

Abstract
Patients with inflammatory bowel disease are at increased risk for colitis-associated colorectal cancer (CAC). Therefore, controlling intestinal inflammation is a key therapeutic strategy for CAC. MicroRNAs (miRNAs or miRs) are a family of small noncoding RNAs that have the capacity to regulate fundamental biological processes. To date, a number of miRNAs have been identified as critical regulators of inflammation. However, the specific role of miR-26a in colonic inflammation and colitis-associated carcinogenesis is still elusive. Here, we generated mice with miR-26a myeloid-cell-specific overexpression to show that miR-26a suppressed the intestinal inflammatory response in macrophages by decreasing nuclear factor κB (NF-κB)/STAT3 activation and interleukin 6 (IL-6) production. At the molecular level, a number of NF-κB regulators, including TLR3, PTEN, and PKCδ, were identified as potential targets of miR-26a. Our results thus identify a novel miRNA-mediated mechanism that suppresses carcinogenic inflammation in the colon.
Keywords: MiR-26a, Intestinal inflammation, Colitis-associated carcinogenesis, NF-κB/STAT3, Macrophage
Graphical abstract
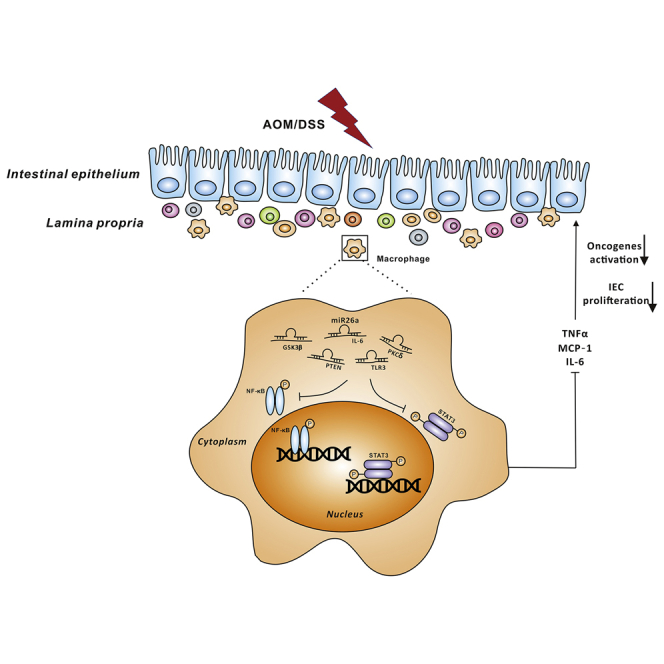
Wendong Huang and colleagues find that miR-26a potently suppresses NF-κB/STAT3 signaling by targeting IL-6, TLR3, PTEN, and PKCδ in macrophages, which is associated with the reduction in colitis-associated tumorigenesis.
Introduction
Inflammatory bowel disease (IBD), including ulcerative colitis (UC) and Crohn’s disease (CD), is a chronic and relapsing inflammatory condition that affects the gastrointestinal tract in millions of people throughout their lives. IBD affected 1 out of 200 individuals in western countries, and the incidence of IBD has been rapidly increasing in South America, Asia, and Africa in the 21st century.1 Because of chronic detrimental damage to the colon and rectum, individuals with either type of IBD are at increased risk of developing colorectal cancer.2,3 The mechanism of colitis-induced tumorigenesis has not been fully investigated; previous study demonstrated that inflammatory responses lead to mutations and epigenetic changes in normal cells. For instance, reactive oxygen species produced by myeloid cells induce mutations in intestinal epithelial cells (IECs).4 In addition, inflammatory responses also contribute to creating transformed and malignant clones. Cytokines produced by macrophages or neutrophils, such as interleukin-1β (IL-1β) and IL-6, activate their receptor-signaling pathways in mutated cells, leading to increased survival and proliferative probability of the transformed clone.5,6 Azoxymethane (AOM) and dextran sodium sulfate (DSS) models have been used to induce colitis-associated colorectal cancer (CAC) in mice, which has two distinct stages of cancer development: initiation and promotion.7 Researchers have found that nuclear factor κB (NF-κB) and STAT3 signaling in lamina propria (LP) immune cells and IECs play central roles in the initiation and promotion of CAC. In unstimulated cells, NF-κB binds with a set of inhibitory proteins named Inhibitor of κB (IκB) family, including IκBα. The first step of NF-κB activation is specifically phosphorylating IκB by IκB kinase (IKK), leading to NF-κB degradation.8 Activation of NF-κB in LP myeloid cells, mainly macrophages, can lead to the production of proinflammatory cytokines to recruit other immune cells and results in intestinal inflammation. Subsequently, the recruited inflammatory cells stimulate the proliferation of premalignant IECs through the secretion of a variety of cytokines, particularly IL-6, IL-11, and IL-22. These cytokines exert their proliferative effect via activation of the STAT3 pathway in IECs, which further synergize with NF-κB to increase the expression of survival and proliferating genes in IECs.9 Therefore, inhibition of NF-κB and STAT3 activation in immune cells or IECs has been suggested as an effective approach to treat CAC.
MicroRNAs (miRNAs) constitute a large class of phylogenetically conserved single-stranded RNA molecules of 19–25 nt that are involved in posttranscriptional gene silencing. miRNAs play important roles in the regulation of cell survival, proliferation, and differentiation as well as angiogenesis, and a growing body of evidence indicates that miRNAs are frequently deregulated in human malignancies and function as either oncogenes or tumor suppressor genes.10 Recently, there has been considerable interest in understanding the mechanism by which miRNAs regulate carcinogenesis, and accumulating evidence suggests that targeting miRNAs may lead to the development of novel cancer therapies.
Among miRNAs, miR-26a is ubiquitously expressed in various tissues, including the thymus, testes, placenta, colon, and small intestine. miR-26a is completely conserved across vertebrates. It plays a dual role in different cancers, as a tumor suppressor in hepatocellular carcinoma, myc-induced lymphoma, breast cancer, nasopharyngeal carcinoma, etc.,11, 12, 13, 14, 15, 16 or as a tumor promoter in glioma and cholangiocarcinoma.17,18 These seemingly conflicting roles suggest that the functional outcome of miR-26a in carcinogenesis may depend on the cellular context of its expression and the array of potential mRNA targets at its disposal.
Other studies have revealed that miR-26a also plays important roles in regulating inflammatory signaling. miR-26a may directly regulate IFN-β expression to modulate the innate immune response to viral infections in human and macaque cells.19 Jiang et al.20 find that miR-26a negatively regulates Toll-like receptor 3 expression in rat macrophages and ameliorates pristane-induced arthritis in rats. Reduced expression of miR-26a in hepatocarcinoma is associated with the activation of signaling pathways related to NF-κB and IL-6.21 Another study reveals that miR-26a is upregulated in UC and CD mucosa,22 indicating that miR-26a plays a potential role in colitis. However, the exact role and mechanism of miR-26a in colonic inflammation and inflammatory-related cancers, such as CAC, is still elusive.
In the present study, we generated transgenic (TG) mice with either global or myeloid-cell-specific miR-26a overexpression. All TG mice showed reduced susceptibility to colitis and CAC. We further demonstrated that the reduction in tumorigenesis was associated with suppression of NF-κB/STAT3 signaling as well as reduced expression of IL-6, TLR3, PTEN, and PKCδ, which were all identified as direct targets of miR-26a. Together, our data identify a potential miRNA-mediated approach for the treatment of colitis and CAC.
Results
miR-26a TG mice are resistant to DSS-induced colitis
To investigate the function of miR-26a in DSS-induced colitis, we generated a miR-26a TG mouse line (miR-26aTg)23 using a gain-of-function approach. We then sought to determine the effect of miR-26a overexpression during the development of experimental colitis in mice. Wild-type (WT) and miR-26aTg mice were challenged with 1% DSS for 7 consecutive days and then sacrificed for measurement of colitis severity. Prior to euthanasia, susceptibility was monitored by measuring body weight and assessing rectal bleeding. Compared with their WT littermates, miR-26aTg mice exhibited significantly decreased body weight loss and rectal bleeding (Figures 1A and 1B). DSS typically causes colon shortening, and this change was also diminished in miR-26aTg mice (Figures 1C and 1D). Histologic analyses showed that inflammation was more severe in WT mice than that in miR-26aTg mice. The colons of miR-26aTg mice contained markedly fewer infiltrating inflammatory cells and displayed significantly reduced colitis severity compared to those of WT mice, with scores of 10.29 ± 3.90 and 6.33 ± 2.07, respectively (p < 0.05) (Figures 1E and 1F).
Figure 1.

miR-26a transgenic mice are resistant to DSS-induced colitis
miR-26aTg mice and their WT littermates were challenged with 1% DSS provided in their drinking water for 7 consecutive days, followed by standard drinking water for 2 days. (A) Body weight was measured every 2 days. (B) Prior to euthanasia, rectal bleeding was assessed in the mice. (C and D) Mice were sacrificed on day 9 for colon length measurement. (E) Representative images of H&E-stained colon sections. (F) Semiquantitative histopathological scoring of colon sections on day 9. Data with error bars are presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
To further confirm the protective role of miR-26a in DSS-induced colitis, we used another approach, locked-nucleic-acid-modified anti-miR-26a (LNA-anti-miR-26a), to inhibit endogenous miR-26a expression in mice. As shown in Figure 2A, the expression of miR-26a in the colon was successfully inhibited by LNA-anti-miR-26a. Pretreatment of mice with the miR-26a inhibitor exacerbated DSS-induced colitis, as reflected by the increases in body weight loss (Figure 2B) and rectal bleeding (Figure 2C), colon shortening (Figures 2D and 2E), and mucosal disruption (Figures 2F and 2G).
Figure 2.

miR-26a inhibitor-treated mice were hypersusceptible to DSS-induced colitis
The miR-26a inhibitor (LNA-anti-miR-26a) was administered to inhibit endogenous miR-26a expression in the DSS-induced colitis model. (A) qPCR analysis of miR-26a expression in LNA-miR-26a-treated and control mice. (B) The body weight was measured every 2 days in the DSS-induced colitis model. (C) Prior to euthanasia, rectal bleeding was assessed in the mice. (D and E) Mice were sacrificed on day 9 for colon length measurement. (F) Representative images of H&E-stained colon sections. (G) Semiquantitative histopathological scoring of colon sections on day 9. Data with error bars are presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
Taken together, these results indicate that miR-26a plays a protective role during the progression of acute colitis. However, miR-26a is globally overexpressed in miR-26aTg mice, and LNA-anti-miR-26a inhibits endogenous miR-26a expression in multiple cell types. Therefore, we next investigated which cell type plays the dominant role in suppressing colitis via miR-26a overexpression.
Overexpression of miR-26a in myeloid cells suppresses DSS-induced colitis
miR-26a has been reported to regulate macrophage polarization,24 migration,25 and responsiveness.26 To determine whether the regulatory effects of miR-26a on macrophages affects the process of colitis and CAC, we crossed LysM-Cre mice with miR-26aTg mice and generated a new mouse line that specifically overexpressed miR-26a in myeloid lineage cells (M-miR26aTg). As shown in Figure 3A, the expression of miR-26a in intraperitoneal macrophages was approximately 6 times higher in M-miR26aTg mice than that in their WT littermates. Mice were challenged with 1% DSS for 7 consecutive days and then sacrificed on day 8 for measurement of colitis severity. Consistent with the results obtained with miR-26aTg mice, overexpression of miR-26a in myeloid cells reduced the severity of colitis, as evidenced by the significantly reduced body weight loss (Figure 3B), colon shortening (Figures 3C and 3D), and histologic scores (Figures 3E and 3F). These results revealed that specific overexpression of miR-26a in myeloid cells is sufficient to protect mice from DSS-induced colitis.
Figure 3.

Overexpression of miR-26a in myeloid cells suppresses DSS-induced colitis
Mice with myeloid cell-specific overexpression of miR-26a (M-miR-26aTg mice) and their WT littermates were challenged with 1% DSS provided in the drinking water for 7 consecutive day, followed by standard drinking water. (A) qPCR analysis of miR-26a in intraperitoneal macrophages of M-miR-26aTg mice and their WT littermates. (B) The body weight was measured every 2 days in the DSS-induced colitis model. (C and D) Mice were sacrificed on day 8 for colon length measurement. (E) Representative images of H&E-stained colon sections. (F) Semiquantitative histopathological scoring of colon sections on day 8. Data with error bars are presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
Overexpression of miR-26a in myeloid cells suppresses colitis-associated tumorigenesis
Colitis is considered a tumor promoter;2,3 therefore, we asked whether the colitis-suppressing ability of miR-26a can decrease the incidence of colitis-induced tumorigenesis.
M-miR26aTg mice and their WT littermates were challenged by treatment with the gene mutation agent AOM (10 mg/kg) followed by three cycles of exposure to 1% DSS to induce colitis-associated cancer (Figure 4A). The tumor burden was assessed 12 weeks after AOM injection. As shown in Figure 4B, M-miR26aTg mice had significantly fewer tumors in the colon; the number of tumors in WT mice (n = 9) and M-miR26aTg mice (n = 9) was 5.2 ± 3.6 and 11.8 ± 6.0, respectively (Figure 4C), and the tumor size distribution was not significantly different (Figure 4D), indicating that miR-26a may suppress AOM/DSS-induced tumor initiation. The decreased tumor number in M-miR26aTg mice was associated with reduced inflammation and a decreased incidence of dysplasia (Figures 4E and 4F). Histological analysis of tumors and adenomatous polyps showed that all WT mice developed tumor-associated dysplasias, 60% of which was classified as adenocarcinoma. By contrast, only 80% of mice in the M-miR26aTg cohort displayed tumor-associated dysplasia in the colon, and the incidence of adenocarcinoma development was approximately 20% in this group (Figure 4F). To reveal the human relevance of miR-26a to suppress colitis and CAC, we sought to measure the expression of miR-26a and its primary miRNA, Pri-miR26a, in colorectal cancer patients. The expression of miR-26a in colorectal cancer and adjacent normal tissues from 40 patients was examined by quantitative polymerase chain reaction (qPCR). As shown in Figure S1A, among the 40 patients, miR-26a was downregulated in 37 tumor samples compared with the adjacent normal tissues. Consistently, the expression level of Pri-miR26a in tumor tissues is significantly lower than adjacent normal tissue (Figure S1B).
Figure 4.

Overexpression of miR-26a in myeloid cells suppresses AOM/DSS-induced CAC
(A) M-miR26aTg mice and their WT littermates were challenged with AOM (10 mg/kg) followed by three cycles of exposure to 1% DSS to induce CAC. (B–D) Mice were sacrificed on day 87 for tumor number and size measurements. (E and F) Colon tissues were stained with H&E and classified as dysplasia or adenocarcinoma. Data with error bars are presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
Collectively, these results indicated that miR-26a overexpression in myeloid cells reduced the initiation of colitis-related tumors. In addition, downregulation of miR-26a is a common feature in human colorectal cancer.
miR-26a suppresses the inflammatory response in myeloid cells as well as oncogene activation in IECs and IEC proliferation at the early stage of CAC
To investigate how myeloid-specific miR-26a overexpression suppresses CAC tumorigenesis, we carefully examined the histopathological changes that occur at the early stage of CAC. Mice were injected with AOM, and 7 days later mice were challenged with 1% DSS provided in their drinking water for 7 consecutive days. Colons were collected on day 21 after AOM injection (Figure 5A). In line with the results in the DSS-induced acute colitis model, the body weight loss (Figure S2A) and colon shortening induced by colitis were significantly reduced in M-miR26aTg mice (Figures S2B and S2C). In addition, histological analysis of colon sections revealed markedly less tissue damage, inflammation, and hyperplasia in M-miR26aTg mice than those in WT mice (Figures S2D and S2E). To further verify whether the decrease in tumorigenesis mediated by miR-26a is associated with inhibition of hyperinflammatory responses, LP cells and IECs were isolated from the distal colon and analyzed separately. LP cells of M-miR26aTg mice exhibited much lower expression levels of proinflammatory cytokines, including MIP2, tumor necrosis factor alpha (TNF-α), IL-1b, ICAM1, KC, and IL-6, than those of WT mice (Figure 5B). Lysates of IECs from M-miR26aTg mice and their WT littermates were subjected to immunoblot analysis. As shown in Figure 5C, IECs of M-miR26aTg mice exhibited lower levels of p-STAT3, P-IκBα, c-myc, and PCNA. Furthermore, we analyzed 5′-bromo-2′-deoxyuridine (BrdU) incorporation in IECs after sacrifice, and these results also demonstrated that miR-26a overexpression in myeloid cells significantly inhibited the proliferation of IECs (Figures 5D and 5E). These results revealed that miR-26a overexpression in myeloid cells reduced the levels of proinflammatory cytokines, resulting in suppression of oncogene activation in IECs and proliferation of IECs.
Figure 5.

miR-26a reduced proinflammatory cytokine production in the LP, resulting in suppression of both oncogene activation in IECs and IEC proliferation
(A) M-miR-26aTg mice and their WT littermates were challenged with AOM (10 mg/kg), and 7 days later, the mice were challenged with 1% DSS provided in the drinking water for 7 consecutive days, followed by standard drinking water for another 7 days. (B) Colons were harvested on day 22, and LP cells were isolated as described in the Materials and methods section. The expression levels of MIP2, TNF-α, IL-1b, ICAM1, KC, and IL-6 were determined by qPCR. (C) Mice were sacrificed on day 22, and the levels of p-STAT3, P-IκBα, c-myc, and PCNA in colonic epithelial cells were determined by immunoblot analysis. (D) Representative images of BrdU-stained colon tissues. (E) Number of BrdU-positive cells on day 22. Data with error bars were presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
miR-26a overexpression leads to decreased proinflammatory cytokine production as well as reduced STAT3 and NF-κB activation in macrophages
The data shown above indicated that LP cells from the colons of M-miR-26aTg mice expressed lower levels of proinflammatory cytokines, which ameliorated colitis and tumor initiation in these mice. However, myeloid cells are a heterogeneous group of immune cells that includes macrophages, dendritic cells, monocytes granulocytes, etc. To obtain more specific evidence for suppression of the inflammatory response by miR-26a in macrophages, bone-marrow-derived macrophages (BMDMs) from WT and M-miR26aTg mice were treated with lipopolysaccharide (LPS) in vitro. qPCR was employed to analyze the expression of proinflammatory genes. The results showed that macrophages from M-miR26aTg mice had reduced expression levels of TNF-α, IL-6, and MCP1 compared with BMDMs from their WT littermates (Figure 6A). Next, we evaluated the activation of NF-κB and STAT3 by western blot analysis. As expected, overexpression of miR-26a in macrophages repressed LPS-induced phosphorylation of IκB-α and STAT3 (Figure 6B). These data indicated a role of miR-26a in negatively regulating NF-κB and STAT3 pathways downstream of inflammatory signals, resulting in lower levels of proinflammatory cytokine expression.
Figure 6.

miR-26a suppresses proinflammatory cytokine expression and NF-κB/STAT3 activation in bone-marrow-derived macrophages (BMDMs)
(A) BMDMs isolated from M-miR-26aTg mice and their WT littermates were stimulated with 10 ng/mL LPS for 6 h, and mRNA was isolated for qPCR analysis of TNF-α, IL-6, and MCP1. (B) LPS-stimulated WT and miR-26a-overexpressing BMDMs were collected, and cell lysates were analyzed by western blotting to determine the levels of p-IκBa, IκBa, p-STAT3 (Tyr705), and STAT3. Data with error bars are presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
miR-26a targets several genes involved in colitis
Previous data have suggested that miR-26a suppresses proinflammatory cytokines by inhibiting the NF-κB/STAT3 pathway, and IL-6 has been reported to be a direct target of miR-26.11 Consistently, our data also indicated that miR-26a significantly reduced the luciferase activity of the IL-6 reporter containing a WT 3′ UTR but did not suppress the activity of the IL-6 reporter with a mutant 3′ untranslated region (UTR) (Figures 7A and 7B). Inhibition of miR-26a by anti-miR-26a increased the luciferase activity of the IL-6 reporter (Figure 7B). These results suggested that IL-6 is a direct target of miR-26a. Next, we attempted to identify other miR-26a targets that are potentially involved in the regulation of macrophage activity, including PTEN,27,28 GSK3β,29 TLR3,30 and PKCδ.31,32 The dual-luciferase reporter assay showed that coexpression of miR-26a significantly suppressed while inhibition of miR-26a increased the activity of firefly luciferase carrying the 3′ UTR of PTEN, GSK3β, TLR3, or PKCδ (Figure 7C), suggesting that miR-26 can bind directly to the sequences in the 3′ UTR of these genes. The effect of miR-26a on the endogenous cellular expression of the abovementioned targets was examined by qPCR. The results showed that introduction of miR-26a decreased the expression of TLR3, PTEN, and PKCδ at the mRNA level (Figure 7D). These results indicated that miR-26a inhibited the NF-κB/IL-6/STAT3 pathway in macrophages by directly targeting IL-6. In addition, we have identified additional miR-26a targets, including PTEN, TLR3, and PKCδ, which also negatively regulate inflammatory responses. To investigate whether PTEN, TLR3, and PKCδ are involved in the anti-tumor effect of miR-26a, qPCR was employed to assess their expression level in colorectal cancer and adjacent normal tissues from 40 patients. The results show that TLR3 and PKCδ were significantly more highly expressed in adjacent normal tissue than tumor (Figures S3A and S3B). However, there was no significant difference in expression level of PTEN between tumor and adjacent normal tissue (Figure S3C).
Figure 7.

miR-26a targets several genes involved in colitis
(A) Putative binding sequence for miR-26a in IL-6. (B) miR-26a significantly reduced the luciferase activity of the IL-6 reporter containing a WT 3′ UTR but not that of the IL-6 reporter containing a mutant 3′ UTR. Inhibition of miR-26a by anti-miR-26a increased the luciferase activity of the IL-6 reporter. (C) Coexpression of miR-26a significantly suppressed while inhibition of miR-26a increased the activity of firefly luciferase carrying the 3′ UTR of PTEN, GSK3β, TLR3, or PKCδ. (D) qPCR was used to analyze endogenous expression of GSK3β, IL-6, TLR3, PTEN, and PKCδ after miR-26a introduction. Data with error bars are presented to indicate the mean ± SD values. ∗p < 0.05, ∗∗p < 0.01.
Taken together, these findings indicate that miR-26 may inhibit multiple proinflammatory genes, resulting in suppression of colitis and CAC in both mice and humans.
Discussion
miR-26a is known to play critical roles in various physiological and pathological processes, including pancreatic cell differentiation,23 liver disorders,33,34 and glucose and lipid metabolism.35 Human cancers often exhibit global underexpression of miRNAs. We have previously demonstrated that miR-26a enhances miRNA biogenesis in diverse cancer cell lines, xenograft tumors, and normal human tissues, thereby providing a novel function by which miR-26a acts as a modulator of miRNA maturation.36
Previous studies also identified miR-26a as a potential tumor suppressor in lymphoma37 and hepatocellular carcinoma (HCC).11,12,38 However, its role in colorectal cancer is still largely unclear. Zeitels et al.39 crossed miR-26a TG mice with APCmin/+ mice to reveal the protective role of miR-26a in colorectal cancer. In addition, APCmin/+ mice overexpressing miR-26a in the intestinal epithelium showed the same result, indicating the ability of miR-26a to suppress the activity of oncogenes in IECs. In the current study, we used miR-26Tg mice and LNA-miR26a to demonstrate a protective role of miR-26a in DSS-induced colitis. Next, M-miR-26aTg mice were employed to reveal that specific overexpression of miR-26 in myeloid cells is sufficient to increase susceptibility to colitis and CAC. At the early stage of CAC, miR-26a suppressed the production of proinflammatory cytokines in the colonic LP, and this ability to suppress colitis inhibited oncogene activation in IECs and reduced the proliferation of IECs, resulting in decreased tumor formation. Our data demonstrated that miR-26a suppressed colitis and CAC by regulating immune cells in the tumor microenvironment rather than by directly affecting IEC proliferation, apoptosis, or migration. In summary, our results provide a more comprehensive understanding of the mechanism by which miR-26a suppresses colon cancer.
Our data showed that BMDMs of M-miR-26aTg mice released lower levels of TNF-α and IL-6 than those of their WT littermates when stimulated by LPS. Furthermore, miR-26a repressed LPS-induced phosphorylation of IκB-α and STAT3, indicating a role of miR-26a in suppressing proinflammatory cytokine production by negatively regulating NF-κB and STAT3 signaling. In addition, a dual-luciferase reporter assay was employed to validate the direct interaction between miR-26a and IL-6. These results revealed that miR-26a suppressed NF-κB and STAT3 signaling in macrophages by targeting IL-6, consistent with a previous report.11 Those data showed that a sequence in miR-26a binds to the 3′ UTR of IL-6 in HCC cells, and this binding sequence was the same sequence identified to bind in our experiments (Figure 7A). In contrast, Chen et al.40 reported that the IL-6 transcript lacks a sequence for direct binding with miR-26a in human BEAS-2B and A549 cells. These opposing results may be due to different cell lines being used. Chen et al.40 demonstrated that although miR-26a lacks a suitable sequence for binding to IL-6, miR-26a indeed suppresses TNF-α-mediated IL-6 activation by downregulating the NF-κB-related factors HMGA1 and MALT1. Taken together, these results indicated that miR-26a indeed downregulated NF-κB and STAT3 signaling as well as IL-6 expression, but the detailed molecular mechanisms may not be identical across different cells.
Jiang et al.20 reported that miR-26a directly interacts with TLR3 and negatively regulates downstream cytokine expression in NR8383 cells, a macrophage cell line established from normal rat alveolar macrophage cells. Zeitels et al.39 reported that miR-26a suppresses colon cancer by repressing PTEN in the intestinal epithelium. Du et al.41 reported that miR-26a inhibits the apoptosis of oral keratinocytes by directly targeting PKCδ. We also validated TLR3, PTEN, and PKCδ as targets of miR-26a by a dual-luciferase reporter assay and found that suppression of TLR3, PTEN, and PKCδ expression increased susceptibility to colitis and CAC.
In summary, we demonstrated that miR-26a directly targets IL-6, TLR3, PTEN, and PKCδ and defined the suppressive role of miR-26a in colitis and CAC. These findings revealed new insights into miRNA-mediated tumor suppression through inhibition of the inflammatory response in immune cells, which may provide novel approaches for the treatment of inflammation-related cancers.
Materials and methods
Mice
As described in a previous study,23 we generated miR-26a TG mice. In brief, a DNA fragment encoding the miR-26a-1 locus was inserted into the Rosa26 locus. The synthetic CAG promoter and a loxP-flanked Neo-STOP cassette were inserted upstream of the miR-26a-1 locus. Targeted embryonic stem cells (ESCs) were injected into blastocysts to generate mice, and a mixed C57BL/6 and 129 background was maintained. Hypoxanthine guanine phosphoribosyltransferase-Cre mice were bred with mice carrying the target allele to delete the Neo-STOP cassette during embryogenesis.42 LysM-Cre mice were bred with mice carrying the target allele to selectively delete the Neo-STOP cassette in macrophages.43 Heterozygous TG mice and their littermate WT mice were used for experiments. All procedures followed the National Institutes of Health guidelines for the care and use of laboratory animals.
Induction of acute colitis
Acute colitis was induced with 1% (w/v) DSS (MP Biomedicals) dissolved in sterile distilled water provided ad libitum for 7 consecutive days, followed by standard drinking water until the end of the experiment.
Induction of colitis-associated cancer
Mice were injected intraperitoneally with 10 mg/kg AOM (Sigma). After 7 days, 1% DSS was provided in their drinking water for 7 consecutive days, followed by regular standard drinking water for 2 weeks. This cycle was repeated three times, and mice were sacrificed 4 weeks after the last DSS cycle.
In vivo delivery of LNA-modified anti-miR-26a
The LNA-anti-miR26a oligonucleotides were purchased from Exiqon (Denmark). Mice were injected intraperitoneally with 10 mg/kg anti-miR-26a or vehicle control every 2 days for a total of four times.
Histological analysis
Formalin-preserved colon sections were processed and embedded in paraffin by standard techniques. Longitudinal sections of 5 μm thickness were stained with hematoxylin and eosin (H&E) and examined by a pathologist blinded to the experimental groups. The extent of inflammation was measured and scored as described previously.44
In situ intestinal proliferation assay
The number of proliferating cells in the intestinal epithelium was determined using the immunoperoxidase staining protocol with the thymidine analog BrdU as described earlier.45 In brief, 1 mg/mL BrdU in PBS was injected intraperitoneally. Three hours later, colon tissue was collected, fixed with 10% neutral buffered formalin, and embedded in paraffin. Immunohistochemistry was performed using an in situ BrdU staining kit (BD Biosciences) according to the manufacturer’s recommendations. Tissues were counterstained with hematoxylin.
Isolation of LP cells and IECs
Colons were dissected, washed with ice-cold PBS supplemented with antibiotics (penicillin plus streptomycin), and cut into small pieces. Colon pieces were then incubated with RPMI medium supplemented with 3% FBS, 0.5 mM DTT, 5 mM EDTA, and antibiotics at 37°C for 30 min with gentle shaking. After removing the epithelial layer, the remaining colon segments were incubated at 37°C with RPMI medium containing 0.5% Collagenase D (Roche) and 0.05% DNase (Roche) for 30 min with gentle shaking. The supernatant was passed through a 70 μm cell strainer to isolate LP cells.
BMDM culture and stimulation
Bone marrow cells were harvested from mouse femurs and filtered through a 70 μm strainer. After red blood cells were lysed with ACK lysis buffer (Thermo Fisher), the cell pellet was washed and suspended in DMEM containing 20% FBS and 10 ng/mL murine macrophage colony-stimulating factor (M-CSF) (Biolegend), and cells were plated in a six-well tissue culture plate at 2 × 106 cells/well and incubated for 7 days. To activate BMDMs, 10 ng/mL LPS (Sigma) was added to the medium, and cells were harvested after 6 h.
miR-26a and LNA miR-26a inhibitor transfections
The miRNA mimic and miRCURY LNA miR-26a inhibitor were purchased from Ambion (Austin, TX, USA) and Exiqon (Vedbaek, Denmark), respectively. Transfections of miRNAs or inhibitors were performed using HiPerfect (QIAGEN, Valencia, CA, USA) according to the manufacturer’s protocol.
Luciferase activity assays
HeLa cells were transfected with 40 nM miRNA precursors (GenePharma, Shanghai, China) and 200 ng of psiCHECK-2.2 (Promega) constructs containing an insert of the 3′ UTR or flanking sequences of seed nucleotides of miR-26a target genes using Attractene (QIAGEN) in 96-well plates. Twenty-four hours after transfection, cells were analyzed with a dual-luciferase reporter assay system (Promega). To generate mutant reporter constructs, the seed sequence in the 3′ UTR (5′-TACTTGA-3′) was mutated to 5′-ATGATGA-3′.
Cell culture and transfection
Cell lines were purchased from American Type Culture Collection. Transfection was performed with Attractene (QIAGEN, Valencia, CA, USA) according to the manufacturer’s instructions. Stable transformants were selected in complete medium containing 500 μg/mL G418 (Sigma, St. Louis, MO, USA).
Real-time PCR analysis
For analysis of miR-26a, reverse transcription was performed with Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Real-time PCR was performed using the Power SYBR Green PCR Master Mix protocol (Applied Biosystems, Foster City, CA, USA). To analyze mRNAs, reverse transcription was performed with Superscript III reverse transcriptase and Oligo(dT)20 primers at 50°C for 1 h. The expression levels of miRNAs were normalized to those of 5S RNA. mRNA expression levels were normalized to those of β-actin (Ambion, Austin, TX, USA). The sequences of the primers used are listed in Table S1.
Immunoblot analysis
Cell lysates were separated by appropriate sodium dodecyl sulfate-polyacrylamide gel electrophoresis methods and transferred to nitrocellulose membranes. After blocking in 5% nonfat milk, membranes were incubated with the following primary antibodies: anti-phospho-STAT3, anti-total STAT3, and anti- IκBα, obtained from Cell Signaling Technology (Danvers, MA, USA) and anti-c-myc, anti-PCNA, and anti-GAPDH, purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Membranes were washed and reacted with peroxidase-conjugated secondary antibodies (Amersham Biosciences, UK).
Statistical analysis
Experiments were repeated at least three times with consistent results. The results were analyzed for statistical significance with Student’s t test for unpaired samples in Excel (Microsoft), and p <0.05 was considered significant.
Acknowledgments
This work is supported by National Natural Science Foundation of China 81972745 and 81372621 (W. Han), the Ministry of Science and Technology of the People's Republic of China 2018ZX09201018-005, and the National Natural Science Foundation of China 81970561 (X.Fu). W. Huang is supported by the US National Cancer Institute 2R01CA139158.
Author contributions
W. Han and W. Huang conceived and designed the study; W.Z. and X.F. performed the experiments; X.F., J.X., and H.P. analyzed the data and prepared the figures; W. Han and W.Z. wrote the manuscript; W. Huang and X.F. revised the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.02.029.
Contributor Information
Xianghui Fu, Email: xfu@scu.edu.cn.
Weidong Han, Email: hanwd@zju.edu.cn.
Wendong Huang, Email: whuang@coh.org.
Supplemental information
References
- 1.Ng S.C., Shi H.Y., Hamidi N., Underwood F.E., Tang W., Benchimol E.I., Panaccione R., Ghosh S., Wu J.C.Y., Chan F.K.L. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390:2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi T., Siegmund B., Le Berre C., Wei S.C., Ferrante M., Shen B., Bernstein C.N., Danese S., Peyrin-Biroulet L., Hibi T. Ulcerative colitis. Nat. Rev. Dis. Primers. 2020;6:74. doi: 10.1038/s41572-020-0205-x. [DOI] [PubMed] [Google Scholar]
- 3.Roda G., Chien Ng S., Kotze P.G., Argollo M., Panaccione R., Spinelli A., Kaser A., Peyrin-Biroulet L., Danese S. Crohn’s disease. Nat. Rev. Dis. Primers. 2020;6:22. doi: 10.1038/s41572-020-0156-2. [DOI] [PubMed] [Google Scholar]
- 4.Greten F.R., Grivennikov S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity. 2019;51:27–41. doi: 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grivennikov S., Karin E., Terzic J., Mucida D., Yu G.Y., Vallabhapurapu S., Scheller J., Rose-John S., Cheroutre H., Eckmann L., Karin M. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15:103–113. doi: 10.1016/j.ccr.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dmitrieva-Posocco O., Dzutsev A., Posocco D.F., Hou V., Yuan W., Thovarai V., Mufazalov I.A., Gunzer M., Shilovskiy I.P., Khaitov M.R. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity. 2019;50:166–180.e7. doi: 10.1016/j.immuni.2018.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell C., McKenney P.T., Konstantinovsky D., Isaeva O.I., Schizas M., Verter J., Mai C., Jin W.-B., Guo C.-J., Violante S. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature. 2020;581:475–479. doi: 10.1038/s41586-020-2193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Q., Lenardo M.J., Baltimore D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell. 2017;168:37–57. doi: 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grivennikov S.I., Karin M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi: 10.1016/j.cytogfr.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Negrini M., Nicoloso M.S., Calin G.A. MicroRNAs and cancer--new paradigms in molecular oncology. Curr. Opin. Cell Biol. 2009;21:470–479. doi: 10.1016/j.ceb.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Yang X., Liang L., Zhang X.F., Jia H.L., Qin Y., Zhu X.C., Gao X.M., Qiao P., Zheng Y., Sheng Y.Y. MicroRNA-26a suppresses tumor growth and metastasis of human hepatocellular carcinoma by targeting interleukin-6-Stat3 pathway. Hepatology. 2013;58:158–170. doi: 10.1002/hep.26305. [DOI] [PubMed] [Google Scholar]
- 12.Kota J., Chivukula R.R., O’Donnell K.A., Wentzel E.A., Montgomery C.L., Hwang H.W., Chang T.C., Vivekanandan P., Torbenson M., Clark K.R. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. doi: 10.1016/j.cell.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X., Lwin T., Zhang X., Huang A., Wang J., Marquez V.E., Chen-Kiang S., Dalton W.S., Sotomayor E., Tao J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia. 2013;27:2341–2350. doi: 10.1038/leu.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang B., Liu X.X., He J.R., Zhou C.X., Guo M., He M., Li M.F., Chen G.Q., Zhao Q. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis. 2011;32:2–9. doi: 10.1093/carcin/bgq209. [DOI] [PubMed] [Google Scholar]
- 15.Li X., Liu L., Shen Y., Wang T., Chen L., Xu D., Wen F. MicroRNA-26a modulates transforming growth factor beta-1-induced proliferation in human fetal lung fibroblasts. Biochem. Biophys. Res. Commun. 2014;454:512–517. doi: 10.1016/j.bbrc.2014.10.106. [DOI] [PubMed] [Google Scholar]
- 16.Yu L., Lu J., Zhang B., Liu X., Wang L., Li S.Y., Peng X.H., Xu X., Tian W.D., Li X.P. miR-26a inhibits invasion and metastasis of nasopharyngeal cancer by targeting EZH2. Oncol. Lett. 2013;5:1223–1228. doi: 10.3892/ol.2013.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huse J.T., Brennan C., Hambardzumyan D., Wee B., Pena J., Rouhanifard S.H., Sohn-Lee C., le Sage C., Agami R., Tuschl T., Holland E.C. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23:1327–1337. doi: 10.1101/gad.1777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang J., Han C., Wu T. MicroRNA-26a promotes cholangiocarcinoma growth by activating β-catenin. Gastroenterology. 2012;143:246. doi: 10.1053/j.gastro.2012.03.045. 56.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witwer K.W., Sisk J.M., Gama L., Clements J.E. MicroRNA regulation of IFN-beta protein expression: rapid and sensitive modulation of the innate immune response. J. Immunol. 2010;184:2369–2376. doi: 10.4049/jimmunol.0902712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang C., Zhu W., Xu J., Wang B., Hou W., Zhang R., Zhong N., Ning Q., Han Y., Yu H. MicroRNA-26a negatively regulates toll-like receptor 3 expression of rat macrophages and ameliorates pristane induced arthritis in rats. Arthritis Res. Ther. 2014;16:R9. doi: 10.1186/ar4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji J., Shi J., Budhu A., Yu Z., Forgues M., Roessler S., Ambs S., Chen Y., Meltzer P.S., Croce C.M. MicroRNA expression, survival, and response to interferon in liver cancer. N. Engl. J. Med. 2009;361:1437–1447. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fasseu M., Tréton X., Guichard C., Pedruzzi E., Cazals-Hatem D., Richard C., Aparicio T., Daniel F., Soulé J.C., Moreau R. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PLoS ONE. 2010;5:e13160. doi: 10.1371/journal.pone.0013160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu X., Jin L., Wang X., Luo A., Hu J., Zheng X., Tsark W.M., Riggs A.D., Ku H.T., Huang W. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc. Natl. Acad. Sci. USA. 2013;110:17892–17897. doi: 10.1073/pnas.1317397110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sahu S.K., Kumar M., Chakraborty S., Banerjee S.K., Kumar R., Gupta P., Jana K., Gupta U.D., Ghosh Z., Kundu M., Basu J. MicroRNA 26a (miR-26a)/KLF4 and CREB-C/EBPβ regulate innate immune signaling, the polarization of macrophages and the trafficking of Mycobacterium tuberculosis to lysosomes during infection. PLoS Pathog. 2017;13:e1006410. doi: 10.1371/journal.ppat.1006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chai Z.T., Zhu X.D., Ao J.Y., Wang W.Q., Gao D.M., Kong J., Zhang N., Zhang Y.Y., Ye B.G., Ma D.N. microRNA-26a suppresses recruitment of macrophages by down-regulating macrophage colony-stimulating factor expression through the PI3K/Akt pathway in hepatocellular carcinoma. J. Hematol. Oncol. 2015;8:56. doi: 10.1186/s13045-015-0150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni B., Rajaram M.V., Lafuse W.P., Landes M.B., Schlesinger L.S. Mycobacterium tuberculosis decreases human macrophage IFN-γ responsiveness through miR-132 and miR-26a. J. Immunol. 2014;193:4537–4547. doi: 10.4049/jimmunol.1400124. [DOI] [PubMed] [Google Scholar]
- 27.Schabbauer G., Matt U., Günzl P., Warszawska J., Furtner T., Hainzl E., Elbau I., Mesteri I., Doninger B., Binder B.R., Knapp S. Myeloid PTEN promotes inflammation but impairs bactericidal activities during murine pneumococcal pneumonia. J. Immunol. 2010;185:468–476. doi: 10.4049/jimmunol.0902221. [DOI] [PubMed] [Google Scholar]
- 28.Sahin E., Haubenwallner S., Kuttke M., Kollmann I., Halfmann A., Dohnal A.M., Chen L., Cheng P., Hoesel B., Einwallner E. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J. Immunol. 2014;193:1717–1727. doi: 10.4049/jimmunol.1302167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H., Kumar A., Lamont R.J., Scott D.A. GSK3β and the control of infectious bacterial diseases. Trends Microbiol. 2014;22:208–217. doi: 10.1016/j.tim.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatematsu M., Seya T., Matsumoto M. Beyond dsRNA: Toll-like receptor 3 signalling in RNA-induced immune responses. Biochem. J. 2014;458:195–201. doi: 10.1042/BJ20131492. [DOI] [PubMed] [Google Scholar]
- 31.Jialal I., Machha A., Devaraj S. Small interfering-RNA to protein kinase C-delta reduces the proinflammatory effects of human C-reactive protein in biobreeding diabetic rats. Horm. Metab. Res. 2013;45:326–328. doi: 10.1055/s-0032-1327643. [DOI] [PubMed] [Google Scholar]
- 32.Loegering D.J., Lennartz M.R. Protein kinase C and toll-like receptor signaling. Enzyme Res. 2011;2011:537821. doi: 10.4061/2011/537821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han W., Fu X., Xie J., Meng Z., Gu Y., Wang X., Li L., Pan H., Huang W. MiR-26a enhances autophagy to protect against ethanol-induced acute liver injury. J. Mol. Med. (Berl.) 2015;93:1045–1055. doi: 10.1007/s00109-015-1282-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu H., Tian Y., Tang D., Zou S., Liu G., Song J., Zhang G., Du X., Huang W., He B. An Endoplasmic Reticulum Stress-MicroRNA-26a Feedback Circuit in Nonalcoholic Fatty Liver Disease. Hepatology. 2020 doi: 10.1002/hep.31428. Published online June 22, 2020. [DOI] [PubMed] [Google Scholar]
- 35.Fu X., Dong B., Tian Y., Lefebvre P., Meng Z., Wang X., Pattou F., Han W., Wang X., Lou F. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J. Clin. Invest. 2015;125:2497–2509. doi: 10.1172/JCI75438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu X., Meng Z., Liang W., Tian Y., Wang X., Han W., Lou G., Wang X., Lou F., Yen Y. miR-26a enhances miRNA biogenesis by targeting Lin28B and Zcchc11 to suppress tumor growth and metastasis. Oncogene. 2014;33:4296–4306. doi: 10.1038/onc.2013.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sander S., Bullinger L., Klapproth K., Fiedler K., Kestler H.A., Barth T.F., Möller P., Stilgenbauer S., Pollack J.R., Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–4212. doi: 10.1182/blood-2008-03-147645. [DOI] [PubMed] [Google Scholar]
- 38.Yang X., Zhang X.F., Lu X., Jia H.L., Liang L., Dong Q.Z., Ye Q.H., Qin L.X. MicroRNA-26a suppresses angiogenesis in human hepatocellular carcinoma by targeting hepatocyte growth factor-cMet pathway. Hepatology. 2014;59:1874–1885. doi: 10.1002/hep.26941. [DOI] [PubMed] [Google Scholar]
- 39.Zeitels L.R., Acharya A., Shi G., Chivukula D., Chivukula R.R., Anandam J.L., Abdelnaby A.A., Balch G.C., Mansour J.C., Yopp A.C. Tumor suppression by miR-26 overrides potential oncogenic activity in intestinal tumorigenesis. Genes Dev. 2014;28:2585–2590. doi: 10.1101/gad.250951.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen C.Y., Chang J.T., Ho Y.F., Shyu A.B. MiR-26 down-regulates TNF-α/NF-κB signalling and IL-6 expression by silencing HMGA1 and MALT1. Nucleic Acids Res. 2016;44:3772–3787. doi: 10.1093/nar/gkw205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du J., Gao R., Wang Y., Nguyen T., Yang F., Shi Y., Liu T., Liao W., Li R., Zhang F. MicroRNA-26a/b have protective roles in oral lichen planus. Cell Death Dis. 2020;11:15. doi: 10.1038/s41419-019-2207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang S.H., Silva F.J., Tsark W.M., Mann J.R. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32:199–202. doi: 10.1002/gene.10030. [DOI] [PubMed] [Google Scholar]
- 43.Clausen B.E., Burkhardt C., Reith W., Renkawitz R., Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 44.Williams K.L., Fuller C.R., Dieleman L.A., DaCosta C.M., Haldeman K.M., Sartor R.B., Lund P.K. Enhanced survival and mucosal repair after dextran sodium sulfate-induced colitis in transgenic mice that overexpress growth hormone. Gastroenterology. 2001;120:925–937. doi: 10.1053/gast.2001.22470. [DOI] [PubMed] [Google Scholar]
- 45.Zaki M.H., Boyd K.L., Vogel P., Kastan M.B., Lamkanfi M., Kanneganti T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.