, 53.97, 50.57, 45.26 (2C), 43.27 (2C), 32.09, 24.87. Anal. C20H21Cl2N3O•2HCl•H2O (C, H, N).
4.1.30. 5-chloro-1-(4-chlorophenyl)pentan-1-one (10a)
The procedure reported by Komissarov, V. V. et al [35] was followed to afford 5-chloro-1-(4-chlorophenyl)pentan-1-one, 10a. Yield 71%. 1H NMR (300 MHz, CDCl3) δ 7.90 (2H, d, J = 9.0 Hz), 7.45 (2H, d, J = 9.0 Hz), 3.59 (2H, t, J = 6.0 Hz), 2.99 (2H, d, J = 6.9 Hz), 1.85–1.91 (4H, m).
4.1.31. 5-chloro-2-(3-chloropropyl)-2,3-dihydro-1H-inden-1-one (10b)
Using 5-chloro-1-(4-chlorophenyl)pentan-1-one 10a, the synthesis of 5-chloro-2-(3-chloropropyl)-2,3-dihydro-1H-inden-1-one 10b followed similar procedure as 6a. Yield 100%. 1H NMR (300 MHz, CDCl3) δ 7.68 (1H, d, J = 8.1 Hz), 7.46 (1H, s), 7.35 (1H, d, J = 8.1 Hz), 3.56–3.61 (1H, m), 3.35 (1H, dd, J = 7.8, 17.4 Hz), 2.77–2.84 (1H, m), 2.68–2.75 (1H, m), 2.00–2.08 (1H, m), 1.90–1.96 (2H, m), 1.66–1.75 (2H, m).
4.1.32. 5-chloro-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (19)
Following the general alkylation reaction described in 4.1.4, compound 19 was prepared in moderate yield (yield 37%) as the hydrobromide salt. 1H NMR (DMSO-d6): 9.70 (2H, brs), 8.14 (1H, d, J = 5.4 Hz), 7.80 (1H, dd, J = 6.9, 8.1 Hz), 7.71 (1H, s), 7.64 (1H, d, J = 8.1 Hz), 7.47 (1H, d, J = 8.1 Hz), 7.15 (1H, d, J = 8.7 Hz), 6.87 (1H, t, J = 6.0 Hz), 4.37 (2H, d, J = 14.7 Hz), 3.61 (2H, d, J = 11.7 Hz), 3.25–3.40 (3H, m), 3.06–3.19 (4H, m), 2.72–2.88 (2H, m), 1.76–1.82 (3H, m), 1.48–1.55 (1H, m) 13C NMR (151 MHz, CD3OD) δ 207.52, 155.75, 153.11, 144.23, 141.34, 138.21, 134.77, 128.01, 126.70, 124.60, 114.61, 112.55, 56.54, 50.57 (2C), 46.53, 43.16 (2C), 32.14, 27.63, 21.40. Anal. C21H24ClN3O•2HBr (C, H, N).
4.1.33. Ethyl 5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (11b)
A modified method described by Paccani and co-workers [42] was followed to access the 5-substituted β-keto ester 11b. In brief, a solution of the 5-chloro indanone 11a, (100 mmol, 1 equiv) in diethyl carbonate (50 mL) was added dropwise to a stirred suspension of NaH (200 mmol, 60% in mineral oil previously washed with hexanes) in diethyl carbonate (DEC) (25 mL) at 0 °C with stirring (note green coloration). When evolution of gas has ceased, the mixture was allowed to stir at room temperature overnight. The mixture was then diluted in CH2Cl2 and treated with aqueous acetic acid solution. The aqueous phase was separated and extracted with CH2Cl2. The combined organic extracts were dried (Na2SO4) and concentrated under reduced pressure to yield the brown thick oil crude. The crude was loaded onto a cartridge and purified using flash chromatography with a gradient elution (up to 10% EtOAc in hexanes) to afford ethyl 5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate, 11b. Needle-like white crystals. Yield: 89%. 1H NMR (300 MHz, CDCl3) δ 10.47 – 10.20 (1H, m), 7.65 – 7.40 (6H, m), 4.31 – 4.13 (4H, m), 3.71 – 3.64 (1H, m), 3.55 – 3.38 (2H, m), 3.37 – 3.22 (2H, m), 1.35 – 1.19 (6H, m). 13C NMR (75 MHz, CDCl3) δ 198.15, 168.59, 155.11, 144.83, 135.83, 134.08, 131.40, 130.78, 130.06, 129.88, 129.80, 127.98, 125.70, 123.71, 121.81, 102.72, 61.83, 60.23, 53.24, 32.40, 29.91, 14.42, 14.17.
4.1.34. Ethyl 5-chloro-2-(4-chlorobutyl)-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (11c).
To a suspension of hexane washed NaH (60% in mineral oil, 12 mmol) in dry DMF was added a solution of ethyl 5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 11b (10 mmol) dropwise with stirring (note the green coloration) over 30 min. After the evolution of gas has ceased, the 1-bromo-4-chlorobutane (20 mmol) was added dropwise and allowed to stir for 18–24 h. After, the reaction was quenched in water (100 mL), shaken with CH2Cl2 (2×100 mL), the organic layers pulled together, washed with brine, and dried over Na2SO4 and concentrated in-vacuo to obtain the crude product which was then used directly in the next step without further purification.
4.1.35. 5-chloro-2-(4-chlorobutyl)-2,3-dihydro-1H-inden-1-one (11d)
Generally, an acid catalyzed decarboxylation of the tertiary β-keto ester was followed under microware heating conditions to afford alkylation agent 11d in good to moderate yield. Briefly, the crude β-keto ester 11c was dissolved in 10 mL of glacial acetic acid and transferred into a 20 mL Biotage microwave vial equipped with a stirrer. To this homogenous solution was added 3 mL of conc. HCl. The vial was sealed and subjected to microwave heating (Biotage, 120°C, 200W, 18 bar) for 30 min. The vial was then allowed to cool to room temperature and internal pressure released prior to opening. The content was neutralized with saturated NaHCO3 and extracted with CH2Cl2 (3 × 50 mL). The organic layers were pulled together, washed with brine, dried (Na2SO4), and solvent removed in-vacuo to produce a thick, oily crude. The crude was purified on flash column chromatography using gradient elution (up to 20% EtOAc in hexanes) to afford the corresponding alkylating agent 11d as a clear yellowish oil. Yield (overall over 2 steps): 51%. 1H NMR (300 MHz, CDCl3) δ 7.63 (1H, dd, J = 8.1, 1.4 Hz), 7.41 (1H, dd, J = 1.9, 1.0 Hz), 7.30 (1H, dd, J = 8.2, 1.8), 3.52 (2H, tt, J = 6.6, 1.0 Hz), 3.29 (1H, ddt, J = 17.4, 7.9, 0.8 Hz), 2.84 – 2.59 (2H, m), 1.99 – 1.85 (1H, m), 1.85 – 1.72 (2H, m), 1.63 – 1.50 (2H, m), 1.50 – 1.39 (1H, m). 13C NMR (75 MHz, CDCl3) δ 205.96, 154.61, 141.28, 134.76, 128.28, 126.65, 124.98, 45.00, 43.03, 34.06, 32.54.
4.1.36. 5-chloro-2-(4-(4-(pyridin-2-yl)piperazin-1-yl)butyl)-2,3-dihydro-1H-inden-1-one (20)
Using 11d as the alkylating agent, compound 20 was prepared under the same general alkylation procedure. The final compound 20 was converted to its HCl salt to obtain a white flaky solid. Yield: 72%, Mp: 233–234 °C. 1H NMR (300 MHz, DMSO-d6) δ 11.63 (2H, s), 8.08 (1H, d, J = 1.5, 5.9 Hz), 7.99 (1H, d, J = 8.0 Hz), 7.66 (1H, s), 7.62 (1H, d, J = 8.1 Hz), 7.45 (1H, dd, J = 1.6, 8.2 Hz), 7.36 (1H, d, J = 9.0 Hz), 6.98 (1H, d, J = 6.4 Hz), 3.73 – 3.49 (4H, m), 3.31 (1H, dd, J = 7.7, 17.5 Hz), 3.10 (4H, q, J = 8.9, 11.0 Hz), 2.91 – 2.65 (2H, m), 1.93 – 1.63 (4H, m), 1.61 – 1.21 (4H, m). 13C NMR (75 MHz, DMSO-d6) δ 207.25, 159.52, 156.23, 147.96, 140.13, 137.85, 135.50, 128.32, 127.35, 125.13, 113.33, 107.43, 58.21, 53.04 (2C), 47.30, 45.11 (2C), 32.53, 30.99, 26.68, 25.10. Anal. C22H26ClN3O.2HCl (C, H, N).
4.1.37. Ethyl 5-chloro-2-methyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate (12a)
To a stirred suspension of NaH (15mmol, 60% in mineral oil previously washed with hexanes) in dry DMF was added a solution of ethyl 5-chloro-1-oxo-2,3-dihydro-1H-indene-2-carboxylate 11b in dry DMF (7.5 mmol) portion-wise under N2. After stirring for 2 h, iodomethane (15 mmol) was added dropwise and the reaction stirred for 24 h. The reaction was quenched with water (50 mL), extracted with CH2Cl2 (2 × 50 mL), the organic layers pulled together was washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure to obtain the crude product which was loaded onto a cartridge and purified by flash chromatography using a gradient of up to 5% EtOAc in hexanes to obtain compound the intermediate compound. Yield: 60%; 1H NMR (300 MHz, CDCl3) δ 7.54 (1H, d, J = 8.1 Hz), 7.34 (1H, s), 7.22 (1H, d, J = 8.5 Hz), 3.99 (2H, q, J = 7.4 Hz), 3.56 (1H, d, J = 17.3 Hz), 2.85 (1H, d, J = 17.3 Hz), 1.36 (3H, s), 1.13 – 0.99 (3H, m).
4.1.38. 5-chloro-2-methyl-2,3-dihydro-1H-inden-1-one (12b)
Ethyl 5-chloro-2-methyl-1-oxo-2,3-dihydro-1H-indene-2-carboxylate, 12a (2.2 mmol) was subsequently hydrolyzed and decarboxylated by dissolving it in acetic acid (5 mL) and concentrated HCl (2 mL) was added and transferred into a 20 mL-microwave vial with a stirrer and tightly sealed. The mixture was heated in the microwave at 80 °C for 1 h, the reaction was allowed to cool to room temperature, basified with NaHCO3 solution and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was loaded onto a cartridge and purified by flash chromatography using a gradient of 5% EtOAc in hexanes to afford 12b. Yield: 79 %; 1H NMR (300 MHz, CDCl3) δ 7.61 (1H, d, J = 8.6 Hz), 7.38 (1H, s), 7.28 (1H, d, J = 8.0 Hz), 3.40 – 3.25 (1H, m), 2.71 – 2.61 (2H, m), 1.26 (3H, d, J = 7.3 Hz).
4.1.39. 6-chloro-2-(3-chloropropyl)-2-methyl-2,3-dihydro-1H-inden-1-one, 12c and 6-chloro-2-(4-chlorobutyl)-2-methyl-2,3-dihydro-1H-inden-1-one (12d)
A solution of the 12b (3.32 mmol) in dry DMF was added dropwise to a stirred suspension of NaH (9.96 mmol, 60% in mineral oil previously washed with hexanes) in dry DMF. After the reaction mixture was stirred for an hour, dihalo-aklylhalide (1-bromo-3-chloropropane for 12c or 1-bromo-4-chlorobutane for 12d) (6.64 mmol) was added dropwise and stirred overnight. The reactions were quenched in water, extracted with CH2Cl2, the organic layer washed with brine, dried over Na2SO4 and concentrated under reduced pressure to obtain the crude product which was used in the next step without further purification.
4.1.40. 6-chloro-2-methyl-2-(3-(4-(pyridin-2-yl)piperazin-1-yl)propyl)-2,3-dihydro-1H-inden-1-one (21)
The crude alkylating agent 12c was reacted with pyridinyl-piperazine under the general N-alkylation reaction conditions described above to afford compound 21 as a solid. Yield: 62 %. Mp: 232–233.6 °C. 1H NMR (300 MHz, CDCl3) δ 8.17 (1H, d, J = 4.7 Hz), 7.67 (1H, d, J = 8.2 Hz), 7.45 (2H, d, J = 8.7 Hz), 7.35 (1H, d, J = 7.9 Hz), 6.62 (2H, d, J = 8.1 Hz), 3.51 (4H, s), 3.11 (1H, d, J = 17.3 Hz), 2.87 (1H, d, J = 17.6 Hz), 2.51 (4H, s), 2.33 (2H, d, J = 7.3 Hz), 2.07 – 1.99 (1H, m), 1.61 (3H, t, J = 7.6 Hz), 1.22 (3H, s). 13C NMR (151 MHz, CD3OD) δ 209.76, 154.72, 152.26, 145.04, 141.56, 136.75, 133.97, 128.15, 126.87, 125.00, 114.45, 113.15, 56.79, 50.37 (2C), 43.25, 43.23 (2C), 39.30, 34.19, 22.84, 19.15. Anal. C22H26ClN3O•2HCl•H2O (C, H, N).
4.1.41. 6-chloro-2-methyl-2-(4-(4-(pyridin-2-yl)piperazin-1-yl)butyl)-2,3-dihydro-1H-inden-1-one (22)
The crude alkylating agent 12d was reacted with pyridinyl-piperazine in a similar manner as described for 21 to afford under the general N-alkylation reaction conditions described 4.1.4 to afford compound 22 as a solid. Yield: 52%. Mp: 214–215.9 °C; 1H NMR (300 MHz, CDCl3) δ 8.20 – 8.13 (1H, m), 7.70 – 7.62 (1H, m), 7.50 – 7.42 (2H, m), 7.38 – 7.22 (1H, m), 6.70 – 6.54 (2H, m), 3.52 (5H, t, J = 4.8 Hz), 3.09 (1H, d, J = 17.5 Hz), 2.85 (1H, d, J = 17.5 Hz), 2.50 (4H, t, J = 5.2 Hz), 2.39 – 2.27 (2H, m), 2.09 – 1.92 (1H, m), 1.68 – 1.47 (4H, m), 1.20 (3H, d, J = 1.8 Hz). 13C NMR (151 MHz, CD3OD) δ 210.38, 154.89, 145.04, 141.43, 136.76, 134.23 (2C), 128.06, 126.77, 124.85, 114.43, 113.15, 56.48, 50.32 (2C), 49.00, 43.24, 43.21.
4.2. Receptor binding affinity studies
Binding affinities reported in Tables 1–3 were conducted by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP). Details of the methods and radioligands used for the binding assays were previously reported [43].
[35S] GTPγS binding
The method from Newman-Tancredi et al, [44] was adapted for assessment of the functional status of compound 21 as follows: To prepare the membranes, five 15 cm plates of HEK-h5HT1A cells, 80–90% confluent, provided enough membranes for one assay plate (4 drug curves). GTPγS assay buffer (20 mM HEPES, pH 7.4, 10 mM MgCl2, 100 mM NaCl, and 0.2 mM DTT) was used throughout the assay. The cells were scraped from the plates into buffer, centrifuged at 200 rpm for 15 min, the supernatant was removed, and the pellet was homogenized in 10 ml buffer/ plate of cells. The homogenate was centrifuged at 17,500 rpm for 15 min, and the resulting pellet was washed 2 times by homogenization in 10 ml buffer and centrifugation to remove serotonin that was present in the growth medium. The supernatant was removed, and the final pellet from 5 plates was resuspended in 10 ml of assay buffer.
Cell membranes (40–75 μg protein) were preincubated (10 min, room temperature) with test compound in duplicate in assay buffer. The reaction was initiated by the addition of GDP (3 μM) and [35S]GTPγS (0.1 nM, 1350 Ci/mmol, Perkin Elmer, ~150,000 cpm) in a final volume of 1 ml. The reaction was incubated for 60 min at room temperature on a rotating platform. Non-specific binding is defined with 1 μM WAY-100635. Agonist efficacy is expressed relative to that of 5-HT tested at a maximally effective concentration (100 nM) in each experiment. A dose response curve with the prototypical full agonist serotonin was conducted in each experiment to identify full and partial agonist compounds. Experiments were terminated by rapid filtration over Filtermat A with ice-cold saline using a Tomtec harvester and radioactivity determined using a Perkin Elmer microbetaplate counter. EC50 values are calculated with GraphPad Prism and maximal effect for a given drug each day is normalized to the maximal effect of serotonin.
HEK-5-HT7A / cAMP Functional Assay
Human embryonic kidney cells were transfected with 10 μg h5-HT7A cDNA using PEI in unsupplemented DMEM. After 5 hr, cells were plated at a density of 200,000 cells per well in 48 well plates in DMEM supplemented with 10% charcoal-stripped FetalClone and pen-strep. The medium was removed about 18 hr later. For agonist assays, 0.9 ml EBSS (116 mM NaCl, 22 mM glucose, 15 mM HEPES, 8.7 mM NaH2PO4, 5.4 mM KCl, 1.3 mM CaCl2, 1.2 mM MgSO4, 1 mM ascorbic acid, 0.5 mM IBMX [3-isobutyl-1-methyl-xanthine] without BCS, pH 7.4 at 37°C) was added. After 20 min, the compound was added in a final volume of 1 ml, and incubated for 20 min. For antagonists, 0.8 ml EBSS is added, cells were incubated for 10 min, the compound was added, and incubated for 10 min after which serotonin (100 nM) was added. For all conditions, after 20 min incubation with agonist, the reaction was terminated by aspiration of the buffer, and 0.1 ml trichloroacetic acid was added. Plates were incubated for 2 hr on a rotator. Adenylate cyclase activity was measured using a cyclic AMP EIA kit (Cayman). Aliquots (40 μl) of each well were diluted to 200 μl with EIA buffer from the kit, and 50 μl of the dilution was added to the EIA plate. After addition of tracer and monoclonal antibody, the EIA plates were incubated for 18 hr at 4°C. The reaction was aspirated, plates were washed 5×300 μl with wash buffer, and Ellman’s reagent was added. After a two-hour incubation in the dark on a rotator, the plates were read at 410 nm. Basal cAMP is subtracted from all values. 5-HT7A agonists stimulate cAMP formation, maximal stimulation was defined with 10 μM serotonin. The maximal drug effect is normalized to maximal serotonin effect in the tables. For antagonists, maximal inhibition of cAMP formation is defined with 10 μM lurasidone.
Data analysis:
For functional assays, GraphPAD Prism was used to calculate either EC50 (agonists) or IC50 (antagonists) values using data expressed as pg cAMP for adenylate cyclase activity, % serotonin-stimulation for GTPγS binding.
Supplementary Material
Highlights.
SYA16263 shows high affinity at 5-HT1AR and moderate affinity at 5-HT7R
Structure-affinity relationship study focused on aryl-alkyl moiety of SYA16263
New dual 5-HT1AR and 5-HT7R ligand, 21 identified
Compound 21 has sub-nanomolar (5-HT1AR) and low nanomolar (5-HT7R) affinity
Compound 21 shows full agonist (for 5-HT1AR) and antagonist ( for 5-HT7R) effects
ACKNOWLEGEMENTS
This work was financially supported by an NIH/NIGMS SCORE grant number 2SC1GM116724 and a Title III Grant to Florida A&M University. We also acknowledge the financial support from Anxiolytech toward the ongoing studies. The work was also supported in part by the NIH Biomedical Endowment Eminent Scholar Chair in Biomedical Sciences research grant to S.Y.A. Ki determinations and receptor binding assays were generously carried out by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271-2013-00017-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. Funding sources acknowledged had no involvement in the study design, data collection, interpretation, article preparation and submission of this manuscript.
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Serretti A, De Ronchi D, Lorenzi C, Berardi D, New antipsychotics and schizophrenia: a review on efficacy and side effects, Curr. Med. Chem 11(3) (2004) 343–358. [DOI] [PubMed] [Google Scholar]
- [2].Mauri MC, Paletta S, Maffini M, Colasanti A, Dragogna F, Di Pace C, Altamura AC, Clinical Pharmacology of Atypical Antipsychotics: An Update, EXCLI J 13 (2014) 1163–1191. [PMC free article] [PubMed] [Google Scholar]
- [3].Attard A, Taylor DM, Comparative effectiveness of atypical antipsychotics in schizophrenia: what have real-world trials taught us?, CNS Drugs 26(6) (2012) 491–508. [DOI] [PubMed] [Google Scholar]
- [4].Palacios JM, Probst A, Cortes R, The distribution of serotonin receptors in the human brain: high density of [3H]LSD binding sites in the raphe nuclei of the brainstem, Brain Res 274(1) (1983) 150–155. [DOI] [PubMed] [Google Scholar]
- [5].Carhart-Harris RL, Nutt DJ, Serotonin and brain function: a tale of two receptors, J. Psychopharmacol 31(9) (2017) 1091–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barnes NM, Sharp T, A review of central 5-HT receptors and their function, Neuropharmacology 38(8) (1999) 1083–1152. [DOI] [PubMed] [Google Scholar]
- [7].Baldwin D, Rudge S, The role of serotonin in depression and anxiety, Int. Clin. Psychopharmacol 9(Suppl 4) (1995) 41–45. [DOI] [PubMed] [Google Scholar]
- [8].Mann JJ, Role of the Serotonergic System in the Pathogenesis of Major Depression and Suicidal Behavior, Neuropsychopharmacology 21(1) (1999) 99–105. [DOI] [PubMed] [Google Scholar]
- [9].Bantick RA, Deakin JF, Grasby PM, The 5-HT1A receptor in schizophrenia: a promising target for novel atypical neuroleptics?, J. Psychopharmacol 15(1) (2001) 37–46. [DOI] [PubMed] [Google Scholar]
- [10].Rollema H, Lu Y, Schmidt AW, Zorn SH, Clozapine increases dopamine release in prefrontal cortex by 5-HT1A receptor activation, Eur. J. Pharmacol 338(2) (1997) R3–5. [DOI] [PubMed] [Google Scholar]
- [11].Ohno Y, New insight into the therapeutic role of 5-HT1A receptors in central nervous system disorders, Cent. Nerv. Syst. Agents Med. Chem 10(2) (2010) 148–157. [DOI] [PubMed] [Google Scholar]
- [12].Ramboz S, Oosting R, Amara DA, Kung HF, Blier P, Mendelsohn M, Mann JJ, Brunner D, Hen R, Serotonin receptor 1A knockout: an animal model of anxiety-related disorder, Proc. Natl. Acad. Sci. U. S. A 95(24) (1998) 14476–14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Heisler LK, Chu HM, Brennan TJ, Danao JA, Bajwa P, Parsons LH, Tecott LH, Elevated anxiety and antidepressant-like responses in serotonin 5-HT1A receptor mutant mice, Proc. Natl. Acad. Sci. U. S. A 95(25) (1998) 15049–15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang S-M, Han C, Lee S-J, Patkar AA, Masand PS, Pae C-U, Vilazodone for the Treatment of Depression: An Update, Chonnam Med. J 52(2) (2016) 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Frampton JE, Vilazodone: in major depressive disorder, CNS Drugs 25(7) (2011) 615–627. [DOI] [PubMed] [Google Scholar]
- [16].Modica MN, Lacivita E, Intagliata S, Salerno L, Romeo G, Pittalà V, Leopoldo M, Structure-Activity Relationships and Therapeutic Potentials of 5-HT(7) Receptor Ligands: An Update, J. Med. Chem 61(19) (2018) 8475–8503. [DOI] [PubMed] [Google Scholar]
- [17].Glass JD, Grossman GH, Farnbauch L, DiNardo L, Midbrain Raphe Modulation of Nonphotic Circadian Clock Resetting and 5-HT Release in the Mammalian Suprachiasmatic Nucleus, J. Neurosci 23(20) (2003) 7451–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hedlund PB, Sutcliffe JG, Functional, molecular and pharmacological advances in 5-HT7 receptor research, Trends Pharmacol. Sci 25(9) (2004) 481–486. [DOI] [PubMed] [Google Scholar]
- [19].Roberts AJ, Hedlund PB, The 5-HT(7) receptor in learning and memory, Hippocampus 22(4) (2012) 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Abbas AI, Hedlund PB, Huang XP, Tran TB, Meltzer HY, Roth BL, Amisulpride is a potent 5-HT7 antagonist: relevance for antidepressant actions in vivo, Psychopharmacology (Berl.) 205(1) (2009) 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Naumenko VS, Popova NK, Lacivita E, Leopoldo M, Ponimaskin EG, Interplay between serotonin 5-HT1A and 5-HT7 receptors in depressive disorders, CNS Neurosci. Ther 20(7) (2014) 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hoyer D, Hannon JP, Martin GR, Molecular, pharmacological and functional diversity of 5-HT receptors, Pharmacol. Biochem. Behav 71(4) (2002) 533–554. [DOI] [PubMed] [Google Scholar]
- [23].Renner U, Zeug A, Woehler A, Niebert M, Dityatev A, Dityateva G, Gorinski N, Guseva D, Abdel-Galil D, Fröhlich M, Döring F, Wischmeyer E, Richter DW, Neher E, Ponimaskin EG, Heterodimerization of serotonin receptors 5-HT1A and 5-HT7 differentially regulates receptor signalling and trafficking, J. Cell Sci 125(Pt 10) (2012) 2486–2499. [DOI] [PubMed] [Google Scholar]
- [24].Bernstein P, Successful Drug Discovery Volume 3. Edited by Fischer János, Klein Christian and Childers Wayne E., ChemMedChem 13(18) (2018) 2011. [Google Scholar]
- [25].Rague A, Tidgewell K, Pharmacophore Comparison and Development of Recently Discovered Long Chain Arylpiperazine and Sulfonamide Based 5-HT7 Ligands, Mini Rev. Med. Chem 18(7) (2018) 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Peprah K, Zhu XY, Eyunni SVK, Setola V, Roth BL, Ablordeppey SY, Multi-receptor drug design: Haloperidol as a scaffold for the design and synthesis of atypical antipsychotic agents, Bioorg. Med. Chem 20(3) (2012) 1291–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Sampson D, Zhu XY, Eyunni SV, Etukala JR, Ofori E, Bricker B, Lamango NS, Setola V, Roth BL, Ablordeppey SY, Identification of a new selective dopamine D4 receptor ligand, Bioorg. Med. Chem 22(12) (2014) 3105–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bricker BA, Peprah K, Kang HJ, Ablordeppey SY, Evaluation of SYA16263 as a new potential antipsychotic agent without catalepsy, Pharmacol. Biochem. Behav 179 (2019) 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ablordeppey SY., Zhu XY., Identification of agents displaying functional activation of dopamine D2 and D4 receptors and methods for treatment of psychosis, Patent WO 2018148529A1, 2018.
- [30].West CT, Donnelly SJ, Kooistra DA, Doyle MP, Silane reductions in acidic media. II. Reductions of aryl aldehydes and ketones by trialkylsilanes in trifluoroacetic acid. Selective method for converting the carbonyl group to methylene, J. Org. Chem 38(15) (1973) 2675–2681. [Google Scholar]
- [31].Kilbourn MR, Welch MJ, Dence CS, Tewson TJ, Saji H, Maeda M, Carrier-added and no-carrier-added syntheses of [18F]spiroperidol and [18F]haloperidol, Int. J. Appl. Radiat. Isot 35(7) (1984) 591–598. [DOI] [PubMed] [Google Scholar]
- [32].Ofori E, Zhu XY, Etukala JR, Bricker BA, Ablordeppey SY, Synthesis and evaluation of the structural elements in alkylated tetrahydroisoquinolines for binding to CNS receptors, Bioorg. Med. Chem 24(22) (2016) 5730–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Peprah K, Zhu XY, Eyunni SV, Etukala JR, Setola V, Roth BL, Ablordeppey SY, Structure-activity relationship studies of SYA 013, a homopiperazine analog of haloperidol, Bioorg. Med. Chem 20(5) (2012) 1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Luche JL, Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones, J. Am. Chem. Soc 100(7) (1978) 2226–2227. [Google Scholar]
- [35].Komissarov VV, Kniazhanskaia ES, Atrokhova AV, Gottikh MB, Kritsyn AM, [The search of novel inhibitors of HIV-1 integrase among 5-(4-halogenophenyl)-5-oxopentyl derivatives of nucleic bases], Bioorg. Khim 40(5) (2014) 578–587. [DOI] [PubMed] [Google Scholar]
- [36].Jordis U, Frohlich J, Treu M, Hirnschall M, Czollner L, Kalz B, Welzig S, Derivatives and analogs of galanthamine, Patent Number US2007027138A1, 2007.
- [37].Onyameh EK, Bricker BA, Ofori E, Ablordeppey SY, Enantioseparation of 5-chloro-2-{2-[3,4-dihydroisoquinoline-2(1H)-yl]ethyl}−2-methyl-2,3-dihydro-1H-inden-1-one (SYA 40247), a high-affinity 5-HT(7) receptor ligand, by HPLC-PDA using amylose tris-(3, 5- dimethylphenylcarbamate) as a chiral stationary phase, Biomed. Chromatogr 33(9) (2019) e4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ofori E, Zhu XY, Etukala JR, Peprah K, Jordan KR, Adkins AA, Bricker BA, Kang HJ, Huang XP, Roth BL, Ablordeppey SY, Design and synthesis of dual 5-HT1A and 5-HT7 receptor ligands, Bioorg. Med. Chem 24(16) (2016) 3464–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang G, Liu C, Yi H, Meng Q, Bian C, Chen H, Jian JX, Wu LZ, Lei A, External Oxidant-Free Oxidative Cross-Coupling: A Photoredox Cobalt-Catalyzed Aromatic C-H Thiolation for Constructing C-S Bonds, J. Am. Chem. Soc 137(29) (2015) 9273–9280. [DOI] [PubMed] [Google Scholar]
- [40].Lackey K, Cory M, Davis R, Frye SV, Harris PA, Hunter RN, Jung DK, McDonald OB, McNutt RW, Peel MR, Rutkowske RD, Veal JM, Wood ER, The discovery of potent cRaf1 kinase inhibitors, Bioorg. Med Chem. Lett 10(3) (2000) 223–226. [DOI] [PubMed] [Google Scholar]
- [41].Rampa A, Piazzi L, Belluti F, Gobbi S, Bisi A, Bartolini M, Andrisano V, Cavrini V, Cavalli A, Recanatini M, Valenti P, Acetylcholinesterase inhibitors: SAR and kinetic studies on omega-[N-methyl-N-(3-alkylcarbamoyloxyphenyl)methyl]aminoalkoxyaryl derivatives, J. Med. Chem 44(23) (2001) 3810–3820. [DOI] [PubMed] [Google Scholar]
- [42].Rossi Paccani R, Donati D, Fusi S, Latterini L, Farina G, Zanirato V, Olivucci M, Toward a stable α-cycloalkyl amino acid with a photoswitchable cationic side chain, J. Org. Chem 77(4) (2012) 1738–1748. [DOI] [PubMed] [Google Scholar]
- [43].Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R, Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology, Neuropsychopharmacology 28(8) (2003) 1400–1411. [DOI] [PubMed] [Google Scholar]
- [44].Newman-Tancredi A, Gavaudan S, Conte C, Chaput C, Touzard M, Verrièle L, Audinot V, Millan MJ, Agonist and antagonist actions of antipsychotic agents at 5HT1A receptors: a [35S]GTPγS binding assay. Eur J Pharmacol 355 (1998) 245–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.