

Abstract
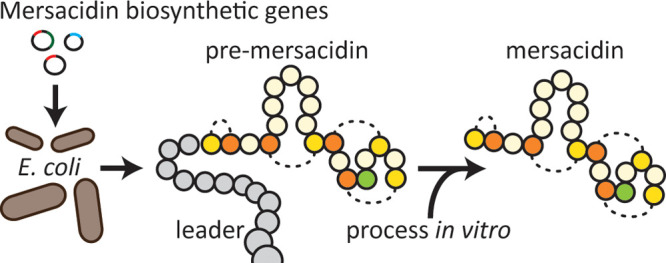
The lanthipeptide mersacidin is a ribosomally synthesized and post-translationally modified peptide (RiPP) produced by Bacillus amyloliquefaciens. It has antimicrobial activity against a range of Gram-positive bacteria, including methicillin-resistant Staphylococcus aureus, giving it potential therapeutic relevance. The structure and bioactivity of mersacidin are derived from a unique combination of lanthionine ring structures, which makes mersacidin also interesting from a lantibiotic-engineering point of view. Until now, mersacidin and its derivatives have exclusively been produced in Bacillus strains and purified from the supernatant in their bioactive form. However, to fully exploit its potential in lanthipeptide-engineering, mersacidin would have to be expressed in a standardized expression system and obtained in its inactive prepeptide form. In such a system, the mersacidin biosynthetic enzymes could be employed to create novel peptides, enhanced by the recent advancements in RiPP engineering, while the leader peptide prevents activity against the expression host. This system would however need a means of postpurification in vitro leader processing to activate the obtained precursor peptides. While mersacidin’s native leader processing mechanism has not been confirmed, the bifunctional transporter MrsT and extracellular Bacillus proteases have been suggested to be responsible. Here, a modular system is presented for the heterologous expression of mersacidin in Escherichia coli, which was successfully used to produce and purify inactive premersacidin. The purified product was used to determine the cleavage site of MrsT. Additionally, it was concluded from antimicrobial activity tests that in a second processing step mersacidin is activated by specific extracellular proteases from Bacillus amyloliquefaciens.
Keywords: mersacidin, lanthipeptides, RiPPs, leader processing, E. coli, heterologous expression, mutagenesis
Introduction
Mersacidin is a ribosomally synthesized and post-translationally modified peptide (RiPP), first described in 1992 as a fermentation product of Bacillus species HIL Y-85,547281 (Figure 1A). It has potent antimicrobial activity against a range of Gram-positive bacteria, including methicillin-resistant strains.1 Its antimicrobial properties come from a singular mode of action; i.e., peptidoglycan biosynthesis is inhibited by binding of mersacidin to the bacterial cell wall precursor Lipid-II at a unique binding site.2,3 Mersacidin has a globular shape4 brought about by its lanthionine ring structures, which give a high stability to the peptide and offer interesting properties from an engineering point of view (Figure 1A). Ring A, for example, could be added to linear peptides to improve their stability. Lanthionine rings can be engineered into linear peptides to increase their stability,5 but to preserve the original function, a minimal change in the peptides’ structure is desirable. Mersacidin’s uniquely small ring A spans no additional amino acids, making the application of this ring an attractive prospect in lanthipeptide engineering of this nature. The other ring structures, with their respective characteristics, might also prove to be useful additions to the lanthipeptide engineering toolbox: Ring B spans seven amino acids, while rings C and D are intertwined with ring D containing a decarboxylated cysteine.
Figure 1.

(A) Amino acid sequence of mersacidin with a schematic overview of its lanthionine rings and decarboxylation site. (B) The mersacidin gene cluster, containing mrsK2R2 (regulation of immunity), mrsFEG (immunity), mrsA (precursor), mrsR1 (regulation of mersacidin production), mrsD (decarboxylation), mrsM (dehydration and cyclization), and mrsT (transport and leader protease).
Like for many lanthipeptides, the production of mersacidin is partially dependent on autoinduction.6 The production of mersacidin-precursor MrsA from its gene cluster (Figure 1B) is mediated by MrsR1, while MrsR2 and MrsK2 regulate the expression of the immunity genes mrsFEG.7 Mersacidin’s four methyl-lanthionine rings are installed by MrsM,8 which is responsible for both dehydration of threonine and serine residues and the subsequent cyclization of the dehydrated threonines with the respective cysteine residues. Decarboxylation of the final cysteine is done by MrsD,9 a LanD enzyme related to the flavoprotein EpiD, which removes a CO2 and two hydrogen atoms leading to a decrease in monoisotopic mass of 46.0055 Da.10,11 When fully modified, premersacidin is exported by the bifunctional transporter MrsT, which contains a proteolytic domain that has been proposed to be involved in leader processing.8,12,13 While this is typical for Class-II lanthipeptides, the conserved recognition site for cleavage by LanT in the mersacidin leader does not precisely align with that of the related peptides (Figure 2).8,14,15 If the proteolytic domain of MrsT would cleave after the conserved glycine–alanine recognition site, part of the leader remains attached to the core peptide, and it has thus been suggested that the final activation of mersacidin is done by extracellular Bacillus proteases.8
Figure 2.

Alignment of several class-II lanthipeptide leaders to their GG-type class-II LanT cleavage site,8 combined with a conservation logo.18 If mersacidin is cleaved after this sequence by MrsT upon export, between positions −7 and −6, six residues of the leader remain attached to the core peptide, likely rendering the exported product inactive.8,12,19−21
Lanthipeptides are partially categorized by their modification system.16 A major interesting difference between different classes is that class I lanthipeptides, like nisin, have two enzymes for maturation, i.e., a LanB for dehydration and a LanC for cyclization. In class II lanthipeptides, like mersacidin, these activities are combined in one enzyme called LanM. The respective specificities of these modification systems underlie separate possibilities regarding ring-structure formation. For a comparison of mersacidin maturation to that of other RiPPs, there are some excellent reviews available.16,17
To date, mersacidin and its derivatives have been exclusively produced by using its full gene cluster in Bacillus species22,23 through the purification of mature mersacidin from the producer supernatant. Although MrsA has been expressed in combination with MrsD in E. coli,9 to study the oxidative decarboxylation reaction, the coexpression of MrsM in this system has not been reported. Thus, no form of premersacidin has been purified to date. Consequently, the suggested mechanism for mersacidin leader processing has not yet been confirmed.
Recently, much progress has been made in the field of lanthipeptide engineering, including the production of hybrid RiPPs, the incorporation of noncanonical amino acids, ring shuffling, and more.17,24 Concurrently, it has become evident that, while many RiPPs have therapeutic applications in theory, these applications are hampered by drawbacks such as in vivo instability.25 Solutions for these drawbacks are being found in the development of novel lanthipeptide engineering tools.17,26 Thus, while mersacidin has good activity against clinically relevant strains, the lack of a concise heterologous expression system excludes it from the many advancements made in lanthipeptide expression in standardized systems like E. coli BL21(DE3). Such a system would thus not only provide opportunities for leader processing studies but also increase the relevance of the mersacidin modification machinery in today’s field of lanthipeptide improvement by engineering. The performance of the heterologous expression of mersacidin in E. coli rather than Bacillus has the extra advantage that E. coli lacks the wide variety of extracellular proteases that Bacillus has. Bacillus species produce a range of proteases,27 which would likely cleave the leader peptide of the produced peptide, activating it during production or purification. In E. coli, novel peptides can be produced without the need for an immunity mechanism against these new-to-nature peptides, as the leader is not likely to be processed by the producer. Effectively, the produced peptide stays inactive until the leader is processed in vitro.
Here, a modular plasmid system is presented for the complete biosynthesis of premersacidin in E. coli. This system employs a minimal gene cluster, achieving full premersacidin biosynthesis by expressing three of the ten genes, mrsAMD, from the mersacidin gene cluster. As a proof of principle, the system was employed to study mersacidin leader processing. The activation of mersacidin by B. amyloliquefaciens supernatant was examined. Additionally, the cleavage site of MrsT was studied through the digestion of premersacidin in vitro with the MrsT proteolytic domain, MrsT150. In addition to the previously mentioned advantages of this system in mersacidin related lanthipeptide engineering, the modular nature of this heterologous expression system allows one to study the effects of modification by MrsM and MrsD alone or simultaneously.
Results
The mersacidin biosynthetic genes mrsAMD were amplified from the genomic DNA of Bacillus amyloliquefaciens strain BH072, which contains the mersacidin gene cluster. The nucleotide sequence of all biosynthetic genes from this gene cluster is identical with that of Bacillus sp. HIL-Y85/547281,8 with the exception of mrsM. The mrsM gene encodes a protein differing 3 amino acids from MrsM in the reported mersacidin producer strain (Figure S1). The amplified biosynthetic genes were placed in compatible E. coli plasmids.
The resulting modular expression system (Figure 3) was employed to achieve heterologous expression of mersacidin in E. coli BL21(DE3). The N-terminally His-tagged mersacidin precursor His-MrsA was expressed without modification enzymes, in combination with MrsM, or in combination with MrsM + MrsD. All samples were purified from the cell pellet through His-tag purification, followed by desalting with an open C18 column. The samples were freeze-dried and dissolved in Milli-Q, after which they were analyzed.
Figure 3.

Plasmids for expression of mersacidin in varying levels of maturation in E. coli.
Analysis by MALDI-TOF (Figure 4, Table 1) showed that the expression of His-MrsA in the presence of MrsM gives a major product resembling fully dehydrated His-MrsA with a theoretical mass of 8018.94 Da. When coexpressed with MrsM and MrsD, the modified His-MrsA approaches the theoretical mass of mature His-premersacidin, 7972.91 Da.
Figure 4.

MALDI-TOF spectra for results from different expression plasmid combinations. These masses approximate their theoretical masses (Table 1), and the masses for His-MrsA + MrsM and His-MrsA + MrsM + MrsD were later determined with higher resolution through LC-MS (Figure S6C,D).
Table 1. Theoretical Average Masses of Different Peptides Produced in This Study, Compared to Those Found in MALDI-TOF and LC-MS Analyses (Table S3).
| peptide | method | theoretical mass (Da) | found mass (Da) | difference | figure |
|---|---|---|---|---|---|
| His-MrsA | MALDI-TOF | 8108.99 | 8113.05 | –4.06 | Figure 4 |
| His-MrsA + MrsM | MALDI-TOF-5H2O | 8018.94 | 8019.42 | –0.48 | |
| LC-MS-5H2O | 8016.65 | 2.29 | Supporting Information | ||
| His-MrsA + MrsM + MrsD (His-premersacidin) | MALDI-TOF-5H2O, -CO2 | 7972.91 | 7977.80 | –4.89 | Figure 4 |
| LC-MS-4H2O, -CO2 | 7990.92 | 7989.66 | –1.26 | Supporting Information | |
| LC-MS-5H2O, -CO2 | 7972.91 | 7970.66 | –2.25 | ||
| His-premersacidin + supernatant BH072 | LC-MS mersacidin | 182622 | 1826.98 | –0.98 | Figure 7 |
| His-premersacidin + MrsT150-His | MALDI-TOF His-premersacidin -6 to 20 -3H2O, -CO2 | 2437.86 | 2439.56 | 1.70 | |
| MALDI-TOF His-premersacidin -6 to 20 -4H2O, -CO2 | 2419.85 | 2420.14 | 0.29 | ||
| MALDI-TOF His-premersacidin -6 to 20 -5H2O, -CO2 | 2401.84 | 2399.55 | –2.29 | ||
| MALDI-TOF His-premersacidin -48 to -6 | 5590.10 | 5588.85 | 1.25 |
To confirm full modification of His-MrsA by MrsM + MrsD, i.e., the production of His-premersacidin, it was subjected to an antimicrobial activity test. As mersacidin has been hypothesized to be activated by extracellular proteases, the isolated prepeptide was incubated with BH072 supernatant sampled from different growth times between 20 and 96 h. All digested samples were active against Micrococcus flavus on a bacterial lawn, indicating that His-premersacidin could be activated by BH072 supernatant from all tested growth times (Figure S2). The BH072 supernatant of all growth times without added His-premersacidin did not produce a halo in the antimicrobial activity tests, indicating the supernatant contained no compounds in concentrations high enough to inhibit cell growth. Furthermore, in MALDI-TOF analysis of the BH072 supernatant alone, mersacidin could not be detected (Figure S5).
The acquired His-premersacidin was further analyzed by HPLC (Table S2), which showed that the major product was separated into two distinguishable peaks (Figure 5). Separated by HPLC, only one of these peaks contained activatable premersacidin, which was identified through an antimicrobial activity test against Micrococcus flavus (Figure S3). The peak with presumed premersacidin was purified by HPLC (Figure S6A) and further analyzed by LC-MS (Figure S6D). The main products from this peak were found to approach the theoretical masses of 7972.91 Da (His-MrsA-5H2O, -CO2) and 7990.92 Da (His-MrsA-4H2O, -CO2). This dehydration state is comparable to that of the main product before purification (Figure 4C), suggesting that the product being divided into an activatable and nonactivatable peak is the result of one or more cyclizations not occurring. The lack of this peak in samples expressed without MrsD (Figures 5 and S6A) points toward the C-terminal ring not being formed. Most importantly, free-cysteine assays of the activatable peak showed that all rings are formed, and it thus proves that the fully dehydrated product is fully mature premersacidin (Figure S6D). LC-MS analysis of His-MrsA + MrsM gave expected mass data but also showed signs of degradation, pointing toward a stability function of the C-terminal decarboxylation performed by MrsD.
Figure 5.
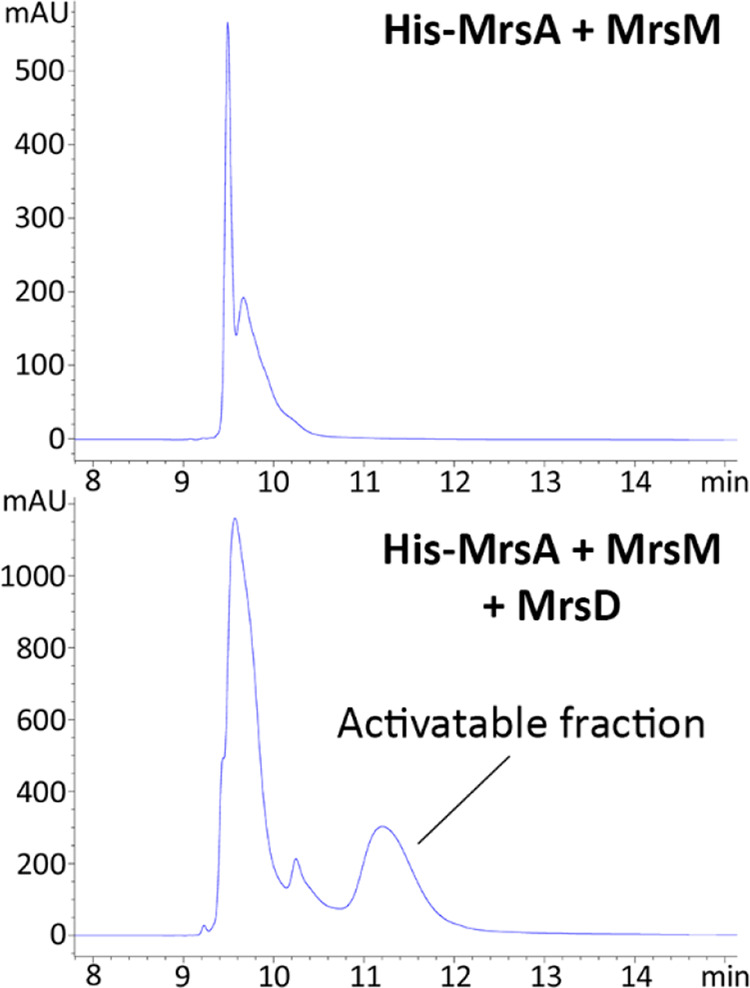
HPLC spectra of His-MrsA + MrsM and His-MrsA + MrsM + MrsD. In the presence of MrsD, there is a clear extra peak, which contains His-premersacidin. As the product in both peaks is decarboxylated, the decarboxylation itself does not cause this shift but rather closes one or more lanthionine rings.
In the initial protocol, the peak containing activatable premersacidin constituted approximately 15% of all the product. This value could be increased to 25–30% by optimizing the expression protocol for maximal antimicrobial activity (Figure S7, Table S4). The protocol was optimized for induction time and temperature and for the amino acid sequence of MrsM. During optimization, it was found that restoring the MrsM amino acid sequence to that published for mersacidin producer Bacillus sp. HIL-Y85/547281,8 did not affect modification efficiency. It was therefore decided to use the mrsM gene from BH072 for further expression purposes. Optimal production of His-premersacidin was reached by inducing the complete system concurrently at 16 °C for 24 h. Using this optimized protocol, a per liter yield could be reached of 2 mg of unmodified His-MrsA, 2.5 mg of His-MrsA + MrsM, and 7.5 mg of His-MrsA + MrsM + MrsD. Since the addition of MrsD requires an extra plasmid and antibiotic resistance marker, the 3-fold increase of production in the complete biosynthetic system is striking. This again points toward the notion that the C-terminal decarboxylation provides stability to the peptide.
With the quantified production yields, a standardized antimicrobial activity test was performed. Each of the prepeptides was spotted against M. flavus with and without the addition of BH072 supernatant (Figure 6). The only sample with antimicrobial activity was His-premersacidin incubated with BH072 supernatant. This indicates that, unsurprisingly, MrsD is crucial for mersacidin activity. A peak with a mass resembling that of mersacidin (1826 Da) was identified upon MALDI-TOF analysis of the activated His-premersacidin (Figure 7A, Table 1)
Figure 6.

Antimicrobial activity of mersacidin at varying levels of maturation with and without BH072 supernatant (SN) against M. flavus. (1) His-MrsA + SN, (2) His-MrsA + MrsM + SN, (3) His-MrsA + MrsM + MrsD + SN, (4) SN, (5) His-MrsA, (6) His-MrsA + MrsM, and (7) His-MrsA + MrsM + MrsD. Only His-MrsA + MrsM + MrsD can be activated.
Figure 7.

Digestion patterns of His-premersacidin in combination with (A) BH072 supernatant, where the expected mass for active mersacidin is found upon digestion with BH072 supernatant, and (B) MrsT150-His, where masses are found complying with the theory that MrsT cleaves the mersacidin leader between positions −7 and −6 (Table 1).
Finally, to assess the proteolytic activity of MrsT, its proteolytic domain was expressed in E. coli with a C-terminal His-tag (MrsT150-His) after the example shown for LagD.28 Upon incubation with His-premersacidin, MrsT150-His cleaves at the theoretically predicted site, leaving six amino acids of the leader attached to the core peptide (Figure 7B, Table 1). As expected, the remaining six amino acids inhibit antimicrobial activity until subsequent cleavage with BH072 supernatant (Figure S4), which demonstrates a two-step activation process to liberate active mersacidin.
Discussion
Using the system described here, fully modified premersacidin was purified from the Gram-negative host E. coli BL21(DE3). The modular plasmid system, employing a minimal gene cluster, allowed for the product to be purified from the producer cell pellet in its inactive prepeptide from. This is in contrast to previously described expression systems, where the complete mersacidin gene cluster was employed in Bacillus sp. to acquire mature mersacidin from the supernatant.22,29 Those systems, while having an advantage when expressing wild-type mersacidin, would experience multiple problems when employing it to produce new-to-nature peptides. The transporter MrsT might not export certain substrates deviating too far from the natural product, which would obstruct purification from the supernatant. Additionally, the production of peptides with an antimicrobial mode of action deviating from that of mersacidin might kill the producer cell upon activation by Bacillus extracellular proteases. Or, when a produced peptide is susceptible to degradation, it would be digested in the supernatant by extracellular proteases before it can be purified. All the previously described drawbacks are circumvented in the system presented here. An additional advantage of production in E. coli is that it is a well-established expression host for which many lanthipeptide engineering methods have been described.16,17 This combination makes the system developed here a useful tool in mersacidin related lanthipeptide engineering. The plasmid system can for example be used to produce RiPP hybrids,30 incorporate nonproteinogenic amino acids,31 or stabilize linear peptides.5
In the antimicrobial activity tests against M. flavus, it was shown that Bacillus amyloliquefaciens supernatant can activate premersacidin. As natural producer strain, HIL Y-85,54728 is reported to start mersacidin production after 48 h;1 the fact that supernatant from a 20 h BH072 culture can activate mersacidin points toward a general protease present in the supernatant rather than a dedicated one. Additionally, MrsT was found to cleave at the predicted site between positions −7 and −6.8 This partial leader cleavage does not lead to activation, and so, an extra processing step is required for activation after export. All these results point strongly toward the conclusion that mersacidin is activated by one or more extracellular Bacillus proteases. Bacillus species are known for their wide range of extracellular proteases with a generally broad substrate specificity.27 Some proteases from Bacillus subtilis, AprE, Bpr, and WprA, were shown to activate presubtilin by cleaving the leader peptide after it was exported out of the cell.32 However, as the first residue of the mersacidin core is part of an unusual ring structure, the number of extracellular proteases able to perform the final leader processing step may be limited. Hence, it is possible that, due to differences between homologous Bacillus proteases, the mersacidin maturing protease is specific to B. amyloliquefaciens. Although the identification of any proteases involved here is not required for this expression system, their identification would allow for interesting insights into the binding pocket properties of proteases capable of cleaving at this unique position. The mechanism of leader processing confirmed here is not unique,33 but the function of partial processing during export is unclear, especially because in this study the preprocessing was found to be unnecessary for mersacidin activation by extracellular proteases and to not increase the rate at which mersacidin is activated after it is transported out of the cell. However, the proteolytic domain is likely to be crucial for leader recognition12 and thus export. Conceivably, direct activation upon export might cause mersacidin to adhere to its producer, decreasing its effectiveness, which additionally could unbalance the autoinduction6 mechanism. Furthermore, the remaining six residues of the leader might increase solubility and facilitate mersacidin diffusion until its maturation by extracellular proteases.
While there already was strong evidence that MrsD is crucial for mersacidin activity,2,9 the observed lack of activity of supernatant-digested His-MrsA + MrsM can be regarded conclusive in this regard. In the HPLC-spectra of His-premersacidin, both the small activatable peak and the large peak are decarboxylated. The masses found in both peaks are similar regardless of activity. This indicates that the shift from the large to the small peak is caused by a specific ring closing that is crucial for activity. As the HPLC spectrum of His-MrsA + MrsM does not contain the small activatable peak, decarboxylation appears to be important for this ring closure. These results essentially point toward the C-terminal decarboxylation drastically improving closing of the final ring. This would explain the lower stability of His-MrsA + MrsM compared to the fully modified premersacidin as well as the lower production of fully modified product when MrsD expression is delayed compared to MrsM (Figure S7).
In conclusion, the system for heterologous expression of mersacidin in E. coli presented here is functional and has already been applied to elucidate mersacidin leader processing. This system can be used to produce mersacidin, mersacidin derivatives, or completely new molecules in E. coli, employing current developments in RiPP engineering.
Materials and Methods
Bacterial Strains and Growth Conditions
Bacillus amyloliquefaciens was grown at 37 °C in LB broth (Formedium) at 225 rpm. E. coli TOP 10 and BL21(DE3) were grown at 37 °C on LB agar (Formedium) plates after transformation or in LB broth at 225 rpm for plasmid isolation or expression unless stated otherwise. E. coli cultures were supplemented with chloramphenicol (when using pACYC, 15 or 10 μg/mL for expression cultures) or ampicillin (when using pBAD, 100 μg/mL). The indicator strain Micrococcus flavus was grown under identical conditions, except in activity test plates, where it was grown in 0.5% agar.
Molecular Cloning
All molecular cloning was done in accordance with previously described methodology,34 supplemented with the protocols of respective reagent manufacturers. Oligonucleotides for insert and vector amplification and adaptation were ordered from Biolegio (Nijmegen, The Netherlands) (Table S1). PCRs were performed using Phusion polymerase (Thermofisher) in reaction conditions recommended by the manufacturer, after which the reactions were cleaned up using a NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel). Subsequent restrictions of the PCR products were done using FastDigest restriction enzymes (Thermo Scientific), after which the DNA was cleaned up as previously mentioned. Ligation of the respective fragments was done with T4 DNA ligase (Thermo Scientific), after which CaCl2 chemically competent E. coli TOP10 was transformed with the resulting ligation mixture. Plasmid DNA was isolated from transformants using a NucleoSpin Plasmid EasyPure kit (Macherey-Nagel), after which correct transformants were selected after DNA sequencing by Macrogen Europe (Amsterdam, The Netherlands). For expression purposes, CaCl2 chemically competent E. coli BL21(DE3) was transformed with sequenced plasmid DNA from TOP10.
In all cases, the original genetic sequence of MrsA, MrsM, MrsT, and MrsD was amplified either directly or indirectly from the B. amyloliquefaciens BH07235 genomic DNA using primers to introduce overhangs for restriction and ligation (Table S1). In all cases, the vector was amplified through PCR, in most cases introducing restriction sites.
To construct pACYC-MrsT150-His, the nucleotide sequence encoding the N-terminal 150 amino acids of MrsT was placed downstream of T7(1) of pACYCDuet-1 using PstI + NcoI for the backbone and PstI + NcoI compatible Eco31I for the inset. The C-terminal His-tag remained from the substrate of which the pACYC backbone was amplified. His-MrsA was cloned into pACYC, and MrsA + MrsM was placed in pACYCDuet-1 behind T7(1) and T7(2), respectively, using suitable Eco31I overhangs. MrsD was cloned into pBAD using PstI + NcoI for the backbone and PstI + Eco31I (NcoI) for the inset. An additional plasmid was created in which the amino acid sequence of MrsM was adjusted to that published for the native producer.8,22 The plasmid pACYC His-MrsA + MrsM2 was created using pACYC His-MrsA + MrsM as a template for three PCRs introducing the required mutations and compatible Eco31I restriction sites, which were ligated using golden gate assembly.
Heterologous Expression
Mersacidin
Expressions of His-MrsA, His-MrsA + MrsM, and His-MrsA + MrsD were optimized to the following protocol. For each, an overnight culture was made from several colonies off a plate of freshly transformed BL21(DE3). The overnight culture was diluted 50 times in fresh medium prewarmed to 37 °C and incubated for 2.5 h at 37 °C and 225 rpm. The culture was then placed at 16 °C and 225 rpm without precooling and directly induced with either 1 mM IPTG (His-MrsA, His-MrsA + MrsM) or 1 mM IPTG + 0.2% Arabinose (His-MrsA + MrsM + MrsD). After induction, the cultures were incubated for 24 h, after which the cells were harvested.
Optimization of Mersacidin Expression
Using HPLC spectra to identify the relative size of the peak containing the activatable premersacidin, in combination with antimicrobial activity tests, the expression protocol was optimized. The effect of expression at 16, 18, 28, or 37 °C was studied as well as the effect of inducing MrsM and MrsD either concurrently or with a 2 h delay to each other. Additional optimization experiments considered the difference between expression for 16 or 24 h and using the originally published amino acid sequence of MrsM from HIL Y-85,54728 (Figure S7, Table S4).
MrsT150-His
The expression of MrsT150-His was done identically to that of mersacidin, until induction. The expression culture was induced with 1 mM IPTG and grown for 4.5 h at 37 °C and 225 rpm, after which the cells were harvested.
Peptide Purification and Analysis
His-tag Purification
The pellet was washed once in binding buffer (20 mM H2NaPO4 (Merck), 0.5 M NaCl (VWR), 20 mM imidazole (Merck), pH 7.4) and then sonicated in 1/100th culture volume binding buffer until visually lysed. The lysate was centrifuged at 10 000g for 60 min and filtered through a syringe filter (0.45 μM). The purification was performed on an open column of 1 mL of Ni-NTA slurry (Qiagen) per 1 L of expression culture volume. Binding and a first wash were done with binding buffer; a second wash was done with wash buffer (20 mM H2NaPO4, 0.5 M NaCl, 20 mM imidazole, pH 7.4), followed by elution using 1.8 mL of elution buffer (20 mM H2NaPO4, 0.5 M NaCl, 20 mM imidazole, pH 7.4) per 1 mL of Ni-NTA slurry.
C18 Purification
The His-tag elution was supplemented with 0.5% trifluoroacetic acid (TFA) (Sigma-Aldrich) until pH 4 and brought onto an open column containing 1.5 mL of 55–105 μm C18 resin (Waters), equilibrated to 0% acetonitrile (ACN) (VWR) + 0.1% TFA. After binding, a first wash was done with 0% ACN + 0.1% TFA and a second wash with 20% ACN + 0.1% TFA. Finally, the sample was eluted in 7.5 mL of 50% ACN + 0.1% TFA and subsequently freeze-dried.
MALDI-TOF and LC-MS Analyses
Freeze-dried samples eluted in Milli-Q water were analyzed by MALDI-TOF or LC-MS as previously described.36 Samples to be analyzed by MALDI-TOF that were cleaved in salt containing buffer were desalted by adding 2 μL of Milli-Q to a dried, spotted sample on the MALDI-TOF plate, which was subsequently resorbed quickly with tissue paper. After this wash step, the matrix was added as described.
Antimicrobial Activity Tests
75 μL of fresh overnight Micrococcus flavus culture was added per 100 mL of hand warm 0.5% LB agar. Of this mixture, 12 mL was added to each 90 mm diameter Petri dish or 25 mL, to each 140 mm diameter Petri dish. All samples contained ca. 30 μg of prepeptide, of which at most 6 μg was active core peptide, and were set to a 10 μL final volume. For all activity tests, a positive control of 9 μL of 25 ng/μL nisin was spotted. All activity tests were performed in triplicate with tested material taken from at least three different expression batches.
In the digests of His-MrsA variants (Figure 6) with BH072 supernatant, samples contained either 3 μL of BH072 supernatant or 3 μL of Milli-Q for the negative control. The samples were incubated at 37 °C for 1 h, after which 9 μL was spotted. For the activity test with MrsT150-His, a His-premersacidin sample containing 1.5 μL of MrsT150-His His-tag elution was incubated for 3 h at 37 °C in duplicate. After incubation, 2 μL of BH072 supernatant was added to one of the duplicates and to a sample containing undigested His-premersacidin. All three samples were incubated for another 30 min at 37 °C, after which 9 μL was spotted.
Supernatant for Activity Tests and Mersacidin Production in BH072
Bacillus amyloliquefaciens BH072 was grown for 96 h, taking samples of the supernatant at 20, 48, 72, and 96 h. The samples were centrifuged, after which the supernatant was filtered (0.2 μM) and stored for activity tests and MALDI-TOF analysis.
Cleavage of Premersacidin with MrsT150-His
The activatable peak was purified by HPLC from an amount of ca. 1 mg of freeze-dried His-MrsA + MrsM + MrsD from C18 purification. The HPLC purified sample was freeze-dried and dissolved in 48 μL of Milli-Q. Of this solution, 2.5 μL was added to 6 μL of Milli-Q and 1.5 μL of MrsT150-His His-tag elution. The mixture was placed at 37 °C for 3 h, after which the sample was analyzed by MALDI-TOF.
Acknowledgments
J.H.V. was funded by The Netherlands Organization for Scientific Research (NWO, ALWOP.214). We thank Rogier van Essen and Emma Michetti for their help in the lab and Fleur Ruijne for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00601.
Figure S1, a comparison of MrsM from HIL-Y85/54728 and BH072; Figures S2–S4, antimicrobial activity tests; Figures S5 and S6, MALDI-TOF and LC-MS analysis data; Figure S7 and Tables S2 and S4, tricine gels, HPLC data, and activity tests of the premersacidin expression protocol optimization; Table S1, a list of primers used; Table S3, theoretical mass tables of mersacidin and its expected fragments (PDF)
Author Present Address
∇ A.H.J.: Department of Environmental Science, Aarhus University, Frederiksborgvej 399, 4000 Roskilde, Denmark.
Author Contributions
The study and experiments were conceived and designed by J.H.V. and O.P.K. and then performed by J.H.V. and A.H.J. Results were analyzed by J.H.V. and O.P.K. The paper was written by JHV. All authors contributed to reading and correcting the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Chatterjee S.; Chatterjee S.; Lad S. J.; Phansalkar M. S.; Rupp R. H.; Ganguli B. N.; Fehlhaber H. W.; Kogler H. (1992) Mersacidin, a New Antibiotic from Bacillus Fermentation, Isolation, Purification and Chemical Characterization. J. Antibiot. 45 (6), 832–838. 10.7164/antibiotics.45.832. [DOI] [PubMed] [Google Scholar]
- Brötz H.; Bierbaum G.; Reynolds P. E.; Sahl H. G. (1997) The Lantibiotic Mersacidin Inhibits Peptidoglycan Biosynthesis at the Level of Transglycosylation. Eur. J. Biochem. 246 (1), 193–199. 10.1111/j.1432-1033.1997.t01-1-00193.x. [DOI] [PubMed] [Google Scholar]
- Brötz H.; Bierbaum G.; Leopold K.; Reynolds P. E.; Sahl H. G. (1998) The Lantibiotic Mersacidin Inhibits Peptidoglycan Synthesis by Targeting Lipid II. Antimicrob. Agents Chemother. 42 (1), 154–160. 10.1128/AAC.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasch T.; Naumann T.; Markert R. L. M.; Sattler M.; Schubert W.; Schaal S.; Bauch M.; Kogler H.; Griesinger C. (1997) Constitution and Solution Conformation of the Antibiotic Mersacidin Determined by NMR and Molecular Dynamics. Eur. J. Biochem. 244 (2), 501–512. 10.1111/j.1432-1033.1997.00501.x. [DOI] [PubMed] [Google Scholar]
- Kuipers A.; Moll G. N.; Wagner E.; Franklin R. (2019) Efficacy of Lanthionine-Stabilized Angiotensin-(1–7) in Type I and Type II Diabetes Mouse Models. Peptides 112, 78–84. 10.1016/j.peptides.2018.10.015. [DOI] [PubMed] [Google Scholar]
- Schmitz S.; Hoffmann A.; Szekat C.; Rudd B.; Bierbaum G. (2006) The Lantibiotic Mersacidin Is an Autoinducing Peptide. Appl. Environ. Microbiol. 72 (11), 7270–7277. 10.1128/AEM.00723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guder A.; Schmitter T.; Wiedemann I.; Sahl H. G.; Bierbaum G. (2002) Role of the Single Regulator MrsR1 and the Two-Component System MrsR2/K2 in the Regulation of Mersacidin Production and Immunity. Appl. Environ. Microbiol. 68 (1), 106–113. 10.1128/AEM.68.1.106-113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altena K.; Guder A.; Cramer C.; Bierbaum G. (2000) Biosynthesis of the Lantibiotic Mersacidin: Organization of a Type B Lantibiotic Gene Cluster. Appl. Environ. Microbiol. 66 (6), 2565–2571. 10.1128/AEM.66.6.2565-2571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer F.; Schmid D. G.; Altena K.; Bierbaum G.; Kupke T. (2002) The Flavoprotein MrsD Catalyzes the Oxidative Decarboxylation Reaction Involved in Formation of the Peptidoglycan Biosynthesis Inhibitor Mersacidin. J. Bacteriol. 184 (5), 1234–1243. 10.1128/JB.184.5.1234-1243.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupke T.; Kempter C.; Gnau V.; Jung G.; Gotz F. (1994) Mass Spectroscopic Analysis of a Novel Enzymatic Reaction. Oxidative Decarboxylation of the Lantibiotic Precursor Peptide EpiA Catalyzed by the Flavoprotein EpiD. J. Biol. Chem. 269 (8), 5653–5659. 10.1016/S0021-9258(17)37510-5. [DOI] [PubMed] [Google Scholar]
- Kupke T.; Kempter C.; Jung G.; Gotz F. (1995) Oxidative Decarboxylation of Peptides Catalyzed by Flavoprotein EpiD. Determination of Substrate Specificity Using Peptide Libraries and Neutral Loss Mass Spectrometry. J. Biol. Chem. 270 (19), 11282–11289. 10.1074/jbc.270.19.11282. [DOI] [PubMed] [Google Scholar]
- Bobeica S. C.; Dong S. H.; Huo L.; Mazo N.; McLaughlin M. I.; Jiménez-Osés G.; Nair S. K.; van der Donk W. A. (2019) Insights into AMS/PCAT Transporters from Biochemical and Structural Characterization of a Double Glycine Motif Protease. eLife 8, e42305. 10.7554/eLife.42305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahl H.-G.; Bierbaum G. (1998) LANTIBIOTICS: Biosynthesis and Biological Activities of Uniquely Modified Peptides from Gram-Positive Bacteria. Annu. Rev. Microbiol. 52 (1), 41–79. 10.1146/annurev.micro.52.1.41. [DOI] [PubMed] [Google Scholar]
- Caetano T.; Krawczyk J. M.; Mösker E.; Süssmuth R. D.; Mendo S. (2011) Heterologous Expression, Biosynthesis, and Mutagenesis of Type II Lantibiotics from Bacillus Licheniformis in Escherichia Coli. Chem. Biol. 18 (1), 90–100. 10.1016/j.chembiol.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Wang J.; Ge X.; Zhang L.; Teng K.; Zhong J. (2016) One-Pot Synthesis of Class II Lanthipeptide Bovicin HJ50 via an Engineered Lanthipeptide Synthetase. Sci. Rep. 6 (March), 38630. 10.1038/srep38630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K.-D.; Fischbach M. A.; Garavelli J. S.; Göransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. N.; Moore B. S.; Müller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Piel J.; Reaney M. J. T.; Rebuffat S.; Ross R. P.; Sahl H.-G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Süssmuth R. D.; Tagg J. R.; Tang G.-L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. (2013) Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep. 30 (1), 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montalbán-López M.; Scott T. A.; Ramesh S.; Rahman I. R.; van Heel A. J.; Viel J. H.; Bandarian V.; Dittmann E.; Genilloud O.; Goto Y.; Grande Burgos M. J.; Hill C.; Kim S.; Koehnke J.; Latham J. A.; Link A. J.; Martínez B.; Nair S. K.; Nicolet Y.; Rebuffat S.; Sahl H.-G.; Sareen D.; Schmidt E. W.; Schmitt L.; Severinov K.; Süssmuth R. D.; Truman A. W.; Wang H.; Weng J.-K.; van Wezel G. P.; Zhang Q.; Zhong J.; Piel J.; Mitchell D. A.; Kuipers O. P.; van der Donk W. A. (2021) New Developments in RiPP Discovery, Enzymology and Engineering. Nat. Prod. Rep. 38, 130–239. 10.1039/D0NP00027B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G.; Hon G.; Chandonia J.; Brenner S. (2004) WebLogo: A Sequence Logo Generator. Genome Res. 14, 1188–1190. 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Van Der Donk W. A. (2016) Structural Characterization and Bioactivity Analysis of the Two-Component Lantibiotic Flv System from a Ruminant Bacterium. Cell Chem. Biol. 23 (2), 246–256. 10.1016/j.chembiol.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadelli M.; Karsioti A.; Anastasiou R.; Georgalaki M.; Tsakalidou E. (2007) Characterization of the Gene Cluster Involved in the Biosynthesis of Macedocin, the Lantibiotic Produced by Streptococcus Macedonicus. FEMS Microbiol. Lett. 272 (1), 75–82. 10.1111/j.1574-6968.2007.00740.x. [DOI] [PubMed] [Google Scholar]
- Wescombe P. A.; Upton M.; Renault P.; Wirawan R. E.; Power D.; Burton J. P.; Chilcott C. N.; Tagg J. R. (2011) Salivaricin 9, a New Lantibiotic Produced by Streptococcus Salivarius. Microbiology 157 (5), 1290–1299. 10.1099/mic.0.044719-0. [DOI] [PubMed] [Google Scholar]
- Herzner A. M.; Dischinger J.; Szekat C.; Josten M.; Schmitz S.; Yakéléba A.; Reinartz R.; Jansen A.; Sahl H.-G.; Piel J.; Bierbaum G. (2011) Expression of the Lantibiotic Mersacidin in Bacillus Amyloliquefaciens FZB42. PLoS One 6 (7), e22389 10.1371/journal.pone.0022389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekat C.; Jack R. W.; Skutlarek D.; Färber H.; Bierbaum G. (2003) Construction of an Expression System for Site-Directed Mutagenesis of the Lantibiotic Mersacidin. Appl. Environ. Microbiol. 69 (7), 3777–3783. 10.1128/AEM.69.7.3777-3783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S.; Montalbán-López M.; Peterhoff D.; Deng J.; Wagner R.; Held M.; Kuipers O. P.; Panke S. (2019) Analysis of Modular Bioengineered Antimicrobial Lanthipeptides at Nanoliter Scale. Nat. Chem. Biol. 15 (5), 437–443. 10.1038/s41589-019-0250-5. [DOI] [PubMed] [Google Scholar]
- Ongey E. L.; Yassi H.; Pflugmacher S.; Neubauer P. (2017) Pharmacological and Pharmacokinetic Properties of Lanthipeptides Undergoing Clinical Studies. Biotechnol. Lett. 39 (4), 473–482. 10.1007/s10529-016-2279-9. [DOI] [PubMed] [Google Scholar]
- Deng J.-J.; Viel J. H.; Kubyshkin V.; Budisa N.; Kuipers O. P. (2020) Conjugation of Synthetic Polyproline Moietes to Lipid II Binding Fragments of Nisin Yields Active and Stable Antimicrobials. Front. Microbiol. 11, 575334. 10.3389/fmicb.2020.575334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest F. G. (1977) Extracellular Enzyme Synthesis in the Genus Bacillus. Bacteriol. Rev. 41 (3), 711–753. 10.1128/BR.41.3.711-753.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havarstein L. S.; Diep D. B.; Nes I. F. (1995) A Family of Bacteriocin ABC Transporters Carry out Proteolytic Processing of Their Substrates Concomitant with Export. Mol. Microbiol. 16, 229–240. 10.1111/j.1365-2958.1995.tb02295.x. [DOI] [PubMed] [Google Scholar]
- Appleyard A. N.; Choi S.; Read D. M.; Lightfoot A.; Boakes S.; Hoffmann A.; Chopra I.; Bierbaum G.; Rudd B. A. M.; Dawson M. J.; Cortes J. (2009) Dissecting Structural and Functional Diversity of the Lantibiotic Mersacidin. Chem. Biol. 16 (5), 490–498. 10.1016/j.chembiol.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart B. J.; Kakkar N.; Hudson G. A.; Van Der Donk W. A.; Mitchell D. A. (2017) Chimeric Leader Peptides for the Generation of Non-Natural Hybrid RiPP Products. ACS Cent. Sci. 3 (6), 629–638. 10.1021/acscentsci.7b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldach F.; Altoma R.; Kuthning A.; Caetano T.; Mendo S.; Budisa N.; Süssmuth R. D. (2012) Congeneric Lantibiotics from Ribosomal in Vivo Peptide Synthesis with Noncanonical Amino Acids. Angew. Chem., Int. Ed. 51 (2), 415–418. 10.1002/anie.201106154. [DOI] [PubMed] [Google Scholar]
- van Tilburg A. Y.; van Heel A. J.; Stülke J.; de Kok N. A. W.; Rueff A.-S.; Kuipers O. P. (2020) MiniBacillus PG10 as a Convenient and Effective Production Host for Lantibiotics. ACS Synth. Biol. 9 (7), 1833–1842. 10.1021/acssynbio.0c00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvey C.; Stein T.; Düsterhus S.; Karas M.; Entian K. D. (2003) Activation of Subtilin Precursors by Bacillus Subtilis Extracellular Serine Proteases Subtilisin (AprE), WprA, and Vpr. Biochem. Biophys. Res. Commun. 304 (1), 48–54. 10.1016/S0006-291X(03)00529-1. [DOI] [PubMed] [Google Scholar]
- Sambrook J., and Russel D. W. (2001) Molecular Cloning: A Laboratory Manual, 4th ed., Cold Spring Harbor Laboratory Press, New York, USA. [Google Scholar]
- Zhao X.; de Jong A.; Zhou Z.; Kuipers O. P. (2015) Complete Genome Sequence of Bacillus Amyloliquefaciens Strain BH072, Isolated from Honey. Genome Announc. 3 (2), e00098-15. 10.1128/genomeA.00098-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Yin Z.; Breukink E.; Moll G. N.; Kuipers O. P. (2020) An Engineered Double Lipid II Binding Motifs-Containing Lantibiotic Displays Potent and Selective Antimicrobial Activity against Enterococcus faecium. Antimicrob. Agents Chemother. 64 (6), e02050-19 10.1128/AAC.02050-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.