

Abstract
Calcium (Ca2+) signals play fundamental roles in immune cell function. The main sources of Ca2+ influx in mammalian lymphocytes following antigen receptor stimulation are Ca2+ release-activated Ca2+ (CRAC) channels. They are formed by ORAI proteins in the plasma membrane and are activated by stromal interaction molecules (STIM) located in the ER. Loss-of-function mutations in ORAI1 and STIM1 that abolish Ca2+ influx cause a unique disease syndrome called CRAC channelopathy in humans, characterized by immunodeficiency, autoimmunity and non-immunological symptoms. Studies in mice lacking Stim and Orai genes have illuminated many cellular and molecular mechanisms by which these molecules control lymphocyte function. CRAC channels are required for the differentiation and function of several T lymphocyte subsets that provide immunity to infection, mediate inflammation and prevent autoimmunity. This review examines new insights into how CRAC channels control T cell-mediated immunity.
Calcium signals control of T cell development and immunity
Dynamic changes in intracellular Ca2+ concentrations ([Ca2+]i) are crucial for the differentiation and effector functions of T cells by controlling the activation of signal transduction molecules and transcription factors [1]. Changes in [Ca2+]i occur upon T cell receptor (TCR) engagement that results in the release of Ca2+ from the ER and the opening of Ca2+ channels in the plasma membrane of mammalian cells. Ca2+ homeostasis in T cells is maintained through a network of Ca2+ channels and pumps as well as Na+ and K+ channels that indirectly control [Ca2+]i; for instance, by controlling the membrane potential [1, 2]. These channels have been reviewed in detail elsewhere [1, 2]. Besides the Ca2+-release activated Ca2+ (CRAC) channel described here [3], other channels have been reported to mediate Ca2+ signals in T cells. These include the transient receptor potential (TRP) channels TRPM2 [4, 5], TRPV1 [6] and TRPM7 [7, 8]. Deletion of TRPM7 in murine all T cells severely impairs T cell development and function [9]. Several purinergic receptors including P2X4 and P2X7 mediate Ca2+ influx upon binding extracellular ATP [10–15]. Moreover, P2X4 can mediate human CD4+ T cell activation at the immunological synapse and regulate T cell migration as shown by high-resolution live cell imaging in vitro [10, 11]. P2X7 inhibits regulatory T cell (Treg) differentiation in favor of proinflammatory T helper (Th) 17 cells [12, 13] and promotes the establishment and maintenance of (tissue-resident) memory CD8+ T cells in mice [14, 15]. Moreover, several studies have implicated voltage-gated Ca2+ channels (VGCC) in T cell function [16, 17] but their role and molecular function remain debated. In this review, we focus on recent work investigating the role of CRAC channels and store-operated Ca2+ entry (SOCE) in T cell-mediated immunity in mice and humans. These data shed light on the complex clinical phenotype caused by loss-of-function mutations in ORAI1 and STIM1 in humans, termed CRAC channelopathy, that is characterized by immunodeficiency, autoimmunity and non-immunological symptoms. After a brief overview of the molecular regulation of CRAC channels by ORAI and STIM proteins, we discuss new data regarding the role of CRAC channels in immunity to infection with a focus on CD8+ T cells and T follicular helper (Tfh) cells. We next discuss how SOCE can regulate immune tolerance and autoimmunity by controlling the function of Treg and Th17 cells. We also discuss the molecular mechanisms by which CRAC channels can control the function of various T cell subsets, and explore the quantitative SOCE requirements of T cell subsets. These findings are relevant as they may bear translational implications when considering CRAC channel inhibition as a putative therapeutic approach to treating certain autoimmune diseases and inflammation.
CRAC channel signaling and function in T cells
The CRAC channel is formed by ORAI1, a tetraspanning plasma membrane protein, and its two homologs ORAI2 and ORAI3 (Figure 1), (named after the three horae (hours) Dyke, Eirine and Eunomia in Greek mythology) [18]. All three homologs form functional CRAC channels when ectopically expressed by themselves, albeit with slightly different biophysical properties [19]. ORAI channels are activated by stromal interaction molecule 1 (STIM1) and STIM2 located in the membrane of the ER [3, 20]. ORAI and STIM proteins interact with each other in ER-plasma membrane junctions where both membranes come into close contact [3, 20]. CRAC channels are activated following TCR stimulation and the generation of the second messenger inositol-1,4,5-trisphosphate (IP3), which binds to IP3 receptors (IP3R) in the ER membrane [3]. IP3Rs are Ca2+ permeable ion channels whose IP3-mediated opening results in Ca2+ efflux from the ER because of the ~ 10,000 fold concentration gradient between Ca2+ in the ER and cytoplasm [2]. Subsequently, IP3R opening causes a transient increase in [Ca2+]i and a simultaneous decrease in ER Ca2+ concentrations; The latter is the trigger for conformational changes of STIM1 and its homolog STIM2 that allow both proteins to bind to and open ORAI channels in the plasma membrane [3]. The resulting Ca2+ influx is called store-operated Ca2+ entry (SOCE) because its activation is regulated by Ca2+ concentrations in ER stores. The molecular regulation of CRAC channels and SOCE has been reviewed in detail elsewhere [3].
Figure 1. Store-operated Ca2+ entry (SOCE) and Ca2+ dependent transcriptional regulation.
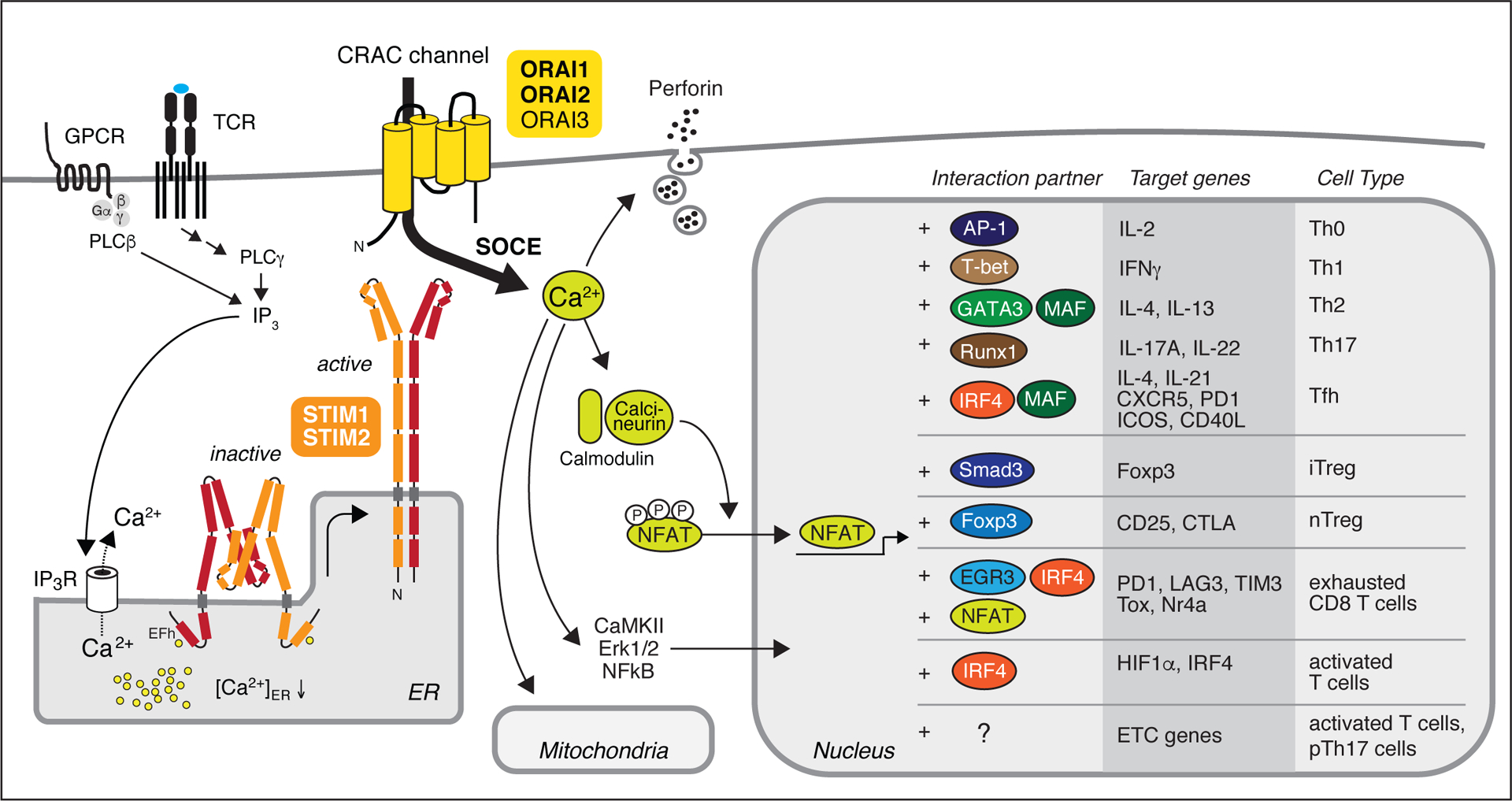
T cell receptor (TCR) and G protein-coupled receptor (GPCR) stimulation of T cells leads to the production of inositol-1,4,5-trisphosphate (IP3) that binds to IP3 receptors (IP3Rs) located in the endoplasmic reticulum (ER) membrane. Opening of IP3R channels results in a transient increase in cytosolic Ca2+ and a decrease in the ER Ca2+ concentration, which activates stromal interaction molecules 1 (STIM1) and STIM2. Both proteins form homodimers that undergo conformational changes and translocate to ER-plasma membrane (PM) junctions, where they bind to ORAI proteins. ORAI1 and its homologues ORAI2 and ORAI3 form hexameric complexes in the PM that constitute the Ca2+ release-activated Ca2+ (CRAC) channel. Ca2+ influx through CRAC channels is called store-operated Ca2+ entry (SOCE). It triggers various processes in T cells including the secretion of cytolytic granules and the activation of Ca2+-dependent enzymes including calcineurin, Erk1/2 and CaMKII, and transcription factors such as NF-kB and NFAT. Dephosphorylation of NFAT results in its translocation to the nucleus where it cooperates with other transcription factors to promote activation, differentiation and effector functions of various T helper (Th) and T regulatory (Treg) cell subsets. NFAT interaction partners and their specific functions in T cells have been reviewed in detail elsewhere [45, 46]. Note that NFAT binding to ETC genes in T cells has not been reported, indicated by “?”. pTh17, pathogenic Th17 cells.
ORAI1 is the most abundantly expressed ORAI homolog in all human T cell subsets and LOF mutations in ORAI1 abolish CRAC currents and SOCE [21–24]. In murine T cells, the three Orai genes are more evenly expressed (Figure 2A); Deletion of Orai1 only partially reduces SOCE, and full suppression of SOCE only occurs in the absence of both Orai1 and Orai2 [25–29] (Figure 2B). Somewhat paradoxically, deletion of Orai2 enhances SOCE in murine T cells (a phenomenon that was also observed in macrophages, astrocytes and mast cells) [27, 30]. The crystal structure of Drosophila sp. Orai shows that the CRAC channel is formed by hexamers of tetraspanning Orai subunits [31]. Given the highly conserved membrane domains and membrane topology of ORAI1, 2 and 3, it is likely that ORAI homologs assemble into heteromeric CRAC channels, although this remains to be tested. Accordingly, ORAI1 and ORAI2 can form heteromeric channels in which ORAI2 attenuates the CRAC channel function of ORAI1, which might explain why Orai2-deficient murine CD4+ and CD8+ T cells have stronger SOCE compared to WT control cells [27, 32]. The stoichiometry of ORAI homologs in native CRAC channel complexes is not known, but may vary between different T cell subsets and other immune cells based on differences in mRNA expression. The formation of heteromeric complexes is a common feature of ion channels such as TRP, GABAA and glutamate receptors. In the case of CRAC channels, it is reasonable to speculate that it might provide a means to fine-tune channel function by adjusting the ORAI subunit composition. Human and mouse T cells express both STIM homologs, but STIM1 mRNA and protein expression amounts are higher than those for STIM2 (Figure 2A) [33]. LOF mutations in STIM1 almost completely abolish SOCE in human T cells, whereas deletion of Stim1 in mouse T cells results in strong but incomplete suppression of SOCE (Figure 2B) [33–37]. Deletion of Stim2 in murine CD4+ T cells only partially reduces CRAC currents and SOCE but prevents sustained Ca2+ influx and activation of the transcription factor nuclear factor of activated T cells (NFAT), which is required for cytokine expression [33, 37]. Nevertheless, STIM2 is essential for murine T cell function because combined deletion of Stim1 and Stim2 genes completely abolishes SOCE, with profound effects on the differentiation and function of several T cell subsets, including Tfh and Treg cells [33, 38–43].
Figure 2. ORAI and STIM expression and SOCE-dependent control of T cell function.
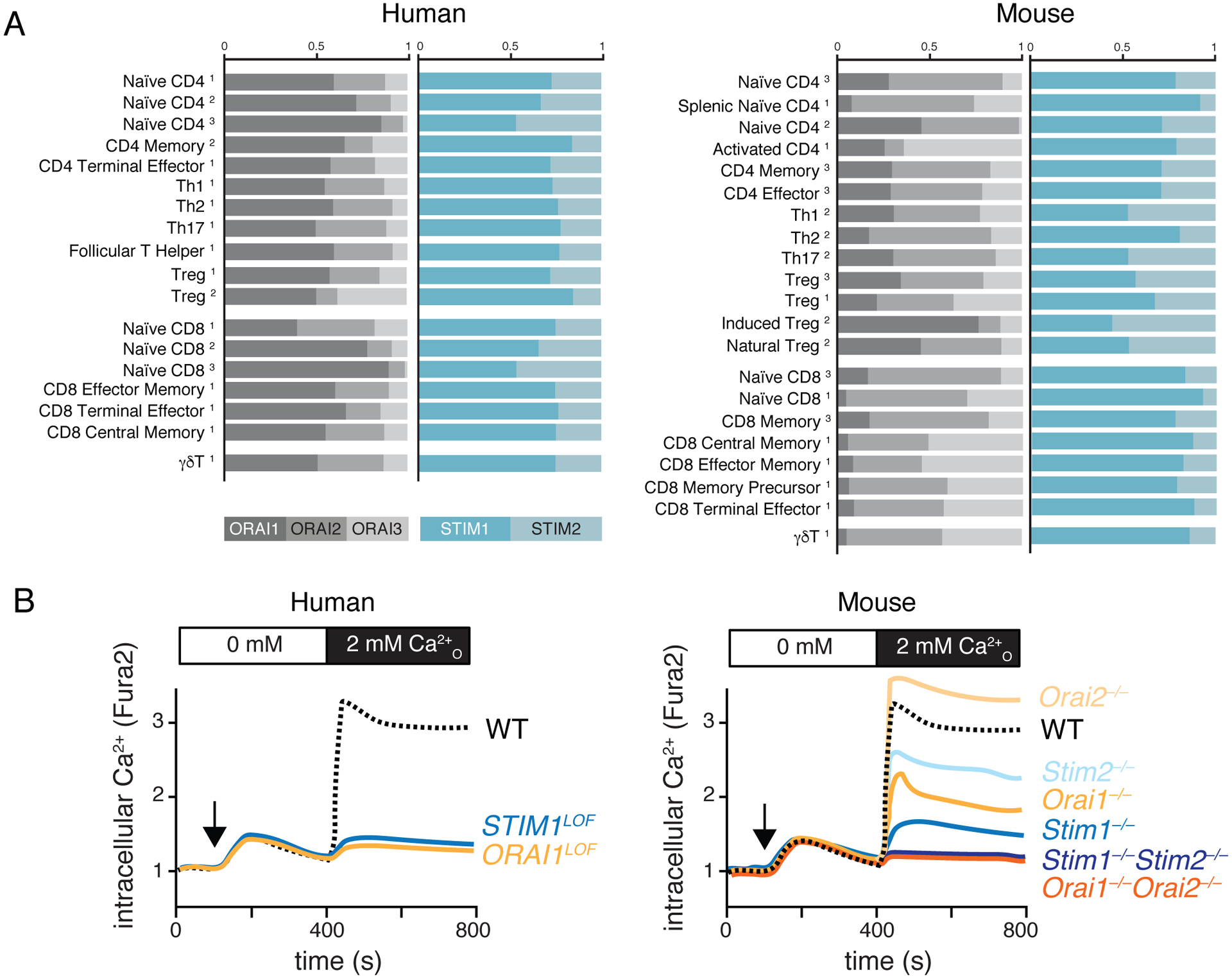
(A) Expression of ORAI and STIM homologs in human and mouse T cells. mRNA expression levels in human T cells from GEO (1, GSE107011), FANTOM5 (2) and Haemopedia (3) databases (left) and mouse T cells from Immgen (1), ArrayExpress (2, E-MTAB-2582) and Haemopedia (3) databases (right). ORAI1 is the most highly expressed ORAI homolog in human but not mouse T cells. (B) Relative contribution of ORAI and STIM proteins to SOCE in human and mouse T cells. Loss-of-function (LOF) mutations of ORAI1 and STIM1 genes all but abolish SOCE in human T cells (left). Combined genetic deletion of Orai1 and Orai2 genes or Stim1 and Stim2 genes in mouse T cells also abolishes SOCE, whereas deletion of individual genes results in partial SOCE defects (or a SOCE increase in the case of Orai2-deficient T cells).
Ca2+ signals through CRAC channels regulate the function of many proteins including the serine/threonine-phosphatase calcineurin, and its main target in T cells, NFAT (Figure 1) [44, 45]. The NFAT family consists of four members of which three, NFATc1 (NFAT2), NFATc2 (NFAT1) and NFATc3 (NFAT4), are expressed in murine and human T cells [46]. Noteworthy, although all three NFAT homologs are regulated by SOCE, the spatiotemporal dynamics of their Ca2+-dependent activation differ [47, 48]. In addition to NFAT, SOCE and calcineurin modulate NF-κB signaling by dephosphorylating Bcl-10 which leads to the assembly of the CARMA1/Bcl-10/MALT1 complex upstream of NF-κB activation in primary human CD4+ T cells and cell lines [49, 50]. Ca2+-bound calmodulin also regulates CaM kinase II and IV function, resulting in phosphorylation of the transcription factor CREB, which regulates the transcription of IL-2 and other genes in murine T cells [51].
Immunity to infection
Immunodeficiency in ORAI1 and STIM1-deficient patients
The importance of SOCE for immune function is evident from patients with LOF mutations in STIM1 and ORAI1 that abolish CRAC channel function (Box 1) [52]. Monogenic diseases associated with mutations in STIM2, ORAI2 or ORAI3 have not been reported yet. Patients lacking ORAI1 or STIM1 function develop combined immunodeficiency (CID) typically in their first year of life, which manifests with recurring severe, often life-threatening infections with DNA viruses (EBV, VZV, CMV, HSV, HHV8 and Adenovirus) and RNA viruses (Influenza virus, Parainfluenza virus, RSV, Rotavirus, and Norovirus) [22–24, 34, 53–57]. Patients are also prone to infections by gram-positive and gram-negative bacteria [22–24, 34, 53–57]. Less frequently, patients develop pulmonary, intestinal, cutaneous or disseminated fungal infections caused by Pneumocystis jirovecii, Candida albicans, or Aspergillus fumigatus (S.K., M.V., S.F., accepted manuscript: DOI: 10.15252/emmm.201911592). The CID associated with impaired SOCE, i.e. CRAC channelopathy, is not primarily due to defective development of immune cells, unlike several other ion channelopathies [58]. Patients with LOF mutations in STIM1 and ORAI1 have largely normal frequencies of lymphoid and myeloid immune cells [58]. Likewise, Stim1fl/fl Stim2fl/fl Cd4Cre and Orai1fl/fl Orai2–/– Cd4Cre mice with combined deletion of Stim1/Stim2 or Orai1/Orai2, respectively, which abolishes SOCE in T cells, exhibit normal populations of conventional αβ T cells [26, 27, 59–61], indicating that CRAC channels are not essential for T cell development. This is unexpected because CRAC channels are the dominant source of Ca2+ influx in mature T cells and for several decades, Ca2+ signals have been reported in thymocytes and postulated to be required for T cell development and selection [62–65]. It is unclear which other Ca2+ channels might be involved in Ca2+ signaling in immature T cells. One candidate may be the IP3 receptor (IP3R), which mediates the release of Ca2+ from ER stores upon TCR stimulation [66]. Mice with conditional deletion of all three Itpr genes in immature T cells have reduced numbers of CD4+ CD8+ (double positive, DP) thymocytes [66], suggesting that IP3R-mediated Ca2+ signaling in response to pre-TCR ligation can regulate the DN to DP transition and the development of αβ T cells. Other Ca2+ channels such as voltage-gated Ca2+ channels may also contribute to T cell development, but the underlying physiological mechanisms by which these channels function in T cells remain mysterious [16, 67]. The only T cell populations whose development is affected by the loss of SOCE are agonist-selected T cells, which escape negative selection in the thymus and are converted into Foxp3+ Treg cells and other unconventional T cell types, such as invariant NK T (iNKT) cells and CD8αα+ intraepithelial lymphocytes (IELs). The numbers of Treg cells, iNKT cells and IELs have been found to be reduced in Stim1fl/fl Stim2fl/fl VaviCre and Stim1fl/fl Stim2fl/fl LckCre mice relative to littermate control animals [59]. Similarly, iNKT and Treg cells are also reduced in some patients with LOF mutations in ORAI1 compared with healthy controls [23].
Box 1: Clinician’s Corner.
CRAC channelopathy is caused by autosomal recessive LOF mutations in ORAI1 (OMIM 612782) or STIM1 (OMIM 612783) genes [52], abolishing Ca2+ influx (SOCE). CRAC channelopathy is clinically defined by (1) combined immunodeficiency (CID) with severe bacterial, viral and fungal infections typically starting in the first year of life; (2) autoimmune hemolytic anemia (AIHA) and thrombocytopenia; (3) muscular hypotonia with atrophy of type 2 muscle fibers; (4) anhidrotic ectodermal dysplasia (EDA) characterized by impaired dental enamel calcification (amelogenesis imperfecta type 3) and eccrine sweat gland function [52, 146]. CID is severe and often requires hematopoietic stem cell transplantation. Noteworthy, autosomal dominant gain-of-function (GOF) mutations in ORAI1 and STIM1, cause tubular aggregate myopathy (TAM) and Stormorken syndrome, but seem not to affect immune function.
CID in ORAI1 and STIM1 mutant patients occurs despite largely normal numbers of immune cells in the blood with the exception of reduced Treg and NKT cell numbers in some patients [23]. Total serum Ig titers are also normal or elevated, but specific antibodies to vaccine antigens or infectious pathogens are typically reduced or absent compared to healthy children. In the absence of SOCE, the proliferation, cytolytic activity and cytokine production of T cells is severely compromised [27, 38, 39, 41, 147]. Many of these findings are recapitulated in knockout mice lacking CRAC channel genes.
The simultaneous presence of CID and autoimmunity is not unique to CRAC channelopathy but is a feature of many primary immunodeficiencies due to mutations that affect the function of both effector T cells and Treg cells, such as AIRE, STAT3 and many others [148, 149]. Mutations in ORAI1 and STIM1 result also in an unbalanced immune response with concurrent defects in effector T cells required for immunity to infection and Treg cells that maintain immunological tolerance. In mice, SOCE was shown to control the development of Treg cells in the thymus (tTreg) and the differentiation of follicular Treg (Tfr) cells, tissue resident Treg cells, and induced Treg (iTreg) cells.
SOCE is essential for the function of effector Th1, Th2 and Th17 cells that mediate inflammation in autoimmune and allergic diseases including multiple sclerosis (MS), Crohn’s disease, rheumatoid arthritis, psoriasis and allergic asthma [2, 92, 147]. Genetic or pharmacological inhibition of CRAC channel attenuates the function of pathogenic Th1 and Th17 cells and ameliorates inflammation and disease in murine models of MS, colitis and graft-versus-host disease [27, 43, 92, 109, 120]. From an immunological perspective, the usefulness of CRAC channel blockers for the treatment of autoimmunity and inflammation depends on whether it is possible to inhibit proinflammatory T cells without interfering with the function of effector T cells, Treg cells and other immune cells that provide immunity to infection and immune homeostasis.
The CID resulting from CRAC channelopathy in patients is best characterized in terms of impaired lymphocyte function. T, B and NK cells of patients with ORAI1 and STIM1 LOF mutations have severely impaired SOCE, which affects many of their functions, including their activation, proliferation and cytokine production [22–24, 34, 36]. By contrast, the impact of human ORAI1 and STIM1 mutations on cells of the innate immune system and the contribution of potential defects to CID in CRAC channelopathy is less well defined. Neutrophils of individual patients with LOF mutations in ORAI1 and STIM1 exhibit almost normal SOCE and functions [55]. The function of other myeloid immune cells in these patients has not been carefully studied. This does not, however, rule out a contribution of defective SOCE in innate immune cells to CID in these patients. Indeed, using mice and immune cell lines, SOCE has been reported to regulate a number of myeloid cell functions. For instance, neutrophils isolated from Stim1fl/fl Stim2fl/fl VaviCre mice, showed impaired the production of cytokines and reactive oxygen species, as well as phagocytosis [68–70]. In mast cells, ORAI1 and CRAC channel function were found to be essential for degranulation and cytokine secretion in vitro and the elicitation of passive cutaneous anaphylaxis in vivo as shown using Orai1–/– mice [28]. One study recently showed that the serine protease allergen Der p3 (present in house dust mite (HDM) extract and a potent allergen), activated CRAC channels by binding to the protease-activated receptor (PAR) 4; Inhibition of Der p3 activity or ORAI1 channels suppressed mast cell activation as shown by biochemical and functional cellular assays in vitro [71]. In other myeloid cell lineages, the role of SOCE is less obvious; for instance, combined deletion of Stim1 and Stim2 in murine bone marrow derived macrophages (BMDM) and dendritic cells (BMDC) generated from BM of Stim1fl/fl Stim2fl/fl VaviCre or Stim1fl/fl Stim2fl/fl MxCre only had moderate or no effects on cell function despite abolished SOCE [72, 73]. One of these studies identified a defect in antigen cross-presentation through MHC class I and migration of BMDCs generated from Stim1fl/fl LysMCre in co-culture assays in vitro and in vivo using adoptive transfer models [73]. Further research using mouse models of innate immune function and primary human myeloid immune cells is necessary to understand the contribution of SOCE to innate immunity to infection. This topic is discussed in more detail in these expert reviews [74, 75].
SOCE-dependent molecular mechanisms of T cell function
The mechanisms by which SOCE regulates T cell function are diverse and have been described in detail elsewhere [1, 76]. Among the better known functions of SOCE are the regulation of T cell proliferation [41], apoptosis [29, 77], secretion of cytotoxic granules by CD8+ T cells [39, 78], production of cytokines and chemokines [79, 80] and migration [81, 82]. TCR induced Ca2+ signals have long been noted to regulate the motility of T cells and their interaction with antigen presenting cells (APC) by reportedly acting as a stop signal [63, 83, 84]. A recent study elegantly showed that it is indeed Ca2+ influx through ORAI1 channels that regulates murine CD4+ T cell motility by monitoring cytosolic [Ca2+]i along with cell motility using intravital microscopy techniques combined with a genetically encoded Ca2+ sensor [85]. A potential mechanism underlying the Ca2+-dependent motility of human T cells is the turnover of LFA-1 adhesions by the Ca2+ regulated enzyme Calpain 2 [86]. The role of Ca2+ as a T cell stop signal is not universal and may depend on the state of T cell activation. A recent study found that murine CD8+ T cells undergo a phase of diminished SOCE ~ 48h after TCR stimulation that correlates with a defect in establishing stable contacts with APCs as demonstrated by intravital 2-photon microscopy [87]. This phenomenon was interpreted as a mechanism to prevent the reengagement of activated T cells by new APCs presenting different antigens. By contrast, in another study, no correlation between Ca2+ signals and motility arrest was observed in Tfh cells [88]. Intravital imaging revealed that, although high affinity interactions of Tfh cells with germinal center (GC) B cells elicited Ca2+ signals in Tfh cells, they remained motile and engaged in short-lived contacts with GC B cells, suggesting that Ca2+ does not act as a stop signal in Tfh cells [88]. Another aspect of T cell motility potentially regulated by SOCE is chemotaxis. Murine CD4+ T cells from Stim1fl/fl Stim2fl/fl Cd4Cre mice exhibit reduced migration to CCL19 and CCL20 gradients in vitro relative to WT controls [43], consistent with a similar defect of human CD4+ T cells lacking SOCE when migrating towards CCL21 [81]. These findings suggest that chemokine-induced SOCE may also be required for T cell homing to secondary lymphoid organs in vivo. A study using in vitro activated human CD4+ T cells expressing a dominant-negative mutant of ORAI1 that abolishes SOCE indeed found a severe defect in the ability of adoptively transferred T cells to home to murine lymph nodes (LN) [81]. By contrast, no defect in homing to LNs was observed when SOCE-deficient naive CD4+ T cells from Stim1fl/fl Stim2fl/fl Cd4Cre mice were transferred into WT host mice [82]. In the same study, T cell motility on lipid bilayers in vitro and in LNs in vivo in mice was comparable to WT T cells, with only a moderate delay in the onset of CD4+ T cell arrest by APCs in vivo. This delay may have implications for the spatiotemporal coordination of antigen recognition and stable interactions with target cells or APCs in specific tissues in situ, which merits further investigation. The cause of the divergent findings regarding the role of SOCE in T cell homing to LNs are not clear, but may be related to the use of naïve vs. in vitro activated effector T cells in these studies, suggesting that SOCE requirements with regard to migration and homing may depend on the differentiation state of T cells.
Metabolic control of lymphocyte function through CRAC channel signaling
One of the most important roles of SOCE in T cells is controlling the function of Ca2+ dependent transcription factors and gene expression. T cells of CRAC channel deficient patients and mice fail to express a broad spectrum of cytokines including IL-2, IL-4, IL-10, IFNγ, TNFα, IL-17A and GM-CSF, a defect that is not limited to specific T helper (Th) subsets [23, 24, 34, 89]. Many of these genes are transcriptionally regulated by NFAT and other Ca2+-dependent transcription factors [45, 90]. Thus, Defective TNFα and IFNγ production by SOCE-deficient CD4+ Th1 cells and CD8+ T cells is likely to contribute to impaired antiviral and antitumor immunity. Beyond cytokines, SOCE controls gene expression in a variety of pathways that determine the differentiation of CD4+ T cells into Tfh cells and certain Treg cell subsets (Figure 3) as well as T cell metabolism, cell cycle, proliferation and apoptosis [27, 29, 40–42, 59, 91, 92]. Indeed, an important, newly defined role of SOCE is the control of T cell metabolism, growth and proliferation through the transcriptional regulation of several metabolic pathways. Using Stim1fl/fl Stim2fl/fl Cd4Cre mice with T cell-specific deletion of both genes, we recently showed that SOCE and calcineurin regulate the expression of multiple glucose transporters and glycolytic enzymes resulting in impaired glucose uptake, glycolytic metabolism and cell cycle entry in vitro and in adoptive animal transfer models [41]. Similar defects were observed in murine and human CD4+ T cells following calcineurin inhibition [41] and in murine CD4+ and CD8+ T cells isolated from Nfatc1fl/fl Cd4cre mice [93]. In both studies, defects in SOCE, calcineurin and NFAT resulted in impaired expression of glycolytic enzymes and a diminished ability of T cells to undergo the metabolic switch from oxidative phosphorylation (OXPHOS) to glycolysis after TCR stimulation with CD3 and CD28 antibodies, which is essential for T cell growth and proliferation (Figure 4). As a consequence, SOCE-deficient T cells from Stim1fl/fl Stim2fl/fl Cd4Cre mice failed to proliferate in vitro and undergo clonal expansion after infection with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV) in vivo [41]. Moreover, bypassing defective SOCE through the expression of a constitutively active NFATc1 partially restored metabolic function and T cell proliferation in these mice [41]. These findings demonstrate that the SOCE-calcineurin-NFAT pathway can control T cell proliferation by regulating T cell metabolism [41, 93]. The data may also contribute to explain the impaired T cell proliferation in response to TCR or mitogen stimulation in samples from CRAC channel-deficient patients [34, 53, 56]. They may also help explain some of the potent immunosuppressive effects of the calcineurin inhibitors tacrolimus and cyclosporin A, which inhibit cytokine production and proliferation of T cells, because they directly interfere with the metabolic activation of lymphocytes.
Figure 3. SOCE controls the development and function of murine conventional and regulatory T cells.
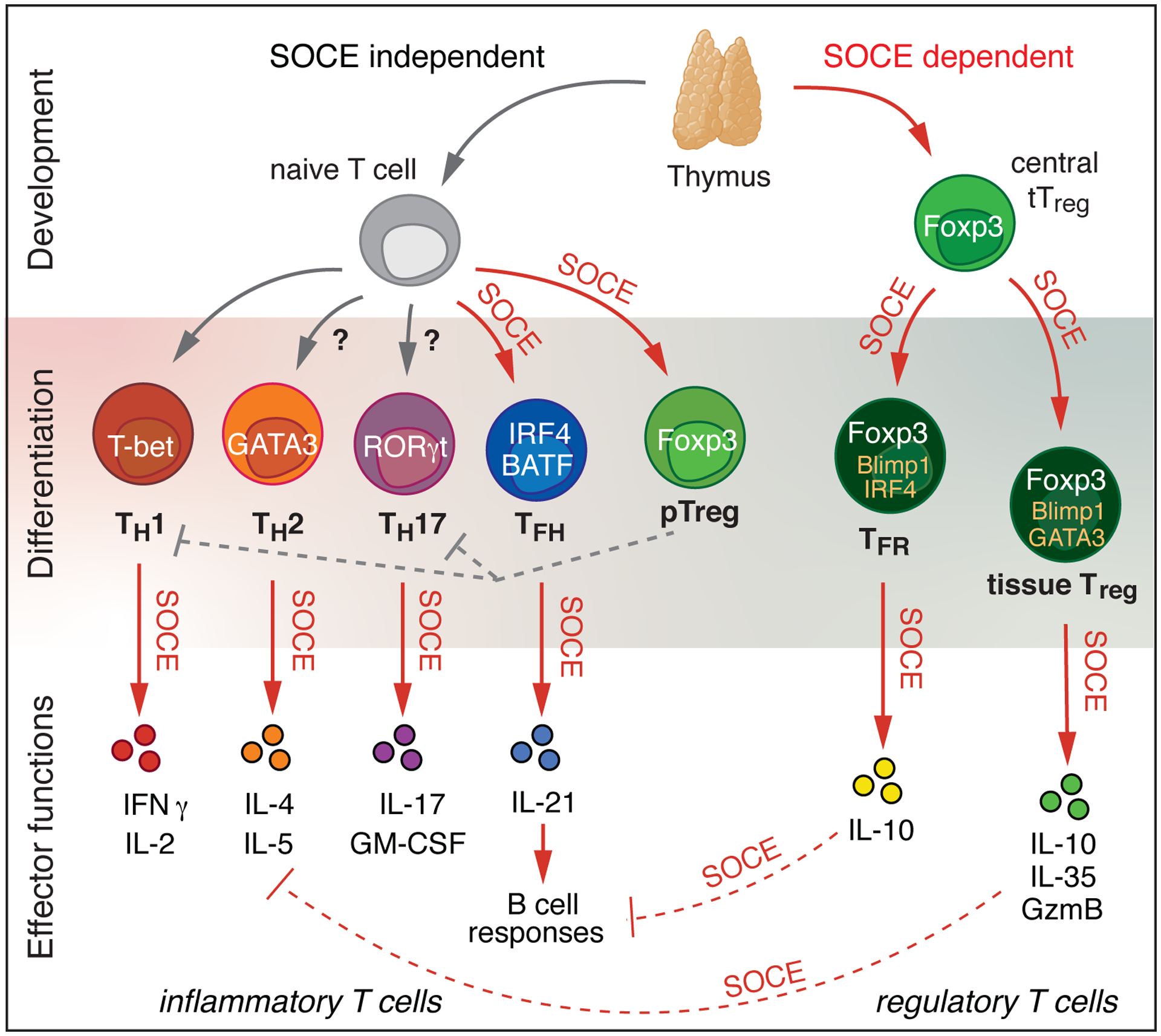
Thymic development of conventional CD4+ and CD8+ αβ T cells is independent of SOCE. By contrast, the development of thymic-derived, agonist-selected regulatory T (Treg) cells (and other unconventional T cell subsets including iNKT cells and CD8αα+ intraepithelial lymphocytes, not shown) requires SOCE. In the periphery, the differentiation of naïve CD4+ T cells into certain T cell subsets like T follicular helper (Tfh) cells and induced Treg (pTreg) cells requires SOCE, whereas the differentiation of Th1 cells appears to be SOCE-independent. The expression of GATA3 by Th2 cells and RORγt by pathogenic Th17 cells is dependent on SOCE, thus influencing the development of these Th subsets [43, 112]. The differentiation of thymus-derived Treg cells (tTreg) into specialized effector Treg subsets, such as T follicular regulatory (Tfr) and tissue-resident Treg cells, which regulate the GC response and maintain tissue-specific organ homeostasis, respectively, is controlled by SOCE. Various effector functions of Th cells including the production of IFNγ, IL-2, IL-4, IL-5, IL-17, GM-CSF and IL-21 cytokines, and the expression of immunosuppressive molecules by Treg cells, such as IL-10, IL-35 and granzyme B, are dependent on SOCE.
Figure 4. SOCE controls the proliferation and clonal expansion of CD4+ and CD8+ T cells.
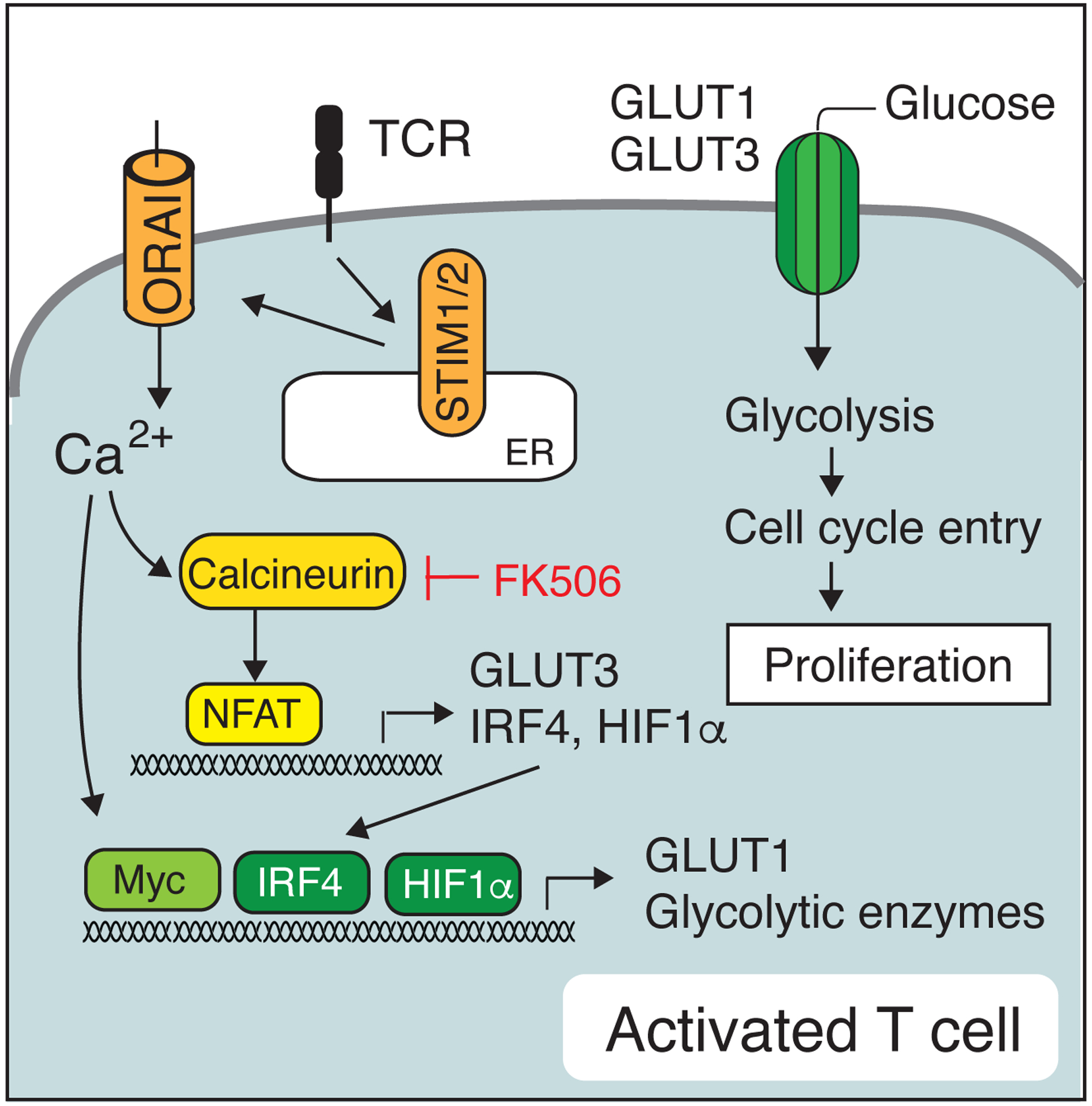
Ca2+ mediated activation of calcineurin and NFAT induces, directly or indirectly, the expression of several transcription factors such as IRF4, Myc and HIF1α, which together with NFAT regulate the expression of glucose transporters and glycolytic enzymes that support the metabolic switch of activated mouse and human T cells, their entry into the cell cycle and proliferation [41].
Besides glycolysis, SOCE regulates OXPHOS, which – although used by all T cells for energy production – is of particular importance for the function of certain T cell subsets such as memory and Treg cells [94]. In addition, a recent study showed that the pathogenic function of Th17 cells required OXPHOS and was regulated by STIM1. Specifically, Stim1fl/fl Cd4cre mice that were crossed to mice with T cell specific expression of a hyperactive form of the transcription factor STAT3 (STAT3C), drives the development of pathogenic Th17 cells, showed reduced expression of electron transport chain (ETC) genes, decreased ATP production and increased concentrations of mitochondrial reactive oxygen species (mROS) in CD4+ T cells relative to control mice that only express STAT3C (Figure 5). As a consequence, Stim1-deficient pathogenic Th17 cells failed to induce inflammation in several tissues including the skin, lung and gut [92]. In non-immune cells, including human and mouse fibroblasts, myocytes and hepatocytes, that lack functional ORAI1 or STIM proteins, defective SOCE resulted in impaired mobilization of fatty acids from lipid droplets, expression of neutral lipases and hence lipolysis, and mitochondrial fatty acid oxidation (FAO), which was at least in part due to the reduced expression of transcriptional regulators of lipid metabolism peroxisome proliferator-activated receptor gamma coactivator α (PGC-1α), and peroxisome proliferator-activated receptor α (PPARα) [91]. Whether SOCE also regulates lipid metabolism in immune cells (in particular T cell subsets that depend on FAO such as memory T cells and Treg cells), remains to be investigated. Collectively, these studies suggest an important role of the SOCE-calcineurin-NFAT pathway in the transcriptional regulation of several metabolic pathways in T cells
Figure 5. SOCE regulates the function of pathogenic Th17 cells.
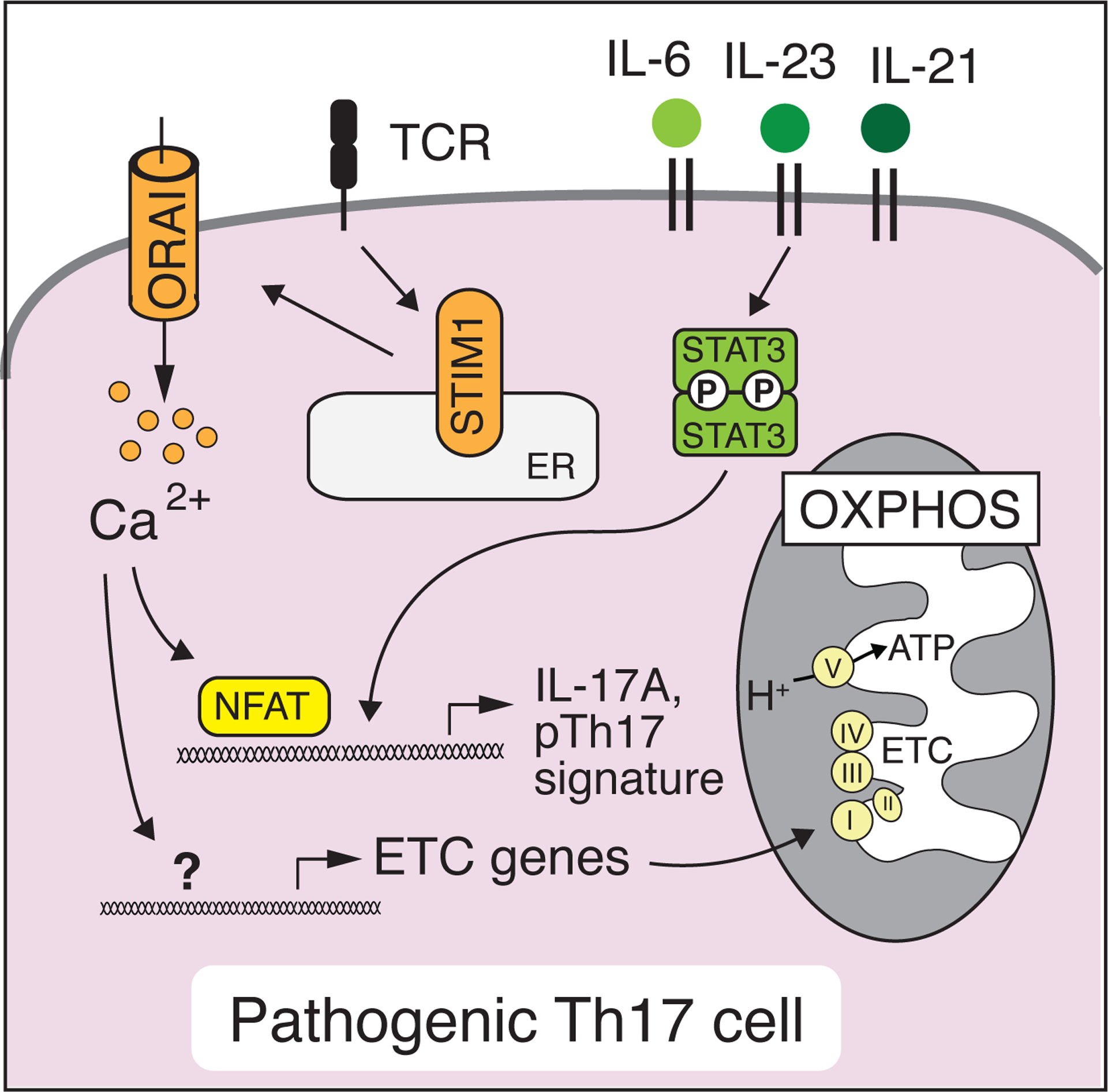
The differentiation of murine Th17 cells depends on cytokine signaling via STAT3 which together with SOCE regulates the expression of Th17 cytokines such as IL-17A, IL-17F, GM-CSF and molecules that are associated with a pathogenic Th17 cell signature [125, 134]. SOCE also controls the expression of nuclear-encoded mitochondrial genes in pathogenic Th17 cells that form the electron transport chain (ETC) and promote oxidative phosphorylation (OXPHOS) [92].
The function of cytotoxic lymphocytes (CTLs) is dependent on SOCE as patients with LOF mutations in ORAI1 or STIM1 can present with impaired NK and CD8+ T cell cytotoxic functions (in at least one reported case) [21, 34, 95, 96]. Others have found that the optimal [Ca2+]i for efficient perforin-dependent cytotoxicity of human CTLs in vitro peaks at 122 – 334 nM, whereas higher [Ca2+]i or lack of SOCE result in suboptimal killing [78]. This is of relevance as Impaired CTL function in SOCE-deficient patients likely contributes to their susceptibility to severe and chronic viral infections and virus-associated tumors such as EBV+ B cell lymphoma and HHV-8+ Kaposi sarcoma [24, 97, 98]. Defects in cytolytic function are also found in CD8+ T cells of SOCE-deficient mice [38, 39]. Deletion of SOCE in T cells of Stim1fl/fl Stim2fl/fl Cd4Cre or Orai1fl/fl Orai2–/– Cd4Cre mice, respectively, results in a failure to control LCMV infection in mice [27, 38, 42] and the growth of tumors [39]. The cytotoxic function of CD8+ T cells depends, at least partially, on the SOCE-mediated expression of death effector molecules such as Fas ligand and on cytotoxic granule release [39]. How SOCE regulates cytotoxic granule secretion is not well understood, but may require the Ca2+ dependent activation of NFAT; indeed, CD8+ T cells from Nfatc1fl/fl Cd4Cre mice exhibit reduced cytotoxic function and impaired recruitment of cytosolic organelles to the immunological synapse when in contact with APCs in vitro [93]. Of note, NK cells from Stim1fl/fl Stim2fl/fl Nkp46Cre mice showed normal degranulation and cytolytic activity in vitro and rejection of MHC class I-deficient RMA-S tumor cells in vivo [99], suggesting that the cytotoxic function of murine NK cells is independent of SOCE. While the above discussed evidence indicates that SOCE is required for the majority of CTL functions in antiviral and antitumor immunity, this role may not be universal and depend on the CTL subset and species.
Mouse models of SOCE in immunity to infection
A key function of CD4+ and CD8+ T cells is their ability to orchestrate immune responses to viruses and other pathogens. SOCE-deficient CD8+ T cells of Stim1fl/fl Stim2fl/fl Cd4Cre mice fail to undergo clonal expansion after LCMV infection and have impaired effector functions, such as cytokine production and cytolytic activity [38, 39] (Figure 6). At later timepoints post-infection, the maintenance of Stim1/Stim2-deficient CD8+ memory T cells and their ability to mount a recall response to reinfection is severely impaired [38]. The latter defect is not intrinsic to CD8+ T cells but due to impaired SOCE in CD4+ T cells. Indeed, In the presence of SOCE-sufficient wildtype CD4+ T cells, memory responses of Stim1/Stim2-deficient CD8+ T cells are restored to normal [38]. SOCE in murine and human CD4+ T cells is required to provide help to CD8+ T cells, for instance by licensing dendritic cells through CD40 ligand expression and the secretion of cytokines such as IL-21 [38, 42]. SOCE in both CD4+ and CD8+ T cells is necessary for sterilizing immunity to LCMV infection because Stim1fl/fl Stim2fl/fl Cd4Cre mice are unable to eliminate the Armstrong strain of LCMV (LMCVARM) resulting in chronic infection with this virus stain that typically only causes an acute infection. Moreover, LCMV-specific CD8+ T cells from Nfatc1fl/fl Nfatc2–/– Cd4cre also fail to control LCMVARM infection in adoptive T cell transfer models [100] suggesting that SOCE-dependent activation of NFAT is required for effective antiviral immunity. This also implies that SOCE-mediated NFAT activation contributes to the increased susceptibility to recurring viral infections seen in patients with ORAI1 and STIM1 LOF mutations. A common feature of CID in these patients is their impaired production of antigen-specific antibodies after infection or vaccination [18, 42]. Seroconversion is impaired despite normal or elevated total serum titers of IgM, IgG and IgA antibodies [23, 24, 36] pointing to a potential role of SOCE in B cells and antibody responses. However, Stim1fl/fl Stim2fl/fl Mb1Cre mice with a conditional deletion of both Stim1 and Stim2 genes in B cells that lack BCR-induced SOCE have normal antibody responses and class switch recombination (CSR) after immunization [101], which is surprising because SOCE-deficient patient almost completely lack antigen-specific antibody responses. By contrast, abolishing SOCE in T cells of Stim1fl/fl Stim2fl/fl Cd4Cre mice strongly impairs the production of high affinity IgG antibodies after immunization or infection with LCMVARM and the clone 13 strain of LCMV (LCMVCL13) that causes chronic infection [42], suggesting that defective antibody responses in human patients lacking SOCE might be due to defective T cell, but not B cell, function. In support of this conclusion, the frequencies of LCMV-specific Tfh cells, which provide cognate help to GC B cells and thus promote their clonal selection, affinity maturation and CSR [102, 103] are strongly reduced in Stim1fl/fl Stim2fl/fl Cd4Cre mice compared to WT littermate controls [42]. As a consequence, GC B cells are strongly reduced in these mice, suggesting that humoral immune responses to viral antigens are dependent on SOCE in T cells [42]. Indeed, SOCE controls the expression of many genes encoding receptors and ligands that initiate Tfh cell differentiation including Icos, Cxcr5, Pdcd1, Btla, Cd40l, Ox40 as well as Il21 and Il4 in murine and human CD4+ T cells [42]. Furthermore, expression of transcription factors IRF4 and BATF that are critical for follicular T cell differentiation are markedly reduced in murine Stim1/Stim2-deficient Tfh cells compared to SOCE-sufficient, WT Tfh cells in mice [42] (Figure 7). At least some of the SOCE effects on gene expression during Tfh cell differentiation are mediated by NFAT. For instance, Expression of the Ca2+-inducible NFATc1/alpha isoform is reduced in Tfh cells isolated from Stim1fl/fl Stim2fl/fl Cd4Cre mice, whereas overexpression of constitutively active NFATc1 partially rescues their Tfh cell differentiation. Consistent with these findings, adoptive transfer of LCMV-specific CD4+ T cells from Nfatc1fl/fl Nfatc2−/−Cd4Cre mice into WT host mice resulted in impaired Tfh cell differentiation and GC reactions following LCMV Armstrong infection [104]. Furthermore, Orai1fl/fl Orai2−/−Cd4Cre mice with combined deletion of Orai1 and Orai2 genes in all T cells and thus, abolished SOCE, also show impaired Tfh cell differentiation, GC responses, and reduced virus-specific IgG production following LCMVARM infection [27]. However, mice with deletion of individual CRAC channel genes such as Stim1fl/fl Cd4Cre, Stim2fl/fl Cd4Cre, Orai1fl/fl Cd4Cre and Orai2−/−mice have normal Tfh cell numbers in secondary lymphoid organs and systemic antibody production after immunization with KLH or infection with LCMVARM [27, 42, 60]. These findings suggest that moderate levels of SOCE are sufficient for Tfh cell function, whereas only complete loss of SOCE impairs humoral immunity in mice, consistent with abolished SOCE and recurrent DNA and RNA virus infections in patients with LOF mutations in STIM1 and ORAI1 [22–24, 34, 53–57].
Figure 6. Immunity to viral infection requires SOCE in both CD8+ and CD4+ T cells.
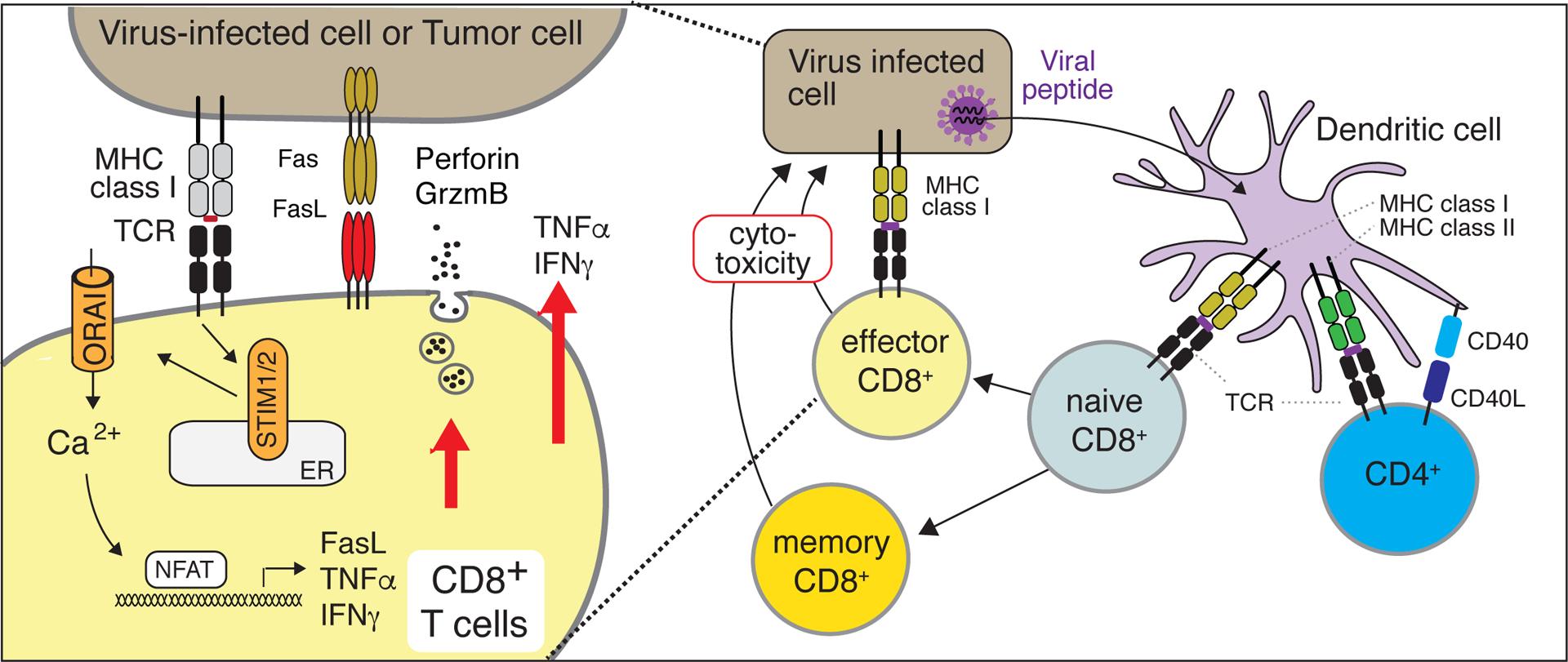
In mouse and human CD8+ T cells, SOCE controls the secretion of cytolytic granules and the expression of Fas ligand (FasL) and inflammatory cytokines in an NFAT-dependent manner. Strong suppression of SOCE results in the impaired differentiation of murine CD8+ cytotoxic effector T cells and interferes with the maintenance of LCMV-specific CD8+ memory T cells. The latter depends on SOCE in CD4+ T cells and their expression of CD40L. In the absence of SOCE, memory CD8+ T cell responses are impaired and acute viral infections become chronic [38].
Figure 7. SOCE in CD4+ T cells controls the germinal center (GC) reaction and humoral immunity.
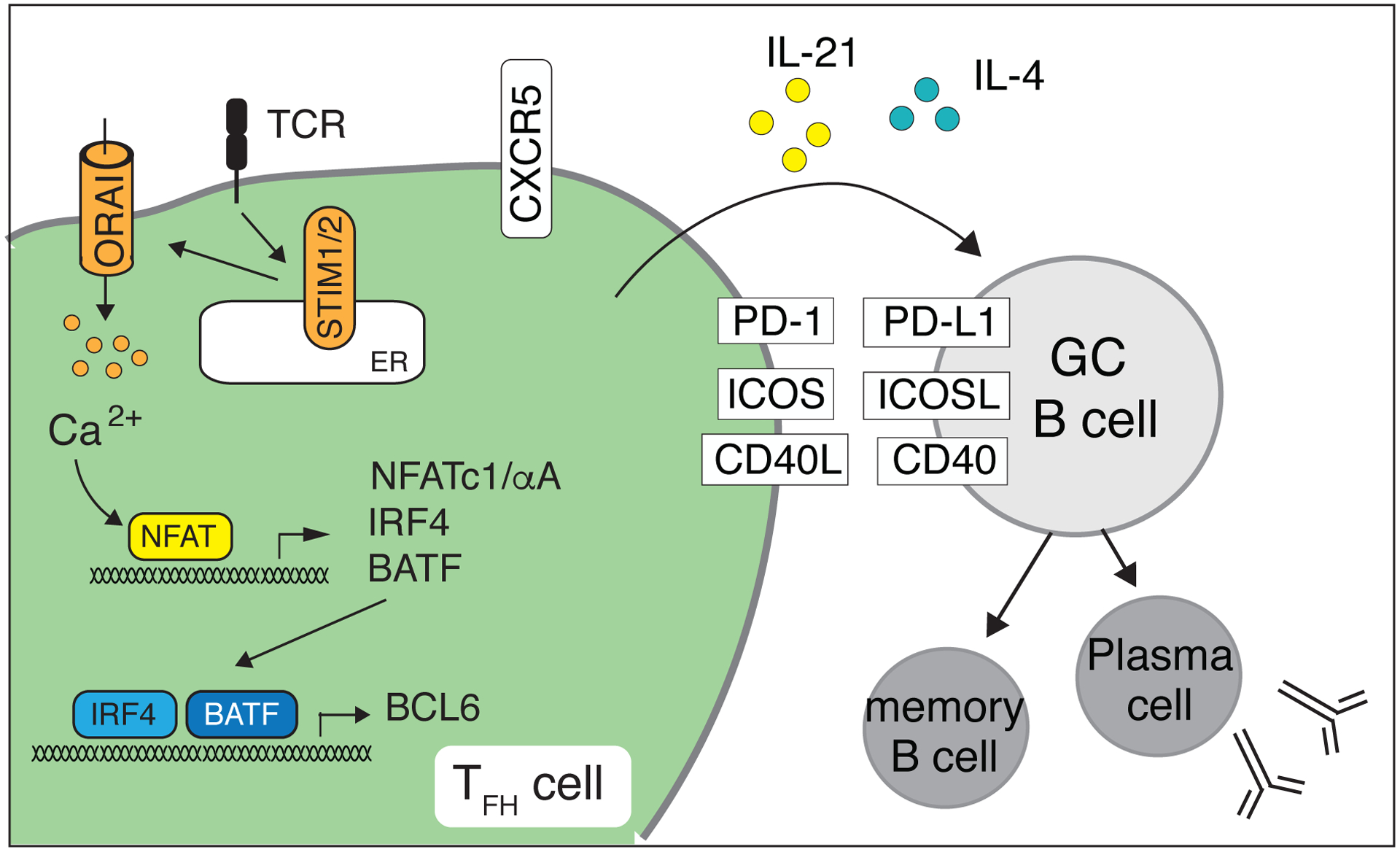
SOCE regulates the expression of IRF4 and BATF that initiate the differentiation of murine CD4+ T cells into T follicular helper (Tfh) cells and their migration into GCs by upregulating CXCR5 expression. NFAT, together with Bcl-6, controls the induction of PD-1, ICOS and CD40L as well as the production of the B cell-stimulating cytokines IL-4 and IL-21 by Tfh cells, which collectively help GC B cells to undergo class-switch recombination and affinity maturation of their antigen receptors and to differentiate into memory B cells and plasma cells [42].
Aside from viral infections, patients with CRAC channelopathy suffer from recurrent and chronic infections with fungal, bacterial and mycobacterial pathogens including Bacillus Calmette-Guérin (BCG), a live attenuated vaccine strain of Mycobacterium bovis [23, 56]. In line with increased susceptibility to mycobacterial infection in patients lacking SOCE, Stim1fl/fl Cd4Cre mice succumbed prematurely to infection with Mycobacterium tuberculosis (Mtb) [77]. Whereas Mtb burdens in Stim1fl/fl Cd4Cre mice were comparable to wildtype mice in the acute phase of infection, they gradually increased during the chronic phase ~ 45–120 days post-infection. This increase in Mtb burden was associated with a hyperinflammatory phenotype in Stim1-deficient mice characterized by elevated concentrations of proinflammatory cytokines such as IFNγ. Whereas the antigen-driven production of IFNγ depends on TCR signaling and SOCE -- and therefore was reduced in T cells of Stim1fl/fl Cd4Cre mice during the acute phase of Mtb infection, IFNγ concentrations were strongly increased during the chronic phase driven by elevated concentrations of IL-12 and/ IL-18, which promoted the SOCE-independent production of IFNγ [77]. The hyperinflammatory response of Stim1fl/fl Cd4Cre mice was further characterized by lymphoproliferation and pulmonary lymphocytosis, which coincided with impaired activation-induced cell death (AICD) (Figure 8) and reduced numbers of Treg cells in the lung compared to littermate control animals [77]. In murine and human T cells, SOCE regulates the expression of proapoptotic factors such as FasL and TRAIL, as well as Bok, Bak and Noxa [29, 77]. Both AICD and Treg cells are essential immune tolerance mechanisms that mediate the clonal deletion and suppression of effector T cells, respectively, and thus, keep immune responses during chronic infection in check and prevent hyperinflammation. A similar observation was made in the context of chronic viral infection. T cell-specific deletion of SOCE in Stim1fl/fl Stim2fl/fl Cd4Cre mice resulted in the death of mice after infection with LCMVCL13 [42]. This result is unexpected because even in T cell-deficient Rag1−/− mice LCMVCL13 infection is not lethal [105], suggesting that the mortality of Stim1fl/fl Stim2fl/fl Cd4Cre mice may not be due to their immunodeficiency, but rather impaired immune tolerance mechanisms and hyperinflammation, which certainly merits further investigation. Together, these findings suggest that SOCE can play a dual role in the infection cycle, although this response may be context-, species- and pathogen-specific. During acute infection, SOCE is required for effector function of T cells to fight infectious pathogens, but at later stages of infection, may become essential for a variety of immune tolerance mechanisms to limit the immune response and prevent hyperinflammation, as well as the associated morbidity.
Figure 8. Apoptosis in T cells is regulated by SOCE.
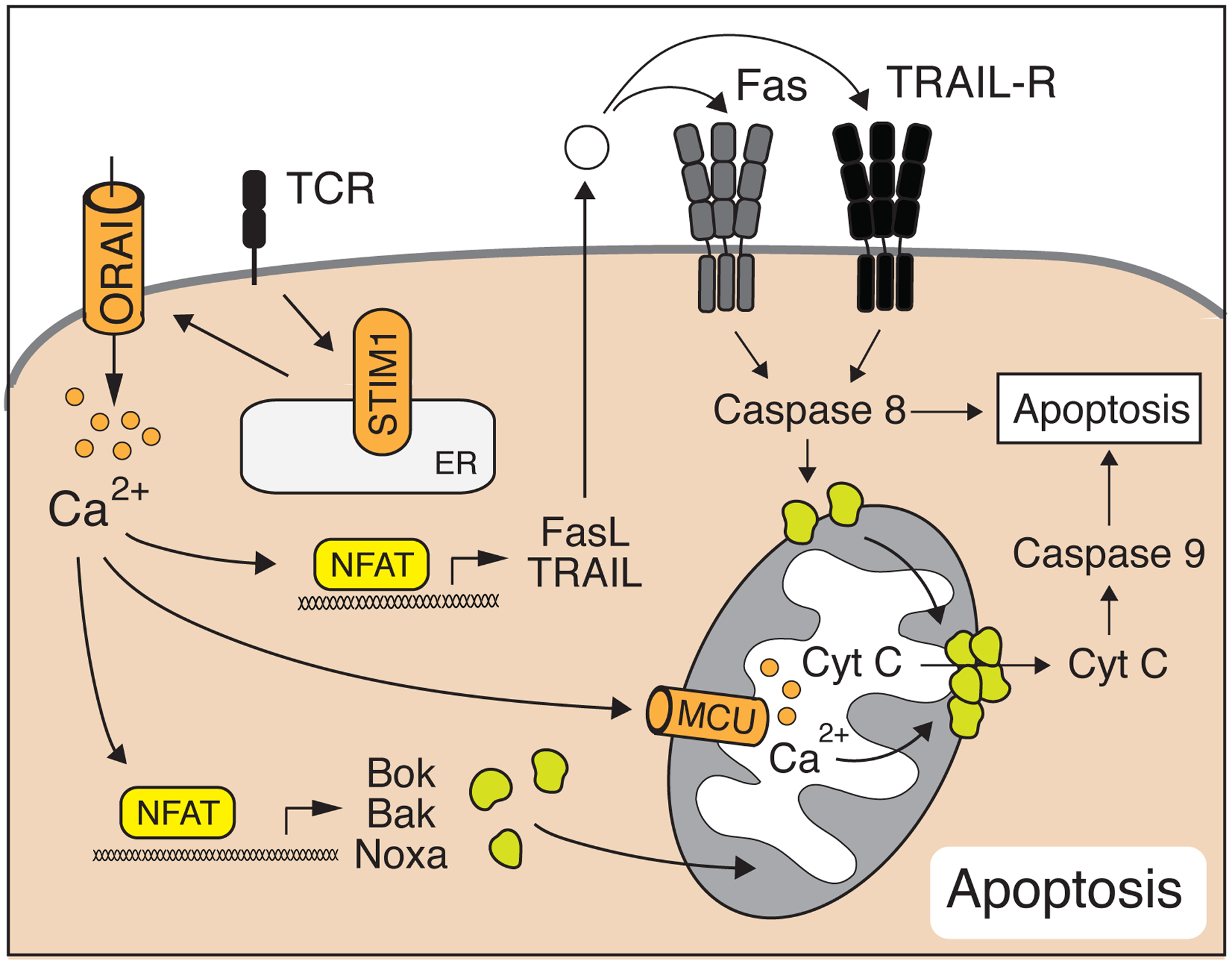
Ca2+ influx triggers activation-induced cell death (AICD) in murine T cells by inducing FasL and TRAIL expression. In addition, SOCE regulates cell-intrinsic apoptosis mechanisms by regulating the expression of the Bcl-2 family members Bok, Bak and Noxa and by facilitating Ca2+-dependent cytochrome C release from mitochondria [29].
SOCE in Autoimmunity and inflammation
An intriguing aspect of SOCE in lymphocytes is that it is essential to maintain immunological tolerance and prevent autoimmunity. Most patients with CRAC channelopathy develop autoantibodies against red blood cells and platelets resulting in autoimmune hemolytic anemia (AIHA) and thrombocytopenia [23, 52]. Other signs of autoimmunity observed in these patients are antinuclear antibodies (ANA) [23, 24], eosinophilic colitis, and Crohn’s disease [52] (Box 1). In Stim1fl/fl Stim2fl/fl LckCre or Stim1fl/fl Stim2fl/fl Cd4Cre deficient mice, the complete loss of SOCE in all T cells results in hepatosplenomegaly, lymphadenopathy and the presence of a variety of auto-antibodies against nuclear antigens (ANA), Ro and La antigens, as well as double stranded DNA [33, 42, 106]. A potential cause of autoimmunity in CRAC channel-deficient patients could be the failed negative defective selection immature T cells in the thymus because of impaired Ca2+ signaling following stimulation of the pre-TCR complex of self-reactive T cells. However, analyses of the TCR Vβ repertoire in STIM1- and ORAI1-deficient patients and TCR Vα and Vβ repertoires in Stim1fl/fl Stim2fl/fl LckCre mice showed that Vβ chains were expressed at normal frequencies in CD4+ T cells of patients and mice [34, 59]. Backcrossing of these mice to HY TCR transgenic mice resulted in only a moderate delay in negative selection relative to littermate controls [59]. Therefore SOCE appears to be dispensable for the thymic development and selection of conventional αβ T cells in both mice and men [34, 59].
SOCE is, however, essential for maintaining peripheral tolerance because of its essential role in the development and function of Foxp3+ Treg cells. In the thymus, SOCE controls the development of natural, thymus-derived Treg cells (tTreg) as Stim1fl/fl Stim2fl/fl LckCre, Stim1fl/fl Stim2fl/fl CD4Cre or Orai1fl/fl Orai2−/− Cd4Cre mice that completely lacked SOCE in all T cells have reduced numbers of thymic and peripheral Treg cells [23, 33, 34, 40, 59, 96] suggesting that SOCE may be required for tTreg development. Similarly, several patients with LOF mutations in ORAI1 and STIM1 have reduced numbers of Treg cells in their blood [23, 33, 34, 40, 59, 96]. The molecular mechanisms by which SOCE regulates this process are not fully understood and remain to be tested. SOCE may promote the maturation of Treg cells indirectly by modulating IL-2 signaling. In Stim1fl/fl Stim2fl/fl LckCre mice, IL-2 production by T cells is substantially reduced and Treg precursor cells show impaired IL-2R expression (CD25 and CD122) [33], indicating that abrogated IL-2 signaling is responsible for the impaired tTreg cell development. However, recombinant IL-2 only partially rescues Foxp3 expression, Treg differentiation, and the defective function Treg cells in vitro [59], suggesting that SOCE may have additional intrinsic functions in tTreg cells. In addition to promoting tTreg cell development in the thymus [40, 59], SOCE is required for the effector differentiation of murine tTreg cells into follicular Treg (Tfr) cells that migrate into the GCs of secondary lymphoid organs where they regulate the affinity maturation of GC B cells and antibody production [107]. Conditional deletion of SOCE in all T cells (i.e. in Stim1fl/fl Stim2fl/fl Cd4Cre mice) or Treg cells (Stim1fl/fl Stim2fl/fl Foxp3Cre mice) results in severely reduced Tfr cell numbers, spontaneous formation of GCs, and production of a plethora of autoantibodies [40, 42] (Figure 9). Tfr cells express many genes characteristic of Tfh cells including the chemokine receptor CXCR5, the costimulatory receptors PD-1 and ICOS and the transcription factor Bcl-6, but also Foxp3 and typical Treg surface markers such as CTLA-4 and GITR [107]. Treg cells of Stim1fl/fl Stim2fl/fl Cd4Cre and Stim1fl/fl Stim2fl/fl Foxp3Cre mice show indeed a reduced or absent expression of many of these molecules compared to littermate controls, contributing to the defective differentiation of Tfr cells [40]. tTreg cells in mice are known to be able to differentiate into tissue-resident Treg cells found in adipose tissue, skeletal muscle and other organs where they can control tissue homeostasis and immune tolerance [40, 107, 108]. The role of SOCE in controlling murine tissue-resident Treg cells became apparent from the analysis of Stim1fl/fl Stim2fl/fl Foxp3Cre mice (in which Stim1 and Stim2 are deleted after Foxp3 expression and Treg development in the thymus) which showed dramatically reduced numbers of tissue-resident Treg cells in the lung, liver, bone marrow and other non-lymphoid organs in these mice, despite normal Treg numbers in the thymus and LNs [40]. Transcriptional analyses of Treg cells from Stim1fl/fl Stim2fl/fl Foxp3Cre mice revealed that SOCE controls a complex gene expression program that regulates lineage identity and differentiation of tissue-resident Treg cells (Figure 9) [40].
Figure 9. SOCE is essential for immune tolerance by controlling the development and function of Treg cells.
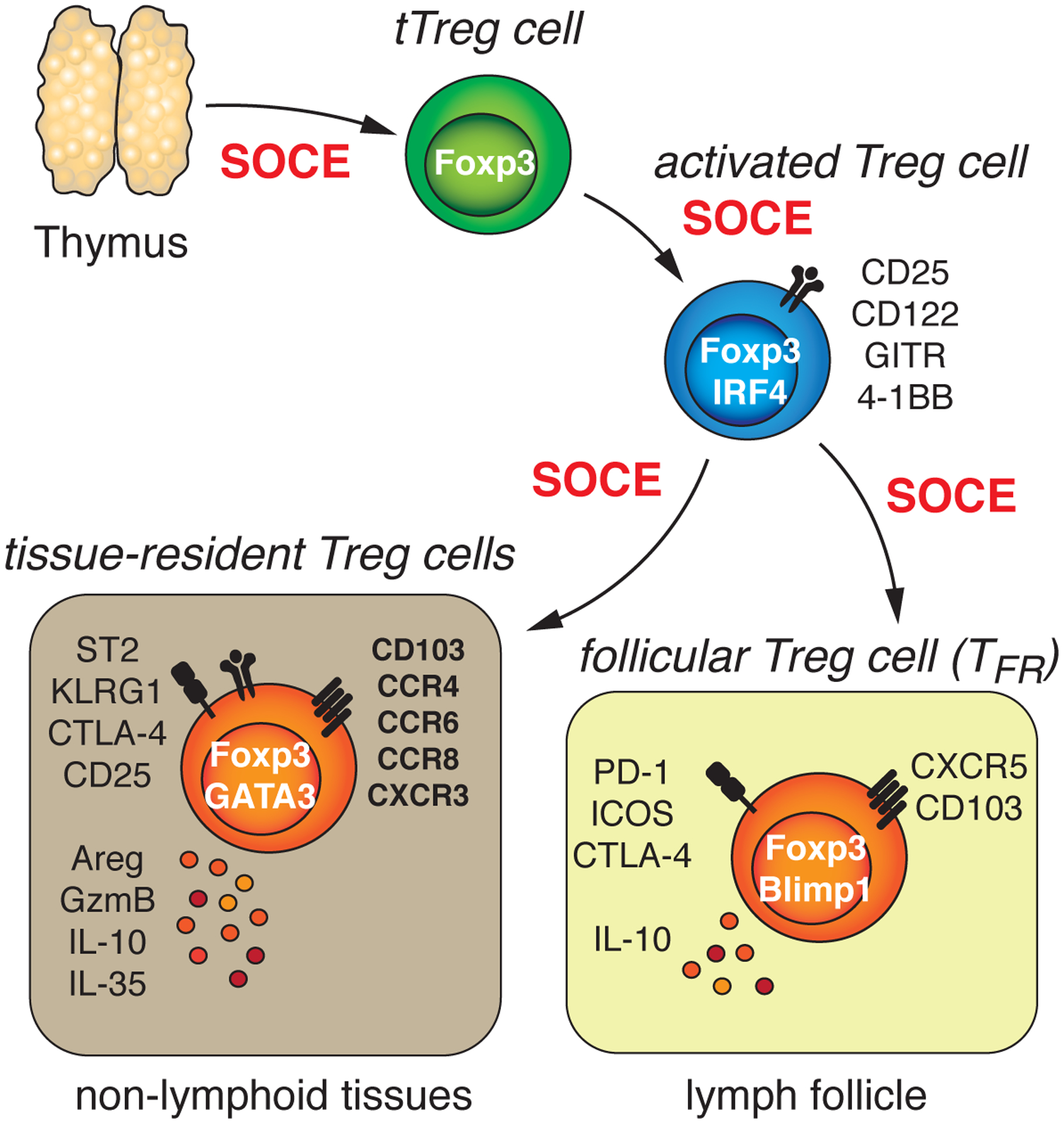
SOCE in T cells is crucial for the generation of ‘agonist-selected’ T regulatory (Treg) cells in mice during their development in the thymus [33, 59]. Inactivation of SOCE in tTreg cells after their thymic development revealed additional roles of CRAC channels in mature Treg cells. Whereas SOCE is largely dispensable for the maintenance of Treg cells in peripheral lymphoid organs, the differentiation tTreg cells into tissue-resident Treg cells and/or their homing into the parenchyma of non-lymphoid organs in mice is strictly dependent on CRAC channels [40]. Similarly, the differentiation of tTreg cells into T follicular regulatory (Tfr) cells and their migration into B cell follicles requires SOCE. Absence of tissue-resident Treg and Tfr cells in mice that completely lack SOCE specifically in Treg cells results in the production of autoantibodies, the accumulation of self-reactive T cells and multiorgan autoimmunity.
Apart from tTreg cells, SOCE controls the differentiation of naive murine CD4+ T cells into induced or peripheral Foxp3+ Treg (iTreg or pTreg) cells [77], which develop during chronic infection, in tumors or in the intestinal mucosae in response to commensal bacteria [108]. pTreg differentiation in vitro can be induced by stimulating naive CD4+ T cells in the presence of TGFβ [77]. This process is dependent on SOCE as CD4+ T cells from Stim1fl/fl Cd4Cre mice show impaired pTreg development in vitro and develop pulmonary hyperinflammation during chronic Mtb infection due to defective pTreg differentiation in vivo [77]. If defects in pTreg differentiation contribute to reduced Treg numbers in patients with ORAI1 and STIM1 mutations is not understood. Finally, SOCE is required for the suppressive function of both tTreg and pTreg cells in mice [33, 40, 77, 109]. Deletion of SOCE in T cells of Stim1fl/fl Cd4Cre, Stim1fl/fl Stim2fl/fl Cd4Cre or Orai1fl/fl Orai2−/− Cd4Cre mice impairs their ability to suppress the proliferation of effector T cells in vitro and in vivo [33, 40, 42, 59, 77, 110]. It is noteworthy that lack of ORAI1 in Orai1−/− or Orai1fl/fl Cd4Cre mice had no effect on the development of tTreg and pTreg cells and their suppressive function [61, 109]. Since Orai1 deficiency in murine T cells results in only partial reduction of SOCE (due to the compensating effects of ORAI2), moderate Ca2+ influx appears to be sufficient for the development and function of tTreg and pTreg cells (Figures 2 and 10).
Figure 10. Distinct SOCE requirements of different T cell subsets and T cell-mediated immune responses.
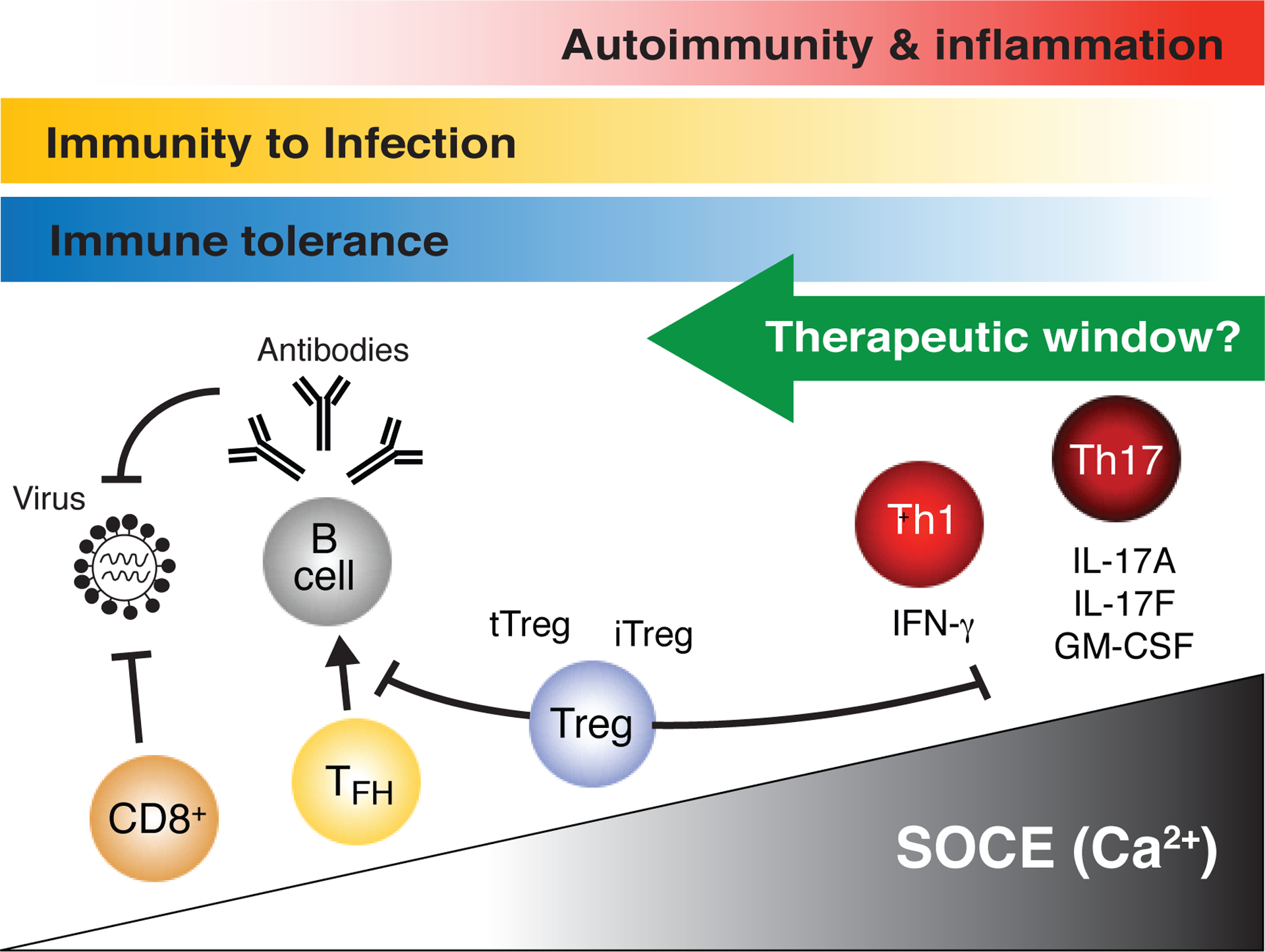
The production of inflammatory cytokines such as IL-17A, IL-17F, GM-CSF and IFNγ by murine pathogenic Th17 and Th1 cells requires strong Ca2+ signals because partial inhibition of SOCE by genetic deletion of Orai1 or Stim1 attenuates cytokine production and Th17/Th1-mediated diseases such as EAE and IBD. By contrast, the function of murine CD4+ Tfh and CD8+ T cells, which are required for humoral and cellular immunity to infection, respectively, are less dependent on SOCE as only combined deletion of Orai1/Orai2 or Stim1/Stim2 (and thus SOCE) abolishes their function. Similarly, effector Treg cell functions are less dependent on SOCE as defects are observed only when SOCE is almost completely abolished. These distinct SOCE requirements of murine T cell subsets provide a compelling rationale to postulate the existence of a therapeutic window for the treatment of Th1/Th17-cell mediated autoimmune diseases in human patients through partial inhibition of SOCE without interfering with the ability of Tfh and CD8+ T cells to provide immunity to infection or Treg cells to maintain immune tolerance.
The consequence of impaired development and function of multiple Treg subsets in mice with either T cell- or Treg-specific deletion of SOCE is multisystem inflammation that is characterized by severe lymphocytic infiltration of the lung, liver, skin, salivary glands and other tissues [33, 106]. Stim1fl/fl Stim2fl/fl Cd4Cre mice furthermore develop humoral autoimmunity characterized by increased numbers of GC B cells, spontaneous formation of GCs and production of autoantibodies against ANA, dsDNA as well as Ro and La antigens, compared to littermate control animals [42, 106]. Spontaneous autoantibody production is a consequence of strongly impaired development and function of Tfr cells which results in Tfh cell accumulation and GC formation [42]. An additional cause of autoantibody production in these mice is the production of IL-4 by Tfh cells, which promotes IgG1 and IgE secretion and Th2 cell-mediated autoimmunity as evidenced by a lymphoproliferative disorder and spontaneous dermal inflammation [110]. The autoimmunity and inflammation of Stim1fl/fl Stim2fl/fl Cd4Cre or Stim1fl/fl Stim2fl/fl LckCre mice is even more pronounced in Stim1fl/fl Stim2fl/fl Foxp3Cre mice, with selective deletion of SOCE in Treg cells, but normal effector T cell functions [40]. These mice develop severe autoimmunity characterized by premature death, production of a broad spectrum of autoantibodies (including ANA, anti-dsDNA and anti-RBC antibodies) and severe systemic inflammation affecting the skin, liver, lung and other organs [33, 40, 42]. Autoantibody production and multiorgan inflammation are associated with the almost complete absence of Tfr cells and tissue resident Treg cells, respectively, in these mice [40]. The main cause of mortality in Stim1fl/fl Stim2fl/fl Foxp3Cre mice appears to be independent of autoantibodies as deletion of B cells only ameliorates their AIHA but does not prolong their survival [40]. Collectively, these data demonstrate that SOCE is essential for Treg development in the thymus, their post-thymic differentiation and function and thus, the ability of Treg cells to maintain immunological self-tolerance.
SOCE is required for the proinflammatory function of T helper cells
An important role of SOCE is to regulate the function of Th1, Th2 and Th17 cells, which provide adaptive immunity to viral, parasitic as well as bacterial and fungal infections. However, pathogenic Th1, Th2 and Th17 cells also mediate inflammation in a wide range of disorders including graft-vs-host disease (GvHD), allergic asthma, and autoimmune diseases such as multiple sclerosis (MS), inflammatory bowel disease (IBD), and rheumatoid arthritis [111]. Less is known about the role of SOCE in Th2 cells and type 2 immune responses. Specifically, the expression of IL-4 has been reported to be markedly reduced in T cells lacking Stim1 [33] or Orai1 expression [25, 27] compared to WT control T cells, and almost absent in Th2 and Tfh cells of Stim1fl/fl Stim2fl/fl Cd4Cre and Orai1fl/fl Orai2–/– Cd4Cre mice [27, 33, 41]. By contrast, ectopic expression of STIM1 or STIM2 elevated IL-4 production in murine CD4+ T cells in vitro [33]. Similarly, the T cells of patients with STIM1 and ORAI1 LOF mutations showed impaired expression of Th2 cytokines IL-4, IL-5 and IL-13 after ex vivo stimulation with PMA/Ionomycin [42, 89]. Furthermore, the Th2 defining transcription factor GATA3 appears to be regulated by SOCE, as its expression was reduced in murine T cells treated with a CRAC channel inhibitor [112]. How STIM and ORAI proteins contribute to type 2 immune responses in diseases such as allergic asthma, requires further study.
Th1 and Th17 cells play crucial roles in GvHD [113, 114], which occurs in the context of allogenic stem cell transplantation and is mediated by alloreactive donor T cells that recognize host tissue. Mouse models have shown an important role of SOCE in T cells in the induction GvHD. Specifically, injection of SOCE-deficient CD4+ and CD8+ T cells from Orai1fl/fl Orai2–/– Cd4Cre mice (on the C57BL/6 background) into MHC-mismatched BALB/c mice fails to induce GvHD, which is accompanied by strongly reduced concentrations of IFNγ and TNFα in the serum of host mice [27]. Transfer of T cells from Stim1–/– or Orai1fl/fl Cd4Cre mice, which have partially reduced SOCE, showed attenuated GvHD severity and serum IFNγ and TNFα concentrations [27, 60]. The role of SOCE in allogenic, proinflammatory T cell responses is further emphasized by a study showing that SOCE-deficient T cells from mice expressing a dominant negative ORAI1R93W mutant protein fail to reject fully MHC mismatched skin allografts resulting in prolonged graft survival and less perivascular leukocyte infiltration compared with respective WT controls [26]. Th1 and Th17 cells are also essential modulators of disease pathology in MS and the experimental autoimmune encephalomyelitis (EAE) mouse model of MS. MS is an autoimmune disease of the CNS in which myelin-reactive T cells differentiate into Th1 and Th17 cells that produce proinflammatory cytokines and cause CNS inflammation [43, 109, 115]. T cells isolated from MS lesions of patients have been found to produce IFNγ and IL-17A [115]. In mice, deletion of the transcription factors T-bet and RORγt, which control the differentiation of Th1 and Th17 cells, or IL-23, which is important for the development of pathogenic Th17 cells, can protect mice from EAE [116–119]. SOCE appears to be essential for T cell function in EAE because lack of SOCE in murine T cells completely protects Stim1fl/fl Cd4Cre and Stim1fl/fl Stim2fl/fl Cd4Cre mice from developing EAE or attenuates the severity of EAE in Orai1fl/fl Cd4Cre and Stim2fl/fl Cd4Cre mice [43, 109, 120]. The degree of protection from EAE correlated with the extent to which SOCE was impaired with strongly reduced or abolished SOCE in T cells lacking Stim1 or Stim1/Stim2 completely preventing EAE, whereas partial inhibition of SOCE in Orai1 and Stim2-deficient T cells attenuating EAE severity [43, 109]. Protection from EAE was associated with decreased production of IL-17A, GM-CSF and IFNγ by myelin-specific T cells [43, 109, 120]. Attenuation of EAE has also been observed in mice with T cell-specific deletion of the CRAC channel regulator 2A (CRACR2A) due to impaired production of Th1 and Th17 cytokines and reduced SOCE [121]. Moreover, induced genetic deletion of Orai1 or Stim1 in adoptively transferred, MOG-specific T cells after the onset of EAE halts disease progression relative to WT controls [109]. A similar disease-modifying effect is observed in wildtype mice with established EAE symptoms that were treated with the selective CRAC channel blocker AMG1 and compound 5 [109, 112], suggesting that SOCE inhibition might potentially be beneficial for the treatment of MS. Another disease in which Th1 and Th17 cells play an important pathogenic role is Crohn’s disease (CD), a severe form of IBD. The numbers of Th1 and Th17 cells are increased in the colon of CD patients and mice with experimentally induced IBD [122]. The importance of SOCE in IBD was demonstrated experimentally by using an adoptive T cell transfer mouse model of IBD. Transfer of naïve, SOCE-deficient CD4+ T cells from mice expressing a non-functional ORAI1R93W channel, Stim1fl/fl Cd4Cre and Orai1fl/fl Orai2−/− Cd4Cre mice into lymphopenic host mice failed to induce colitis, which was associated with defective production of IL-17A, IFNγ, and TNFα in SOCE-deficient cells isolated from mesenteric lymph nodes of host mice compared to WT T cells [26, 27].
SOCE regulates encephalitogenic and colitogenic Th17 cells by several mechanisms including their ability to produce cytokines such as IL-17A, IL-17F and GM-CSF [23, 27, 43, 92, 109]. Mice with genetic deletion of Orai1 or Stim1 have marked reduced expression of these cytokines relative to WT littermate controls, and are also protected from EAE and IBD [26, 43, 109, 112, 120]. This is in line with reduced IL-17A, IL-17F and GM-CSF concentrations and ameliorated severity of EAE [123] and IBD [124] in Nfatc1fl/fl Nfatc2−/− Cd4Cre mice with T cell-specific ablation of NFATc1 and NFATc2. Besides controlling cytokine expression, SOCE also regulates the differentiation of Th17 cells, potentially by controlling the expression of RORγt. The levels of this Th17 lineage-defining transcription factor were reduced in Th17 cells of Orai1−/− mice compared with WT control T cells, potentially due to the observed impaired NFAT binding to the RORγt promoter in Orai1−/− T cells [112]. In other studies, however, the mRNA and protein expression of RORγt was normal in Th17 cells of Stim1fl/fl Cd4Cre and Orai1fl/fl Cd4Cre mice with conditional deletion of either Stim1 or Orai1 in T cells [43, 109]. A likely explanation for these apparently discrepant results may be found in the slightly different conditions under which SOCE-deficient Th17 cells were polarized (IL-6, TGFβ and IL-23 by [112] vs. IL-6 and TGFβ by [43, 109]). Addition of IL-23 to differentiating Th17 cell cultures not only induces a different gene expression profile in Th17 cells [125] but is also essential for the pathogenicity of Th17 cells as assessed by their ability to induce inflammation in disease models such as EAE and IBD [126]. The expression of IL-23R is SOCE-dependent [43] and CD4+ T cells from Stim1fl/fl Cd4Cre mice that were polarized into pathogenic Th17 cells in vitro in the presence of IL-1β, IL-6, IL-23 had impaired RORγt expression [92]. These findings suggest that SOCE might indirectly regulate the expression of RORγt and pathogenic Th17 cell differentiation through IL-23R signaling, although this warrants further investigation. Of note, The IL-23R signals through signal transducer and activator of transcription 3 (STAT3) [127]. CD4+ T cells of mice with T cell-specific expression of a hyperactive form of STAT3 (STAT3C) spontaneously differentiate into pathogenic Th17 cells and induce severe pulmonary inflammation and psoriasis-like cutaneous inflammation [128]. Of relevance, the function of STAT3C-expressing Th17 cells depends critically on SOCE because T cell-specific deletion of Stim1 in STAT3C expressing mice completely prevents pulmonary and skin inflammation [92]. This is intriguing because pathogenic Th17 cells and STAT3 function have been implicated in inflammatory disorders such as steroid resistant asthma and psoriasis [128–133], suggesting that inhibition of SOCE may be a new approach to the treatment of these diseases. Deletion of SOCE in Th17 cells was reported to protect from inflammation due to reduced production of Th17 effector cytokines and marked diminished expression of signature genes associated with pathogenic Th17 cells from Stim1fl/fl Cd4Cre mice compared to WT control T cells [125, 134]. SOCE also regulates mitochondrial function of STAT3C-expressing pathogenic Th17 cells. Th17 cells of Stim1fl/fl Stat3CLSLCd4Cre mice express STAT3C but lack SOCE and have reduced expression of electron transport chain (ETC) genes and impaired oxidative phosphorylation (OXPHOS) [92]. In this study, Stim1-deficient pathogenic Th17 cells produced higher concentrations of mitochondrial reactive oxygen species (mROS) causing DNA damage and cell death [92]. The cause of increased mROS production remains to be studied further, but may be due to an increased electron leak from the ETC due to altered expression of ETC genes and impaired formation of ETC supercomplexes required for efficient electron transport. Collectively, these studies suggest that SOCE is particularly important for the differentiation of pathogenic Th17 cells and their function beyond just the regulation of cytokine production, which has important implications for the role pathogenic Th17 cells play in certain autoimmune and inflammatory diseases, and certainly deserving further investigation.
Concluding remarks
Since the discovery of STIM1 [135, 136] and ORAI1 [22, 137, 138], we have learned a lot about the role of CRAC channels and SOCE in T cell immune responses in mice and humans. There are, however, a number of questions that remain, the answers to some of which bear important clinical implications (see outstanding questions). Much work has focused on the calcineurin and NFAT as downstream mediators of SOCE. In some cases, the effects of blocking SOCE and calcineurin activity on T cell function are similar. Well documented examples are the expression of cytokines [47, 79], or more recently, the regulation of aerobic glycolysis and proliferation by the SOCE-calcineurin-NFAT pathway [41, 93, 139]. It is important to emphasize, however, that SOCE regulates many more Ca2+ dependent signaling molecules and transcription factors besides calcineurin and NFAT. Such molecules, which translate Ca2+ signals into gene expression and thus determine the differentiation and effector programs of T cells, include the enzymes AMPK, Akt, CaMKII or Erk1/2 [41, 140, 141] and transcription factors CREB and NF-KB (Figure 1) [51, 142]. A role for SOCE in regulating NF-kB activation was recently shown in Jurkat T cells [142] and primary mouse B cells, where it is involved in metabolic reprogramming and survival of B cells after BCR crosslinking [143]. Furthermore, calcineurin was shown to have many other targets besides NFAT [44], which in murine T cells includes the Src kinase Lck [144]. The SOCE-calcineurin-NFAT pathway therefore is not as linear as commonly assumed and has many more branch points. For this reason, defects in SOCE are not necessarily equivalent to those in calcineurin function or NFAT activity. This conclusion has clinical significance because it affects how we assess the benefits and adverse effects of CRAC channel blockers compared to calcineurin inhibitors such as cyclosporin A and tacrolimus.
Because SOCE is a major signaling pathway in T cells and defects in SOCE cause a wide array of immunological dysfunctions, the question arises as to which T cell functions, subsets and immune responses are most dependent on SOCE. Several aspects of T cell biology are not regulated by SOCE, including the development of αβ and γδ T cells and their selection in the thymus, the differentiation of some Th cell subsets (for instance Th1 cells) and potentially, T cell migration, as discussed above. Furthermore, T cell functions resulting from cytokine receptor activation, which does not induce Ca2+ signals, is independent of SOCE, as demonstrated by IL-12 and IL-18-induced IFNγ production by Stim1-deficient murine and human CD8+ T cells [77] and IL-2 and IL-7-induced activation of metabolic pathways and proliferation of CD4+ and CD8+ T cells from Stim1fl/fl Stim2fl/fl Cd4Cre mice [41]. These TCR and SOCE-independent signaling pathways likely explain why patients and mice lacking CRAC channel function maintain normal T cell numbers. A related question is whether all T cells require the same quantitative level of SOCE for their function. Studies in mice with single or combined deletion of Orai and Stim genes and pharmacological CRAC channel inhibition revealed important quantitative differences regarding the role of SOCE in Th1, Th17, Tfh, Treg and CD8+ T cells. Deletion of Orai1 or Stim1 in mouse T cells reduces SOCE by ~ 30–80% (Figure 2B), which is sufficient to attenuate the pathogenic function of Th1 and Th17 cells and their ability to cause EAE, IBD, pulmonary and skin inflammation and GvHD [27, 43, 92, 109, 112, 120]. This level of SOCE suppression does not, however, diminish cellular or humoral immune responses by CD4+ and CD8+ T cells to viral infections and tumors [27, 38, 39, 42, 78]. Likewise, immune tolerance mechanisms, including the development and function of Treg cells, are normal in Orai1 or Stim1-deficient mice, whereas complete suppression of SOCE in Stim1fl/fl Stim2fl/fl Cd4Cre or Orai1fl/fl Orai2−/− Cd4Cre mice and ORAI1 or STIM1-deficient patients results in impaired Treg numbers and function [23, 33, 36, 40, 59]. Together, these studies demonstrate that distinct CD4+ and CD8+ T cell subsets appear to have different SOCE requirements for their differentiation and function (Figure 10). Given the importance of SOCE for the function of several proinflammatory T cell subsets such as Th1 and Th17 cells the question arises if CRAC channel inhibition can be used for the treatment of autoimmune and inflammatory diseases such as MS, IBD and psoriasis, in which these T cells play an important pathophysiological role. Several phase I and II clinical trials have been conducted or are under way to test CRAC channels inhibitors in various autoimmune and inflammatory diseases such as plaque psoriasis, acute pancreatitis, chronic asthma as well as relapsing or refractory non-Hodgkin’s lymphoma, which have been reviewed in detail elsewhere [145]. A related question is whether the anti-inflammatory effects of CRAC channel inhibition can be achieved without causing undue susceptibility to infections, compromising antitumor immunity or impairing the tolerogenic function of Treg cells. Given the different SOCE requirements of Th1 and Th17 cells compared to CD8+ and Treg cells discussed above, a therapeutic window for SOCE inhibition may indeed exist (Figure 10). How these insights gained largely from studies in mice translate to human immune responses remains to be investigated. Nonetheless, we have come almost full circle from identifying patients with CID due to mutations in ORAI1 and STIM1 that abolish CRAC channel function to characterizing the role of SOCE in immunity to infection, inflammation and autoimmunity by using mice with targeted deletion of CRAC channel genes and developing CRAC channel inhibitors that have therapeutic potential in several immune mediated disorders.
ACKNOWLEDGEMENTS.
The authors thank Anthony Tao for the analysis of ORAI and STIM gene expression shown in Figure 2. This work was funded by NIH grants AI097302, AI130143 and AI137004 and an Irma T. Hirschl career scientist award to S.F.
GLOSSARY
- Activation-induced cell death (AICD)
A form of regulated cell death that plays an important role in peripheral T cell tolerance
- Affinity maturation
Process by which B cells increase their affinity to an antigen through repetitive cellular selection and somatic hypermutation in the GC
- Autoimmune hemolytic anemia (AIHA)
Autoimmune disease in which autoantibodies bind and destroy erythrocytes
- Ca2+ release-activated Ca2+ (CRAC) channel
Ca2+ selective ion channel formed by ORAI1, ORAI2 and ORAI3 proteins in the plasma membrane and activated by STIM1 and STIM2
- Channelopathies
Group of diseases caused by defective ion channel function
- Class-switch recombination (CSR)
Genetic rearrangement that changes the heavy chain segments of the immunoglobulin genes
- Combined Immunodeficiency (CID)
A form of primary immunodeficiency resulting from defects in T and B cell function
- Experimental autoimmune encephalomyelitis (EAE)
Murine model of multiple sclerosis (MS)
- Germinal center (GC)
Microstructure within the follicles of secondary lymphoid organs where B cells undergo affinity maturation, class-switch recombination (CSR) and differentiation into plasma cells
- Glycolysis
Metabolic pathway that catabolizes glucose
- Graft-versus-host disease (GvHD)
Immune disease resulting from an incompatible allogeneic hematopoietic stem cells transplant
- HY-transgenic mice
Mouse model to study positive and negative T cell selection
- Immunological Synapse
Temporary interface between an antigen presenting cell (APC) and a T cell
- Inflammatory bowel disease (IBD)
Crohn’s disease and ulcerative colitis in human patients are forms of IBD characterized by chronic inflammation of the gastrointestinal tract
- Intraepithelial lymphocytes (IEL)
Specific subset of lymphocytes that are found within the epithelial layers of mucosal tissues
- Lipid bilayers
Phospholipid membranes that are used to study membrane dynamics, ion channel functions and transport processes across membranes
- Lymphocytic choriomeningitis virus (LCMV)
Virus widely used to study adaptive immune responses in mice
- Membrane potential
Electrochemical gradient across membranes maintained by ATP-dependent ion pumps
- Negative selection
Elimination of self-reactive T cells clones during thymic development
- MOG-specific T cells
T cell clones that express a (transgenic) TCR that recognizes the myelin oligodendrocyte glycoprotein (MOG) antigens
- Nuclear factor of activated T cells (NFAT)
A family of four Ca2+ and calcineurin-regulated transcription factors
- ORAI1, ORAI2, ORAI3
Genes encoding the pore-forming subunit of the CRAC channel
- Oxidative phosphorylation (OXPHOS)
A metabolic process in mitochondria that uses the oxidation of nutrients to produce ATP
- Regulatory T cells (Tregs)
A subset of Foxp3+ CD4+ T cells that maintain immune tolerance by suppressing the function of other immune cells. tTreg cells develop in the thymus, whereas pTreg cells are derived from naive CD4+ T cells in the periphery
- Ro/La antigens
Antigenic targets of autoantibodies that are typical for different autoimmune diseases, such as Sjögren’s syndrome, rheumatoid arthritis and systemic lupus erythematosus
- STIM1, STIM2
ER membrane proteins that sense the ER Ca2+ concentration and activate CRAC channels
- Store-operated Ca2+ entry (SOCE)
A form of Ca2+ influx mediated by CRAC channels
- Stormorken syndrome
Autosomal dominant genetic disorder that is characterized by defective platelet function, increased bleeding tendency, thrombocytopenia, anemia and tubular aggregate myopathy
- TCR Vα and Vβ repertoire
The variable (V) regions of the TCR α and β chains that represent the entire TCR diversity
- T follicular helper (Tfh) and regulatory (Tfr) cells
CD4+ T cell subsets characterized by the expression of CXCR5 and Bcl-6 that localize to germinal centers
- T helper (Th) 17 cells
CD4+ T cell subset characterized by expression of the transcription factor RORγt and production of IL-17. Th17 cells are involved in immunity to bacteria and fungi, but also induce inflammation in many autoimmune diseases
Footnotes
CONFLICT OF INTEREST DISCLOSURE. S.F. is a scientific cofounder of Calcimedica. M.V. and S.K. have no competing interests.
REFERENCES
- 1.Trebak M and Kinet JP (2019) Calcium signalling in T cells. Nat Rev Immunol 19 (3), 154–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feske S et al. (2015) Ion channels in innate and adaptive immunity. Annu Rev Immunol 33, 291–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prakriya M and Lewis RS (2015) Store-Operated Calcium Channels. Physiol Rev 95 (4), 1383–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beck A et al. (2006) Nicotinic acid adenine dinucleotide phosphate and cyclic ADP-ribose regulate TRPM2 channels in T lymphocytes. FASEB J 20 (7), 962–4. [DOI] [PubMed] [Google Scholar]
- 5.Melzer N et al. (2012) TRPM2 cation channels modulate T cell effector functions and contribute to autoimmune CNS inflammation. PLoS One 7 (10), e47617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bertin S et al. (2014) The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells. Nat Immunol 15 (11), 1055–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beesetty P et al. (2018) Inactivation of TRPM7 kinase in mice results in enlarged spleens, reduced T-cell proliferation and diminished store-operated calcium entry. Sci Rep 8 (1), 3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romagnani A et al. (2017) TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat Commun 8 (1), 1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin J et al. (2008) Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 322 (5902), 756–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ledderose C et al. (2018) Purinergic P2X4 receptors and mitochondrial ATP production regulate T cell migration. J Clin Invest 128 (8), 3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Woehrle T et al. (2010) Pannexin-1 hemichannel-mediated ATP release together with P2X1 and P2X4 receptors regulate T-cell activation at the immune synapse. Blood 116 (18), 3475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenk U et al. (2011) ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal 4 (162), ra12. [DOI] [PubMed] [Google Scholar]
- 13.Schenk U et al. (2008) Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal 1 (39), ra6. [DOI] [PubMed] [Google Scholar]
- 14.Borges da Silva H et al. (2018) The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8(+) T cells. Nature 559 (7713), 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stark R et al. (2018) T RM maintenance is regulated by tissue damage via P2RX7. Sci Immunol 3 (30). [DOI] [PubMed] [Google Scholar]
- 16.Omilusik K et al. (2011) The Ca(v)1.4 calcium channel is a critical regulator of T cell receptor signaling and naive T cell homeostasis. Immunity 35 (3), 349–60. [DOI] [PubMed] [Google Scholar]
- 17.Wang H et al. (2016) Low-Voltage-Activated CaV3.1 Calcium Channels Shape T Helper Cell Cytokine Profiles. Immunity 44 (4), 782–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feske S (2019) CRAC channels and disease - From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 80, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lis A et al. (2007) CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol 17 (9), 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feske S et al. (2012) Ion channels and transporters in lymphocyte function and immunity. Nat Rev Immunol 12 (7), 532–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chou J et al. (2015) A novel mutation in ORAI1 presenting with combined immunodeficiency and residual T-cell function. J Allergy Clin Immunol 136 (2), 479–482 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feske S et al. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441 (7090), 179–85. [DOI] [PubMed] [Google Scholar]
- 23.Lian J et al. (2018) ORAI1 mutations abolishing store-operated Ca(2+) entry cause anhidrotic ectodermal dysplasia with immunodeficiency. J Allergy Clin Immunol 142 (4), 1297–1310 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarl CA et al. (2009) ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol 124 (6), 1311–1318 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gwack Y et al. (2008) Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol 28 (17), 5209–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCarl CA et al. (2010) Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. J Immunol 185 (10), 5845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vaeth M et al. (2017) ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat Commun 8, 14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vig M et al. (2008) Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol 9 (1), 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim KD et al. (2011) ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J Immunol 187 (7), 3620–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsvilovskyy V et al. (2018) Deletion of Orai2 augments endogenous CRAC currents and degranulation in mast cells leading to enhanced anaphylaxis. Cell Calcium 71, 24–33. [DOI] [PubMed] [Google Scholar]
- 31.Hou X et al. (2012) Crystal structure of the calcium release-activated calcium channel Orai. Science 338 (6112), 1308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inayama M et al. (2015) Orai1-Orai2 complex is involved in store-operated calcium entry in chondrocyte cell lines. Cell Calcium 57 (5–6), 337–47. [DOI] [PubMed] [Google Scholar]
- 33.Oh-Hora M et al. (2008) Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol 9 (4), 432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs S et al. (2012) Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol 188 (3), 1523–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maus M et al. (2015) Missense mutation in immunodeficient patients shows the multifunctional roles of coiled-coil domain 3 (CC3) in STIM1 activation. Proc Natl Acad Sci U S A 112 (19), 6206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picard C et al. (2009) STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med 360 (19), 1971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diercks BP et al. (2018) ORAI1, STIM1/2, and RYR1 shape subsecond Ca(2+) microdomains upon T cell activation. Sci Signal 11 (561). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw PJ et al. (2014) CD4(+) and CD8(+) T cell-dependent antiviral immunity requires STIM1 and STIM2. J Clin Invest 124 (10), 4549–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weidinger C et al. (2013) STIM1 and STIM2-mediated Ca(2+) influx regulates antitumour immunity by CD8(+) T cells. EMBO Mol Med 5 (9), 1311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vaeth M et al. (2019) Tissue resident and follicular Treg cell differentiation is regulated by CRAC channels. Nat Commun 10 (1), 1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaeth M et al. (2017) Store-Operated Ca(2+) Entry Controls Clonal Expansion of T Cells through Metabolic Reprogramming. Immunity 47 (4), 664–679 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaeth M et al. (2016) Store-Operated Ca(2+) Entry in Follicular T Cells Controls Humoral Immune Responses and Autoimmunity. Immunity 44 (6), 1350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma J et al. (2010) T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur J Immunol 40 (11), 3028–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roy J and Cyert MS (2020) Identifying New Substrates and Functions for an Old Enzyme: Calcineurin. Cold Spring Harb Perspect Biol 12 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaeth M and Feske S (2018) NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Res 7, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hermann-Kleiter N and Baier G (2010) NFAT pulls the strings during CD4+ T helper cell effector functions. Blood 115 (15), 2989–97. [DOI] [PubMed] [Google Scholar]
- 47.Kar P and Parekh AB (2015) Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol Cell 58 (2), 232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin YP et al. (2019) Selective recruitment of different Ca(2+)-dependent transcription factors by STIM1-Orai1 channel clusters. Nat Commun 10 (1), 2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frischbutter S et al. (2011) Dephosphorylation of Bcl-10 by calcineurin is essential for canonical NF-kappaB activation in Th cells. Eur J Immunol 41 (8), 2349–57. [DOI] [PubMed] [Google Scholar]
- 50.Palkowitsch L et al. (2011) The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J Biol Chem 286 (9), 7522–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barton K et al. (1996) Defective thymocyte proliferation and IL-2 production in transgenic mice expressing a dominant-negative form of CREB. Nature 379 (6560), 81–5. [DOI] [PubMed] [Google Scholar]
- 52.Lacruz RS and Feske S (2015) Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 1356, 45–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le Deist F et al. (1995) A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood 85 (4), 1053–62. [PubMed] [Google Scholar]
- 54.Partiseti M et al. (1994) The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem 269 (51), 32327–35. [PubMed] [Google Scholar]
- 55.Elling R et al. (2016) Preserved effector functions of human ORAI1- and STIM1-deficient neutrophils. J Allergy Clin Immunol 137 (5), 1587–1591 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feske S et al. (1996) Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur J Immunol 26 (9), 2119–26. [DOI] [PubMed] [Google Scholar]
- 57.Klemann C et al. (2017) Hemophagocytic lymphohistiocytosis as presenting manifestation of profound combined immunodeficiency due to an ORAI1 mutation. J Allergy Clin Immunol 140 (6), 1721–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vaeth M and Feske S (2018) Ion channelopathies of the immune system. Curr Opin Immunol 52, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh-Hora M et al. (2013) Agonist-selected T cell development requires strong T cell receptor signaling and store-operated calcium entry. Immunity 38 (5), 881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beyersdorf N et al. (2009) STIM1-independent T cell development and effector function in vivo. J Immunol 182 (6), 3390–7. [DOI] [PubMed] [Google Scholar]
- 61.Jin S et al. (2013) Natural regulatory T cells are resistant to calcium release-activated calcium (CRAC/ORAI) channel inhibition. Int Immunol 25 (9), 497–506. [DOI] [PubMed] [Google Scholar]
- 62.Aifantis I et al. (2001) Constitutive pre-TCR signaling promotes differentiation through Ca2+ mobilization and activation of NF-kappaB and NFAT. Nat Immunol 2 (5), 403–9. [DOI] [PubMed] [Google Scholar]
- 63.Bhakta NR et al. (2005) Calcium oscillations regulate thymocyte motility during positive selection in the three-dimensional thymic environment. Nat Immunol 6 (2), 143–51. [DOI] [PubMed] [Google Scholar]
- 64.Nakayama T et al. (1992) In vivo calcium elevations in thymocytes with T cell receptors that are specific for self ligands. Science 257 (5066), 96–9. [DOI] [PubMed] [Google Scholar]
- 65.Melichar HJ et al. (2013) Distinct temporal patterns of T cell receptor signaling during positive versus negative selection in situ. Sci Signal 6 (297), ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ouyang K et al. (2014) Loss of IP3R-dependent Ca2+ signalling in thymocytes leads to aberrant development and acute lymphoblastic leukemia. Nat Commun 5, 4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jha A et al. (2015) Essential roles for Cavbeta2 and Cav1 channels in thymocyte development and T cell homeostasis. Sci Signal 8 (399), ra103. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H et al. (2014) STIM1 calcium sensor is required for activation of the phagocyte oxidase during inflammation and host defense. Blood 123 (14), 2238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clemens RA et al. (2017) STIM1 and STIM2 cooperatively regulate mouse neutrophil store-operated calcium entry and cytokine production. Blood 130 (13), 1565–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Demaurex N and Saul S (2018) The role of STIM proteins in neutrophil functions. J Physiol 596 (14), 2699–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lin YP et al. (2018) The Allergen Der p3 from House Dust Mite Stimulates Store-Operated Ca(2+) Channels and Mast Cell Migration through PAR4 Receptors. Mol Cell 70 (2), 228–241 e5. [DOI] [PubMed] [Google Scholar]
- 72.Vaeth M et al. (2015) Ca2+ Signaling but Not Store-Operated Ca2+ Entry Is Required for the Function of Macrophages and Dendritic Cells. J Immunol 195 (3), 1202–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nunes-Hasler P et al. (2017) STIM1 promotes migration, phagosomal maturation and antigen cross-presentation in dendritic cells. Nat Commun 8 (1), 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clemens RA and Lowell CA (2019) CRAC channel regulation of innate immune cells in health and disease. Cell Calcium 78, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Demaurex N and Nunes P (2016) The role of STIM and ORAI proteins in phagocytic immune cells. Am J Physiol Cell Physiol 310 (7), C496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feske S (2007) Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol 7 (9), 690–702. [DOI] [PubMed] [Google Scholar]
- 77.Desvignes L et al. (2015) STIM1 controls T cell-mediated immune regulation and inflammation in chronic infection. J Clin Invest 125 (6), 2347–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou X et al. (2018) A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J Physiol 596 (14), 2681–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Feske S et al. (2001) Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol 2 (4), 316–24. [DOI] [PubMed] [Google Scholar]
- 80.Negulescu PA et al. (1994) Intracellular calcium dependence of gene expression in single T lymphocytes. Proc Natl Acad Sci U S A 91 (7), 2873–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Greenberg ML et al. (2013) Orai1 function is essential for T cell homing to lymph nodes. J Immunol 190 (7), 3197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Waite JC et al. (2013) Interference with Ca(2+) release activated Ca(2+) (CRAC) channel function delays T-cell arrest in vivo. Eur J Immunol 43 (12), 3343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Negulescu PA et al. (1996) Polarity of T cell shape, motility, and sensitivity to antigen. Immunity 4 (5), 421–30. [DOI] [PubMed] [Google Scholar]
- 84.Donnadieu E et al. (1994) Antigen recognition by helper T cells elicits a sequence of distinct changes of their shape and intracellular calcium. Curr Biol 4 (7), 584–95. [DOI] [PubMed] [Google Scholar]
- 85.Dong TX et al. (2017) Intermittent Ca(2+) signals mediated by Orai1 regulate basal T cell motility. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Svensson L et al. (2010) Calpain 2 controls turnover of LFA-1 adhesions on migrating T lymphocytes. PLoS One 5 (11), e15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bohineust A et al. (2018) Termination of T cell priming relies on a phase of unresponsiveness promoting disengagement from APCs and T cell division. J Exp Med 215 (5), 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shulman Z et al. (2014) Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science 345 (6200), 1058–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feske S et al. (2000) The duration of nuclear residence of NFAT determines the pattern of cytokine expression in human SCID T cells. J Immunol 165 (1), 297–305. [DOI] [PubMed] [Google Scholar]
- 90.Hermann-Kleiter N et al. (2012) Nuclear orphan receptor NR2F6 directly antagonizes NFAT and RORgammat binding to the Il17a promoter. J Autoimmun 39 (4), 428–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maus M et al. (2017) Store-Operated Ca(2+) Entry Controls Induction of Lipolysis and the Transcriptional Reprogramming to Lipid Metabolism. Cell Metab 25 (3), 698–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaufmann U et al. (2019) Calcium Signaling Controls Pathogenic Th17 Cell-Mediated Inflammation by Regulating Mitochondrial Function. Cell Metab 29 (5), 1104–1118 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klein-Hessling S et al. (2017) NFATc1 controls the cytotoxicity of CD8(+) T cells. Nat Commun 8 (1), 511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Geltink RIK et al. (2018) Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu Rev Immunol 36, 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Maul-Pavicic A et al. (2011) ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A 108 (8), 3324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schaballie H et al. (2015) A novel hypomorphic mutation in STIM1 results in a late-onset immunodeficiency. J Allergy Clin Immunol 136 (3), 816–819 e4. [DOI] [PubMed] [Google Scholar]
- 97.Byun M et al. (2010) Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med 207 (11), 2307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sahin G et al. (2010) Classic Kaposi sarcoma in 3 unrelated Turkish children born to consanguineous kindreds. Pediatrics 125 (3), e704–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Freund-Brown J et al. (2017) Cutting Edge: Murine NK Cells Degranulate and Retain Cytotoxic Function without Store-Operated Calcium Entry. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xu T et al. (2019) NFAT1 and NFAT2 Differentially Regulate CTL Differentiation Upon Acute Viral Infection. Front Immunol 10, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Matsumoto M et al. (2011) The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity 34 (5), 703–14. [DOI] [PubMed] [Google Scholar]
- 102.Crotty S (2011) Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29, 621–63. [DOI] [PubMed] [Google Scholar]
- 103.Ma CS et al. (2012) The origins, function, and regulation of T follicular helper cells. J Exp Med 209 (7), 1241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Martinez GJ et al. (2016) Cutting Edge: NFAT Transcription Factors Promote the Generation of Follicular Helper T Cells in Response to Acute Viral Infection. J Immunol 196 (5), 2015–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maltby S et al. (2018) Osteoblasts Are Rapidly Ablated by Virus-Induced Systemic Inflammation following Lymphocytic Choriomeningitis Virus or Pneumonia Virus of Mice Infection in Mice. J Immunol 200 (2), 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cheng KT et al. (2012) STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjogren’s syndrome. Proc Natl Acad Sci U S A 109 (36), 14544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xie MM and Dent AL (2018) Unexpected Help: Follicular Regulatory T Cells in the Germinal Center. Front Immunol 9, 1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Harrison OJ and Powrie FM (2013) Regulatory T cells and immune tolerance in the intestine. Cold Spring Harb Perspect Biol 5 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaufmann U et al. (2016) Selective ORAI1 Inhibition Ameliorates Autoimmune Central Nervous System Inflammation by Suppressing Effector but Not Regulatory T Cell Function. J Immunol 196 (2), 573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oh-Hora M et al. (2019) Stromal Interaction Molecule Deficiency in T Cells Promotes Spontaneous Follicular Helper T Cell Development and Causes Type 2 Immune Disorders. J Immunol 202 (9), 2616–2627. [DOI] [PubMed] [Google Scholar]
- 111.Yasuda K et al. (2019) The pathogenicity of Th17 cells in autoimmune diseases. Semin Immunopathol 41 (3), 283–297. [DOI] [PubMed] [Google Scholar]
- 112.Kim KD et al. (2014) Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J Immunol 192 (1), 110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yu Y et al. (2011) Prevention of GVHD while sparing GVL effect by targeting Th1 and Th17 transcription factor T-bet and RORgammat in mice. Blood 118 (18), 5011–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wu T et al. (2013) Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J Immunol 191 (1), 488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lock C et al. (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8 (5), 500–8. [DOI] [PubMed] [Google Scholar]
- 116.Bettelli E et al. (2004) Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 200 (1), 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lovett-Racke AE et al. (2004) Silencing T-bet defines a critical role in the differentiation of autoreactive T lymphocytes. Immunity 21 (5), 719–31. [DOI] [PubMed] [Google Scholar]
- 118.Cua DJ et al. (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421 (6924), 744–8. [DOI] [PubMed] [Google Scholar]
- 119.Ivanov II et al. (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126 (6), 1121–33. [DOI] [PubMed] [Google Scholar]
- 120.Schuhmann MK et al. (2010) Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol 184 (3), 1536–42. [DOI] [PubMed] [Google Scholar]
- 121.Woo JS et al. (2018) CRACR2A-Mediated TCR Signaling Promotes Local Effector Th1 and Th17 Responses. J Immunol 201 (4), 1174–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ahern PP et al. (2008) The interleukin-23 axis in intestinal inflammation. Immunol Rev 226, 147–59. [DOI] [PubMed] [Google Scholar]
- 123.Dietz L et al. (2015) NFAT1 deficit and NFAT2 deficit attenuate EAE via different mechanisms. Eur J Immunol 45 (5), 1377–89. [DOI] [PubMed] [Google Scholar]
- 124.Vaeth M et al. (2012) Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A 109 (40), 16258–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lee Y et al. (2012) Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13 (10), 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gaffen SL et al. (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14 (9), 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Korn T et al. (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27, 485–517. [DOI] [PubMed] [Google Scholar]
- 128.Fogli LK et al. (2013) T cell-derived IL-17 mediates epithelial changes in the airway and drives pulmonary neutrophilia. J Immunol 191 (6), 3100–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McKinley L et al. (2008) TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol 181 (6), 4089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Al-Ramli W et al. (2009) T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol 123 (5), 1185–7. [DOI] [PubMed] [Google Scholar]
- 131.Gavino AC et al. (2016) STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 71 (12), 1684–1692. [DOI] [PubMed] [Google Scholar]
- 132.Halwani R et al. (2017) Th-17 regulatory cytokines IL-21, IL-23, and IL-6 enhance neutrophil production of IL-17 cytokines during asthma. J Asthma 54 (9), 893–904. [DOI] [PubMed] [Google Scholar]
- 133.Calautti E et al. (2018) Psoriasis: A STAT3-Centric View. Int J Mol Sci 19 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gaublomme JT et al. (2015) Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell 163 (6), 1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liou J et al. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 15 (13), 1235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roos J et al. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol 169 (3), 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vig M et al. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312 (5777), 1220–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhang SL et al. (2006) Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc Natl Acad Sci U S A 103 (24), 9357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Riella LV and Alegre ML (2018) Novel Role of Calcineurin Inhibitors in Curbing T Cells’ Sweet Tooth. Am J Transplant 18 (1), 3. [DOI] [PubMed] [Google Scholar]
- 140.Limnander A et al. (2011) STIM1, PKC-delta and RasGRP set a threshold for proapoptotic Erk signaling during B cell development. Nat Immunol 12 (5), 425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tamas P et al. (2006) Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J Exp Med 203 (7), 1665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu X et al. (2016) T Cell Receptor-induced Nuclear Factor kappaB (NF-kappaB) Signaling and Transcriptional Activation Are Regulated by STIM1- and Orai1-mediated Calcium Entry. J Biol Chem 291 (16), 8440–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Berry CT et al. (2020) BCR-Induced Ca(2+) Signals Dynamically Tune Survival, Metabolic Reprogramming, and Proliferation of Naive B Cells. Cell Rep 31 (2), 107474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dutta D et al. (2017) Recruitment of calcineurin to the TCR positively regulates T cell activation. Nat Immunol 18 (2), 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stauderman KA (2018) CRAC channels as targets for drug discovery and development. Cell Calcium 74, 147–159. [DOI] [PubMed] [Google Scholar]
- 146.Concepcion AR et al. (2016) Store-operated Ca2+ entry regulates Ca2+-activated chloride channels and eccrine sweat gland function. J Clin Invest 126 (11), 4303–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shaw PJ and Feske S (2012) Physiological and pathophysiological functions of SOCE in the immune system. Front Biosci (Elite Ed) 4, 2253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Grimbacher B et al. (2016) The crossroads of autoimmunity and immunodeficiency: Lessons from polygenic traits and monogenic defects. J Allergy Clin Immunol 137 (1), 3–17. [DOI] [PubMed] [Google Scholar]
- 149.Schmidt RE et al. (2017) Autoimmunity and primary immunodeficiency: two sides of the same coin? Nat Rev Rheumatol 14 (1), 7–18. [DOI] [PubMed] [Google Scholar]