

Abstract
Gene-environment interactions (GxE) are often suggested to play an important role in the aetiology of psychiatric phenotypes, yet so far, only a handful of genome-wide environment interaction studies (GWEIS) of psychiatric phenotypes have been conducted. Representing the most comprehensive effort of its kind to date, we used data from the UK Biobank to perform a series of GWEIS for neuroticism across 25 broadly conceptualised environmental risk factors (trauma, social support, drug use, physical health). We investigated interactions on the level of SNPs, genes, and gene-sets, and computed interaction-based polygenic risk scores (PRS) to predict neuroticism in an independent sample subset (N = 10,000). We found that the predictive ability of the interaction-based PRSs did not significantly improve beyond that of a traditional PRS based on SNP main effects from GWAS, but detected one variant and two gene-sets showing significant interaction signal after correction for the number of analysed environments. This study illustrates the possibilities and limitations of a comprehensive GWEIS in currently available sample sizes.
Subject terms: Genetics, Psychiatric disorders
Introduction
Neuroticism is a personality trait that is characterised by emotion dysregulation and negative affect. It has been thought to confer a general susceptibility to mental health problems, resulting in the frequent experience of negative emotions such as worry, sadness, self-consciousness, or anger1–3. High neuroticism is associated with increased psychiatric comorbidity, and there is a substantial overlap between neuroticism and a wide range of psychiatric disorders, particularly depression and anxiety4–6. The associated societal costs of neuroticism are substantial7, leading to increased use of both mental and physical health services due to poorer overall health and quality of life8.
Twin studies have estimated the heritability of neuroticism to be around 40%, with the rest typically attributed to non-shared environmental factors9–13. In recent years, the genetic aetiology of neuroticism has been studied using large-scale genome-wide association studies (GWAS) which have uncovered more than a hundred genomic loci that point towards genes and pathways involved in brain functioning14,15.
In the epidemiological literature, neuroticism and related phenotypes have been linked with a range of different environmental factors, with traumatic events, childhood maltreatment, and social support receiving the greatest attention16–23. Despite such studies consistently implicating environments that are shared within families, twin studies tend to assign very little or no proportion of variance to shared environmental factors:10–13 a phenomenon called the ‘the shared environment paradox’24.
It has been hypothesised that shared environments simply do not matter as much as do non-shared environments25, a notion which has been related to the distinction between the ‘objective’ and ‘effective’ environments26. That is, while an environment may ‘objectively’ be shared between family members, their ‘effective’ environment, i.e., the environment as they experience it, is nevertheless unique; as is then also the resulting impact of that environment on each individual.
More recently, Uher and Zwicker proposed that the most parsimonious explanation for this shared environment paradox is the presence of gene-environment interactions (GxE). They argue that GxE would lead monozygotic twins to respond more similarly to shared environmental exposures than dizygotic twins and that GxE should therefore result in a substantial proportion of the shared environmental influences being wrongly attributed to genetic factors, causing an inflation of the heritability estimate instead24.
From a biological perspective, GxE can be seen as the process by which environmental influences are moderated by genetic factors (or vice versa). GxE has been speculated to play an integral role in the aetiology of psychiatric phenotypes for a long, as it provides an explanation for why some develop psychiatric symptoms after particular risk exposures while others do not24,27–30. Though neuroticism has traditionally been viewed as a relatively stable trait, a more dynamic aetiology has been proposed whereby it is continuously influenced by ongoing gene-environment interactions throughout the life span31.
To date, however, there have been few truly genome-wide GxE studies (GWEIS) of psychiatric phenotypes, and the majority of molecular GxE research has been limited to candidate genes29,32–36. It is only quite recently that the available data and computational resources have begun to allow for the conduction of GWEIS, but as interactions may require larger sample sizes to detect effects of similar magnitude as main effects, sample size requirements may be even greater for GWEIS than for GWAS37,38.
To overcome this, some have reduced the multiple testing burden by pre-selecting variants based on main effects from GWAS39,40. While these two-stage approaches could potentially yield more significant SNPs, individual SNP effects are unlikely to yield insight into the higher-order biological mechanisms underlying GxE (as is the case for GWAS41), and the lack of genome-wide GxE data limits the opportunity for follow-up analyses such as gene-set analysis, which could elucidate the function of GxE effects42. In addition, since interacting SNPs may not display strong main effects, this approach could also lead to potential key interactions going undetected40,43. Another option may be to model interactions of individual variants with multiple environments simultaneously44, though this is also at the cost of environmental specificity which could complicate the interpretation of any functional follow-up analysis.
Alternatively, global GxE effects across the entire genome may be investigated by estimating the proportion of variance explained by GxE effects45, or by modelling interactions with polygenic risk scores constructed using SNP main effects from GWAS46–49. But while such approaches may indicate the presence of GxE, they cannot determine which SNPs or genes are driving the interactions. For the purposes of gaining relevant biological information from the GxE analyses, we, therefore, considered GWEIS to be the most suitable approach.
Beyond issues with power, GWEIS requires particular consideration regarding the control of error rate inflation, as it is particularly vulnerable to the effects of heteroscedastic residuals50. While this can be resolved with the use of heteroscedasticity consistent, or so-called robust, standard errors51,52, these are not currently available in software optimised for large-scale genetic analysis like PLINK53, and researchers have had to implement this them themselves34,54. Interaction effects may also be confounded by covariate-SNP and covariate-environment interaction effects unless these are accounted for55, but doing so can dramatically increase the number of variables analysed and add further computational constraints to this already intensive analysis.
To our knowledge, there have only been three GWEIS of psychiatric phenotypes to date, all of which have focused on depressive symptoms and used some composite measure of stressful life events as environment32–34. These studies have found few significant interactions, though only one of these studies featured a sample size close to 100,000 individuals (the rest fewer than 10,000). As such, it is evident that there is a substantial gap in the available genome-wide evidence for GxE in mental health phenotypes in general, including neuroticism for which there are currently none.
To address this, we used data from the UK Biobank56 to perform a series of GWEIS for neuroticism, with a total of 25 broadly defined environmental variables (N = 84,711–313,339; Table 1). While ensuring proper control for inflation and confounding as mentioned above, we first explored SNP-environment interactions between all 25 environmental variables and a total of 8,614,007 SNPs genome-wide.
Table 1.
Overview of environmental factors.
| Category | UKB ID | Full name | Short name | N | Type | Levels | Range |
|---|---|---|---|---|---|---|---|
| Physical health | 2188 | Long-standing illness, disability, or infirmity | Disability/infirmity | 308,892 | Ordinal | 2 | Yes–No |
| Physical health | 23,104 | Body mass index (BMI) | BMI | 308,303 | Continuous | – | 12.8–68.4 |
| Physical health | 100,048 | Pain types experienced for 3+ months* | Chronic pain | 313,219 | Count | 4 | 0–3 |
| Physical health | 4548 | Health satisfaction | Health satisf. | 123,307 | Ordinal | 6 | Extremely unhappy–…–Extremely happy |
| Physical health/Trauma | 20,528 | Diagnosed with a life-threatening illness | Terminal illness | 106,089 | Ordinal | 3 | Never–Yes, but not in the last 12 months–Yes, within the last 12 months |
| Trauma | 20,531 | Victim of sexual assault | Sexual assault | 105,362 | Ordinal | 3 | Never–Yes, but not in the last 12 months–Yes, within the last 12 months |
| Trauma | 20,529 | Victim of physically violent crime | Physical assault | 106,257 | Ordinal | 3 | Never–Yes, but not in the last 12 months–Yes, within the last 12 months |
| Trauma | 6145 | Illness, injury, bereavement, the stress in last 2 years | Multiple stress | 312,278 | Count | 7 | Serious illness, injury, or assault to yourself–…–Death of a close relative–…–None of the above |
| Trauma/Social support | 20,488 | Physically abused by family as a child | Child. physical abuse | 106,240 | Ordinal | 5 | Never true–…–Sometimes true– ...–Very often true |
| Trauma/Social support | 20,487 | Felt hated by a family member as a child | Felt hated | 106,183 | Ordinal | 5 | Never true–…–Sometimes true– ...–Very often true |
| Trauma/Social support | 20,489 | Felt loved as a child | Felt loved | 106,098 | Ordinal | 5 | Never true–…–Sometimes true– ...–Very often true |
| Social support | 20,522 | Been in a confiding relationship as an adult | Adult confiding rel. | 104,073 | Ordinal | 5 | Never true–…–Sometimes true– ...–Very often true |
| Social support | 1031 | Frequency of friend/family visits | Friend/family visits | 312,492 | Ordinal | 7 | No friends/family outside household–…–Almost daily |
| Social support | 6160 | Leisure/social activities | Social activities | 312,940 | Count | 6 | Sports club–…–Pub or social club –…–None of the above |
| Social support | 2110 | Able to confide | Able to confide | 307,308 | Ordinal | 6 | Never or almost never–…– Almost daily |
| Social support | 4559 | Family relationship satisfaction | Family satisf. | 122,578 | Ordinal | 6 | Extremely unhappy–…– Extremely happy |
| Social support | 4570 | Friendships satisfaction | Friendship satisf. | 122,423 | Ordinal | 6 | Extremely unhappy–…– Extremely happy |
| Sociodemographic | 4537 | Work/job satisfaction | Work satisf. | 84,711 | Ordinal | 6 | Extremely unhappy–…– Extremely happy |
| Sociodemographic | 4581 | Financial situation satisfaction | Financial satisf. | 123,172 | Ordinal | 6 | Extremely unhappy–…– Extremely happy |
| Sociodemographic | 189 | Townsend deprivation index at recruitment | TDI | 313,108 | Continuous | – | −6.26–11 |
| Sociodemographic/Education | 845 | Age completed full-time education | Y/o schooling | 203,479 | Continuous | – | 5–35 |
| Cognitive function | 20,016 | Fluid intelligence score | Intelligence | 120,422 | Continuous | – | 0–13 |
| Sleep | 1200 | Sleeplessness/insomnia | Insomnia | 313,321 | Ordinal | 3 | Never/rarely–Sometimes– Usually |
| Substance use | 1558 | Alcohol intake frequency | Alcohol intake | 313,339 | Ordinal | 6 | Never–…–Daily or almost daily |
| Substance use | 20,116 | Smoking status | Smoking | 312,638 | Ordinal | 3 | Never–Previous–Current |
628* = item constructed using multiple variables, the UKB ID is the UK Biobank category ID (see “Methods” section).
Given that conceptually meaningful interaction effects may not be evident on the level of individual SNPs, whose effects are likely small in magnitude, we sought to elucidate relevant biological mechanisms that might govern GxE by testing whether single SNP-environment interaction effects were over-represented within particular genes, tissues, or gene-sets. We also evaluated the predictive ability of SNP-interaction effects across the genome by constructing interaction-based polygenic scores (iPRSGxE) for each environment and used these to predict neuroticism in an independent subset of the UKB sample (N = 10,000).
In parallel with all interaction-based analyses, we performed a traditional neuroticism GWAS in the same sample to evaluate the concordance between the top interaction effects and corresponding main effects, as well as to allow the predictive power of the iPRSs to be contrasted to that of a traditional main effect PRS constructed from the GWAS results (see Fig. 1 for an overview of the analysis workflow).
Fig. 1. Overview of the analysis workflow.
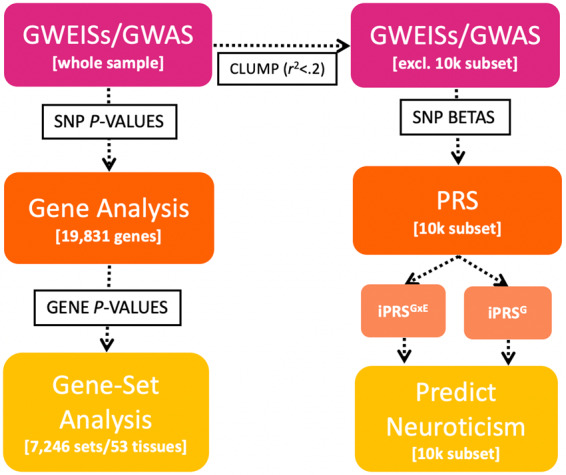
We first obtained SNP-environment interaction effect estimates for the 25 environments (GWEISs), as well as SNP main effects (GWAS). Results from these analyses were used to perform gene and gene-set analyses. The effect of SNPs, genes, and gene-sets that reached standard genome-wide significance (i.e., not corrected for the 25 environments) in the interaction-based analyses were compared with their corresponding main effects. Interaction-based polygenic risk scores (iPRSs) were constructed in an independent subset of the sample to predict neuroticism: this was done by modelling the interactions between each environment and a polygenic risk score constructed based on either the SNP-environment interaction effect from GWEIS (iPRSGxE), as well as the SNP main effect from GWAS (iPRSG) as a comparison.
The selection of environmental variables was based on the epidemiological literature, and consisted primarily of variables relating to trauma and social support, but also to physical health, socioeconomic status, cognitive function, sleep, and substance use (Table 1; see “Methods” section). While some of these environmental variables are not traditionally seen as ‘environments’ (such as cognitive function, insomnia, BMI), we decided to include these here anyway as they have often been highlighted as risk factors in epidemiological studies in the past57–59. Given that many of these environments are themselves heritable, it is thus possible that some interactions we observe could reflect gene-gene interactions (GxG) rather than pure GxE (see Plomin et al.60 and Vinkhuyzen et al.61 for discussions about the heritability of the environment). Though our rationale for including these is that any potential interactions, be it GxE or GxG, may nonetheless highlight relevant biological mechanisms that contribute to neuroticism. For reasons of convenience, we chose to retain the general term ‘GxE’ throughout the paper, but acknowledge that the term ‘Gene x Trait’ interaction is more suitable.
We note that although a potential correlation between the genetic influences on the environment and the outcome phenotype has been a cause for concern for the estimation of GxE in twin studies62, we would not expect this to lead to spurious detection of interaction effects in a GWEIS setting, since the linear regression model allows the SNP and environment main effects to be modelled simultaneously, and can thus account for any correlation that exists between these, as well as with the interaction term.
Results
Interacting SNPs implicated by GWEIS
Due to the risk of inflation of the GWEIS test statistics that were mentioned previously50,54, we analysed SNP-environment interactions in a linear regression framework in R, computing t-statistics for the interaction coefficients using robust standard errors in the form of the Huber-White sandwich estimator51,52 (see “Methods” section for more detail, and Suppl. Info (A): ‘Heteroscedasticity and Spurious Inflation of GWEIS Test Statistics’ for comparison with traditional, model-based standard errors). In order to account for any potentially confounding covariate interactions55, we also included covariate-SNP and covariate-environment interaction effects in the model in addition to covariate main effects (see “Methods” section).
We analysed the single SNP-by-environment interactions between each of the 25 environments (N = 84,711–313,339; Table 1) and a total of 8,614,007 SNPs (minor allele frequency >0.01; imputation quality >0.9; see “Methods” section), from which we identified 8 independent SNPs (r2 < 0.8) for 7 environments that showed interaction effects at the standard genome-wide significance threshold of p < 5e−8 (Table 2; Suppl. Figures). Of these, one intergenic SNP on chromosome 6 remained significant after applying further Bonferroni correction for the number of environments analysed (p < 5e−8/25 = 2e−9; rs115385310, ‘felt hated as a child’); an SNP which was also suggestively significant for ‘childhood physical abuse’ (p = 6.93e−7).
Table 2.
SNPs-environment interactions detected in the GWEIS.
| Environment | SNP | Chr | BP | Gene | PGWEIS | PGWAS | MAF |
|---|---|---|---|---|---|---|---|
| Felt hated | rs115385310 | 6 | 6,721,120 | — | 6.09e−10* | 0.454 | 0.02 |
| Able to confide | rs874616 | 15 | 87,901,482 | — | 8.57e−9 | 0.310 | 0.66 |
| Work satisf. | rs4461224 | 2 | 23,485,507 | — | 1.44e−8 | 0.223 | 0.10 |
| TDI | rs11700517 | 21 | 33,273,542 | HUNK | 1.76e−8 | 0.195 | 0.16 |
| Adult confiding rel. | rs12649942 | 4 | 38,261,175 | — | 2.86e–8 | 0.996 | 0.58 |
| Social activities | rs111497581 | 4 | 91,033,882 | CCSER1** | 3.98e−8 | 0.756 | 0.02 |
| Terminal illness | rs5928040 | X | 32,623,390 | DMD | 4.97e−8 | 0.705 | 0.19 |
| Terminal illness | rs185839186 | 5 | 7,409,995 | ADCY2 | 4.98e−8 | 0.842 | 0.02 |
SNPs from all 25 GWEIS analyses with a p-value below the standard genome-wide significance threshold (p < 5e−8), i.e., not corrected for the number of environments. * = survived Bonferroni correction for the 25 environments (p < 5e−8/25 = 2e−9); ** = SNP located within 15 kb upstream of the transcription start site.
These results are in stark contrast to a traditional GWAS on neuroticism performed using the same main effect covariates as the GWEISs (see “Methods” section), for which 103 independent significant SNPs were detected. For the 8 SNPs that showed significant interactions at the standard genome-wide significance threshold (p < 5e−8), we did not find any evidence of a significant main effect in the GWAS (p > 0.05; Table 2).
Genes implicated by SNP-environment interactions
To facilitate functional interpretation of GWEIS results, we sought to determine whether SNP-environment interaction effects across the genome tended to congregate within particular genes. Although any direction of effect is inevitably lost when aggregating the effects from multiple SNPs, this analysis nonetheless provides information about whether variants in certain genes could moderate the effect of specific environmental exposures on the phenotype.
We thus performed 25 gene-based tests for 19,831 protein-coding genes in MAGMA using the interaction p-values from the GWEISs as input (see “Methods” section). From these analyses, we found a total of 10 genes from 7 environments that reached standard genome-wide significance, correcting only for the number of genes analysed (p < 2.52e−6 (0.05/19,831); Table 3; Suppl. Tables 1a–1y); though none survived further correction for the number of environments (p < 2.52e−6/25).
Table 3.
Genes detected in the MAGMA gene analyses.
| Environment | Gene | Chr | BPSTART | BPSTOP | PGWEIS | PGWAS |
|---|---|---|---|---|---|---|
| Alcohol intake | CLDN4 | 7 | 73,213,872 | 73,247,014 | 1.16e−7 | 0.190 |
| WBSCR27 | 7 | 73,248,920 | 73,256,865 | 1.44e−6 | 0.093 | |
| Chronic pain | VPS9D1 | 16 | 89,773,542 | 89,787,394 | 5.59e−7 | 0.532 |
| FANCA | 16 | 89,803,957 | 89,883,065 | 1.30e−6 | 0.016 | |
| Intelligence | EIF5A | 12 | 7,210,318 | 7,215,774 | 8.43e−7 | 0.116 |
| POLE | 12 | 133,200,348 | 133,263,951 | 1.18e−6 | 0.080 | |
| Family satisf. | CWC27 | 5 | 64,064,757 | 64,314,590 | 1.22e−6 | 0.054 |
| Sexual assault | FHIT | 3 | 59,735,036 | 61,237,133 | 1.15e−6 | 1e−4 |
| Social activities | ZSWIM3 | 20 | 44,486,256 | 44,507,761 | 1.85e−6 | 0.097 |
| Y/o schooling | TUSC5 | 17 | 1,182,957 | 1,204,281 | 2.14e−6 | 0.702 |
Results from MAGMA gene analysis of 19,831 genes, using the GWEIS interaction p-values as input (p < 0.05/19,831 = 2.52e−6); No gene survived additional Bonferroni correction for the number of environments analysed (p < 2.52e−6/25 = 1.01e−7).
Similar to the SNP-level results, the concordance between suggestive main and interaction effects on the gene-level was low, and only one gene (FHIT, ‘sexual assault’) reached suggestive significance in the main effects gene analysis for neuroticism (p = 1e−4; Table 3; Suppl. Table 1z).
Gene sets enriched by the most interactable genes
In order to determine whether the most strongly associated genes for any environment (including sub-significant ones) tended to be overrepresented within particular pathways, cellular locations, or implicated in particular tissue-specific gene expression patterns, we performed competitive gene-set and gene-property analyses in MAGMA using the results from the 25 GWEIS-based gene analyses as input. These analyses concerned 7426 gene sets (MSigDB) and 53 tissues (GTEx; see “Methods” section).
At a p-value threshold of 6.85e−6 (.05/(7246 + 53)), 12 gene sets from 7 environments were significant (Table 4), but no tissues (Suppl. Tables 2a–2y and 3a–3y). Of these 12 gene sets, two survived the additional correction for the number of environments analysed (p < 6.85e-6/25): ‘nucleotide transmembrane transporter activity’ (terminal illness), and ‘glucose binding’ (insomnia).
Table 4.
Gene-sets detected in the MAGMA gene-set analyses.
| Environment | Pathway/Tissue | PGWEIS | PGWAS |
|---|---|---|---|
| Insomnia | Glucose binding | 1.44e–8* | 0.095 |
| Terminal illness | Nucleotide transmembrane transport | 1.61e–8* | 0.151 |
| Nucleotide transmembrane transporter activity | 1.09e–6 | 0.091 | |
| Nucleotide transport | 3.40e–6 | 0.011 | |
| Sexual assault | Telomerase pathway | 1.60e–6 | 0.219 |
| RNA-dependent DNA biosynthetic process | 3.68e–6 | 0.572 | |
| Friendship satisf. | Positive regulation of ion transport | 1.70e–6 | 0.763 |
| Regulation of metal ion transport | 1.71e–6 | 0.360 | |
| Regulation of calcium ion transport | 4.41e–6 | 0.354 | |
| Child. physical abuse | PKA mediated phosphorylation of CREB | 4.03e–6 | 0.645 |
| Physical assault | Negative regulation of DNA metabolic process | 4.73e–6 | 0.254 |
| Family satisf. | Advanced glycosylation endproduct receptor signalling | 6.13e–6 | 0.807 |
Results from MAGMA analysis of 7,246 MSigDB gene sets and 53 gene expression patterns from GTEx (p < 0.05/(7,246 + 53) = 6.85e–6). * = gene sets that survived Bonferroni correction for the number of analysed environments (p < 6.85e–6/25 = 2.74e–7).
Again, none of the interacting gene-sets showed evidence of a significant, or even suggestively significant, the main effect in neuroticism (Table 4; Suppl. Tables 2z and 3z).
Interaction-based polygenic risk scores (iPRS)
To evaluate the predictive accuracy of our SNP-environment interactions, we constructed interaction-based polygenic risk scores (iPRSGxE)—taking the sum of effect alleles weighted by the interaction beta and the environment—and used these to model neuroticism in an independent subset of the UKB sample (N = 10,000; see “Methods” section). An alternative to GWEIS mentioned earlier is to model the interaction between a traditional main effect PRS from GWAS and an environmental variable of interest (henceforth: iPRSG). Since the iPRSG is more widely accessible than the iPRSGxE (as it does not require existing GWEISs), we also computed iPRSGs for each environment as a comparison.
The variance explained by each iPRS was evaluated by comparing the fit between a full model containing the iPRS and covariates to that of a covariate only model (here, the environment main effect and the main effect PRS were included in addition to the standard covariates used for the GWEIS/GWAS, as well as the interactions between these and the standard covariates; see “Methods” section). This was done using the anova() function in R.
For any of the 25 environments, neither the iPRSGxE nor the iPRSG provided a significant increase in model fit (p < 0.05/25/2; see “Methods” section) above that of the covariates only model, with the attributable variance reaching a maximum of .04% for the iPRSGxEs and .03% for the iPRSGs (Fig. 2). This contrasts with the traditional main effect PRS, which explained 2.06% of the variance in neuroticism beyond standard covariates (p = 6.31e−49; see “Methods” section).
Fig. 2. Prediction accuracy of interaction-based polygenic risk scores.

Interaction-based polygenic risk scores (iPRS) were computed for an independent subset of 10,000 individuals, using the sum of risk alleles weighted by the SNP betas and the environment. For the iPRSGxEs the SNP-environment interaction beta was used as a weight, whereas for the iPRSGs, the SNP main effect betas from GWAS was used instead. The Y-axis reflects the ∆r2, i.e., the difference in adjusted r2 between models containing the iPRSs and all covariates to a covariates-only model. The differences in model fit between the full model and the covariate only model were evaluated using anova in R. Only environments with a ∆r2 greater than zero are shown; none of the iPRSGs or iPRSGxEs explained a significant proportion of variance in neuroticism beyond covariates (p < 0.05/25/2).
We, therefore, conclude that based on the environments and sample population analysed here, there is currently limited evidence that genome-wide GxE effects in the form of iPRSs can improve prediction accuracy in neuroticism beyond what can already be achieved using SNP and environment main effects.
Discussion
In this study, we have investigated genome-wide gene-environment interactions in neuroticism across a total of 25 different environmental variables previously associated with mental health outcomes. From all SNP, gene, and gene-set based analyses, we detected one SNP (rs115385310 for ‘felt hated as a child’) and two gene-sets (‘glucose binding’ for ‘insomnia’ and ‘nucleotide transmembrane transport’ for ‘terminal illness’) that survived Bonferroni-correction for the number of environments analysed.
Although multiple interactions were found at standard genome-wide significance thresholds (i.e., not correcting for the number of environments), they were substantially fewer than that detected in a traditional GWAS on neuroticism, in which we identified just over 100 independent significant SNPs. This is in line with the notion that the power to detect interactions is lower than that of main effects, and suggests that even larger data sets will be required before we can uncover a more considerable fraction of relevant interactions. The lack of predictive value for interaction-based polygenic risk scores (iPRSs) echoed this further.
A GWEIS analysis will naturally suffer more from an increased multiple testing burden compared to, for example, two-stage GxE approaches which pre-select genetic variants based on their observed main effects. In this study, however, we found that none of the interacting SNPs identified at standard genome-wide significance thresholds (i.e., uncorrected for the number of environments) showed any evidence of even suggestive main effects in the GWAS—the same was largely true the gene and gene-set level results—implying that preselection based on main effects could result in key interactions being overlooked. In addition, as individual SNP interaction effects might themselves not yield notable insight into the biological mechanisms that govern GxE (as is typically the case with single SNP analyses41), the genome-wide nature of GWEIS is vital as it allows for follow-up analyses, such as gene-set analysis, which can elucidate the function of GxE effects.
Although the multiple testing burden was further exacerbated by the analysis of multiple environments here, we argue that this approach could enable the identification of common patterns across environments and further strengthen the evidence for any particular gene or pathway (particularly when restricted to environments already thought to be implicated). While a systematic investigation of shared GxE effects was not conducted here due to the lack of power even when not correcting for the environments, we hope that our results may prove useful for researchers conducting similar studies in the future, for example, as a basis for replication or meta-analysis. As increased sample sizes lead to the detection of more reliable SNP-environment interactions, we expect that results from GWEIS and related functional follow-up analyses will become valuable for our understanding of the biological mechanisms that underlie GxE.
In this study, we selected neuroticism as our phenotype of interest due to its significant public health impact7,8 and widespread links with several clinical psychiatric disorders3–6. Although evidence suggests that neuroticism is more dynamic than traditionally thought31, as a personality trait, it may nonetheless be more stable than some clinical phenotypes, such as depression or alcohol use disorder63–66, and could also be comparatively less sensitive to GxE. In addition, it should be noted that the UKB sample analysed here consists of a relatively older population, and since the influence of GxE may be more pronounced at an earlier stage of development67, the age of this sample might have affected our power to detect certain GxE effects.
Here, we chose to model the relationships between all variables as linear (and thus, treating ordinal environments as continuous), but there is a possibility that some interactions may have a more complex, non-linear form. For instance, for the ordinal environmental variable ‘physical assault’ we now assumed that having been assaulted recently versus in the past results in a similar change in the SNP effect as having been assaulted in the past compared to never. While this may not be a fully accurate representation of the data, we expected that the increased multiple testing resulting from analysing the levels of each ordinal variable separately would nevertheless have had a more severe impact on power.
Finally, we wish to reiterate a key limitation regarding the interpretation of our results in the context of heritable environments. The environmental component in GxE is sometimes seen as an independent force that regulates the penetrance of genetic effects (or vice versa), while in practice, any environmental measure obtained in a cross-sectional design is unlikely to be free from genetic influence60,61. Although there have been efforts to distinguish GxG from GxE in the twin modelling literature62,68, doing so in this setting is not uncomplicated, and simply conditioning on heritable components could induce collider bias69.
In this study, we chose to be particularly lenient with what we considered ‘environment’ in favour of covering as broad a range of relevant variables as possible. Based on these results alone, it is therefore not possible to determine whether any interaction detected here represents one with the environmental components directly (GxE) or with some heritable component thereof (GxG). If well-powered, however, we argue that GWEISs of heritable environments are still useful as they could elucidate important sources of aetiological heterogeneity which can be followed up in greater depth using experimental or more controlled observational designs in the future.
Representing the largest effort of its kind to date, we used a total of 25 environmental variables to investigate gene-environment interactions in neuroticism. Although power is low compared to GWAS, we detected one variant and two gene sets that showed significant interaction after correction for the number of environments analysed. Larger sample sizes are, however, needed to obtain more reliable estimates of relevant SNP-environment interaction effects, which will be required in order to understand the molecular mechanisms that govern gene-environment interactions in neuroticism.
Methods
Genotype data and quality control
All genotype and phenotype data were obtained from the UK Biobank56 (release 3, March 2018), and this study was conducted under the UK Biobank application 16406. Data collection, primary quality control, and imputation of the genotype data were performed by the UK Biobank itself, the full details of which have been described elsewhere70. We applied further quality control in order to ensure the inclusion only of high-quality variants. This entailed filtering SNPs with a minimum info score of .9 (HRC panel imputed), maximum missingness of 5%, and a minor allele frequency of at least 1%, resulting in a total of 8,614,007 SNPs for the analysis.
We used only European, unrelated samples with concordant sex (see Suppl. Info (A): UK Biobank Sample Information and Quality Control). Thirty principal components (computed with FlashPCA71) were included as covariates in all analyses to control for population stratification. To ensure that the selection of SNPs remained constant across environments, quality control and filtering were performed on the full subset of individuals with complete neuroticism data (see below), and it is, therefore, possible that exact minor allele frequencies and call rates may vary slightly between the sample subsets for each environment.
Phenotype
Neuroticism was measured using the Eysenck Personality Questionnaire (Revised Short Form72), which contains 12 dichotomous items asking participants to indicate whether they agree with statements such as “Do you worry too long after an embarrassing experience?”, or “Do you ever feel ‘just miserable' for no reason?”. An individual’s level of neuroticism was quantified as the sum of items with which they agreed, ranging from 0 and 12. We included only individuals who had provided complete responses to all items (thus performing no imputation of missing values), resulting in 313,467 samples. To ensure that neuroticism and each environment had been measured simultaneously, we used data collected from the first visit only.
Environmental factors
We considered broadly as ‘environment’ a wide range of variables available from the UKB Biobank that have been associated with neuroticism and related mental health phenotypes in the literature. This included primarily those relating to trauma exposure16–20 and social support21–23, but also socioeconomic deprivation73,74, education75 and cognitive ability57,76, substance use77,78, sleep58,79,80, and physical health (overweight/obesity59,81, physical disability82,83, chronic pain84,85). We gathered all available variables that related to any of these categories, limiting the final selection to a subset of 25. We selected variables as such that there was at least one variable from each category, then giving preference to those with larger total sample sizes and less skew in relation to the remaining variables. Given their central role in the literature, we prioritised a wider selection of items related to trauma and social support but sought to include at least one item related to all other domains. Here, we refer to these variables as ‘environments’ as that is their role in the current analyses while acknowledging that many of the selected environments have a (sometimes considerable) heritable component.
The majority of environments were ordinal, consisting of responses such as ‘never true’ to ‘very often true’, or ‘never or almost never’ to ‘almost daily’ (see Table 1). There were two categorical environments that allowed endorsement of multiple answer options: ‘social activities’ and ‘multiple stress’, which we converted to sum scores representing the number of endorsed options. ‘Chronic pain’ was constructed using a collection of pain items that indicated whether participants had experienced pain in multiple regions for three months or more (category ID: 100048). Scores on this variable reflect the sum of regions in which participants experienced pain for 3+ months, with a maximum score of 3. Indicating no pain or pain for less than 3 months in any number of regions gave a score of 0. Indicating chronic pain in one region gave a score of 1, in two regions a score of 2, and indicating pain all over the body, or pain in three or more regions for 3+ months, gave a score of 3. The reason for this truncation was to allow the inclusion of pain all over the body without making strong assumptions about the severity compared to multiples of separate areas.
To ensure that neuroticism and all environmental measures were measured at the same time point, we analysed data from the first visit only. All environments were analysed as continuous, and as with neuroticism, we performed no imputation of missing responses for any of the environments.
GWEIS
SNP-environment interactions were analysed in a linear interaction model in R (v3.2.1). As have been shown previously50,54, GWEIS test statistics are particularly susceptible to spurious inflation of test statistics due to heteroscedasticity of the residuals. To deal with this, we relied on Huber-White estimated standard errors, also known as a sandwich estimator. Unlike model-based standard errors, which are computed using a single residual variance term for all observations, the sandwich estimator allows a unique residual variances term across observations, approximated using the squared residuals51,52.
Our script is an adaptation of a PLINK R plugin originally developed by Almli et al.54, which performs a joint test of SNP and SNP-environment interaction effects (https://epstein-software.github.io/robust-joint-interaction). Beyond run-time optimisation, we computed p-values for the gene-environment interaction (rather than the joint test of SNP main and interaction effects, as done initially), and included covariate-SNP and covariate-environment interactions in addition to covariate main effects. As has been shown55, covariate main effects alone do not effectively control for potentially confounding interactions of the covariate with the SNP or the environment, and unless controlled for, such interactions may be captured in the SNP-environment interaction term. We thus implemented the following linear regression model for every SNP and environment:
where Yi represents the phenotype measure for any individual i, Gi the SNP allele count, and Ei the environmental measure. Ci is a k × 1 vector of covariates, with k equalling the total number of covariates, and ϵi the residual, and ′ denotes the transpose. The intercept (β0) and betas for the SNP (βG), environment (βE), and SNP-environment interaction term (βGxE) are all scalars, while the betas for the covariate-environment (βCxE) and covariate-SNP (βCxG) interactions are k × 1 vectors. The parameter of interest here is βGxE: the beta for the SNP-environment interaction.
As covariates, we included age, sex, 30 PCs, and all assessment centres with N > 10,000. As recommendations or standards regarding the number of PCs that should be included typically concern main effects analyses, we could not exclude the possibility that potentially more complex confounding effects of ancestry might arise when analysing interactions, and therefore chose a more cautious approach of including as many as 30 PCs.
For the analysis, PLINK formatted genotype data was read into R (v3.2.1) using the read.plink() function from the snpStats package (see the Suppl. Info (B)–Analysis script for the full R script). As per the snpStats default settings, autosomal SNPs were coded as 0, 1, and 2, representing the homozygous minor, heterozygous, or homozygous major genotypes, respectively. On the X chromosome, male genotypes were coded as 0 and 2, representing single copies of the minor or major alleles.
GWAS
We conducted two GWASs of neuroticism in PLINK v.2.053 using the same set of covariates as in the GWEIS: one using the full neuroticism sample (N = 313,467), done with the purpose of determining whether interacting SNPs, genes, or gene-sets displayed any main effects on neuroticism, and one that excluded a test set of 10,000 individuals done for the purpose of constructing a main effect polygenic risk score.
Gene analyses
To investigate whether SNP-environment interaction signals tended to congregate within genic regions, we performed genome-wide gene analyses with MAGMA (v1.07b)42 using the p-values from the GWEIS as input. Gene locations for 20,260 protein-coding genes were obtained from Ensembl (GRCh37, p13, v96), of which 19,831 contained at least one SNP in our data. To allow the inclusion of nearby, potentially regulatory SNPs, we used windows of 2 kb upstream and 1 kb downstream of the transcription start and stop sites, respectively. For computational efficiency, a random subset of 10,000 individuals from the UKB data set was used as a reference for the estimation of LD.
As an aggregation method for the SNP effects, we employed the ‘multi model’ which is a hybrid between the commonly used ‘mean model’, which simply averages the SNP effects across the gene, and the ’top model’, which uses the lowest SNP p-value corrected for gene size. In essence, the ‘multi model’ applies both the ‘mean’ and ‘top’ models and selects the one with the best fit.
Gene-set and gene property analyses
Competitive gene-set and gene-property analyses were performed for all GWEISs and the GWAS using MAGMA (v1.07b)42. A total of 7246 gene set definitions were obtained from MsigDB (v6.2), including gene ontology (GO) terms, cellular locations, and biological pathways from multiple sources (e.g., KEGG, Reactome, BioCarta). These were analysed in a competitive framework (as is the default in MAGMA), testing whether the average association with genes within a gene set is greater than that of genes outside the gene set, while correcting for LD.
To test for tissue specificity of associated genes, we used the recently implemented conditional gene property analysis in MAGMA. In this framework, any given tissue can be conceptualised as a gene-set, where gene mRNA expression levels represent the continuous gene-set membership for any given gene, with its mean gene expression level across tissues included as a covariate. For this analysis, we used the mean log-transformed gene mRNA expression profiles in 53 different tissues obtained from GTEx (v7).
Polygenic risk scores (PRS)
In order to evaluate the predictive ability of our GWEIS results, we constructed interaction-based polygenic risk scores (iPRSGxE) using the SNP-environment interaction effects from each of the 25 GWEISs. As a comparison, we created alternative iPRSs representing the interactions between a standard main effect PRS and each environment (iPRSG). We evaluated the predictive accuracy of all iPRSs (i.e., the iPRSGxEs and iPRSGs) against that of a traditional main effect PRS.
To obtain GWEIS and GWAS effect sizes for these PRS analyses, we excluded a hold-out sample of 10,000 individuals (to be used for prediction) and re-analysed all main and interaction effects as described previously. For each analysis, we extracted the independent significant SNPs using clumping in PLINK53 (r2 < 0.2; 250kb), and used these SNPs to construct PRSs for every individual in the hold-out sample.
The different PRS scores were defined as follows. The standard main effect PRS was computed as for each individual i, with Gij their genotype value for SNP j (k being the number of SNPs used), and the GWAS effect size. For environment E, the iPRSG score was computed as , and the iPRSGxE as , with the GWEIS interaction effect size of SNP j.
A possible alternative to how we computed the iPRSGxE here may be to include the SNP main effect from the interaction analyses in the iPRSGxE itself, i.e., . As we are interested in determining the extent to which GxE predicts neuroticism beyond any gene and environment main effects, however, we constructed our using only the interaction terms, and instead included PRSi as a covariate to account for the genetic main effect.
The PRS scores were constructed using SNPs significant at different p-value thresholds (.001, .05, .1, .2, …, .8, .9, 1). For each PRS score, we then fit a linear regression in the hold-out sample with neuroticism as an outcome, with the PRS score and a set of covariates as predictors. An estimate of the predictive ability of the PRS score was then computed as the difference between the adjusted r2 for this model and the corresponding covariate-only model. Here, we chose to use the adjusted r2, rather than the full r2, as this provides an unbiased estimation of the population explained variance in models with multiple predictors. For the main effect PRS, as well as for both the iPRSGxE and iPRSG for each environment, we selected the PRS based on the p-value threshold for which the predictive ability was greatest.
As covariates, we used the same base covariates as in the GWEIS/GWAS analyses (age, sex, array, and all assessment centres with N > 50). For the traditional PRS, the covariates only model was , with Yi the neuroticism score for any one individual i in the holdout sample, Ci the 1 × k vector of base covariates (with ‘ denoting the transpose, and k the number of covariates), β0 the intercept, βC the covariate effect sizes, and ϵi the residual. The full model including the PRS is then , with PRSi representing the main effect PRS for that individual, and βPRS the beta coefficient for the PRS on neuroticism in the hold-out sample.
For the iPRSGxE and iPRSG scores, however, we also included the relevant environment and the main effect PRS as covariates, as well interaction between these and the base covariates (similar to the GWEIS setup). Thus, the covariate only model used for any iPRS with environment E is:
with PRSi and Ei representing the traditional main effect PRS and the environment, respectively (with βPRS and βE their effect on the neuroticism), and the interaction between the main effect PRS and the covariates (with related effect size βPxC), and the covariate-environment interaction (with effect size βExC). The full model for any iPRS would then also contain the term for the iPRS and its effect on neuroticism in addition to all variables in the null model, i.e.:
The reason why we include the main effect PRS derived from the GWAS as a representation of the SNP main effects, rather than simply a PRS constructed from the SNP main effect from the GWEIS, is because the GWEIS PRSs will have been pruned based on the interaction effects, and will thus underestimate the total amount of variance contributed by SNP main effects across the genome. Since we are specifically interested in how much the iPRSs contribute above and beyond what can be obtained using a simple main effect PRS from GWAS, the SNP main effects as obtained in the GWEIS would not have been appropriate.
Supplementary information
Acknowledgements
This work was funded by COSYN (Comorbidity and Synapse Biology in Clinically Overlapping Psychiatric Disorders: Horizon 2020 Program of the European Union under RIA grant agreement 667301 to D.P.), a European Research Council advanced grant (Grant no, ERC-2018-AdG GWAS2FUNC 834057 [to DP]), and the Netherlands Organization for Scientific Research (NWO: VICI 435-14-005). The analyses were carried out on the Genetic Cluster Computer, which is financed by the Netherlands Organization for Scientific Research (NWO: 480-05-003), by the VU University (Amsterdam, The Netherlands) and the Dutch Brain Foundation, hosted by the Dutch National Computing and Networking Services SurfSARA. This research was conducted using the UK Biobank Resource (application number 16406), and we would like to thank all participants who consented to participate in this research, as well as the researchers involved in the collection of the data.
Author contributions
D.P. conceived of the study. J.W. performed the analyses and drafted the manuscript. J.W., S.v.d.S., D.P., and C.d.L. participated in the interpretation of the results and editing of the manuscript. All authors provided meaningful contributions at all stages of the project.
Data availability
Summary statistics from all the 25 GWEISs, as well as the neuroticism GWAS can be downloaded from the website of the Department of Complex Trait Genetics, CNCR (http://ctg.cncr.nl). Summary statistics from the gene, gene-set, and gene-property analyses are available in Suppl. Tables 1–3.
Code availability
The full R script used to perform the GWEIS analyses can be accessed in Suppl. Info (B)–Analysis script. Analysis scripts used for the PRS, gene-, gene-set, or gene-property analyses are available from the authors upon request.
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
5/13/2022
A Correction to this paper has been published: 10.1038/s41398-022-01974-2
Change history
4/8/2021
A Correction to this paper has been published: 10.1038/s41398-021-01334-6
Contributor Information
Josefin Werme, Email: j.werme@vu.nl.
Christiaan A. de Leeuw, Email: c.a.de.leeuw@vu.nl
Supplementary information
The online version contains supplementary material available at 10.1038/s41398-021-01288-9.
References
- 1.Eysenck HJ. Biological basis of personality. Nature. 1963;199:1031–1034. doi: 10.1038/1991031a0. [DOI] [PubMed] [Google Scholar]
- 2.Costa PT, McCrae RR. Four ways five factors are basic. Pers. Individ. Dif. 1992;13:653–665. [Google Scholar]
- 3.Ormel J, Rosmalen J, Farmer A. Neuroticism: a non-informative marker of vulnerability to psychopathology. Soc. Psychiatry Psychiatr. Epidemiol. 2004;39:906–912. doi: 10.1007/s00127-004-0873-y. [DOI] [PubMed] [Google Scholar]
- 4.Eysenck HJNeuroticism. Anxiety and depression. Psychol. Inq. 1991;2:75–76. [Google Scholar]
- 5.Griffith JW, et al. Neuroticism as a common dimension in the internalizing disorders. Psychol. Med. 2010;40:1125–1136. doi: 10.1017/S0033291709991449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malouff, J. M., Thorsteinsson, E. B. & Schutte, N. S. The relationship between the five-factor model of personality and symptoms of clinical disorders: a meta-analysis. J. Psychopathol. Behav. Assess. 27, 101–114 (2005).
- 7.Lahey BB. Public health significance of neuroticism. Am. Psychol. 2009;64:241–256. doi: 10.1037/a0015309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuijpers P, et al. Economic costs of neuroticism: a population-based study. Arch. Gen. Psychiatry. 2010;67:1086. doi: 10.1001/archgenpsychiatry.2010.130. [DOI] [PubMed] [Google Scholar]
- 9.Vukasovic T, Bratko D. Heritability of personality: a meta-analysis of behavior genetic studies. Psychol. Bull. 2015;141:769–785. doi: 10.1037/bul0000017. [DOI] [PubMed] [Google Scholar]
- 10.Jang KL, Livesley WJ, Vemon PA. Heritability of the big five personality dimensions and their facets: a twin study. J. Pers. 1996;64:577–592. doi: 10.1111/j.1467-6494.1996.tb00522.x. [DOI] [PubMed] [Google Scholar]
- 11.Jardine R, Martin NG, Henderson AS, Rao DC. Genetic covariation between neuroticism and the symptoms of anxiety and depression. Genet. Epidemiol. 1984;1:89–107. doi: 10.1002/gepi.1370010202. [DOI] [PubMed] [Google Scholar]
- 12.Lake RIE, Eaves LJ, Maes HHM, Heath AC, Martin NG. Further evidence against the environmental transmission of individual differences in Neuroticism from a collaborative study of 45,850 twins and relatives on two continents. Behav. Genet. 2000;30:223–233. doi: 10.1023/a:1001918408984. [DOI] [PubMed] [Google Scholar]
- 13.Viken RJ, Rose RJ, Kaprio J, Koskenvuo M. A developmental genetic analysis of adult personality: extraversion and neuroticism from 18 to 59 years of age. J. Pers. Soc. Psychol. 1994;66:722–730. doi: 10.1037//0022-3514.66.4.722. [DOI] [PubMed] [Google Scholar]
- 14.Nagel M, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018;50:920–927. doi: 10.1038/s41588-018-0151-7. [DOI] [PubMed] [Google Scholar]
- 15.Luciano M, et al. Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat. Genet. 2018;50:6–11. doi: 10.1038/s41588-017-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jaffee SR. Child maltreatment and risk for psychopathology in childhood and adulthood. Annu. Rev. Clin. Psychol. 2017;13:525–551. doi: 10.1146/annurev-clinpsy-032816-045005. [DOI] [PubMed] [Google Scholar]
- 17.Allen B, Lauterbach D. Personality characteristics of adult survivors of childhood trauma. J. Trauma. Stress. 2007;20:587–595. doi: 10.1002/jts.20195. [DOI] [PubMed] [Google Scholar]
- 18.Mineka S, Zinbarg R. A contemporary learning theory perspective on the etiology of anxiety disorders: It’s not what you thought it was. Am. Psychol. 2006;61:10–26. doi: 10.1037/0003-066X.61.1.10. [DOI] [PubMed] [Google Scholar]
- 19.Bunce SC, Larson RJ, Peterson C. Life after trauma: personality and daily life experiences of traumatized people. J. Pers. 1995;63:165–188. doi: 10.1111/j.1467-6494.1995.tb00806.x. [DOI] [PubMed] [Google Scholar]
- 20.Roy A. Childhood trauma and neuroticism as an adult: possible implication for the development of the common psychiatric disorders and suicidal behaviour. Psychol. Med. 2002;32:1471–1474. doi: 10.1017/s0033291702006566. [DOI] [PubMed] [Google Scholar]
- 21.Santini ZI, Koyanagi A, Tyrovolas S, Mason C, Haro JM. The association between social relationships and depression: a systematic review. J. Affect. Disord. 2015;175:53–65. doi: 10.1016/j.jad.2014.12.049. [DOI] [PubMed] [Google Scholar]
- 22.Wang J, Mann F, Lloyd-Evans B, Ma R, Johnson S. Associations between loneliness and perceived social support and outcomes of mental health problems: a systematic review. BMC Psychiatry. 2018;18:156. doi: 10.1186/s12888-018-1736-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gariépy G, Honkaniemi H, Quesnel-Vallée A. Social support and protection from depression: systematic review of current findings in western countries. Br. J. Psychiatry. 2016;209:284–293. doi: 10.1192/bjp.bp.115.169094. [DOI] [PubMed] [Google Scholar]
- 24.Uher R, Zwicker A. Etiology in psychiatry: embracing the reality of poly-gene-environmental causation of mental illness. World Psychiatry. 2017;16:121–129. doi: 10.1002/wps.20436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Plomin R, Daniels D. Why are children in the same family so different from one another? Int. J. Epidemiol. 2011;40:563–582. doi: 10.1093/ije/dyq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turkheimer E. Three laws of behavior genetics and what they mean. Curr. Dir. Psychol. Sci. 2000;9:160–164. [Google Scholar]
- 27.Rutter M. Gene-environment interdependence. Dev. Sci. 2007;10:12–18. doi: 10.1111/j.1467-7687.2007.00557.x. [DOI] [PubMed] [Google Scholar]
- 28.Cicchetti, D. Resilience under conditions of extreme stress: a multilevel perspective. World Psychiatry9, 145 (2010). [DOI] [PMC free article] [PubMed]
- 29.Assary E, Vincent JP, Keers R, Pluess M. Gene-environment interaction and psychiatric disorders: review and future directions. Semin. Cell Dev. Biol. 2018;77:133–143. doi: 10.1016/j.semcdb.2017.10.016. [DOI] [PubMed] [Google Scholar]
- 30.Uher R. Gene-environment interactions in severe mental illness. Front. Psychiatry. 2014;5:48. doi: 10.3389/fpsyt.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barlow DH, Ellard KK, Sauer-Zavala S, Bullis JR, Carl JR. The origins of neuroticism. Perspect. Psychol. Sci. 2014;9:481–496. doi: 10.1177/1745691614544528. [DOI] [PubMed] [Google Scholar]
- 32.Dunn EC, et al. Genome-Wide Association Study (GWAS) and Genome-Wide Environment Interaction Study (GWEIS) of Depressive Symptoms in African American and Hispanic/Latina Women. Depress Anxiety. 2016;33:265–280. doi: 10.1002/da.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Otowa T, et al. The first pilot genome-wide gene-environment study of depression in the Japanese population. PLoS ONE. 2016;11:e0160823. doi: 10.1371/journal.pone.0160823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arnau-Soler A, et al. Genome-wide by environment interaction studies of depressive symptoms and psychosocial stress in UK Biobank and Generation Scotland. Transl. Psychiatry. 2019;9:1–13. doi: 10.1038/s41398-018-0360-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duncan LE, Keller MC. A critical review of the first 10 years of candidate gene-by-environment interaction research in psychiatry. Am. J. Psychiatry. 2011;168:1041–1049. doi: 10.1176/appi.ajp.2011.11020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nugent NR, Tyrka AR, Carpenter LL, Price LH. Gene-environment interactions: early life stress and risk for depressive and anxiety disorders. Psychopharmacology. 2011;214:175–196. doi: 10.1007/s00213-010-2151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClelland GH, Judd CM. Statistical difficulties of detecting interactions and moderator effects. Psychol. Bull. 1993;114:376–390. doi: 10.1037/0033-2909.114.2.376. [DOI] [PubMed] [Google Scholar]
- 38.Smith PG, Day NE. The design of case-control studies: the influence of confounding and interaction effects. Int. J. Epidemiol. 1984;13:356–365. doi: 10.1093/ije/13.3.356. [DOI] [PubMed] [Google Scholar]
- 39.Børglum AD, et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol. Psychiatry. 2013;18:20. doi: 10.1038/mp.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winham SJ, Biernacka JM. Gene-environment interactions in genome-wide association studies- current approaches and new directions. J. Child Psychol. Psychiatry. 2013;38:319–335. doi: 10.1111/jcpp.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gallagher MD, Chen-Plotkin AS. The Post-GWAS era: from association to function. Am. J. Hum. Genet. 2018;102:717–730. doi: 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas D. Gene–environment-wide association studies: emerging approaches. Nat. Rev. Genet. 2010;11:259–272. doi: 10.1038/nrg2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore R, et al. A linear mixed-model approach to study multivariate gene–environment interactions. Nat. Genet. 2019;51:180–186. doi: 10.1038/s41588-018-0271-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kerin M, Marchini J. Inferring gene-by-environment interactions with a Bayesian whole-genome regression model. Am. J. Hum. Genet. 2020;107:698–713. doi: 10.1016/j.ajhg.2020.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peyrot WJ, et al. Effect of polygenic risk scores on depression in childhood trauma. Br. J. Psychiatry. 2014;205:113–119. doi: 10.1192/bjp.bp.113.143081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Musliner KL, et al. Polygenic risk, stressful life events and depressive symptoms in older adults: a polygenic score analysis. Psychol. Med. 2015;45:1709–1720. doi: 10.1017/S0033291714002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mullins N, et al. Polygenic interactions with environmental adversity in the aetiology of major depressive disorder. Psychol. Med. 2016;46:759–770. doi: 10.1017/S0033291715002172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trotta A, et al. Interplay between Schizophrenia polygenic risk score and childhood adversity in first-presentation psychotic disorder: a pilot study. PLoS ONE. 2016;11:e0163319. doi: 10.1371/journal.pone.0163319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Voorman A, Lumley T, McKnight B, Rice K. Behavior of QQ-plots and genomic control in studies of gene-environment interaction. PLoS ONE. 2011;6:e19416. doi: 10.1371/journal.pone.0019416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayes AF, Cai L. Using heteroskedasticity-consistent standard error estimators in OLS regression: an introduction and software implementation. Behav. Res. Methods. 2007;39:709–722. doi: 10.3758/bf03192961. [DOI] [PubMed] [Google Scholar]
- 52.White H. A heteroskedasticity-consistent covariance matrix estimator and a direct test for heteroskedasticity. Econometrica. 1980;48:817. [Google Scholar]
- 53.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Almli LM, et al. Correcting systematic inflation in genetic association tests that consider interaction effects: application to a genome-wide association study of posttraumatic stress disorder. JAMA Psychiatry. 2014;71:1392–1399. doi: 10.1001/jamapsychiatry.2014.1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keller M. Gene-by-environment interaction studies have not properly controlled for potential confounders: the problem and the (simple) solution. Acc. Chem. Res. 2008;45:788–802. doi: 10.1016/j.biopsych.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sudlow C, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cederblad M, Dahlin L, Hagnell O, Hansson K. Intelligence and temperament as protective factors for mental health. A cross-sectional and prospective epidemiological study. Eur. Arch. Psychiatry Clin. Neurosci. 1995;245:11–19. doi: 10.1007/BF02191539. [DOI] [PubMed] [Google Scholar]
- 58.Hertenstein E, et al. Insomnia as a predictor of mental disorders: a systematic review and meta-analysis. Sleep. Med. Rev. 2019;43:96–105. doi: 10.1016/j.smrv.2018.10.006. [DOI] [PubMed] [Google Scholar]
- 59.Avila C, et al. An overview of links between obesity and mental health. Curr. Obes. Rep. 2015;4:303–310. doi: 10.1007/s13679-015-0164-9. [DOI] [PubMed] [Google Scholar]
- 60.Plomin R, DeFries JC, Knopik VS, Neiderhiser JM. Top 10 replicated findings from behavioral genetics. Perspect. Psychol. Sci. 2016;11:3–23. doi: 10.1177/1745691615617439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vinkhuyzen AAE, van der Sluis S, de Geus EJC, Boomsma DI, Posthuma D. Genetic influences on ‘environmental’ factors. Genes Brain Behav. 2010;9:276–287. doi: 10.1111/j.1601-183X.2009.00554.x. [DOI] [PubMed] [Google Scholar]
- 62.Purcell S. Variance components models for gene–environment interaction in twin analysis. Twin Res. 2002;5:554–571. doi: 10.1375/136905202762342026. [DOI] [PubMed] [Google Scholar]
- 63.Robins RW, Fraley RC, Roberts BW, Trzesniewski KH. A longitudinal study of personality change in young adulthood. J. Pers. 2001;69:617–640. doi: 10.1111/1467-6494.694157. [DOI] [PubMed] [Google Scholar]
- 64.Baca-Garcia E, et al. Diagnostic stability of psychiatric disorders in clinical practice. Br. J. Psychiatry. 2007;190:210–216. doi: 10.1192/bjp.bp.106.024026. [DOI] [PubMed] [Google Scholar]
- 65.Seeley JR, Farmer RF, Kosty DB, Gau JM. Prevalence, incidence, recovery, and recurrence of alcohol use disorders from childhood to age 30. Drug Alcohol Depend. 2019;194:45–50. doi: 10.1016/j.drugalcdep.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Prenoveau JM, et al. Are anxiety and depression just as stable as personality during late adolescence? Results from a three-year longitudinal latent variable study. J. Abnorm. Psychol. 2011;120:832–843. doi: 10.1037/a0023939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heim C, Binder EB. Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012;233:102–111. doi: 10.1016/j.expneurol.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 68.Rathouz PJ, Van Hulle CA, Rodgers JL, Waldman ID, Lahey BB. Specification, testing, and interpretation of gene-by-measured-environment interaction models in the presence of gene-environment correlation. Behav. Genet. 2008;38:301–315. doi: 10.1007/s10519-008-9193-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Day FR, Loh P-R, Scott RA, Ong KK, Perry JRB. A robust example of collider bias in a genetic association study. Am. J. Hum. Genet. 2016;98:392–393. doi: 10.1016/j.ajhg.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abraham G, Inouye M. Fast principal component analysis of large-scale genome-wide data. PLoS ONE. 2014;9:e93766. doi: 10.1371/journal.pone.0093766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eysenck SBG, Eysenck HJ, Barrett P. A revised version of the psychoticism scale. Pers. Individ. Dif. 1985;6:21–29. [Google Scholar]
- 73.Lorant V, et al. Socioeconomic inequalities in depression: a meta-analysis. Am. J. Epidemiol. 2003;157:98–112. doi: 10.1093/aje/kwf182. [DOI] [PubMed] [Google Scholar]
- 74.Ribeiro WS, et al. Income inequality and mental illness-related morbidity and resilience: a systematic review and meta-analysis. Lancet Psychiatry. 2017;4:554–562. doi: 10.1016/S2215-0366(17)30159-1. [DOI] [PubMed] [Google Scholar]
- 75.Bjelland I, et al. Does a higher educational level protect against anxiety and depression? The HUNT study. Soc. Sci. Med. 2008;66:1334–1345. doi: 10.1016/j.socscimed.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 76.Opitz PC, Lee IA, Gross JJ, Urry HL. Fluid cognitive ability is a resource for successful emotion regulation in older and younger adults. Front. Psychol. 2014;5:609. doi: 10.3389/fpsyg.2014.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conway KP, et al. Co-occurrence of tobacco product use, substance use, and mental health problems among adults: Findings from Wave 1 (2013-2014) of the Population Assessment of Tobacco and Health (PATH) Study. Drug Alcohol Depend. 2017;177:104–111. doi: 10.1016/j.drugalcdep.2017.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saban A, et al. The association between substance use and common mental disorders in young adults: results from the South African Stress and Health (SASH) Survey. Pan Afr. Med. J. 2014;17:11–18. doi: 10.11694/pamj.supp.2014.17.1.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Biddle DJ, Kelly PJ, Hermens DF, Glozier N. The association of insomnia with future mental illness: is it just residual symptoms? Sleep. Heal. 2018;4:352–359. doi: 10.1016/j.sleh.2018.05.008. [DOI] [PubMed] [Google Scholar]
- 80.Pigeon WR, Bishop TM, Krueger KM. Insomnia as a precipitating factor in new onset mental illness: a systematic review of recent findings. Curr. Psychiatry Rep. 2017;19:44. doi: 10.1007/s11920-017-0802-x. [DOI] [PubMed] [Google Scholar]
- 81.Rajan TM, Menon V. Psychiatric disorders and obesity: a review of association studies. J. Postgrad. Med. 2017;63:182–190. doi: 10.4103/jpgm.JPGM_712_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Turner RJ, Lloyd DA, Taylor J. Physical disability and mental health: an epidemiology of psychiatric and substance disorders. Rehabil. Psychol. 2006;51:214–223. [Google Scholar]
- 83.Turner RJ, Noh S. Physical disability and depression: a longitudinal analysis. J. Health Soc. Behav. 1988;29:23–37. [PubMed] [Google Scholar]
- 84.Fishbain DA, Cutler R, Rosomoff HL, Rosomoff RS. Chronic pain-associated depression: antecedent or consequence of chronic pain? A review. Clin. J. Pain. 1997;13:116–137. doi: 10.1097/00002508-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 85.Charles, S., Carayannopoulos, A. G. & Pathak, S. Anxiety and depression in patients with chronic pain. In Deer’s Treatment of Pain (eds Deer, T. R., Pope, J. E., Lamer, T. J. & Provenzano, D.) 125–129 (Springer International Publishing, 2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics from all the 25 GWEISs, as well as the neuroticism GWAS can be downloaded from the website of the Department of Complex Trait Genetics, CNCR (http://ctg.cncr.nl). Summary statistics from the gene, gene-set, and gene-property analyses are available in Suppl. Tables 1–3.
The full R script used to perform the GWEIS analyses can be accessed in Suppl. Info (B)–Analysis script. Analysis scripts used for the PRS, gene-, gene-set, or gene-property analyses are available from the authors upon request.