

Abstract
Lonafarnib (Zokinvy™) is an orally active farnesyltransferase inhibitor developed by Eiger BioPharmaceuticals under license from Merck & Co. for the treatment of hepatitis D virus (HDV) infections, and progeria and progeroid laminopathies. The drug was originally discovered by Merck & Co as an investigational drug in oncology. In progeria, lonafarnib inhibits farnesyltransferase to prevent farnesylation and subsequent accumulation of progerin and progerin-like proteins in the nucleus and cellular cytoskeleton. In November 2020, lonafarnib received its first approval in the USA to reduce the risk of mortality in Hutchinson-Gilford Progeria Syndrome (HGPS) and for the treatment of processing-deficient progeroid laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations) in patients ≥ 12 months of age with a body surface area (BSA) of ≥ 0.39 m2. Lonafarnib is under regulatory review in the European Union. Clinical development for the treatment of HDV infections is underway in multiple countries. This article summarizes the milestones in the development of lonafarnib leading to this first approval.
| Digital Features for this AdisInsight Report can be found at 10.6084/m9.figshare.13571219. |
Lonafarnib (Zokinvy™): Key points
| An orally active farnesyltransferase inhibitor being developed by Eiger BioPharmaceuticals under license from Merck & Co. for the treatment of progeria and progeroid laminopathies. |
| Received its first approval on 20 November 2020 in the USA. |
| Approved to reduce the risk of mortality in HGPS and for the treatment of processing-deficient progeroid laminopathies in patients ≥ 12 months of age with a BSA of ≥ 0.39 m2. |
Introduction
Hutchinson-Gilford progeria syndrome (HGPS) is an extremely rare (prevalence 1 in 20 million births), autosomal dominant genetic disorder characterized by premature aging [1]. It is caused by a de novo mutation in the LMNA gene which encodes A-type lamins (mainly lamins A and C). A-type lamins are inner nuclear membrane proteins that play crucial roles in nuclear structure and shape, chromatin organization and nuclear pore and cytoskeleton organization [1]. Lamin A is synthesized as a precursor (prelamin A), which undergoes sequential post-translational processing, including cysteine farnesylation by farnesyltransferase (FTase) and later a second cleavage by the metallopeptidase ZMPSTE24 [2]. An autosomal 1824 C to T mutation in LMNA results in a constitutively farnesylated lamin A (progerin), which accumulates in the nuclear envelope and severely disrupts nuclear architecture and cellular function, thereby causing premature replicative senescence [3, 4]. It was hypothesized that farnesyltransferase inhibitors (FTIs) blocks the accumulation of progerin and decrease the amount of this aberrant protein, potentially improving disease status in HGPS and processing-deficient progeroid laminopathies [5].
Lonafarnib (Zokinvy™) is an orally active FTase inhibitor developed by Eiger BioPharmaceuticals, under license from Merck & Co, for the treatment of hepatitis D virus (HDV) infections, and progeria and progeroid laminopathies.

Key milestones in the development of lonafarnib for the treatment of progeria and progeroid laminopathies. EAP expanded access program, MAA marketing authorization application
The drug was originally discovered by Merck & Co. as an investigational drug in oncology. Development of lonafarnib for oncology has been discontinued due to lack of efficacy. In progeria, lonafarnib inhibits farnesyltransferase to prevent farnesylation and subsequent accumulation of progerin and progerin-like proteins in the nucleus and cellular cytoskeleton. [6]. On 20 November 2020 [7], lonafarnib received its first approval in the USA to reduce the risk of mortality in HGPS and for the treatment of processing-deficient progeroid laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations) in patients ≥ 12 months of age with a body surface area (BSA) of ≥ 0.39 m2 [6]. The recommended dosage of lonafarnib is based on BSA. The starting dosage of lonafarnib for patients with a BSA of ≥ 0.39 m2 is 115 mg/m2 twice daily taken with morning and evening meals to reduce the risk of gastrointestinal (GI) adverse reactions; after 4 months of treatment, lonafarnib dosage is to be increased to 150 mg/m2 twice daily with morning and evening meals. The dosage of lonafarnib dosage should be rounded to the nearest 25 mg increment. An appropriate dosage strength of lonafarnib is not available for patients with a BSA < 0.39 m2 [6]. The Marketing Authorization Application for lonafarnib is under regulatory review in the European Union. Clinical development for the treatment of HDV infections and progeria is underway in multiple countries. Development of lonafarnib in several indications, including solid tumours, myelodysplastic syndromes and leukaemia, has been discontinued due to lack of efficacy.
Company Agreements
In May 2018, Eiger expanded its licensing agreement with Merck to include rights to develop lonafarnib for the treatment of HGPS [8]. Under the terms of agreement, Eiger has commercial and distribution rights to lonafarnib across the licensed and approved indications in the future. Eiger is responsible for regulatory execution, commercialization and distribution activities of lonafarnib for Progeria. Concurrently, Eiger announced that it had completed a collaboration agreement with the Progeria Research Foundation (PRF). Under the terms of agreement, Eiger, at its sole cost and expense, will provide lonafarnib for ongoing clinical trials and expanded access in progeria. Eiger is responsible for the New Drug Application for the progeria indication based on data produced by PRF, Boston Children's Hospital and Eiger [8].
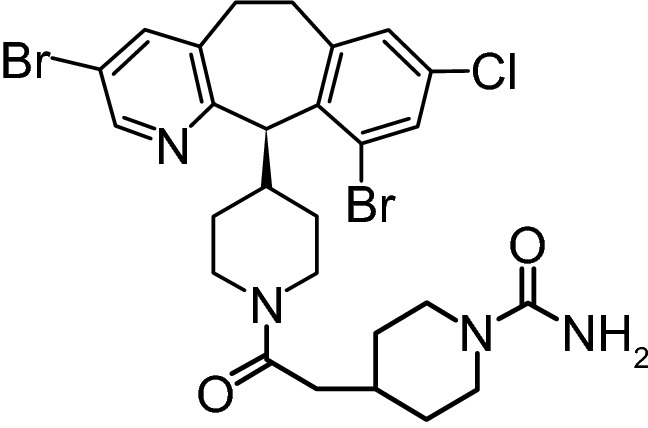
Chemical structure of lonafarnib
Scientific Summary
Pharmacodynamics
Lonafarnib (SCH 66336) is a nonpeptidic, CAAX-competitive, selective FTase inhibitors (half maximal inhibitory concentration 1.9 nmol/L) [9]. In an in vitro study, blocking farnesylation of progerin-transfected cells with lonafarnib restored normal nuclear architecture, and treatment of human HGPS fibroblasts with lonafarnib resulted in significant (p < 0.001) reduction in nuclear blebbing. These results suggest that treatment with lonafarnib represents a potential therapy for patients with HGPS [5].
Coadministration of lonafarnib and sulforaphane (an antioxidant derived from cruciferous vegetables) in HGPS fibroblast cultures exerted a synergistic and additive positive effect on autophagy activity [10]. However, combination treatment activated autophagy to levels that become toxic, as indicated by the increased cell death. On the other hand, intermittent treatment with lonafarnib followed by sulforaphane separately and in repeated cycles ameliorated the HGPS cellular phenotype [10]. Another in vitro study showed that lonafarnib and transient expression of human telomerase promoted HGPS cell proliferation in vitro [3].
Pharmacokinetics
Following oral administration of lonafarnib 115 and 150 mg/m2 twice daily with food in patients with HGPS, the median time to peak plasma concentration of lonafarnib at steady state was 2 and 4 h, respectively [6]. The absolute bioavailability of lonafarnib after oral administration has not been assessed. In healthy subjects, following a single oral dose of lonafarnib 75 mg with a high-fat meal (≈ 43% fat of the total 952 calories), the lonafarnib peak plasma concentration (Cmax) and the area under the concentration-time curve (AUC) decreased by 55% and 29%, respectively, compared with fasted conditions. When taken with a low-fat meal (≈ 12% fat of the total 421 calories), the Cmax and AUC values of lonafarnib decreased by 25% and 12%, respectively, relative to fasted conditions [6].
In vitro protein binding of lonafarnib over the concentration range of 0.5–40.0 μg/mL was ≥ 99% [6]. At steady state, the apparent volumes of distribution of lonafarnib were 87.8 L and 97.4 L following oral administration of lonafarnib 100 and 75 mg twice daily, respectively, in healthy subjects [6].
Lonafarnib is largely metabolized by CYP3A and to a lesser extent by CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19 and CYP2E1 in in vitro studies [6, 11]. Following a single oral dose of radiolabelled lonafarnib 104 mg under fasted condition in healthy subjects, ≈ 62% of the total dose was recovered in faeces and < 1% was recovered in the urine ≤ 240 h post-dose [6]. The two main metabolites of lonafarnib (HM17 and an active metabolite HM21) accounted for 15% and 14% of plasma radioactivity, respectively. The mean elimination half-life of lonafarnib was ≈ 4–6 h following oral lonafarnib 100 mg twice daily in healthy subjects [6].
Gender and age did not affect the pharmacokinetics of lonafarnib to a clinically meaningful extent [6]. The effect of renal or hepatic impairment on the pharmacokinetics of lonafarnib has not been determined. Coadministration of lonafarnib with a strong CYP3A4 inhibitor increases lonafarnib AUC and Cmax (potentially increasing the risk of adverse reactions) and coadministration with a strong CYP3A4 inducer decreases lonafarnib Cmax and AUC (potentially reducing lonafarnib efficacy). Coadministration of lonafarnib with a CYP2C9 inhibitor may also increase lonafarnib AUC and Cmax, potentially increasing the risk of adverse reactions. Lonafarnib is a strong CYP3A mechanism-based inhibitor (increases the AUC and Cmax of the CYP3A substrate), a weak inhibitor of P-gp and strong inhibitor of CYP3A, a moderate CYP2C19 inhibitor and a weak P-gp inhibitor. Coadministration of these agents with lonafarnib increases the AUC and Cmax of these agents [6].
Given the potential for drug-drug interactions, use of lonafarnib with strong or moderate CYP3A inhibitors, strong or moderate CYP3A inducers, select HMG CoA reductase inhibitors (statins) and midazolam is contraindicated [6]. Coadministration of lonafarnib with CYP2C9 inhibitors, weak CYP3A4 inhibitors, CYP2C19 substrates or sensitive CYP3A4 substrates should be avoided; if coadministration is unavoidable, lonafarnib dosage reductions and/or monitoring of patients for adverse reactions may be required [6].
Features and properties of lonafarnib
| Alternative names | EBP 994; MK-6336; Sarasar; SCH 066336; SCH 66336; Zokinvy™ |
|---|---|
| Class | Amides; antineoplastics; antivirals; benzene derivatives; halogenated hydrocarbons; piperidines; pyridines; small molecules |
| Mechanism of Action | Inhibits farnesyltransferase to prevent farnesylation and subsequent accumulation of progerin and progerin-like proteins in the inner nuclear membrane |
| Route of Administration | Oral |
| Pharmacodynamics | Blocked farnesylation of progerin-transfected cells and restored normal nuclear architecture |
| Reduced nuclear blebbing in human HGPS fibroblasts | |
| Pharmacokinetics | Median tmax at steady state 2 and 4 h after 115 and 150 mg/m2 twice daily with food in patients with HGPS |
| Mean half-life ≈ 4–6 h after 100 mg twice daily in healthy subjects | |
| Most frequent adverse events | Vomiting, diarrhoea, infection, nausea, decreased appetite, fatigue, upper respiratory tract infection |
| ATC codes | |
| WHO ATC code | J05A-X (other antivirals); L01 (antineoplastic agents); N07 (other nervous system drugs) |
| EphMRA ATC code | J5B1 (viral hepatitis products); L1 (antineoplastics); N7 (other CNS drugs) |
| Chemical Name | 4-[2-[4-[(11R)-3,10-dibromo-8-chloro-6,11-dihydro-5Hbenzo[1,2] cyclohepta [2,4-b]pyridin-11-yl]piperidin-1-yl]-2- oxoethyl] piperidine-1-carboxamide |
HGPS Hutchinson-Gilford Progeria Syndrome, tmax time to peak plasma concentration
Therapeutic Trials
Hutchinson-Gilford Progeria Syndrome and Progeroid Laminopathies
Two phase 2, single-arm studies showed that treatment with oral lonafarnib increased the average lifespan of patients with HGPS compared with matched, untreated patients from a natural history study [6, 12]. Study 1 (NCT00425607) included 28 patients who received the recommended regimen of lonafarnib for 24–30 months. Of these patients, 26 had classic HGPS, one had non-classic HGPS and one had processing-deficient progeroid laminopathy with LMNA heterozygous mutation with progerin-like protein accumulation [6]. Of the patients who completed Study 1, 26 enrolled in the ongoing Study 2 (NCT00916747), which consisted of two parts. In part 1 of Study 2, patients received lonafarnib with pravastatin and zoledronic acid for ≈ 5 year [6]; however, no additional benefit was seen with triple drug therapy compared with available lonafarnib monotherapy data and therefore treatment with pravastatin and zoledronic acid was discontinued [13]. Triple therapy recipients were permitted to enter part 2 of the study. In part 2 of Study 2 patients received lonafarnib 150 mg/m2 twice daily for ≤ 3 years. This part of Study 2 included 35 treatment-naïve patients (34 with classic HGPS and one patient with non-classic HGPS, with a median age of 6 years and BSA range 0.42–0.90 m2) who [6].
A retrospective survival analysis was conducted based on mortality data from 62 lonafarnib treated HGPS patients, including 27 treatment-naïve patients from study 1 (median age at treatment initiation 7.5 years; BSA range 0.38–0.75 m2) and 35 treatment-naïve HGPS patients from study 2, and data from matched, untreated patients in a separate natural history cohort survival study [6]. Results showed that relative to no treatment, lonafarnib treatment increased the mean lifespan of patients with HGPS by an average of 3 months through the first 3 years of follow-up and increased the mean lifespan by 2.5 years through the last follow-up time (11 years). In the lonafarnib treated versus untreated patients, the mean survival time over the first 3 years of follow-up was 2.8 versus 2.6 years [hazard ratio for the risk of death (HR) 0.30; 95% CI 0.10–0.89], and over 11 years’ follow-up the mean survival time was 8.0 versus 5.5 years (HR 0.40; 95% CI 0.21–0.77). Over the first 3 years of follow-up, 8.1% (5/62) of treated versus 19.4% (12/62) of untreated patients died, and over 11 years’ follow-up, 33.9% (21/62) versus 40.3% (25/62) of patients died [6].
Additionally, study 1 showed that treatment with lonafarnib for ≥ 2 years increased the rate of weight gain, and decreased vascular stiffness, bone structure and audiological status in some children with HGPS, with all patients improving in at least one of these outcomes [14]. Of the 25 patients assessed, nine patients experienced a ≥ 50% increase in the annual rate of weight gain (primary endpoint), six experienced a ≥ 50% decrease, and 10 remained stable. Lonafarnib treatment, decreased arterial pulse wave velocity (by a median of 35%; p = 0.0001 vs. pretherapy) and carotid artery echodensity (p < 0.05 vs pretherapy) and increased skeletal rigidity (by 40–229% depending on the skeletal site) and sensorineural hearing (p ≤ 0.002 vs pretherapy) [14].
Results from study 1 also suggest that lonafarnib reduces the prevalence of stroke and transient ischemic attack, and the frequency and prevalence of headache during 24–29 months of treatment [15]. Prior to treatment, eight of 13 patients with previous neuroimaging data had evidence of prior infarction (four patients each had clinical history of stroke and clinically silent strokes), 15 of 26 patients had experienced headaches (of migraine quality; average frequency of 0.9/week), and four patients had a history of seizures (three of whom also had a history of stroke). During treatment with lonafarnib, only one of four patients with a known history of clinical stroke had a new stroke and the number of patients reporting headaches decreased from 15 at baseline to seven during treatment. The frequency of headaches also decreased to an average of 0.37/week (41% decrease from baseline) [15].
Key clinical trials of lonafarnib
| Drug(s) | Indication | Phase | Status | Location(s) | Identifier | Sponsor |
|---|---|---|---|---|---|---|
| Lonafarnib | Progeria | 2 | Completed | USA | NCT00425607; 07-01-007; P05009; ProLon1 | Boston Children's Hospital |
| Lonafarnib, zoledronic acid, pravastatin | Progeria | 2 |
Enrolling by invitation |
USA | NCT00916747; ProLon2 | Boston Children's Hospital |
| Lonafarnib, zoledronic acid, pravastatin | Progeria | 2 | Completed | USA | NCT00879034; 09-02-0074; P06087 | Boston Children's Hospital |
| Lonafarnib, everolimus | Progeria | 1/2 |
Enrolling by invitation |
USA | NCT02579044; P00017505 | Boston Children's Hospital |
| Lonafarnib | Progeria | EAP | Available | Not available | NCT03895528 | Eiger BioPharmaceuticals |
| Lonafarnib, ritonavir, PEG IFN-alfa-2a, placebo | HDV | 3 | Recruiting | Multinational | NCT03719313; D-LIVR; EIG-LNF-011 | Eiger BioPharmaceuticals |
| Lonafarnib, PEG IFN-alpha, ritonavir | HDV | 2 | Completed | Turkey | NCT02430181; LOWR HDV-1 | Eiger BioPharmaceuticals |
| Lonafarnib, PEG IFN-alpha, ritonavir | HDV | 2 | Completed | Turkey | NCT02430194; LOWR HDV-2; | Eiger BioPharmaceuticals |
| Lonafarnib, ritonavir, placebo | HDV | 2 | Completed | USA | NCT02511431; LOWR HDV-3; 150170; 15-DK-0170 | NIDDK |
| Lonafarnib, ritonavir | HDV | 2 | Completed | Germany | NCT02527707; LOWR 4; EIG-LNF-002 | Eiger BioPharmaceuticals |
| Lonafarnib, PEG IFN-lambda, ritonavir | HDV | 2 | Completed | USA | NCT03600714; LIFT HDV; 180123; 18-DK-0123 | NIDDK |
| Lonafarnib, placebo | HDV | 2 | Completed | USA | NCT01495585; 120046; 12-DK-0046 | NIDDK |
CML Chronic Myelogenous Leukaemia, EAP expanded access program; EORTC European Organisation for Research and Treatment of Cancer, HDV hepatitis D virus infection, NIDDK National Institute of Diabetes and Digestive and Kidney Diseases
Hepatitis Delta Virus Infection
The phase 2 proof-of-concept study (NCT01495585) demonstrated an antiviral dose response with two different twice-daily doses of lonafarnib monotherapy [16].
The phase 2 LOWR-HDV-1 study (NCT02430181; n = 20 enrolled) showed that addition of the CYP3A4 inhibitor ritonavir to lonafarnib allows for a lower dose of lonafarnib to be used while achieving higher concentrations of post-absorption lonafarnib, thereby resulting in better antiviral responses with improved tolerability [17]. Addition of ritonavir 100 mg once daily to a lonafarnib 100 mg twice daily regimen resulted in a mean reduction from baseline to week 4 in HDV RNA of 2.4 versus 2.0 log with lonafarnib 300 mg twice daily monotherapy. Addition of pegylated interferon alfa (PEG-IFNα) 180 μg once-weekly to lonafarnib 100 mg twice daily also resulted in more substantial and rapid HDV-RNA reduction relative to historical responses with PEG-IFNα alone [17].
The phase 2 LOWR-HDV-2 study (NCT02430194) showed that 24 weeks’ treatment with lonafarnib-based regimens achieved virologic responses in a majority of patients with HDV infections [18]. Response rates were assessed with lonafarnib plus ritonavir or lonafarnib plus ritonavir and PEG-IFNα. At 24 weeks, 21 of 33 (64%) patients across all treatment groups were responders (patients achieving > 2 log drop in HDV RNA or below the limit of quantitation for patients with baseline viral load < 2 log). The response rate for patients with baseline viral load of > 4 log and < 4 log was 57% (12/21) and 75% (9/12), respectively [18].
In the randomized, double-blind, placebo-controlled, phase 2a LOWR HDV-3 study (NCT02511431), treatment with lonafarnib 50–100 mg once daily plus ritonavir 100 mg once daily for 24 weeks (n = 12 patients), or 12 weeks of placebo followed by lonafarnib 50–100 mg once daily plus ritonavir 100 mg once daily for 12 weeks (n = 9) demonstrated antiviral activity in patients with chronic HDV infection [19]. In patients receiving 24 weeks’ lonafarnib therapy, HDV RNA levels were significantly lower than with placebo (p = 0.04). Six patients achieved ≥ 2 log decrease in HDV RNA, one patient had undetectable HDV RNA level, three patients had RNA levels of < 14 IU/mL and two patients had RNA levels of < 250 IU/mL; alanine aminotransferase (ALT) levels were normalized in four of six subjects (66%) [19].
Triple combination therapy with lonafarnib 50 and ritonavir 100 mg twice daily plus peginterferon lambda-1a 180 μg weekly for 24 weeks demonstrated antiviral efficacy in patients with chronic HDV infection participating in a phase 2a, open-label, first-in-humans LIFT HDV study (NCT03600714; n = 26) [20, 21]. Twenty-two of 26 patients completed 24 weeks of treatment [21]. At Week 24 (end of treatment), by per-protocol analysis, 17 of 22 patients (77%) achieved the primary endpoint of > 2 log decline in HDV RNA, 11 of 22 patients (50%) were either HDV RNA below limit of quantitation (BLOQ) or HDV RNA was undetectable, and median HDV RNA decline was 3.2 log IU/mL (p < 0.0001). At Week 48 (24 weeks post-treatment), 5 of 22 patients (23%) maintained HDV RNA BLOQ or were HDV RNA undetectable and 6 of 20 patients (30%) achieved the secondary endpoint of > 2 point improvement in histology activity index (HAI) [21].
Adverse Events
Lonafarnib is generally well tolerated in patients with HGPS or processing-deficient progeroid laminopathies, based on 128 patient-years of treatment exposure in the two pivotal, phase 2 studies (NCT00425607 and NCT00916747) [6]. A total of 63 patients in the two studies received lonafarnib for a median duration of 2.2 years, with ≈ 1.9 years at the recommended dose of 150 mg/m2 twice daily. The most common adverse reactions (incidence > 50%) with lonafarnib were vomiting (90%), diarrhoea (81%), infection (78%), nausea (56%), decreased appetite (53%), fatigue (51%) and upper respiratory tract infection (51%) [6].
Gastrointestinal, mostly mild or moderate in severity, occurred most commonly in patients receiving lonafarnib. Of the 57 patients who experienced vomiting, 30 (53%) had mild, 26 (46%) had moderate and one (2%) patient had severe vomiting [6]. Of the 35 patients who experienced nausea, 34 (97%) had mild and one (3%) patient had moderate nausea. Diarrhoea occurred in 51 patients, with mild diarrhoea reported in 38 (75%) of patients, moderate diarrhoea in 9 (18%) of patients and severe diarrhoea in four (8%) of patients. In study 1, 19 of 28 (68%) of patients had vomiting, 10 (36%) patients had nausea and 23 (82%) patients had diarrhoea during the first 4 months of treatment. By the end of treatment, four patients who were still receiving lonafarnib required antiemetics or anti-nauseants. Four patients discontinued lonafarnib therapy, largely because of nausea or vomiting. By the end of therapy, 3 (11%) patients had diarrhoea; 12 (43%) patients were treated with loperamide [6].
Increased ALT levels were reported in 17 (27%) patients in studies 1 and 2, with 14 (82%) patients having mild elevations, one (6%) patient had moderate elevation and two (12%) patients had severe elevation of ALT [6]. Increased aspartate aminotransferase (AST) elevations occurred in 22 (35%) patients, with 21 (95%) patients experiencing mild increases and one (5%) patient experiencing severe increase in AST. One patient with ALT and AST elevations also experienced hypertriglyceridaemia and hyperglycaemia leading to discontinuation of lonafarnib treatment. Treatment with lonafarnib has also been associated with increased blood pressure [6].
Ongoing Clinical Trials
In addition to the ongoing study 2 (NCT00916747), a phase 1/2 dose-escalation study (NCT02579044) is assessing the combination of lonafarnib and everolimus in patients with HGPS or progeroid laminopathies. Recruitment of an estimated 80 patients (aged 18 months to 25 years) is underway and the primary objective of the study is to determine the maximum tolerated dose of everolimus in combination with lonafarnib. Lonafarnib treatment is also available via an Expanded Access Program (NCT03895528) for patients aged ≥ 12 months.
Recruitment of an estimated 400 patients is underway in a randomized, partially double-blind, phase 3 study (NCT03719313) that will evaluate the efficacy and safety of lonafarnib plus ritonavir with or without PEG IFN-α2a in patients chronically infected with HDV. The primary outcome measure is a composite virologic and biochemical response rate.
Current Status
Lonafarnib received its first approval on 20 November 2020 in the USA [7] to reduce the risk of mortality in Hutchinson-Gilford Progeria Syndrome (HGPS) and for the treatment of processing-deficient progeroid laminopathies (with either heterozygous LMNA mutation with progerin-like protein accumulation, or homozygous or compound heterozygous ZMPSTE24 mutations) in patients ≥ 12 months of age with a body surface area (BSA) of ≥ 0.39 m2 [6].
Declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and Conflict of interest
During the peer review process the manufacturer of the agent under review was offered an opportunity to comment on the article. Changes resulting from any comments received were made by the authors on the basis of scientific completeness and accuracy. Sohita Dhillon is a contracted employee of Adis International Ltd/Springer Nature and declares no relevant conflicts of interest. All authors contributed to the review and are responsible for the article content.
Ethics approval, Consent to participate, Consent to publish, Availability of data and material, Code availability
Not applicable.
Footnotes
This profile has been extracted and modified from the AdisInsight database. AdisInsight tracks drug development worldwide through the entire development process, from discovery, through pre-clinical and clinical studies to market launch and beyond.
The original version of this article was revised due to a retrospective Open Access order.
Change history
3/23/2021
A Correction to this paper has been published: 10.1007/s40265-021-01500-6
References
- 1.Guilbert SM, Cardoso D, Lévy N, et al. Hutchinson-Gilford progeria syndrome: rejuvenating old drugs to fight accelerated ageing. Methods. 2020. 10.1016/j.ymeth.2020.04.005 [DOI] [PubMed]
- 2.Harhouri K, Frankel D, Bartoli C, et al. An overview of treatment strategies for Hutchinson-Gilford progeria syndrome. Nucleus. 2018;9(1):246–257. doi: 10.1080/19491034.2018.1460045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Zhou G, Bruno IG, et al. Transient introduction of human telomerase mRNA improves hallmarks of progeria cells. Aging Cell. 2019;18(4):e12979. doi: 10.1111/acel.12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rampogu S, Baek A, Son M, et al. Discovery of lonafarnib-like compounds: pharmacophore modeling and molecular dynamics studies. ACS Omega. 2020;5(4):1773–1781. doi: 10.1021/acsomega.9b02263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Capell BC, Erdos MR, Madigan JP, et al. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2005;102(36):12879–12884. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eiger BioPharmaceuticals Inc. ZOKINVYTM (lonafarnib) capsules, for oral use [US Prescribing Information]. 2020. https://www.zokinvy.com/pdf/ZOKINVY_US_prescribing_information.pdf. Accessed 2020.
- 7.US Food & Drug Administration. FDA Approves First Treatment for Hutchinson-Gilford progeria syndrome and some progeroid laminopathies. [media release]. 20 Nov 2020. https://www.fda.gov/news-events.
- 8.Eiger BioPharmaceuticals Inc. Eiger announces expanded license agreement with merck for investigational candidate lonafarnib and collaboration with the progeria research foundation (PRF). [media release]. 16 May 2018. http://www.eigerbio.com.
- 9.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47(1):15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Gabriel D, Shafry DD, Gordon LB, et al. Intermittent treatment with farnesyltransferase inhibitor and sulforaphane improves cellular homeostasis in Hutchinson-Gilford progeria fibroblasts. Oncotarget. 2017;8(39):64809–64826. doi: 10.18632/oncotarget.19363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghosal A, Chowdhury SK, Tong W, et al. Identification of human liver cytochrome P450 enzymes responsible for the metabolism of lonafarnib (Sarasar) Drug Metab Dispos. 2006;34(4):628–635. doi: 10.1124/dmd.105.007906. [DOI] [PubMed] [Google Scholar]
- 12.Gordon LB, Massaro J, D'Agostino RB, Sr, et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. 2014;130(1):27–34. doi: 10.1161/CIRCULATIONAHA.113.008285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.US Food & Drug Administration. ZOKINVY (lonafarnib): integrated review. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/213969Orig1s000IntegratedR.pdf. Accessed 12 Jan 2021
- 14.Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2012;109(41):16666–16671. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ullrich NJ, Kieran MW, Miller DT, et al. Neurologic features of Hutchinson-Gilford progeria syndrome after lonafarnib treatment. Neurology. 2013;81(5):427–430. doi: 10.1212/WNL.0b013e31829d85c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koh C, Canini L, Dahari H, et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: a proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect Dis. 2015;15(10):1167–1174. doi: 10.1016/S1473-3099(15)00074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yurdaydin C, Keskin O, Kalkan Ç, et al. Optimizing lonafarnib treatment for the management of chronic delta hepatitis: the LOWR HDV-1 study. Hepatology. 2018;67(4):1224–1236. doi: 10.1002/hep.29658. [DOI] [PubMed] [Google Scholar]
- 18.Yurdaydin C, Kalkan C, Karakaya F, et al. Subanalysis of the LOWR HDV-2 study reveals high response rates to lonafarnib in patients with low viral loads. J Hepatol. 2018;68(Suppl 1):S89. doi: 10.1016/S0168-8278(18)30397-0. [DOI] [Google Scholar]
- 19.Koh C, Surana P, Han T, et al. A phase 2 study exploring once daily dosing of ritonavir boosted lonafarnib for the treatment of chronic delta hepatitis: end of study results from the LOWR HDV-3 study. J Hepatol. 2017;66(1 Suppl 1):S101–S2.
- 20.Koh C, Hercun J, Rahman F, et al. A phase 2 study of peginterferon lambda, lonafarnib and ritonavir for 24 weeks: end-of-treatment results from the LIFT HDV Study. J Hepatol. 2020;73(Supplement 1):S130. doi: 10.1016/S0168-8278(20)30774-1. [DOI] [Google Scholar]
- 21.Koh C, et al. A phase 2 study of peginterferon lambda, lonafarnib and ritonavir for 24 weeks: end-of-study results from the LIFT HDV study [abstract and presentation]. In: 13-16 Nov AASLD The Liver Meeting Digital Experience. 2020.