

Abstract
Locus coeruleus (LC) neurodegeneration is associated with cognitive deterioration during the transition from normal cognition to mild cognitive impairment (MCI) and Alzheimer disease (AD). However, the extent to which LC degenerative processes differentiate cognitively normal, “resilient” subjects bearing a high AD pathological burden from those with MCI or AD remains unclear. We approached this problem by quantifying the number of LC neurons and the percentage of LC neurons bearing AT8 tau pathology, TDP-43 pathology, or a marker for DNA/RNA oxidative damage, in well-characterized subjects diagnosed as normal cognition-low AD pathology (NC-LP), NC-high AD pathology (NC-HP), MCI, or mild/moderate AD. In addition, the severity of pontine arteriolosclerosis in each subject was compared across the groups. There was a trend for a step-wise ∼20% loss of LC neuron number between the NC-LP, NC-HP and MCI subjects despite a successive, significant ∼80%–100% increase in tau pathology between these groups. In contrast, increasing pontine arteriolosclerosis severity scores and LC oxidative stress burden significantly separated the NC-LP/HP and MCI/AD groups via comparative, correlation, and regression analysis. Pontine perfusion, as well as LC neuronal metabolic and redox function, may impact noradrenergic LC modulation of cognition during the preclinical and prodromal stages of AD.
Keywords: Alzheimer disease, Arteriolosclerosis, Locus coeruleus, Mild cognitive impairment, Oxidative stress, Pons, Resilience
INTRODUCTION
Locus coeruleus (LC) projection neurons, which transmit norepinephrine to mediate cognitive functions such as vigilance and attention, memory storage and retrieval, executive function, and affect (1–3) undergo extensive degeneration in the putative prodromal stage of Alzheimer disease (AD) termed mild cognitive impairment (MCI) (4–6), supporting the longstanding hypothesis that LC degeneration is a key feature of AD pathogenesis (4–10). Previously, we demonstrated that the extent of LC neuron loss observed postmortem is associated with a worsening of antemortem performance in multiple cognitive domains (e.g. episodic memory, working memory, semantic memory, perceptual speed) (4), similar to data showing that higher LC neuronal density is associated with slower rates of antemortem cognitive decline (11). Recent MRI advancements in in vivo LC imaging have underscored this concept by noting that: 1) higher educational attainment and verbal intelligence correlate with stronger LC signal (12), 2) older adults displaying LC volumetric integrity similar to younger adults perform better on learning and memory tasks (13), and 3) LC MRI findings may serve as a biomarker for AD progression (14–18). Taken together, these data suggest that noradrenergic LC projection system integrity may have a large role in mediating the changes from normal cognition (NC) to cognitive deterioration during AD. However, the extent to which LC neurodegeneration influences this conversion and the abetting mechanistic factors remain unclear.
To explore these issues further, we characterized LC and pontine pathology in postmortem brain tissue acquired from University of Kentucky Alzheimer’s Disease Research Center (UK-ADRC) participants who died with NC and with a low AD pathological burden (NC-LP), participants who died with NC yet displayed a relatively high AD pathological burden consistent with cognitive “resilience” (NC-HP) (19, 20), or those who died with clinical pathologic diagnoses of MCI or mild/moderate AD. Stereologic methods were used to quantify LC neuron number within a rigorously defined rostrocaudal counting frame, as well as the proportion of LC neurons bearing markers of tau pathology, TDP-43 pathology, or oxidative stress. Finally, adjacent sections were analyzed to estimate the extent of pontine arteriolosclerosis during disease progression.
MATERIALS AND METHODS
UK-ADRC Subjects and Case Selection
This study was determined to be exempt from human subjects review by the University of Kentucky and Michigan State University Institutional Review Boards. All procedures were performed in accordance with the ethical standards of the institutions and with the 1964 Helsinki declaration and its later amendments. Formalin-fixed, paraffin-embedded pontine tissue blocks were obtained from participants in the Healthy Brain Aging Volunteers cohort, under the purview of the National Institute of Aging (NIA)-funded UK-ADRC (21). Antemortem clinical evaluations for each subject followed the “Alzheimer’s Disease Centers’ Uniform Data Set,” as required by NACC (22, 23). Known history of substance abuse is an up-front exclusion criterion for this cohort. Pontine tissue blocks were selected from 4 distinct groups by applying the following inclusion and exclusion criteria: The first group consisted of subjects who died with NC and with low AD pathological burden (NC-LP), displaying Braak NFT stages 0–II and minimal or no plaque deposition (according the Consortium to Establish a Registry for Alzheimer Disease [CERAD] neuritic density plaque scores). The second group also consisted of NC subjects but they died with higher AD pathological burden (NC-HP), displaying Braak NFT stages III–V and minimal to severe plaque deposition. In addition, we selected tissue samples from subjects who died with a summary clinical pathologic diagnosis of MCI or mild/moderate AD (MCI separated from AD; Table 1). The diagnosis of dementia or AD met recommendations by the joint working group of the National Institute of Neurologic and Communicative Disorders and Stroke/AD and Related Disorders Association (24). The MCI subjects exhibited age- and education-adjusted impairments upon neuropsychological testing but did not meet the criteria for AD or dementia (21, 25).
TABLE 1.
Clinical, Demographic, and Neuropathological Characteristics by Diagnosis Category
| Diagnosis |
p value | Pair-wise comparison | ||||
|---|---|---|---|---|---|---|
| NC-LP (n = 8) | NC-HP (n = 7) | MCI (n = 7) | AD (n = 7) | |||
| Age (years) at death: | ||||||
| Mean ± SD (range) | 85.8 ± 5.2 (72–90) | 89.1 ± 6.2 (84–100) | 87.1 ± 3.8 (44–95) | 81.7 ± 9.1 (69–93) | 0.2* | — |
| Number (%) of males: | 5 (63%) | 3 (43%) | 3 (43%) | 5 (71%) | 0.6† | — |
| Years of education: | ||||||
| Mean ± SD (range) | 16.0 ± 2.6 (12–20) | 16.1 ± 0.9 (15–18) | 17.1 ± 1.1 (16–18) | 16.0 ± 2.3 (12–20) | 0.9* | — |
| Number (%) with ApoE ε4 allele | 1 (13%) | 1 (14%) | 3 (43%) | 4 (57%) | 0.06† | — |
| Number (%) with hypertension | 5 (63%) | 4 (57%) | 4 (57%) | 4 (57%) | 1.0† | — |
| MMSE | ||||||
| Mean ± SD (range) | 29.1 ± 0.8 (28–30) | 28.9 ± 1.5 (26–30) | 26.0 ± 1.9 (23–28) | 19.4 ± 4.1 (11–25) | 0.0001* | (NC-LP, NC-HP) > AD |
| Postmortem interval (hours) | ||||||
| Mean ± SD (range) | 3.8 ± 2.6 (1.8–10.0) | 3.8 ± 3.2 (1.3–10.8) | 3.4 ± 1.4 (1.2–6.0) | 4.4 ± 2.1 (2.7–8.5) | 0.6* | — |
| Identified microinfarcts | ||||||
| Mean ± SD (range) | 0.2 ± 0.4 (0–1) | 0.6 ± 0.8 (0–2) | 0.6 ± 0.8 (0–7) | 0.7 ± 1.0 (0–2) | 0.1* | — |
| Distribution of Braak scores: | ||||||
| 0 | 2 | 0 | 0 | 0 | ||
| I/II | 6 | 0 | 3 | 1 | 0.0005* | NC-LP < (NC-HP, AD) |
| III/IV | 0 | 7 | 4 | 3 | ||
| V/VI | 0 | 0 | 0 | 3 | ||
| NIA Reagan diagnosis(likelihood of AD) | ||||||
| No AD | 2 | 1 | 1 | 0 | ||
| Low | 5 | 1 | 3 | 1 | 0.02* | NC-LP < AD |
| Intermediate | 1 | 5 | 3 | 3 | ||
| High | 0 | 0 | 0 | 3 | ||
| CERAD diagnosis | ||||||
| No AD | 4 | 1 | 2 | 0 | ||
| Possible | 1 | 1 | 2 | 0 | 0.07* | — |
| Probable | 2 | 3 | 3 | 5 | ||
| Definite | 1 | 2 | 0 | 2 | ||
AD, Alzheimer disease; CERAD, Consortium to Establish a Registry for Alzheimer disease; MCI, mild cognitive impairment; NC-LP, normal cognition-low AD pathology; NC-HP, normal cognition-high AD pathology.
Kruskal-Wallis test, with Dunn’s test for multiple comparisons.
Fisher exact test, with Bonferroni correction for multiple comparisons.
Exclusion criteria included the presence of impactful non-AD neuropathological processes (e.g. frank hippocampal sclerosis, Lewy bodies), significant cerebrovascular pathology (e.g. large strokes or lacunes), the presence of comorbid neurologic disease or psychiatric disorders, chronic infectious disease, head injury, and/or brain cancer (21). Finally, pontine blocks with discernable oblique cuts or tissue blocks missing a significant extent of the LC based on the operational landmarks as outlined below were excluded from analysis.
Tissue Processing, Histology, and Operational Criteria for LC Morphometric Analyses
Tissue blocks from cases meeting the criteria described above were sectioned at 20-μm thickness on a HM 355S automatic rotary microtome (Thermo Scientific, Waltham, MA) and mounted onto charged slides. Prior to analysis, a 1:12 series of slides were heated, deparaffinized in xylenes, and rehydrated in an ethanol series. Slides were then stained for 5 minutes in hematoxylin solution (Harris’ Modified, Thermo) and differentiated in acid alcohol and ammonia water, with rinses in water between each incubation, followed by immersion in eosin (Eosin Y, Thermo) solution for 3 minutes then subsequently dehydrated in an ethanol series and cover slipped with Cytoseal (Thermo). Slides were evaluated to determine whether the decussation of cranial nerve IV was visible in the vellum and that the mesencephalic tract of V was dorsolateral to the neuromelanin-pigmented neurons of the LC (Fig. 1). These operational criteria allowed us the opportunity to include a maximal number of available cases in each diagnostic group while ensuring a standardized counting frame for each tissue block. To ensure rigor and promote reproducibility, all subsequent analyses were performed using randomization and blinding.
FIGURE 1.
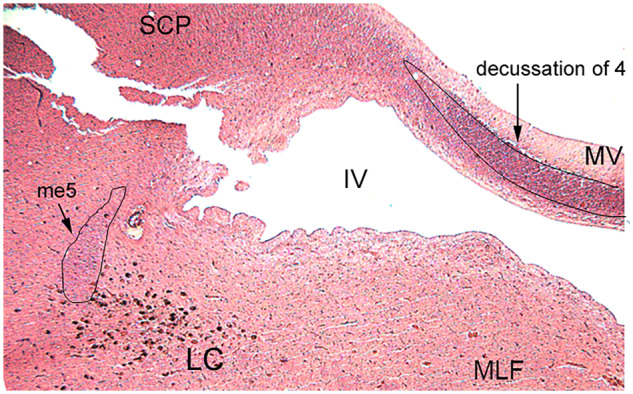
Operational definition of locus coeruleus (LC) counting frame. Hematoxylin and eosin stain shows the requisite landmarks in a representative rostral pons tissue section at the start of the counting frame. The decussation of 4 (trochlear nerve) was visible in the medullary vellum (MV) and the mesencephalic tract of 5 (trigeminal nerve, me5) was dorsolateral to the pigmented LC neuron field. Identification of these anatomical relationships was used as inclusion criteria in case selection and to normalize the rostrocaudal extent of the LC analyzed by stereology across the cases. Abbreviations: SCP, superior cerebellar peduncle; MLF, medial longitudinal fasciculus; IV, fourth ventricle.
Immunohistochemistry
Slides representing the 1:12 series for each analysis and each case were deparaffinized and rehydrated, washed in Tris-buffered saline ([TBS]; pH 7.4) for 10 minutes, incubated at 65°C for 20 minutes in preheated citric acid buffer (pH 6.0), and then cooled to room temperature. Slides were washed in TBS + 0.5% Triton X-100 (TBS-TX) for 10 minutes followed by a 10-minute incubation in 10% hydrogen peroxide (H2O2) in methanol to quench endogenous peroxidase activity. Slides were then washed in three 10-minute exchanges of TBS-TX and blocked in TBS-TX/10% goat serum/2% bovine serum albumin at room temperature for 1 hour. Slides were dried, and a hydrophobic barrier drawn with a PAP pen. Primary antibody was aliquoted directly onto the slides and incubated in a humidified chamber overnight at 4°C. The primary antibodies used in this study were rabbit antityrosine hydroxylase (TH) antiserum (Abcam no. Ab112, Cambridge, UK, 1:500) to label noradrenergic LC neurons, the mouse anti-AT8 monoclonal antibody (Thermo no. MN1020, 1:1000) to label tau pathology, rabbit antiserum to TDP-43 (ProteinTech no. 10782, Rosemont, IL, 1:100) to evaluate aberrant cytoplasmic mislocalization, and the mouse monoclonal anti-oh8G/dG antibody (QED Biosciences no. 12501, San Diego, CA, 1:250) to label DNA/RNA oxidative damage. Following primary incubation, sections were incubated in biotinylated anti-rabbit or antimouse secondary IgG (Vector Laboratories, Burlingame, CA, 1:500) at room temperature for 2 hours, followed by Vector ABC detection kit using horseradish peroxidase (Vector Laboratories). Antibody labeling was visualized using the Vector SG chromogen (Vector Laboratories) or 0.5 mg/ml 3,3′ diaminobenzidine with nickel II sulfate enhancement and 0.03% H2O2 in TBS. Control reactions included deletion of the primary antibodies and preadsorption with recombinant protein (4, 26, 27).
Estimation of LC Neuron Number and Pathology
Quantitative analysis was performed for TH-immunopositive or neuromelanin-positive LC neurons in each case using a Nikon Eclipse 80i microscope (Nikon Instruments, Melville, NY) hard-coupled to a MAC5000 computer-controlled x-y-z motorized stage (Ludl Electronic Products, Hawthorne, NY) and a Retiga 4000 R camera (Qimaging, Surrey, BC Canada), and equipped with StereoInvestigator v9.0 software (MBF Bioscience Williston, VT). Briefly, the operator drew a closed contour around the LC and used the StereoInvestigator Meander Scan function to count labeled cells via total enumeration within the contour on every 12th section, with 10–14 slides per case. Raw cell counts were by multiplied by the sampling fraction to extrapolate the population estimate. The same procedures were used to quantify neurons bearing AT8 tau or RNA/DNA oxidative damage. However, these markers were expressed as the ratio of neuromelanin-positive LC cells for each case.
Histological Assessment of Pontine Vascular Pathology
Semiquantitative assessment of pontine arteriolosclerosis at the level of the LC was examined and scored for severity using a 1:12 series of hematoxylin and eosin-stained slides. Tissue was rated for the extent of arteriolosclerosis in each case in a randomized and blinded manner, as previously described (28, 29). Briefly, the StereoInvestigator Meander Scan function was employed to identify and score the first 6 arterioles coursing perpendicular to the plane of each tissue section in a random fashion (12 sections/case = 72 microvessels/case). The severity was graded on a 4-tier semiquantitative scale: 0 (normal appearing); 1 (mild, with mild thickening of the vessel wall and modest disruption of vessel wall and neuropil), 2 (moderate, with moderate thickening of the vessel wall and often tortuous vessel contours), and 3 (severe, with marked dysmorphic changes including evidence for vessel occlusion) (28).
Power Analysis, Sample Size, and Statistics
Based on stereologic data of LC neuron number from our previous study (4) and α = 0.05, a sample size of 6/group was determined have 80% power to detect an effect size of 1.25 standard deviations between NC-LP and MCI. Having applied the inclusion/exclusion criteria for case selection, we identified the following number of cases for each diagnostic group: NC-LP (n = 8), NC-HP (n = 7), MCI (n = 7), and mild/moderate AD (n = 7; total n = 29). Normality of cohort demographic, clinical, and pathologic characteristics as well as experimental outcomes was tested using the Shapiro-Wilk test (30). Data displaying Gaussian distribution were analyzed by 1 or 2-way ANOVA with Bonferroni correction; otherwise, data were analyzed by Kruskal-Wallis with Dunn’s post hoc testing. Associations between outcome variables and were tested using Spearman rank correlations. Significantly correlated associations were analyzed further by linear or logistic regression. The level of statistical significance was set at p < 0.05.
RESULTS
Demographic, Clinical, and Neuropathological Characteristics
Table 1 summarizes the demographic, clinical, and neuropathological characteristics of the 29 cases examined. There were no significant differences in age, sex, education level, ApoE4 status, comorbid hypertension, postmortem interval (PMI), microinfarcts, or CERAD neuritic plaque scores across the groups examined.
The AD subjects performed significantly worse on the Mini-Mental State Examination (MMSE) compared with the NC-LP (p = 0.0004) and NC-HP (p = 0.001) cases. As expected, Braak NFT stages were also significantly different across the clinical groups. The NC-LP cases displayed significantly lower Braak NFT stages than the NC-HP (p = 0.0045) and AD (p = 0.001) groups. NC-LP cases were classified as Braak NFT stages 0 (25%), I/II (75%), III/IV (0%), or V/VI (0%). The NC-HP cases were Braak NFT stages III/IV (100%). The MCI cases were Braak NFT stages I/II (43%), III/IV (57%), and V/VI (0%), and the AD cohort was classified as Braak stages I/II (14%) III/IV (43%) or V/VI (43%). As expected, the NIA-Reagan diagnosis for likelihood of AD differentiated NC-LP from AD subjects (p = 0.01).
LC Neuron Number Was Decreased in MCI and AD and Displayed Topographical Differences among the NC-LP, NC-HP, and MCI Groups
The mean estimated number of TH-positive LC neurons in the NC-LP group was (17712 ± 4782; range = 12 432–24 516), whereas neuron number was progressively decreased in the NC-HP (13 959± 6024; range = 6240–21 522), MCI (9859± 4384; range = 2940–15 152) and AD (1620± 5213; range = 1620–15 900) groups (Fig. 2A, B). There was a trend noted (∼22%) for a decrease in LC neuron number in NC-HP compared with NC-LP subjects (p = 0.25), whereas a significant ∼40% decrease was observed in the number of LC neurons in MCI compared with NC-LP cases (p = 0.03). An additional ∼10%–15% decrease in LC neuron number was quantified in AD compared with NC-LP resulting in a ∼50%–55% loss of LC neurons in the AD group compared with NC-LP (p = 0.006). Notably, LC neuron number in the NC-HP group was not statistically different from either MCI (p = 0.43) or AD (p = 0.16; Fig. 2B). A comparison of total estimated TH+ LC neuron numbers to total estimated neuromelanin+ LC neuron numbers did not reveal significant differences across the groups (p = 0.8 via 2-way ANOVA with Bonferroni correction), suggesting a preservation of noradrenergic tone in surviving LC neurons during disease progression. Finally, there were no differences in total TH+ LC neuron number between males and females in the diagnostic groups (p = 0.25 via 2-way ANOVA with Bonferroni correction).
FIGURE 2.

Locus coeruleus (LC) neuron loss and topography of degeneration across the diagnostic groups. (A) Representative tyrosine hydroxylase (TH)-immunostained sections showing step-wise LC neuron loss across the diagnostic groups. (B) TH+ cell counts revealed a significant ∼40% decrease in the number of LC neurons in mild cognitive impairment (MCI) compared with normal cognition-low Alzheimer disease (AD) pathology (NC-LP) cases and a ∼50%–55% loss of LC neurons in the AD group compared with NC-LP. There was a nonsignificant ∼22% decrease in LC neuron number in normal cognition-high AD pathology (NC-HP) compared with NC-LP subjects. The ∼18% reduction in LC neuron number between MCI and NC-HP was not significant, nor was the ∼30% difference in cell numbers between AD and NC-HP groups. *p < 0.05; **p < 0.01 via 1-way ANOVA with Bonferroni correction. (C) LC neuron numbers in each case were distributed by rostrocaudal topography. Linear regression revealed that the rostrocaudal extent of LC neuron loss cell loss was significantly different among all cases (p < 0.001), with regression curves maintaining significant differences between NC-LP and NC-HP (p = 0.006) and between NC-HP and MCI (p < 0.001). LC neuron numbers differed significantly between NC-LP and NC-HP within slides 3, 4, 7–11. *p < 0.05 via 2-way ANOVA with Bonferroni correction. Scale bar = 100 μm.
Although the number of TH-positive neurons in the LC of NC-HP individuals did not differ significantly in comparison to NC-LP or MCI groups, the topographical pattern of LC degeneration in NC-HP did differ from NC-LP (Fig. 2C). Since the rostral boundary of the LC for each tissue series was operationally defined (see “Materials and Methods”), TH-positive neuron counts on each slide were compared in a rostrocaudal manner among the groups. Rostrally, there was clear separation between higher LC cell counts in NC-LP and NC-HP compared with MCI and AD, and regression analysis indicated significantly different patterns of rostral to caudal LC number across the groups (p < 0.001), wherein the topography of LC cell loss in NC-HP more closely mirrored NC-LP in the rostral LC, yet aligned with MCI and AD in the more caudal portions (Fig. 2C).
LC Neuron Number Was Associated With Global Cognitive Performance
Estimated TH-positive LC neuronal counts were correlated with demographic data, antemortem cognitive test performance, and postmortem neuropathologic variables. Neuron number was not associated with age (r = 0.23, p = 0.22) or PMI (r = 0.12, p = 0.5). However, lower LC neuron numbers and MMSE scores displayed a significant positive association (r = 0.5379, p = 0.0026). With respect to neuropathological diagnostic criteria, a reduction in TH-positive LC neuron numbers was not correlated with increasing measures of neuropathology as characterized by Braak NFT stages (r = 0.283, p = 0.21), NIA-Reagan (r = −0.10, p = 0.60), or CERAD (r = −0.024, p = 0.9) staging.
LC TDP-43 Pathology in the Diagnostic Groups
TDP-43 proteinopathy was observed in only 1 of the AD cases, reflecting the inclusion/exclusion criteria.
LC Tau Pathology Was Increased in NC-HP and MCI and Correlated With Clinical Pathologic Variables
LC tau pathology across the groups was evaluated using the ratio of AT8-positive to neuromelanin-positive LC neurons in each case (Fig. 3). There was a significant ∼8-fold increase in LC neuronal tau pathology in NC-HP compared with o NC-LP subjects (p = 0.0005), in contrast to an equivalent amount of tau pathology between the NC-HP and MCI groups (p = 0.43; Fig. 3A, B). AT8 burden in LC neurons was associated with reductions in TH-positive LC neuron number (r = −0.67, p < 0.0001), higher Braak stage (r = 0.46, p = 0.012), and lower MMSE scores (r = −0.5, p = 0.008) across subjects.
FIGURE 3.

Locus coeruleus (LC) tau pathology across the diagnostic groups. (A) Representative AT8-immunostained sections showing a step-wise increase in the number of neuromelanin+ LC neurons bearing neurofibrillary tangles (arrows). Note that in later disease stages AT8 labeling is seen in neurons devoid of neuromelanin (arrowheads). (B) The ratio of neuromelanin-positive LC neurons expressing AT8+ tau pathology was significantly increased ∼3-fold in NC-HP cases and ∼4- to 5-fold in MCI and AD cases compared with NC-LP. **p < 0.01; ****p < 0.0001 via 1-way ANOVA with Bonferroni correction. Scale bar = 50 μm.
LC Neuronal Oxidative Stress Was Increased in MCI and AD
LC neuronal oxidative stress pathology across the groups was evaluated using the ratio of oh8G/dG-positive (i.e. neurons expressing DNA/RNA oxidative modifications) in neuromelanin-positive LC neurons in each case (Fig. 4). Although ∼80% of LC neurons in NC-LP and NC-HP subjects displayed evidence of substantial oxidative stress (Fig. 4A, B), levels of the oxidative damage marker were significantly higher in LC neurons in MCI (p = 0.0017) and AD (p < 0.0001) subjects compared with NC-HP subjects (Fig. 4B). Furthermore, increased LC neuronal oxidative stress marker was significantly correlated with increased LC tau pathology (r = 0.48, p < 0.006), decreased LC cell number (r = −0.46, p = 0.012), and lower MMSE score (r = −0.68, p < 0.0001).
FIGURE 4.

Accrual of LC neuronal oxidative stress across the diagnostic groups. (A) Representative oh8G/dG-immunostained sections show prominent nucleic acid oxidative damage in all diagnostic groups (arrows), with evidence for increasing oxidative stress burden in later disease stages. Note that almost all LC neurons were labeled with the DNA/RNA oxidative marker in AD. (B) The ratio of LC neurons expressing oh8G/dG+ oxidative stress pathology was high (see Y-axis) but similar in NC-LP and NC-HP cases. In contrast, LC neuronal oxidative damage was significantly increased in MCI and AD cases compared with NC-LP, as well as NC-HP. *p < 0.05, **p < 0.01; ****p < 0.0001 via 1-way ANOVA with Bonferroni correction. Scale bar = 50 μm.
Pontine Arteriolosclerosis Severity Was Increased in MCI and AD
Arteriolosclerosis severity scoring (28, 29) of arterioles in stage 0 (Fig. 5A), stage 1 (Fig. 5B), stage 2 (Fig. 5C), or stage 3 (Fig. 5D) was performed in the pons at the level of the LC. Severity ratings revealed no differences between NC-LP and NC-HP subjects (p = 0.7; Fig. 5E). In contrast, there was significantly greater arteriole pathology in MCI (p = 0.0053) and AD (p < 0.0001) compared with NC-HP (Fig. 5E). Arteriolosclerosis severity was not associated with TH-positive LC neuron number in the subjects (r = −0.3, p = 0.11) or global cerebral arteriolosclerosis severity (r = −0.21, p = 0.32). However, the degree of pontine arteriole pathology significantly correlated with LC neuronal oxidative stress (r = 0.62, p = 0.0004), LC neuronal tau pathology (r = 0.61, p = 0.0004), Braak stage (r = 0.47, p < 0.01), and individual MMSE scores (r = −0.8, p < 0.0001).
FIGURE 5.

Pontine arteriolosclerosis severity across the diagnostic groups. Representative images of vessels graded as stage 0 (A), stage 1 (B), stage 2 (C), or stage 3 (D), according to arteriolosclerosis severity by an observer blinded to diagnosis, as described in “Materials and Methods.” (E) Pontine arteriolosclerosis severity was similar in NC-LP and NC-HP cases. In contrast, pontine arteriolosclerosis was significantly more severe in MCI and AD cases compared with NC-LP, as well as NC-HP. *p < 0.05, **p < 0.01; ***p < 0.001; ****p < 0.0001 via 1-way ANOVA with Bonferroni correction. Scale bars = 50 μm (stage 0), 70 μm (stage 1), 100 μm (stage 2), and 90 μm (stage 3).
Regression Analysis of LC Variables Across Diagnostic Groups
Linear regression revealed that indices of pontine arteriolosclerosis, LC neuronal oxidative stress, and LC neuronal tau pathology, but not age or PMI, were predictors of LC neuron number (Table 2). Additional analyses showed that pontine arteriolosclerosis, LC neuronal oxidative stress, LC neuronal tau pathology, and LC neuron number also predicted MMSE scores. Finally, these variables as well as MMSE score and Braak NFT stage predicted the diagnostic categorization of NC-LP, NC-HP, MCI, or AD (Table 2).
TABLE 2.
Linear Regression Analysis of Postmortem Variables and Cohort Characteristics
| LC neuron number | MMSE score | Diagnosis | |
|---|---|---|---|
| Pontine arteriosclerosis | r2 = 0.4 | r2 = 0.45 | r2 = 0.63 |
| p = 0.0002 | p < 0.0001 | p < 0.0001 | |
| LC oxidative damage | r2 = 0.3 | r2 = 0.47 | r2 = 0.55 |
| p = 0.002 | p < 0.0001 | p < 0.0001 | |
| LC tau pathology | r2 = 0.4 | r2 = 0.38 | r2 = 0.65 |
| p = 0.0002 | p = 0.0004 | p < 0.0001 | |
| LC neuron number | X | r2 = 0.4 | r2 = 0.38 |
| p = 0.0002 | p = 0.0003 | ||
| Age | r 2 = 0.05 | r2 = 0.19 | r 2 = 0.04 |
| p = 0.22 | p = 0.02 | p = 0.27 | |
| PMI | r 2 = 0.02 | N/A | N/A |
| p = 0.5 | |||
| MMSE | r2 = 0.24 | X | r2 = 0.57 |
| p = 0.007 | p < 0.0001 | ||
| Braak stage | r 2 = 0.1 | r 2 = 0.13 | r2 = 0.39 |
| p = 0.09 | p = 0.054 | p = 0.0003 | |
| Reagan diagnosis | r 2 = 0.01 | r 2 = 0.11 | r 2 = 0.12 |
| p = 0.6 | p = 0.07 | p = 0.06 | |
| CERAD diagnosis | r 2 = 0.06 | r2 = 0.16 | r 2 = 0.12 |
| p = 0.2 | p = 0.03 | p = 0.06 |
CERAD, Consortium to Establish a Registry for Alzheimer Disease; LC, Locus coeruleus; MMSE, Mini-Mental State Examination. Boldface indicates significant differences.
Next, logistic regression was performed to understand which variables might influence whether the subject was NC-HP or MCI (i.e. relatively high AD pathology but unimpaired vs cognitively impaired). Not surprisingly, MMSE score predicted this diagnostic differentiation; however, in the multivariable model Braak NFT stage, Reagan AD criteria-based diagnosis, and CERAD neuritic plaque score did not (Table 3). Of all other variables examined, only the severity of pontine arteriolosclerosis (r2 = 0.4, p = 0.01, area under the curve [AUC] = 0.85) and LC neuronal oxidative damage (r2 = 0.5, p = 0.003, AUC = 0.9) predicted whether the subject was classified as NC-HP or MCI (Table 3).
TABLE 3.
Logistic Regression Analysis of Variables Differentiating the NC-HP and MCI Groups
| LC Neuron Number | LC tau Pathology | LC Oxidative Damage | Pontine Arteriosclerosis | Age | MMSE | Infarcts | Braak | Reagan | CERAD | |
|---|---|---|---|---|---|---|---|---|---|---|
| r 2 | 0.14 | 0.14 | 0.51 | 0.41 | 0.09 | 0.48 | 0.20 | 0.22 | 0.08 | 0.06 |
| p | 0.22 | 0.17 | 0.003 | 0.01 | 0.25 | 0.004 | 0.15 | 0.1 | 0.3 | 0.4 |
| AUC | 0.70 | 0.71 | 0.90 | 0.85 | 0.68 | 0.89 | 0.72 | 0.75 | 0.66 | 0.64 |
AUC, area under the curve; LC, locus coeruleus; MCI, mild cognitive impairment; NC-HP, normal cognition-high AD pathology. Boldface indicates significant differences.
DISCUSSION
LC neuronal degeneration during the incipient stages of AD has been reported by our group and others (4–6, 9, 17), and this study supports prior concepts related to the relationship between LC cell loss and cognitive status during the progression of AD (4, 6). Given the physiological role of forebrain noradrenergic signaling in sleep/wake cycles and affective responses such as mood and stress (1–3, 31–34), which are often disturbed even in the AD prodrome (35–38), early LC cell loss could also impact these emerging symptoms in addition to cognitive decline. In this study, we explored potential differences in LC status between unimpaired subjects with low (NC-LP) versus high (NC-HP) AD pathology vs those with MCI and AD, using the NC-HP group as a biological contrast between NC-LP subjects and elderly people with increasingly severe pathological and clinical disease. Quantitative analysis of multiple pathological markers showed that the NC-HP and MCI groups can be differentiated by the relative severity of pontine arteriolosclerosis and LC oxidative stress burden in the subjects examined and, moreover, that these 2 markers were highly predictive for discriminating between the 2 groups.
NIA workshop reports on the neuropathologic assessment (39, 40) and the preclinical definition (20, 41) of AD acknowledged that people who died with a NC-HP classification may “never manifest clinical symptoms in their lifetime” (20). However, from an operational perspective, this group provides a critical window into the mechanistic progression from the “normal” biology of the aged brain to biological processes associated with prodromal AD (19), and may provide new insights into novel mechanisms linking pathology and dementia. Hence, we also tested whether there were specific measures of LC pathology that differentiated NC-HP and MCI subjects to define potential factors influencing the transition from “resilience” to cognitive impairment.
Quantification of LC neuron numbers revealed a step-wise loss of LC neurons between the NC-LP and NC-HP stage (∼22%) and further neuronal loss between NC-HP and MCI (∼18%; Fig. 2). These differences were not statistically significant, due perhaps to the sample size of subjects who met the inclusion/exclusion criteria within each group—a limitation of the study. However, there was a significant ∼40% loss between the NC-LP and MCI groups that contrasts with our previous study quantifying a ∼30% loss of LC neurons between cognitively unimpaired and MCI subjects (4). This discrepancy could be related to cohort group classifications, tissue preparation (e.g. present use of paraffin-embedded vs free-floating sections) or quantification methods (e.g. present use of total enumeration vs the optical disector probe for cell counting). In any event, these results expand upon and offer new insights into LC degeneration in the earliest stages of AD. For example, Theofilas and colleagues reported that LC volume decreases ∼8.4% with each successive Braak stage, resulting in a significant ∼25% loss of LC volume in control (CDR 0) cases neuropathologically diagnosed postmortem as Braak NFT stage III compared with stage 0 (5). All of the NC-HP cases in the present study were classified as Braak NFT stages III/IV, so the ∼22% decrease in LC neuron number observed here aligns well with these volumetric measures. Interestingly, this previous study also noted that rates of change in the total number of neurons (±neuromelanin) within the LC became significant only in subjects with Braak NFT stages III–VI. When compared with our present results counting TH-labeled LC neurons, it is possible that the relatively constant rate of volume loss observed in this prior study was being driven by phenotypically noradrenergic LC neuron loss (5).
In contrast, Arendt et al. (6) previously used stereology to demonstrate 1) a nonsignificant ∼3% loss of neuromelanin-positive LC neurons in subjects classified as preclinical AD (CDR 0 who also displayed “low” amyloid-Braak-CERAD [ABC] diagnostic scores [42]) compared with those classified as controls (CDR 0 and “not” ABC score), and 2) a significant ∼13% loss of LC neurons in subjects classified as MCI/prodromal AD (CDR 0.5 who also displayed “low” to “intermediate” ABC scores [42]) compared with controls. Whereas the cases in the present study were not assigned an ABC score, the Braak stage and CERAD scores suggest that the NC-HP and MCI individuals predominantly displayed an “intermediate” AD neuropathologic changes. Finally, our estimate of total TH-positive LC cell number in NC-LP (17 712 ± 4782) was similar to other LC morphometric studies (43–45) and, more specifically, to Arendt et al.’s (6) unbiased estimate of neuromelanin-positive neurons (17 487 ± 2736) for control subjects. Moreover, we report a rostrocaudal gradient of topographical LC cell loss (Fig. 2), consistent with the topography of LC volume and cell loss reported in AD (5, 46). Although there was considerable variability across the cases in the present study, we offer new perspectives on this concept by showing that there was a clear separation between the relative preservation of rostral, forebrain-projecting LC neurons (47) in the NC-LP and NC-HP cases compared with the apparent degeneration of these same neurons in the MCI and AD cases. It was also interesting to note the relatively steep rostrocaudal gradient of LC cell loss in the NC-HP cases, which aligned more closely with the NC-LP group rostrally but more closely with the AD group caudally. These observations suggest that analyzing discrete topographical differences in rostral LC neuron loss—and their respective projection sites—between NC-HP and MCI individuals may inform the anatomical basis for how the LC forebrain projection system vulnerability affects the onset of cognitive decline (13).
An additional morphological property that might impact the transition from NC-HP to MCI is the extent of the LC dendritic fields between the 2 groups. The LC displays a complex dendritic arborization pattern (48) receiving prominent afferent inputs including reciprocal glutamatergic innervation from prefrontal and anterior cingulate cortices (49, 50). Interestingly, Boros and colleagues recently demonstrated similar spine densities in prefrontal cortex of NC-LP and NC-HP subjects compared with AD subjects (51), whereas spine head diameter was inversely correlated with MMSE scores (52). Although we were unable to reliably visualize TH-positive neurites for evaluation in the paraffin-embedded sections, it is possible that the structural integrity of the dendritic architecture of the LC neurons is more stable and intact among the remaining LC neurons in the NC-HP patients compared with MCI and AD.
With respect to pathologic changes that might differentiate NC-LP, NC-HP, and MCI, we quantified the percentage of neuromelanin-positive neurons LC neurons bearing markers for tau pathology (AT8), TDP-43 pathology, or oxidative stress (Oh8G/dG as a marker for DNA/RNA oxidative modifications), as well as the severity of pontine arteriolosclerosis. TDP-43 pathology was seen only very rarely in this sample, due to the study’s exclusion criteria. In contrast, AT8 tau pathology—which spans early and late-stage tangles (53)—was progressively increased in NC-HP, MCI, and AD cases compared with NC-LP (Fig. 3), and was significantly associated with reductions in TH-positive LC neuron number, higher Braak NFT stage, and lower MMSE scores. While these observations support Braak’s revised “pretangle” stage of AD progression (54), the lack of significant differences between AT8-positive neurons in NC-HP compared with MCI suggests that LC tau pathological load does not necessarily impact whether a person is cognitively normal or impaired.
On the other hand, there was a significant increase in the ratio of LC neurons bearing oxidative damage in MCI and AD individuals compared with NC-LP or NC-HP individuals. Previously, we reported a downregulation of specific transcripts involved in mitochondrial (e.g. cytochrome C1 and nuclear respiratory factor 1) and redox (eg superoxide dismutase 2 and glutathione peroxidase) function within individual LC neurons from amnestic MCI compared with cognitively intact subjects; moreover, reduced levels of these transcripts correlated with worsening antemortem cognitive variables (4). We used the Oh8G/dG marker as a proxy for cellular oxidative stress in this study, but our findings more specifically suggest that DNA and RNA oxidative damage may contribute to the dysregulation of these key functional classes of genes, which in turn promotes the high oxidative environment within aged LC neurons and the significant increases observed in MCI and AD. Such a deleterious environment may correspond with reduced cellular energy charge driving synaptic dysfunction in projection zones (55, 56).
The source of this high level of oxidative damage within noradrenergic LC neurons and the significant increase in the level of oxidative stress between NC-HP and MCI is unclear, yet a novel observation from our study was that the severity of pontine arteriolosclerosis was also significantly increased during the transition from HC-HP to MCI. The vascular bed of the LC is supplied by the penetrating pontine arteries and is very dense, displaying an estimated ∼1250 mm length per cubic millimeter of tissue in capillaries alone (57). This vascular density was hypothesized to be greater than any other nucleus in the CNS except the supraoptic nucleus (57). Interestingly, polymorphisms in certain AD risk genes (APOE4, CR1, BIN1, and PICALM) contribute both to an earlier time of onset of AD and to the development of coronary artery disease or stroke (58–61), whereas radiographic studies have demonstrated reduced basilar artery wall sheer stress in MCI and AD that was significantly associated with MMSE scores (62). The extent of hypertension was equivalent across the groups (Table 1) suggesting a myriad of potential sources for differential cerebrovascular disease in the subjects, but the present findings suggest that arteriolar pathology within the LC could lead to hypoperfusion and hypoxic conditions (63), resulting in LC neuronal oxidative and metabolic stress, as noted above (4), as well as LC toxicant burden (64). In this regard, Pamphlett et al. (65, 66) have used mass spectrometric analysis and X-ray fluorescence microscopy to demonstrate the presence neurotoxic mercury as well as cadmium, silver, lead, iron, copper, bromine, rubidium, selenium, and nickel in LC neurons in aged subjects. Given additional evidence that these metals contribute to cellular oxidative damage (67, 68) and that heavy metals in neuromelanin-containing organelles can result in neurodegeneration (69), LC neurons in the aged brain may be primed for further toxicant damage during AD. Since LC projection system axons innervate microvessels in the CNS (70–73), regulate blood flow, vessel diameter, and blood volume changes in regions of oxygen demand (74–77), and disrupt several markers of cerebrovascular function upon LC axonal lesions in rat models of AD (78, 79), it is tempting to consider a model wherein LC degenerative processes due to local aberrant perfusion triggers feed-forward vascular pathology in forebrain projection zones.
To identify relationships among LC pathologic variables and clinical neuropathologic criteria among the subjects examined, we used regression analyses. These produced models to understand which variables might more directly influence whether the subject was cognitively impaired. We focused on the conversion between NC-HP and MCI. Linear regression showed that LC neuron number predicted MMSE scores and diagnostic category, whereas LC tau pathology, LC oxidative stress, and pontine arteriolosclerosis severity were all predictors of LC neuron number, MMSE score, and diagnostic category. However, logistic regression showed that only LC oxidative stress, pontine arteriolosclerosis severity and, predictably, MMSE scores were able to discriminate between subjects with relatively high AD pathology who were unimpaired (NC-HP) versus those who were cognitively impaired (MCI).
In summary, we explored mechanisms underlying the transitions along the spectrum from normal/minimal pathology to impaired/with AD-type pathology. The observation for incremental LC neuron loss across this clinical-pathological spectrum, and the relative preservation of rostral LC neurons in NC-HP compared with MCI cases, suggest that there is a critical threshold of rostral, forebrain-projecting neuron loss that contributes to a loss of resilience and onset of cognitive decline and potentially comorbid behavioral disturbances in wakefulness and affect. This threshold may not be driven primarily by the accumulation of LC neuronal tau pathology since there was a striking and equivalent increase of tau pathology in LC neurons from NC-HP and MCI compared with NC-LP subjects. However, the observation that LC neuronal oxidative stress and pontine arteriolosclerosis differentiate HC-NP and MCI subjects indicates that these 2 factors may play a hitherto underappreciated role in this transition. We posit that pathology in the LC vascular bed, subsequent oxidative and metabolic stress, and LC neuronal dysfunction in cognitive efferent regions contribute to the onset of cognitive impairment and dementia and represent rational therapeutic targets for disease modification.
ACKNOWLEDGMENT
We thank the UK-ADRC participants for their altruism and support of brain research in healthy aging and disease.
This study was supported by NIH R01 AG060731, P01 AG014449, P30 AG053760, and P30 AG028383, as well as donors to the BrightFocus Foundation (ADR20201187S), the Saint Mary’s Foundation, and Miles for Memories of Battle Creek, MI.
The authors have no duality or conflicts of interest to declare.
REFERENCES
- 1. Berridge CW, Waterhouse BD.. The locus coeruleus-noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 2003;42:33–84 [DOI] [PubMed] [Google Scholar]
- 2. Counts SE, Mufson EJ, Locus Coeruleus. In: Mai JK, Paxinos G, eds. The Human Nervous System, 3rd edition. London: Academic Press; 2012:425–38 [Google Scholar]
- 3. Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nat Rev Neurosci 2009;10:211–23 [DOI] [PubMed] [Google Scholar]
- 4. Kelly SC, He B, Perez SE, et al. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol Commun 2017;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Theofilas P, Ehrenberg AJ, Dunlop S, et al. Locus coeruleus volume and cell population changes during Alzheimer's disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement 2017;13:236–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arendt T, Bruckner MK, Morawski M, et al. Early neurone loss in Alzheimer’s disease: Cortical or subcortical? Acta Neuropathol Commun 2015;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan-Palay V, Asan E.. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J Comp Neurol 1989;287:373–92 [DOI] [PubMed] [Google Scholar]
- 8. Davies P. Neurotransmitter-related enzymes in senile dementia of the Alzheimer type. Brain Res 1979;171:319–27 [DOI] [PubMed] [Google Scholar]
- 9. Grudzien A, Shaw P, Weintraub S, et al. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging 2007;28:327–35 [DOI] [PubMed] [Google Scholar]
- 10. Mann DM, Lincoln J, Yates PO, et al. Changes in the monoamine containing neurones of the human CNS in senile dementia. Br J Psychiatry 1980;136:533–41 [DOI] [PubMed] [Google Scholar]
- 11. Wilson RS, Nag S, Boyle PA, et al. Neural reserve, neuronal density in the locus coeruleus, and cognitive decline. Neurology 2013;80:1202–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Clewett DV, Lee TH, Greening S, et al. Neuromelanin marks the spot: Identifying a locus coeruleus biomarker of cognitive reserve in healthy aging. Neurobiol Aging 2016;37:117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dahl MJ, Mather M, Duzel S, et al. Rostral locus coeruleus integrity is associated with better memory performance in older adults. Nat Hum Behav 2019;3:1203–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Betts MJ, Kirilina E, Otaduy MCG, et al. Locus coeruleus imaging as a biomarker for noradrenergic dysfunction in neurodegenerative diseases. Brain 2019;142:2558–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braun DJ, Van Eldik LJ.. In vivo brainstem imaging in Alzheimers disease: Potential for biomarker development. Front Aging Neurosci 2018;10:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu KY, Kievit RA, Tsvetanov KA, Cam-CAN, et al. Noradrenergic-dependent functions are associated with age-related locus coeruleus signal intensity differences. Nat Commun 2020;11:1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Olivieri P, Lagarde J, Lehericy S, et al. Early alteration of the locus coeruleus in phenotypic variants of Alzheimer’s disease. Ann Clin Transl Neurol 2019;6:1345–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peterson AC, Li CR.. Noradrenergic dysfunction in Alzheimer’s and Parkinson’s diseases-an overview of imaging studies. Front Aging Neurosci 2018;10:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montine TJ, Cholerton BA, Corrada MM, et al. Concepts for brain aging: Resistance, resilience, reserve, and compensation. Alzheimers Res Ther 2019;11:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:280–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmitt FA, Nelson PT, Abner E, et al. University of Kentucky Sanders-Brown healthy brain aging volunteers: Donor characteristics, procedures and neuropathology. Curr Alzheimer Res 2012;9:724–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beekly DL, Ramos EM, Lee WW, et al. ; NIA Alzheimer’s Disease Centers. The National Alzheimer’s Coordinating Center (NACC) database: The Uniform Data Set. Alzheimer Dis Assoc Disord 2007;21:249–58. [DOI] [PubMed] [Google Scholar]
- 23. Weintraub S, Salmon D, Mercaldo N, et al. The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): The neuropsychologic test battery. Alzheimer Dis Assoc Disord 2009;23:91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–44 [DOI] [PubMed] [Google Scholar]
- 25. Abner EL, Kryscio RJ, Cooper GE, et al. Mild cognitive impairment: Statistical models of transition using longitudinal clinical data. Int J Alzheimers Dis 2012;2012:291920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Counts SE, Chen EY, Che S, et al. Galanin fiber hypertrophy within the cholinergic nucleus basalis during the progression of Alzheimer’s disease. Dement Geriatr Cogn Disord 2006;21:205–14 [DOI] [PubMed] [Google Scholar]
- 27. Counts SE, Nadeem M, Lad SP, et al. Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J Neuropathol Exp Neurol 2006;65:592–601 [DOI] [PubMed] [Google Scholar]
- 28. Neltner JH, Abner EL, Baker S, et al. Arteriolosclerosis that affects multiple brain regions is linked to hippocampal sclerosis of ageing. Brain 2014;137:255–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Blevins BL, Vinters HV, Love S, et al. Brain arteriolosclerosis. Acta Neuropathol 2021;141:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghasemi A, Zahediasl S.. Normality tests for statistical analysis: A guide for non-statisticians. Int J Endocrinol Metab 2012;10:486–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aston-Jones G, Bloom FE.. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci 1981;1:876–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foote SL, Aston-Jones G, Bloom FE.. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc Natl Acad Sci U S A 1980;77:3033–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McCall JG, Al-Hasani R, Siuda ER, et al. CRH engagement of the locus coeruleus noradrenergic system mediates stress-induced anxiety. Neuron 2015;87:605–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Poe GR, Foote S, Eschenko O, et al. Locus coeruleus: a new look at the blue spot. Nat Rev Neurosci 2020;21:644–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cortes N, Andrade V, Maccioni RB.. Behavioral and neuropsychiatric disorders in Alzheimer’s disease. J Alzheimers Dis 2018;63:899–910 [DOI] [PubMed] [Google Scholar]
- 36. Lanctot KL, Amatniek J, Ancoli-Israel S, et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: New treatment paradigms. Alzheimers Dement (N Y) 2017;3:440–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oh J, Eser RA, Ehrenberg AJ, et al. Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimers Dement 2019;15:1253–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhao QF, Tan L, Wang HF, et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: Systematic review and meta-analysis. J Affect Disord 2016;190:264–71 [DOI] [PubMed] [Google Scholar]
- 39. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association Guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 2012;8:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jicha GA, Abner EL, Schmitt FA, et al. Preclinical AD Workgroup staging: Pathological correlates and potential challenges. Neurobiol Aging 2012;33:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sperling RA, Karlawish J, Johnson KA.. Preclinical Alzheimer disease-the challenges ahead. Nat Rev Neurol 2013;9:54–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montine TJ, Phelps CH, Beach TG, et al. ; Alzheimer’s Association. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. German DC, Walker BS, Manaye K, et al. The human locus coeruleus: Computer reconstruction of cellular distribution. J Neurosci 1988;8:1776–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ohm TG, Busch C, Bohl J.. Unbiased estimation of neuronal numbers in the human nucleus coeruleus during aging. Neurobiol Aging 1997;18:393–9 [DOI] [PubMed] [Google Scholar]
- 45. Tomonaga M. Neuropathology of the locus ceruleus: a semi-quantitative study. J Neurol 1983;230:231–40 [DOI] [PubMed] [Google Scholar]
- 46. German DC, Manaye KF, White CL 3rd, et al. Disease-specific patterns of locus coeruleus cell loss. Ann Neurol 1992;32:667–76. [DOI] [PubMed] [Google Scholar]
- 47. Szot P, White SS, Greenup JL, et al. Compensatory changes in the noradrenergic nervous system in the locus ceruleus and hippocampus of postmortem subjects with Alzheimer’s disease and dementia with Lewy bodies. J Neurosci 2006;26:467–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arendt T, Bruckner MK, Bigl V, et al. Dendritic reorganisation in the basal forebrain under degenerative conditions and its defects in Alzheimer’s disease. III. The basal forebrain compared with other subcortical areas. J Comp Neurol 1995;351:223–46 [DOI] [PubMed] [Google Scholar]
- 49. Arnsten AF, Goldman-Rakic PS.. Selective prefrontal cortical projections to the region of the locus coeruleus and raphe nuclei in the rhesus monkey. Brain Res 1984;306:9–18 [DOI] [PubMed] [Google Scholar]
- 50. Porrino LJ, Goldman-Rakic PS.. Brainstem innervation of prefrontal and anterior cingulate cortex in the rhesus monkey revealed by retrograde transport of HRP. J Comp Neurol 1982;205:63–76 [DOI] [PubMed] [Google Scholar]
- 51. Boros BD, Greathouse KM, Gentry EG, et al. Dendritic spines provide cognitive resilience against Alzheimer’s disease. Ann Neurol 2017;82:602–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Boros BD, Greathouse KM, Gearing M, et al. Dendritic spine remodeling accompanies Alzheimer’s disease pathology and genetic susceptibility in cognitively normal aging. Neurobiol Aging 2019;73:92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Braak E, Braak H, Mandelkow EM.. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol 1994;87:554–67 [DOI] [PubMed] [Google Scholar]
- 54. Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011;70:960–9 [DOI] [PubMed] [Google Scholar]
- 55. Butterfield DA, Halliwell B.. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 2019;20:148–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tonnies E, Trushina E.. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J Alzheimers Dis 2017;57:1105–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Finley KH, Cobb S.. The capillary bed of the locus coeruleus. J Comp Neurol 1940;73:49–58 [Google Scholar]
- 58. Boiocchi C, Zorzetto M, Sbarsi I, et al. CR1 genotype and haplotype involvement in coronary artery disease: The pivotal role of hypertension and dyslipidemia. Int J Mol Med 2009;24:181–7 [DOI] [PubMed] [Google Scholar]
- 59. Kosunen O, Talasniemi S, Lehtovirta M, et al. Relation of coronary atherosclerosis and apolipoprotein E genotypes in Alzheimer patients. Stroke 1995;26:743–8 [DOI] [PubMed] [Google Scholar]
- 60. Schaefer EJ, Lamon-Fava S, Jenner JL, et al. Lipoprotein(a) levels and risk of coronary heart disease in men. The lipid Research Clinics Coronary Primary Prevention Trial. JAMA 1994;271:999–1003 [DOI] [PubMed] [Google Scholar]
- 61. Juul Rasmussen I, Tybjærg-Hansen A, Rasmussen KL, et al. Blood-brain barrier transcytosis genes, risk of dementia and stroke: A prospective cohort study of 74,754 individuals. Eur J Epidemiol 2019;34:579–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van Es AC, van der Flier WM, Box FM, et al. Carotid and basilar artery wall shear stress in Alzheimer’s disease and mild cognitive impairment. Dement Geriatr Cogn Disord 2009;28:220–4 [DOI] [PubMed] [Google Scholar]
- 63. Li B, Lu X, Moeini M, et al. Atherosclerosis is associated with a decrease in cerebral microvascular blood flow and tissue oxygenation. PLoS One 2019;14:e0221547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pamphlett R. Uptake of environmental toxicants by the locus ceruleus: A potential trigger for neurodegenerative, demyelinating and psychiatric disorders. Med Hypotheses 2014;82:97–104 [DOI] [PubMed] [Google Scholar]
- 65. Pamphlett R, Bishop DP, Kum Jew S, et al. Age-related accumulation of toxic metals in the human locus ceruleus. PLoS One 2018;13:e0203627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pamphlett R, Mak R, Lee J, et al. Concentrations of toxic metals and essential trace elements vary among individual neurons in the human locus ceruleus. PLoS One 2020;15:e0233300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bonda DJ, Lee HG, Blair JA, et al. Role of metal dyshomeostasis in Alzheimer’s disease. Metallomics 2011;3:267–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Greenough MA, Camakaris J, Bush AI.. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 2013;62:540–55 [DOI] [PubMed] [Google Scholar]
- 69. Zucca FA, Segura-Aguilar J, Ferrari E, et al. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol 2017;155:96–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Edvinsson L, Lindvall M, Nielsen KC, et al. Are brain vessels innervated also by central (non-sympathetic) adrenergic neurones? Brain Res 1973;63:496–9 [DOI] [PubMed] [Google Scholar]
- 71. Hartman BK, Zide D, Udenfriend S.. The use of dopamine -hydroxylase as a marker for the central noradrenergic nervous system in rat brain. Proc Natl Acad Sci U S A 1972;69:2722–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kalaria RN, Andorn AC, Harik SI.. Alterations in adrenergic receptors of frontal cortex and cerebral microvessels in Alzheimer’s disease and aging. Prog Clin Biol Res 1989;317:367–74 [PubMed] [Google Scholar]
- 73. Kalaria RN, Stockmeier CA, Harik SI.. Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci Lett 1989;97:203–8 [DOI] [PubMed] [Google Scholar]
- 74. Bekar LK, Wei HS, Nedergaard M.. The locus coeruleus-norepinephrine network optimizes coupling of cerebral blood volume with oxygen demand. J Cereb Blood Flow Metab 2012;32:2135–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hartman BK, Swanson LW, Raichle ME, et al. Central adrenergic regulation of cerebral microvascaulr permeability and blood flow: Anatomic and physiologic evidence. In: Eisenberg HM, ed. The Cerebral Microvasculature. New York: Plenum Press; 1980:113–26 [DOI] [PubMed] [Google Scholar]
- 76. Raichle ME, Hartman BK, Eichling JO, et al. Central noradrenergic regulation of cerebral blood flow and vascular permeability. Proc Natl Acad Sci U S A 1975;72:3726–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Toussay X, Basu K, Lacoste B, et al. Locus coeruleus stimulation recruits a broad cortical neuronal network and increases cortical perfusion. J Neurosci 2013;33:3390–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kalinin S, Feinstein DL, Xu HL, et al. Degeneration of noradrenergic fibres from the locus coeruleus causes tight-junction disorganisation in the rat brain. Eur J Neurosci 2006;24:3393–400 [DOI] [PubMed] [Google Scholar]
- 79. Kelly SC, McKay EC, Beck JS, et al. Locus coeruleus degeneration induces forebrain vascular pathology in a transgenic rat model of Alzheimer’s disease. J Alzheimers Dis 2019;70:371–88 [DOI] [PMC free article] [PubMed] [Google Scholar]