

Abstract
Cell-based sensors are useful for many synthetic biology applications, including regulatory circuits, metabolic engineering, and diagnostics. While considerable research efforts have been made toward recognizing new target ligands and increasing sensitivity, the analysis and optimization of turn-on kinetics is often neglected. For example, to our knowledge there has been no systematic study that compared the performance of a riboswitch-based biosensor versus reporter for the same ligand. In this study, we show the development of RNA-based fluorescent (RBF) biosensors for guanidine, a common chaotropic agent that is a precursor to both fertilizer and explosive compounds. Guanidine is cell permeable and nontoxic to E. coli at millimolar concentrations, which in contrast to prior studies enabled direct activation of the riboswitch-based biosensor and corresponding reporter with ligand addition to cells. Our results reveal that the biosensors activate fluorescence in the cell within 4 min of guanidine treatment, which is at least 15 times faster than a reporter derived from the same riboswitch, and this rapid sensing activity is maintained for up to 1.6 weeks. Together, this study describes the design of two new biosensor topologies and showcases the advantages of RBF biosensors for monitoring dynamic processes in cell biology, biotechnology, and synthetic biology.
In vivo sensing and quantification of cellular metabolites, ions and other biological small molecules are important to understand cellular signaling pathways and other physiological processes in cell biology.1 The detection of metabolites or small molecules in cells has also been employed within diverse industries such as biomedicine, food processing, environmental pollutant tracing, and forensics.1 Moreover, synthetic biology applications such as metabolite engineering, disease diagnostics, and theranostics, rely on tracking dynamic changes of target molecules in living cells in order to evaluate the function of the engineered biological system.2,3 Therefore, developing in vivo biosensors that enable monitoring of cellular target molecules in real time is highly beneficial to cell biology, biotechnology, and synthetic biology research.
Genetically encodable riboswitches that undergo conformational changes upon recognizing a target molecule have been considered as attractive tools for analyte sensing.4 Riboswitch reporters, where the riboswitch is inserted upstream of a reporter gene, and aptazymes, where a riboswitch is fused to a ribozyme, have been very useful for in vivo validation of riboswitches as well as development of small molecule-responsive gene circuits.5−10 These riboswitch-based tools mainly rely on expressions of various reporter genes such as beta-galactosidase for colorimetric measurements,5,7 GFP for fluorescence,8,11 and luciferases for luminescence.12,13
A critical aspect of the in vivo application of biosensing systems is their activation rate to monitor sensitive and dynamic changes on a cellular time scale. For instance, if a biological process takes place on the order of minutes, but a sensor fully activates within several hours, it cannot accurately capture the dynamics of that process. Considerable progress has been made in developing new biosensors for interesting and relevant ligands, but their activation rate is often not optimized. Reporter systems that rely on gene expression often require a longer time to produce a detectable signal. For example, riboswitch reporters have been observed to respond in the time range of 3–24 h.7,14,15 A time-dependent study of a riboswitch-based dual-color sensor that changes color upon ligand binding revealed that the minimum time required for response is 4 h.16 Engineered aptazymes show fast cleavage kinetics in vitro in the presence of the target ligands,6,17 but similarly long incubation times (20–72 h) have been reported to see changes in aptazyme-controlled GFP and luciferase reporters in bacterial or mammalian cells.6,11,18
Recently, genetically encodable RNA-based fluorescent (RBF) biosensors composed of a ligand-binding riboswitch aptamer fused to an in vitro selected fluorogenic aptamer have emerged as a promising alternative tool.9,19,20 Ligand binding induces aptamer folding, which further facilitates a specific dye to bind the fluorogenic aptamer and exhibit fluorescence turn-on. For example, fluorogenic aptamers such as Spinach or Spinach2, which fluoresce after binding to profluorescent dyes such as 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI) or its derivatives have been fused to ligand-recognizing aptamers in detecting various target ligands including metabolites,21−24 signaling molecules,25−28 and neurotransmitter precursors.29 As the signal turn-on of RBF biosensors does not depend on reporter gene expression, they are expected to respond faster than riboswitch or aptazyme reporters.
In prior studies of RBF biosensors, it was not possible to accurately assess in vivo turn-on kinetics because enzyme activity was required to produce the specific target molecule in the cell, yielding an additional step to biosensor activation. For example, the S-adenosylmethionine (SAM) biosensor was observed to respond in E. coli with a detectable signal in 15 min and maximal signal in 3 h upon the addition of methionine, the SAM precursor.21 In another study, the thiamine pyrophosphate biosensor showed a detectable fluorescence microscopy signal after 1 h and a maximal signal after 3 h of thiamine addition.23 In an earlier report from our group, the cyclic di-GMP biosensor detected dynamic changes of cyclic di-GMP levels in cells within 15–30 min of zinc depletion, which activates a diguanylate cyclase to produce cyclic di-GMP.30
In contrast, when the target molecule can be introduced into cells directly, the signal turn-on time is observed to be shorter. A 5-hydroxytryptophan (5HTP) biosensor composed of a modified guanine riboswitch and Broccoli fluorogenic RNA aptamer provided a distinguishable signal in E. coli within 15 min of 5HTP addition.29 More recently, an RNA integrator constructed by fusing a target binding aptamer, hammerhead ribozyme, and Broccoli provided an observable signal change in E. coli after 10 min of target molecule addition.31
These studies show that RBF biosensors generally work within minutes to hours in cells, while riboswitch and aptazyme reporters generally require several hours to a day. However, to our knowledge there has been no systematic study that has compared cellular turn-on kinetics for a riboswitch-based biosensor against a reporter for the same ligand. This comparison would ameliorate differences between cell permeability, transport, and enzymatic processing of different compounds used as ligands in these studies.
To investigate the real-time kinetics of RNA-based biosensor activation in vivo, we developed a new biosensor for the compound guanidine, which has been shown to be cell permeable. The ykkC type riboswitch is observed to bind guanidine and regulates the gene expression of proteins involved in detoxification of this compound.32 The prior study also showed for the first time that guanidine can be produced in bacteria under normal growth conditions. However, the pathways toward guanidine accumulation and utilization in bacteria are still largely unknown. Beyond its biological importance, guanidine and its close derivatives are found as environmental pollutants or evidence of explosives.33,34 As a contaminant, guanidine has been detected through spectroscopic methods such as UV and HPLC but these only work for in vitro samples.33,35 Therefore, the development of a guanidine biosensor that can work both in the cell-free and cellular context will be significant for both environmental and biological applications.
Here we describe the development of guanidine-responsive RBF biosensors by screening and optimizing two different topological designs connecting the guanidine-I riboswitch with the fluorogenic Spinach2 aptamer. The resulting biosensors selectively turn on fluorescence in the presence of guanidine both in vitro and in live cells. These biosensors enabled in vivo kinetic studies to compare the response times of riboswitch-based biosensors and reporters to the same small molecule ligand. RBF biosensors exhibited a fast response, with a detectable signal within 4 min of analyte addition and a maximal signal within 15–35 min depending on the biosensor variant. The reporter, on the other hand, required nearly an hour before response was detectable. To our knowledge, this is the first report of functional biosensors for guanidine and the first systematic study directly comparing in vivo activation of a riboswitch-based biosensor against a corresponding reporter. Together, these results show that riboswitches can be adapted to provide faster warning via fluorescent biosensors, then trigger gene expression to provide an ameliorating response.
Results and Discussion
Design of Guanidine Biosensors with Two New Topologies
Four different riboswitch classes with unique folds so far have been discovered to bind the ligand guanidine.32,36−39 Of these riboswitches, the guanidine-I class appeared to have structural advantages for biosensor development, as it uses only one out of three helical stem loops to form a ligand binding pocket. In comparison, the guanidine-II riboswitch utilizes both pairing stems loops to bind guanidine,40,41 and the guanidine-III riboswitch possesses a complex pseudoknot involving its terminal ends and stem loop that is involved in RNA triplex formation to bind the ligand.42 The guanidine-IV riboswitch was reported very recently.38,39 In-line probing assays revealed that the guanidine-I class has higher selectivity and more sequence representatives with stronger affinity toward guanidine than the other three classes.32,36−39 Thus, we selected the guanidine-I class riboswitch as the best candidate for biosensor development.
Analysis of the S. acidophilus guanidine-I riboswitch X-ray crystal structure (PDB 5T83) revealed two key challenges for biosensor design.40 First, the 5′ and 3′ ends of this riboswitch are quite distant, spanning almost 26 Å from one another and not engaged in a terminal pairing stem (Figure 1a). This presents an issue because RBF biosensors typically are constructed by fusing the riboswitch aptamer through the terminal pairing stem to the fluorogenic aptamer. Second, key tertiary interactions need to be maintained between the P1 and P3 helices as they were shown to be crucial for riboswitch folding and guanidine recognition. However, we noticed that the P1 stem-loop is not directly involved in this interaction and devised an approach based on this finding.
Figure 1.

Design of guanidine biosensors with two different topologies. (a) X-ray crystal structure of the guanidine-bound S. acidophilus guanidine-I riboswitch aptamer (PDB 5T83) showing the distance between the 5′ and 3′ ends. (b) Design of two different biosensor topologies utilizing a circularly permuted riboswitch with a linker (L) or an artificial 4-way junction (J).
One strategy that our group developed to overcome a terminal pseudoknot riboswitch topology was to create a circular permutation of the fluorogenic aptamer (cpSpinach2) and insert it within a nonterminal riboswitch stem.22 However, applying this strategy to the guanidine-I riboswitch would leave the terminal ends loose and susceptible to both unfolding and cleavage. Instead, two new biosensor topologies termed “Junction (J)” and “Linker (L)” were designed that enable fusion to the Spinach2 aptamer for in vitro testing and cloning into a tRNA scaffold43 for in vivo testing (Figure 1b).
The linker design utilizes an approach we previously reported44 that connects the 5′ and 3′ termini to generate a circularly permuted riboswitch that can be fused to Spinach2. We hypothesized that this approach would maintain the tertiary interactions between P1 and P3 helices necessary for guanidine binding and would result in fluorescence activation of this biosensor topology. While prior cpRiboswitch designs involved closing a terminal pairing stem with a stem loop, in this case a flexible linker was employed as the terminal ends of guanidine-I are much farther apart. A variable poly-Adenosine (poly-A) linker was used (Figure 2a), which was inspired by an approach used to span a 30–40 Å gap and tether functional ribosomal subunits together.45
Figure 2.

Secondary structure model and initial screen of linker and junction biosensors. (a) Design of initial junction and linker biosensor library. (b) The representative secondary structure of a junction biosensor Kpn J 2,2-A. The name denotes the length of the adenosine spacers and the identity of the P0 stem. Residues in red circles undergo direct contact with the ligand, guanidine. M4 mutant with the single nucleotide mutation shown is denoted as Kpn J 2,2-M. (c) In vitro fluorescence response of 18 junction and 4 linker biosensors to the ligand, guanidine. Biosensors with greater than 1.2x fluorescence enhancement at 1 mM guanidine are indicated in red. Data shown are average with standard error of the mean for two replicates.
Alternatively, the junction design involves adding an artificial transducer stem to create an architecture reminiscent of a three- or four-way junction. Three-way junctions are commonly observed in natural riboswitches such as the cyclic-di-GMP or guanine classes.46,47 We hypothesized that transducer stems from other RBF biosensors our lab has developed would retain their native switching properties and form the basis of an artificial P0 stem that communicates with the fluorogenic aptamer Spinach2. Variable adenosine spacers were added before the P0 stem to span the gap.
Screening of Functional Elements in Junction and Linker Designs
The first biosensor designs were developed using an experimentally validated guanidine-I riboswitch sequence from Klebsiella pneumoniae (Kpn).32 A total of 18 junction biosensors were designed that incorporate nine different adenosine spacers and two P0 stems taken from previously designed cyclic di-GMP biosensors.28 In addition, four linker biosensors were designed with four different poly-A tethers. For the linker designs, the circularly permuted (cp) P1 stem was truncated by nine base pairs and nine single bases to fuse with Spinach2. These biosensors were synthesized from assembled DNA templates by in vitro transcription and then screened for fluorescence activation in response to guanidine (Figure 2).
The initial screen was performed at 28 °C with 10 mM Mg2+, which matches in-line probing conditions used to analyze riboswitch aptamers32 and, in our experience, tends to be more permissive for biosensor constructs to fold and bind a target ligand with higher affinities than physiological conditions. We chose a high, saturating concentration (1 mM) and a low concentration (10 μM) to attempt to distinguish biosensor candidates with tighter ligand affinity. Although no response was observed at lower guanidine concentration (10 μM), five junction biosensors did exhibit >1.2-fold fluorescence activation at 1 mM guanidine. The linker biosensors notably displayed constitutively higher fluorescence signal than parent aptamer Spinach2 and did not respond to guanidine. This result suggested that the P1(cp) stem for the linker design was stably forming even in the absence of guanidine.
On the basis of these results, a second round of biosensor design was carried out. For junction biosensors, three more P0 stems28,44 were tested in combination with four spacer lengths that showed function in the previous screen (Figure 3a). Three out of the 12 junction biosensors exhibited greater than 1.4-fold fluorescence activation (Figure 3b). For linker biosensors, seven truncated P1(cp) stems were designed and tested with A4 and A5 linkers (L-4 and L-5). The truncations involved deleting nucleotides and base-pairs in the Spinach2 aptamer rather than P1(cp), which makes tertiary interactions necessary for guanidine binding (Figure 3c). Five out of 14 truncated linker biosensors exhibited >1.2-fold activation and stems Tr-2, -4, and -5 gave ligand responses (Figure 3d).
Figure 3.

Second round designs of junction and truncated linker guanidine biosensors. (a) Screening of 12 junction biosensors with three additional artificial stem sequences and four variable adenosine spacer lengths. (b) In vitro fluorescence response of biosensors to guanidine. Biosensors with a greater than 1.4× fluorescence increase at 1 mM guanidine are indicated in red. Nomenclature of the biosensors denotes the number of adenosines in the sequence and the identity of the P0 stem. (c) Screening of linker biosensors with seven transducer stem truncations and two variable linker lengths. (d) In vitro fluorescence response of optimized linker biosensors to guanidine. The nomenclature for linker (L) biosensors denotes the number of adenosines in the linker region and the identity of the stem truncation. Biosensors with greater than 1.2× fluorescence increase at 1 mM guanidine are indicated in red. Data shown are average with standard error of the mean for two replicates.
The functionality of the two biosensor topologies was compared by measuring the binding affinities of two representative constructs, Kpn J 2,2-A and L-5 Tr-4, to guanidine. A much lower dissociation constant (Kd) was measured for J 2,2-A (∼60 μM) compared to L-5 Tr-4 (∼7 mM), indicating that biosensor topology has a considerable effect on binding affinity to guanidine (Figure S1a). The Kd value determined for guanidine binding to Kpn J 2,2-A is close to the Kd value of the natural riboswitch aptamer (∼20 μM) determined by in-line probing experiments, which are performed at high magnesium concentrations.32
These results point out that the linker design, in contrast to the junction design, either failed to allosterically regulate dye binding to the Spinach2 domain, hampered guanidine binding to the riboswitch domain, or both. The transducer stem truncation was designed to destabilize the stem but may have compromised ligand affinity. Other aspects of the linker design that could contribute to affinity loss include use of the unstructured poly-A tether or the circular permutation of the riboswitch aptamer. However, we previously showed for another four-way junction riboswitch that circular permutations and connections of terminal ends with a stem loop improved ligand affinity,44 so the circular permutation strategy is not always detrimental to biosensor function.
A point mutation in the riboswitch aptamer called M4 was previously shown to disrupt function of the guanidine-I riboswitch.32 To show that fluorescence activation required the functional riboswitch, we generated the same G-to-C mutation in the junction biosensor Kpn J 2,2-M (Figure 2b). The mutant biosensor displayed no fluorescence activation in the presence of 10 mM guanidine (Figure S1b).
Phylogenetic Screen and Characterization of Guanidine Biosensors
We previously showed that representative riboswitches from diverse phylogeny are useful to generate efficient, highly fluorescent, and well-folded RNA biosensors.28 A phylogenetic library was developed based on functional elements from the two design rounds combined with five guanidine-I riboswitches from Bacillus clausii (Bcl), Desulfotomaculum ruminis (Dru), Bacillus subtilis (Bsu), Pseudomonas aeruginosa (Pae), and Pseudomonas fluorescens (Pfl). The phylogenetic junction library consists of the combination of five riboswitch sequences, three functional transducer stems (A, C, D), and four spacer lengths, which generate a total of 60 constructs. The phylogenetic linker library consists of the combination of five riboswitch sequences, two stem truncations (Tr-4 and Tr-5), and two poly-A linker lengths (L-4 and L-5), which provide 20 additional constructs.
All 80 phylogenetic biosensor candidates were synthesized and screened in vitro for response to guanidine at 37 °C with 3 mM Mg2+ to model physiological conditions (Figure S2). The majority of junction biosensor constructs showed some fluorescence activation in response to 1 mM guanidine (Figure S2) and 13 met our criteria of fold-activation greater than 1.4 (Figure 4a), which corresponds to a 21.6% hit rate. Interestingly, the most active constructs were generated from just two of the five phylogenetic sequences. Transducer stems A or D and spacer length 2, 3 were commonly found in these functional designs. In contrast, the majority of truncated linker biosensor constructs showed no fluorescence activation and only one linker biosensor construct met the fold-activation criteria in response to 25 mM guanidine (Figure S2). The poor response of the linker designs again could be due to stem truncation or lack of structural changes in P1 upon guanidine binding, which highlights the utility of exploring different sensor topologies.
Figure 4.

Phylogenetic junction biosensor hits and their proposed activation mechanism. (a) Phylogenetic junction biosensor hits with over 1.4× fold activation at concentrations of 1 mM guanidine. Data shown are the average with standard error of the mean for three replicates. (b) Proposed mechanism of junction biosensors which utilizes an artificial transducer stem (orange) which can assemble in the presence of ligand and induce DFHBI (yellow) fluorescence turn-on.
Previous in-line probing and structural analysis of the guanidine-I riboswitch indicates that ligand binding stabilizes the P3 stem, whereas P1 and P2 stems are mostly preformed.32 However, the P3 stem and loop regions are involved in tertiary contacts that preclude their use as the transducer stem. Instead, we introduced a P0 stem to form an artificial 4-way junction, which acts as a biosensor as P3 helix formation upon guanidine binding apparently stabilizes P0 and the DFHBI binding pocket in Spinach2 (Figure 4b).32,40 Interestingly, P0 stems A through D, which are derived from c-di-GMP riboswitches that form 3-way junctions, worked robustly in biosensor constructs, but not P0 stem E, which is derived from a SAM-I riboswitch that forms a 4-way junction. These results together demonstrate that the artificial junction design strategy developed based on the K. pneumoniae riboswitch structure is transferrable to riboswitches from other bacteria and can generate additional functional biosensors.
The ligand affinity and selectivity of three junction biosensors, Kpn J 2,2-A, Dru J 2,2-A, and Dru J 2,2-D, were compared at near physiological conditions (37 °C and 3 mM MgCl2) in preparation for in vivo studies. Under these more stringent conditions, the Kpn biosensor exhibited a 2.3-fold reduction in binding affinity (Kd ∼ 139 μM). The two Dru biosensors had similar binding affinities toward guanidine (Figure 5a) and both showed higher fold-activation (5- and 4-fold for A and D, respectively) than the Kpn biosensor (2-fold). In addition, all three biosensors showed good selectivity for guanidine over related analogues (Figure 5b). These results indicate that the three junction biosensors should selectively respond to guanidine in vivo.
Figure 5.

Sensitivity and selectivity of select guanidine biosensors. (a) Apparent dissociation constant (Kd) of guanidine for biosensors Kpn J 2,2-A, Dru J 2,2-A and Dru J 2,2-D. (b) In vitro fluorescence of Dru J 2,2-A, Dru J 2,2-D and Kpn J 2,2-A with no ligand (NL), guanidine (G), and other structural analogues at 1 mM concentrations. Data shown are the average with standard deviation of two replicates.
Given their similar binding affinities, use of the same dye-binding aptamer, Spinach2, and use of the same P0 stem, the observed difference in fold-activation and maximal fluorescence for Kpn and Dru biosensors is not due to changes in binding equilibria (Figure S3a). Rather, we expect that the riboswitch aptamer sequence is affecting overall folding efficiency of biosensor constructs. Thus, the maximal fluorescence for a given biosensor will be proportional to the percent that folds into a binding-competent state (Figure S3b). We have found for both RNA-based and protein-based biosensors that “bioprospecting” through phylogenetic libraries is an efficient way to identify well-folding variants.28,48 In some cases, the riboswitch sequence even improved folding efficiency such that our biosensors had higher maximal fluorescence than Spinach2 itself.28
Turn-on Kinetics of Guanidine Biosensors in Live Cells
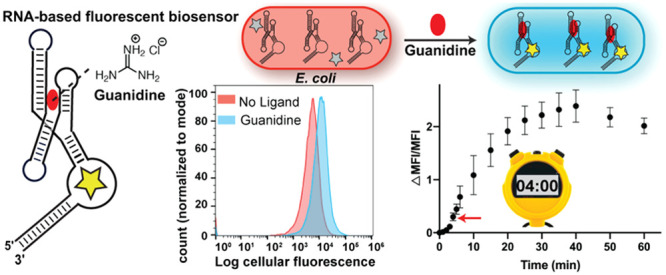
Riboswitch reporters for guanidine were reported to show a distinguishable change in reporter gene expression after overnight treatment with 1–3 mM of guanidine.32 In contrast, our biosensors display significant turn-on with 500 μM of guanidine within a much shorter time. For in vivo testing, the three biosensors and related controls were cloned into a tRNA scaffold, which previously has been shown to favor homogeneous expression of the biosensor and improve stability of RNA constructs against RNases in the cells (Table S3).25,43,49 After biosensor overexpression in BL21(DE3) Star E. coli cells grown in autoinduction media, the cells were diluted and incubated with 50 μM of DFHBI-1T in PBS buffer for 10 min to enable the fluorescent dye to passively enter the cells. Guanidine (500 μM) or water (no ligand control) then was added and mean cellular fluorescence was measured after 15 min at room temperature using flow cytometry (Figure 6a).
Figure 6.

In vivo analysis of guanidine biosensors. (a) Schematic representation of the in vivo assay for RBF biosensors using flow cytometry. Cells transformed with plasmids encoding biosensors were inoculated 24 h in NI (noninducing) media followed by 18 h in AI media at 37 °C to express the RNA biosensors. Cells were diluted in 1X PBS containing DFHBI-1T and incubated for 10 min to allow the dye to diffuse into cells. Mean fluorescence intensity (MFI) values were determined by analyzing 30 000 cells per sample after 15 min incubation with guanidine (500 μM). (b) Representative flow cytometry histograms of biosensor Dru J 2,2-D and Spinach2 in the presence or absence of guanidine. (c) MFI values of the RBF biosensors and Spinach2 in the presence and absence of guanidine. Data shown are the average with standard deviation of four biological replicates. p-Values from Student’s t test for biosensors, Kpn J 2,2-A, Dru J 2,2-A and Dru J 2,2-D are <0.004 and the p-values for Spinach2 and Kpn J 2,2-M are >0.05.
Even with this short exposure to guanidine, cells expressing the biosensors exhibited robust fluorescence activation responses (1.7 to 2.2-fold) (Figure 6b,c). In contrast, cells expressing Spinach2 and mutant Kpn J 2,2-M displayed no significant change in fluorescence in response to 500 μM of guanidine, although a nonspecific response was observed when 10 mM guanidine was used (Figure S4). The cells expressing the biosensors also exhibited minimal change in response to guanidine analogues at 500 μM concentration (Figure S5). Furthermore, we analyzed the stability of the biosensor activity by storing cells expressing the biosensor in noninducing (NI) media at 4 °C and repeating the fluorescence measurements over the course of multiple days. Excitingly, after its expression in E. coli, the guanidine biosensor maintains robust sensing activity for up to 11 days or 1.6 weeks (Figure S6).
The in vivo turn-on kinetics of the biosensors were determined using flow cytometry by analyzing time points before and after guanidine addition. Interestingly, without guanidine being added, we can observe the uptake, export, and subsequent equilibration of the fluorescent dye DFHBI-1T in these experiments for both the biosensor and Spinach2 control (Figure 7a,b). Equilibration occurs within 25 min of dye addition for the biosensor.
Figure 7.

In vivo response kinetics of guanidine biosensors. Plot of average MFI values over time for cells expressing (a) Dru J 2, 2-D biosensor or (b) Spinach2. Other biosensor plots are shown in Figure S7. Water or guanidine was added at time 0 (indicated by red arrow). (c) Plot of ΔMFI/MFI values over time for cells expressing guanidine biosensors or Spinach2. The enlarged portion of the plot displays the turn-on response at earlier time points. Data shown are the average with a standard deviation of 2–3 biological replicates.
The representative graphs show that guanidine addition leads to clear fluorescence turn-on for Dru J 2,2-D, whereas no change is observed for the control Spinach2 (Figure 7a,b, and Figure S7 for other biosensors). To account for the difference in background fluorescence over time, we determined and plotted the ΔMFI/MFI values for all three biosensors and control Spinach2 (Figure 7c). This analysis revealed that the Kpn biosensor displays a much more modest fold-activation in fluorescence than the two Dru biosensors but is faster to reach a maximal fluorescence response in vivo (∼15 min versus ∼30 min). Since the timing of the experiment shown in Figure 6 is 15 min after guanidine addition, this result explains why the Dru biosensors did not exhibit as high a fold turn-on in that experiment as seen in vitro (Figure 5b) and now seen at 30 min after guanidine addition in vivo (3-fold, see Figure 7 and Figure S7).
The difference in biosensor kinetics in vivo does not appear to be due to different expression levels, as all three biosensors were at similar levels as analyzed by RT-qPCR (Figure S8). This result also prompted us to examine the in vitro kinetics of the tRNA-scaffolded biosensors (Table S4). Maximal fluorescence response in vitro was observed in 12 and 18 min after guanidine addition, respectively, for Kpn and Dru biosensors (Figure S9). This means that all three biosensors are slower in vivo than in vitro. Common differences between the cellular and in vitro experiments include the cell membrane and molecular crowding slowing diffusion of guanidine and dye, as well as the biosensor concentrations being different when expressed in the cell compared to in vitro.
Although the maximal fluorescence response ranged from 15 to 30 min, significant signal over background can be observed within 4–5 min of guanidine addition for all three biosensors (Figure 7c). This is due to compensating effects, as the Dru biosensors have slower activation kinetics that are balanced by their higher fold turn-on relative to Kpn. Taken together, these results reveal that riboswitch-based fluorescent biosensors are capable of responding quickly in vivo and in vitro, with significant fluorescent signal over background within as little as 4 min of ligand addition.
Turn-on Kinetics of Guanidine Reporter
A major motivation of our study was to compare cellular turn-on kinetics for a riboswitch-based biosensor versus reporter for the same ligand, which to our knowledge has not been done. To construct the Kpn guanidine reporters, the synthetic promoter BBa_J23100 and the 5′ untranslated region of the K. pneumoniae tauA gene containing the guanidine riboswitch32 (with or without M4 mutation) (Figure 2b) were fused upstream of the lacZ reporter gene (Figure 8a). Notably, the reporter uses the same riboswitch aptamer sequence as the Kpn biosensor. The reporter gene chosen, lacZ, is most commonly used in riboswitch assays and encodes the highly active β-galactosidase enzyme that increases sensitivity of the assay due to lower background than fluorescent protein reporters.
Figure 8.

Analysis of guanidine sensing by a riboswitch lacZ reporter. (a) Schematic representation of the assay for the riboswitch lacZ reporter using the standard Miller assay, which detects expression of β-galactosidase using the colorimetric substrate ortho-nitrophenyl-β-galactoside (ONPG). (b) β-Galactosidase reporter gene expression in Miller Units (MU) as controlled by Kpn-WT and Kpn-M4 riboswitches with different concentrations of guanidine. (c) Plot of guanidine reporter response (ΔMU/MU) over time after addition of 50 mM guanidine at time 0. Data shown are the average with a standard deviation of three biological replicates.
The Kpn-WT and Kpn-M4 mutant reporter plasmids were transformed into BW25113 E. coli cells and riboswitch-regulated gene expression with different guanidine concentrations was quantified using a standard Miller assay.50 Increasing activation of the Kpn-WT reporter expression was observed after 5 h of incubation with 25 mM and higher concentrations of guanidine, whereas the Kpn-M4 reporter was insensitive to guanidine, as expected (Figure 8b). At this time point, no detectable change in reporter expression was observed with 10 mM or lower concentrations of guanidine (data not shown). In fact, the K1/2 of the Kpn-WT reporter was found to be between 50 and 70 mM (Figure S10), whereas 100 mM of guanidine inhibited cell growth.
To analyze the in vivo turn-on kinetics of the reporter in a similar way as the biosensors, the ΔMU/MU values were measured and plotted for time points after the addition of 50 mM guanidine. With this reporter, a maximal signal was observed in 2.5 h, and a significant signal over background can be observed within 1 h of guanidine addition (Figure 8c, Figure S11). Thus, this direct comparison between a biosensor and reporter based on the same riboswitch reveals that the biosensor provides 15 times faster response (4 versus 60 min) even when exposed to 1/100 the concentration of the target analyte (0.5 versus 50 mM).
Conclusions
This study demonstrates the first biosensor developed for guanidine, a commonly used chaotropic agent that is also an explosives precursor, fertilizer component, and recently identified metabolite in bacteria. Unlike most riboswitch ligands that have been targeted for biosensing, guanidine is freely diffusible into cells and does not require further enzymatic processing to yield the target ligand. These properties enabled us to directly compare the in vivo turn-on kinetics of a biosensor and reporter derived from the same riboswitch aptamer sequence in response to the same ligand. These head-to-head clocking experiments demonstrate that the Kpn biosensor is at least 15 times faster than the Kpn reporter and obtains a maximal signal in 15 min, which is considerably before the reporter gives any significant signal over the background. The slower but higher turn-on Dru biosensors are at least 12 times faster and obtain maximal signal in 30 min, which is still before the reporter gives a reliable signal. We state “at least”, because the reporter showed no change in signal after 5 h incubation with 500 μM guanidine, which is the concentration detected by the biosensors. Changes in reporter expression were observed previously upon overnight treatment with 1–3 mM guanidine.32 This reporter used the lysC promoter, but otherwise was the same as the one used in our study. Assuming that overnight treatment is 12–16 h and gives maximal reporter signal, the biosensor maximal response may be actually 48–64 times faster than the reporter.
The observed difference in response times is expected because the reporter must go through additional steps after guanidine binding induces structural changes to the riboswitch, namely transcription and translation of the lacZ gene, which is 3057 base pairs long. These processes are both time-consuming and resource-intensive for the cell. In contrast, the biosensor is approximately the size of the riboswitch and directly binds fluorogenic dye after guanidine-induced structural changes. A recently published study with the fluoride ion-sensing riboswitch analyzed the turn-on of two types of fluorescent reporters in cell-free reactions. A GFP reporter detected 3.5 mM sodium fluoride in 30 min with maximal signal in 8 h, whereas a fluorogenic aptamer reporter (the riboswitch controls expression of the RNA aptamer) provided a detectable signal in 12 min with maximal signal in 35 min.51 This study further enforces that translation may be rate-limiting, although it should be noted that the reporter genes are different lengths, and GFP requires an extra chromophore maturation step. One benefit of cell-free reactions is that conditions such as magnesium concentrations can be optimized to improve performance; the cell-free reporter kinetics were obtained with 12 mM Mg2+, which is above normal physiological concentrations. Since our results show that the fluorescent biosensors exhibit similar binding kinetics in vitro (at 3 mM Mg2+) and in vivo, we expect that they also would function well in cell-free reactions.
One notable aspect of the biosensor kinetics is that these results were achieved with an artificial junction. The guanidine-I riboswitch topology and the large distance between terminal ends posed special challenges to biosensor design that were met by rigorous testing of two distinct topological designs and optimization of several parameters, including transducer stem sequence, adenosine spacer length, and riboswitch aptamer sequence. We found that the first two parameters introduced additional variables that increased library size and limited the number of phylogenetic variants that were assessed. For c-di-GMP biosensors, which only required optimization of the riboswitch aptamer sequence, a larger sampling of phylogenetic diversity identified several biosensor sequences with half-maximal activation (t1/2) in 1–1.5 min.28 On the basis of our current study, we expect that these biosensors would exhibit similar binding kinetics in vivo, and so likely are limited only by the activation kinetics of signaling enzymes that produce or degrade c-di-GMP.
Finally, one underappreciated advantage of RBF biosensors is that their activation mechanism is the same in vitro and in vivo. Thus, after expression in cells, these biosensors possess both reasonably fast response kinetics (minutes, Figure 7) and functional longevity (weeks, Figure S6). In addition, we show that construct improvements obtained from in vitro screening efforts can be directly translated in vivo. For example, higher signal and fold-activation for Dru biosensors were recapitulated in vivo. Ongoing work focuses on establishing in vitro screening methods to make faster RBF biosensors that break the speed limits set by natural riboswitch sequences and further enable real-time sensing for cell biology, biotechnology, and synthetic biology applications.
Material and Methods
Reagents and Oligonucleotides
DNA oligonucleotides used for biosensor constructs and cloning were purchased either from Integrated DNA Technologies (Chicago, IL) or from the University of Utah HSC Core facility. Guanidine hydrochloride and all guanidine analogues were purchased from Sigma-Aldrich (St Louis, MO). DFHBI and DFHBI-1T was synthesized following previously described protocols52,53 and was stored as a 10 mM stock in DMSO at −20 °C. Chemically competent BL21 (DE3) Star cells were purchased from Life Technologies (Carlsbad, CA). BW25113 competent cells were prepared using standard protocols.54
Junction biosensors (Table S1) were constructed by obtaining each phylogenetic riboswitch sequence as an ultramer (IDT) and then performing two sequential PCRs to produce the full-length biosensor sequence. The first PCR utilizes a primer pair that anneals to the 5′ and 3′ ends of the riboswitch sequence with overhangs containing the desired P0 stem, adenosine spacer length, and part of the Spinach2 aptamer. The second PCR utilizes a primer pair that recognizes the partial Spinach2 sequence and possesses overhangs for the remainder of the Spinach2 aptamer. The PCR products after each step were purified either by a 96-well format ZR-96 DNA clean-up kit (Zymo Research) for screening or by QIAquick PCR purification kit (Qiagen) for characterization.
Linker biosensors (Table S2) were constructed by ordering a truncated riboswitch biosensor sequence with the desired poly-A linker length and part of the Spinach2 aptamer as an ultramer (IDT). PCR was performed with primers that recognize the partial Spinach2 sequence on the 5′ and 3′ ends and possess overhang remainders of the Spinach2 aptamer. PCR products were purified either by a 96-well format ZR-96 DNA clean-up kit (Zymo Research) for screening or by QIAquick PCR purification kit (Qiagen) for characterization.
In Vitro Transcription
DNA templates for in vitro transcription were prepared by PCR amplification using Phusion DNA polymerase (NEB) from ultramer oligonucleotides for screening or sequence-confirmed plasmids for analytical experiments. The forward primer introduced an extended T7 promoter sequence at the 5′ end. PCR products were purified either by a 96-well format ZR-96 DNA clean-up kit (Zymo Research) for screening or by QIAquick PCR purification kit (Qiagen) for analytical experiments. RNA was transcribed from DNA templates using T7 RNA polymerase in 40 mM Tris- HCl, pH 8.0, 6 mM MgCl2, 2 mM spermidine, and 10 mM DTT. RNAs were either purified by a 96-well format ZR-96 Clean & Concentrator (Zymo Research) for screening or by denaturing (7.5 M urea) 6% PAGE for analytical experiments. RNAs purified by PAGE were visualized by UV shadowing and extracted from gel pieces using Crush Soak buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl and 1 mM EDTA, pH 8.0). Purified RNAs were precipitated with ethanol, dried, and then resuspended in water. Accurate RNA concentrations were determined by measuring the absorbance at 260 nm after performing a hydrolysis assay to eliminate the hypochromic effect due to an RNA secondary structure.55
General Procedure for in Vitro Fluorescence Assays
In vitro fluorescence assays were carried out with 100 nM RNA in binding buffer containing 10 μM DFHBI, 40 mM HEPES (pH 7.5), 125 mM KCl, 0 or 200 mM NaCl, and 3 or 10 mM MgCl2 as indicated in the figures. Other conditions, including temperature (28 or 37 °C) and concentration of ligands, were varied as indicated in the figures. The biosensor RNA (1 μM) was renatured by heating at 72 °C for 3 min in binding buffer then cooling to an ambient temperature for 10 min prior to addition into the reaction solution. DFHBI was added to the solution containing buffer and RNA, and then the ligand (or water for no ligand control) was added before fluorescence measurement. Binding reactions were performed in 50 μL volumes and fluorescence emission was recorded at the indicated temperature in a Greiner Bio-One 384-well black plate using a SpectraMax i3x plate reader (Molecular Devices) for 60 min. The fluorescence emission was calculated as an average of the values measured between 30 to 60 min with the following instrument parameters: 448 nm excitation, 506 nm emission. Fluorescence turn-on was calculated by dividing the fluorescence in the presence of guanidine by fluorescence in the absence of the guanidine.
Binding Affinity Analysis of Guanidine Biosensors
The binding affinities of guanidine biosensors were measured with 100 nM RNA in binding buffer containing 10 μM DFHBI, 40 mM HEPES (pH 7.5), 125 mM KCl, 0 or 200 mM NaCl, and 3 or 10 mM MgCl2. The guanidine concentration was varied from 10 nM to 10 mM. The fluorescence of the sample with DFHBI but no RNA was subtracted as background to determine relative fluorescence units. The dissociation constant (Kd) for each binding event was calculated from the concentration-dependent fluorescence curves by fitting the normalized fluorescence intensity (FN) versus log of guanidine concentration plot to a nonlinear regression (log (agonist) vs response (three parameter)) using Prism 8 software. FN was calculated as (Fi – F0)/(Fs – F0), where Fi is fluorescence intensity at each ligand concentration, F0 is fluorescence intensity without ligand, and Fs is fluorescence intensity at the saturation point.
In Vitro Fluorescence Turn-on Kinetics
The biosensor RNA in tRNA scaffold (Table S4) (100 nM) was renatured in binding buffer (40 mM HEPES (pH 7.5), 125 mM KCl, 200 mM NaCl, 3 mM MgCl2) as previously mentioned, then pre-equilibrated with 10 μM DFHBI (final concentration) for 15 min at 37 °C. Guanidine (500 μM final concentration) was added using the automated injector module of the SpectraMax i3x plate reader (Molecular Devices) at the 30 s mark of the 20 min measurement period. Kinetic experiments were performed in 100 μL reaction volumes in CORNING Costar 96-well black with clear flat bottom plates due to the use of bottom reads in this mode. Fluorescence measurements were taken every 0.5 s for 20 min total.
Biosensor and Reporter Cloning
For in vivo assays, biosensor sequences were appended with a tRNA scaffold (Table S3) through overhang addition by PCR, and the resulting products were subcloned into the pET31b plasmid using a double restriction digest and ligation with BglII and XhoI restriction sites.43 The Kpn riboswitch reporter includes a synthetic promoter BBa_J23100, obtained from the iGEM Registry of Standard Biological Parts (http://parts.igem.org/Promoters/Catalog/Constitutive), the 5′ untranslated region of the K. pneumoniae tauA gene containing the guanidine riboswitch (with or without M4 mutation), and the coding region of the lacZ reporter gene. The reporter was constructed by cloning the 5′ untranslated region into a modified pRS414 vector containing the BBa_J23100 promoter and lacZ reporter using Gibson Assembly,56 for which both linear backbone and insert fragments were amplified by PCR with Phusion. The 5′ untranslated region harboring the Kpn riboswitch was amplified from the original K. pneumoniae reporter construct received from Prof. Ronald Breaker at Yale University.32
In Vivo Fluorescence Assay Using Flow Cytometry
E. coli BL21 (DE3) Star cells were transformed with 10 ng of plasmid containing biosensor-tRNA construct and cells were plated on LB/carbenicillin plates (Carb: 50 μg/mL). Four single colonies for each construct were inoculated in 0.5 mL noninducing media (NI) containing carbenicillin (50 μg/mL) in a 96 deep-well plate (2.2 mL/well) and grown at 37 °C in an incubator with shaking for 24 h until an OD600 of 3–4 was reached. The NI culture was diluted 50× into ZYP-5052 autoinduction media (AI) containing carbenicillin (50 μg/mL) in a 14 mL polystyrene culture tube and grown for 18 h at 37 °C in an incubator with shaking to express the biosensors.
For end point flow cytometry assays, 2 μL of the AI culture was diluted into 96 μL of PBS buffer containing DFHBI-1T (50 μM) in a 96 well plate (330 μL/well) and incubated for 10 min to allow DFHBI-1T to diffuse into cells. To achieve a final guanidine concentration of 500 μM, 2 μL of 50 mM guanidine was added to the samples and incubated at room temperature for 15 min. Single-cell fluorescence was measured using an Attune NxT flow cytometer (Life Technologies) using the following settings: excitation laser, 488 nm; emission channel, GFP; cell counts for each measurement, 30 000. The data were analyzed using FlowJo software.
For checking the stability of biosensor activity, 3 mL of the AI culture was centrifuged at 4500 rpm for 10 min at 4 °C, the AI media was decanted, and the cell pellet was resuspended in 3 mL of NI media containing carbenicillin (50 μg/mL) and stored at 4 °C. The sensing activity of the biosensor was measured using flow cytometry as described above for up to 11 days. After 11 days, the experiment was concluded as a significant population of nonfluorescent cells was observed.
For kinetic flow cytometry assays, 2 μL of the AI culture was diluted in 96 μL of PBS containing 50 μM DFHBI-1T in a 96 well plate (330 μL/well) and single-cell fluorescence was measured at 0.5, 5, and 10 min to monitor DFHBI-1T diffusion into cells. To achieve a final guanidine concentration of 500 μM, 2 μL of 50 mM guanidine was added and single-cell fluorescence was measured in 1 min time points over 6 min followed by 5 min time points over 60 min using the flow cytometer. For each time point, ΔMFI/MFI was calculated as MFIguanidine – MFIwater/MFIwater, and p-values were calculated using Student’s t test to determine the minimum time required for significant turn-on. p-Values < 0.05 were considered as significant turn-on.
Quantitative RT-PCR (qRT-PCR)
Total RNA samples were isolated by using QIAGEN RNeasy kit (catalog no. 74104) from BL21(DE3) Star E. coli cells transformed with the respective biosensor plasmid and grown in NI media (24 h) followed by grown in AI media (18 h) supplemented with 50 μg/mL carbenicillin. The integrity of the total RNA was analyzed by 1% agarose gel electrophoresis and by using the Agilent 4200 TapeStation system. One step quantitative RT-PCR was performed using NEB Luna Universal One-Step RT-qPCR Kit (catalog no. E3005S) with total RNA samples and appropriate primers. One set of primers was specific to a portion of the tRNA and Spinach2 sequence and was common for all three biosensors. Another set of primers was designed for the endogenous 5S rRNA. For both sets of primers, the amplicon length was 68 nt. The reactions were performed on a 96-well reaction plate using a BioRad CFX96 real time system monitoring by SYBR fluorescence. The thermal cycling conditions used were 10 s at 95 °C, then 30 s at 59 °C. CT (cycle threshold) values were determined using CFX Manager Software with automatic baseline and threshold determination. All reactions were performed in triplicate.
Expression levels of the biosensors were quantified using the relative standard curve method described in the Applied Biosystems guide, “Guide to Performing Relative Quantitation of Gene Expression Using Real-Time Quantitative PCR”. Real-time PCR standard curves were generated by performing RT-PCR with serial dilutions of the total RNA samples (1:1000 to 1:32000) (Figure S9c). PCR efficiency for each set of primers was calculated using the equation E = (10(−1/slope) – 1) × 100. RT-qPCR was performed with the 1:8000 dilution of total RNA samples using both sets of primers (biosensor-specific and 5s rRNA-specific primers). The relative expression level of each biosensor was quantified by dividing the average CT value for each biosensor RNA with the respective CT value for the control, 5S rRNA.
Miller Assay
Chemically competent E. coli BW25113 cells were transformed with 10 ng of the reporter plasmid and plated on LB/carbenicillin (Carb) plates (Carb: 50 μg/mL). Three single colonies were inoculated separately in minimal media containing 1× M9 salts (22 mM KH2PO4, 8.55 mM NaCl, 50 mM Na2HPO4, and 18.7 mM NH4Cl) supplemented with MgSO4 (2 mM), CaCl2 (100 μM), and glucose (0.4%), with the addition of carb (50 μg/mL), and grown at 37 °C in an incubator with shaking overnight. The overnight culture was diluted 200× in minimal media with carb (50 μg/mL) and grown either in the presence (5–90 mM) or absence of guanidine for ∼5 h until the OD reached 0.6. The cell suspension was then analyzed using the colorimetric Miller assay with the β-galactosidase substrate ONPG, and Miller Units were calculated following the previous report.57
For kinetic assays, the overnight cultures were diluted 200× into 3 mL of minimal media with carb (50 μg/mL) and grown until the OD reached 0.6 (∼5 h). Guanidine (50 mM final concentration) was added to the cultures and 500 μL aliquots were taken every 30 min over 5 h to perform the Miller assay. For each time point, ΔMU/MU was calculated as (MUguanidine – MUwater)/MUwater.
Acknowledgments
This work was supported in part by Office of Naval Research Grant N000141912043 to M.C.H. and NIH Grant R01 GM124589 to M.C.H. We thank Prof. Ronald Breaker at Yale University for generously sharing the Kpn riboswitch lacZ reporter plasmid. We thank Prof. James Gagnon at the University of Utah for allowing us to use the TapeStation instrument.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00583.
Supplementary Figures S1–S11 show phylogenetic screen, mechanisms for fluorescence activation, biosensor selectivity, longevity of biosensor signal, RT-qPCR, in vitro turn-on kinetics of biosensors, and in vivo response kinetics of reporter; Table S1–S4 show biosensor sequences (PDF)
Author Contributions
# S.M. and J.T. are considered as joint first authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Sun Z.; Nguyen T.; McAuliffe K.; You M. (2019) Intracellular Imaging with Genetically Encoded RNA-based Molecular Sensors. Nanomaterials 9, 233. 10.3390/nano9020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S.; Ang E. L.; Zhao H. (2018) In vivo biosensors: mechanisms, development, and applications. J. Ind. Microbiol. Biotechnol. 45, 491–516. 10.1007/s10295-018-2004-x. [DOI] [PubMed] [Google Scholar]
- Courbet A.; Renard E.; Molina F. (2016) Bringing next-generation diagnostics to the clinic through synthetic biology. EMBO Mol. Med. 8, 987–991. 10.15252/emmm.201606541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serganov A.; Nudler E. (2013) A decade of riboswitches. Cell 152, 17–24. 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stav S.; Atilho R. M.; Mirihana Arachchilage G.; Nguyen G.; Higgs G.; Breaker R. R. (2019) Genome-wide discovery of structured noncoding RNAs in bacteria. BMC Microbiol. 19, 66. 10.1186/s12866-019-1433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stifel J.; Spöring M.; Hartig J. S. (2019) Expanding the toolbox of synthetic riboswitches with guanine-dependent aptazymes. Synthetic Biology 4, 22. 10.1093/synbio/ysy022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lünse C. E., and Mayer G. (2017) Reporter Gene-Based Screening for TPP Riboswitch Activators, In Antibiotics: Methods and Protocols (Sass P., Ed.), pp 227–235, Springer New York, New York, NY. [DOI] [PubMed] [Google Scholar]
- Yagur-Kroll S.; Lalush C.; Rosen R.; Bachar N.; Moskovitz Y.; Belkin S. (2014) Escherichia coli bioreporters for the detection of 2,4-dinitrotoluene and 2,4,6-trinitrotoluene. Appl. Microbiol. Biotechnol. 98, 885–895. 10.1007/s00253-013-4888-8. [DOI] [PubMed] [Google Scholar]
- Hallberg Z. F.; Su Y.; Kitto R. Z.; Hammond M. C. (2017) Engineering and In Vivo Applications of Riboswitches. Annu. Rev. Biochem. 86, 515–539. 10.1146/annurev-biochem-060815-014628. [DOI] [PubMed] [Google Scholar]
- Chakraborty S.; Mehtab S.; Krishnan Y. (2014) The Predictive Power of Synthetic Nucleic Acid Technologies in RNA Biology. Acc. Chem. Res. 47, 1710–1719. 10.1021/ar400323d. [DOI] [PubMed] [Google Scholar]
- Zhong G.; Wang H.; Bailey C. C.; Gao G.; Farzan M. (2016) Rational design of aptazyme riboswitches for efficient control of gene expression in mammalian cells. eLife 5, e18858 10.7554/eLife.18858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachter A.; Tunc-Ozdemir M.; Grove B. C.; Green P. J.; Shintani D. K.; Breaker R. R. (2007) Riboswitch control of gene expression in plants by splicing and alternative 3′ end processing of mRNAs. Plant Cell 19, 3437–3450. 10.1105/tpc.107.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khani A.; Popp N.; Kreikemeyer B.; Patenge N. (2018) A Glycine Riboswitch in Streptococcus pyogenes Controls Expression of a Sodium:Alanine Symporter Family Protein Gene. Front. Microbiol. 9, 200. 10.3389/fmicb.2018.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler C. C., and Li Y. (2014) Construction and Application of Riboswitch-Based Sensors That Detect Metabolites Within Bacterial Cells, In Therapeutic Applications of Ribozymes and Riboswitches: Methods and Protocols (Lafontaine D., and Dubé A., Eds.) pp 177–197, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Gao X.; Dong X.; Subramanian S.; Matthews P. M.; Cooper C. A.; Kearns D. B.; Dann C. E. 3rd (2014) Engineering of Bacillus subtilis strains to allow rapid characterization of heterologous diguanylate cyclases and phosphodiesterases. Appl. Environ. Microbiol. 80, 6167–6174. 10.1128/AEM.01638-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbaugh S. V.; Goodson M. S.; Dillon K.; Zabarnick S.; Kelley-Loughnane N. (2017) Riboswitch-Based Reversible Dual Color Sensor. ACS Synth. Biol. 6, 766–781. 10.1021/acssynbio.6b00199. [DOI] [PubMed] [Google Scholar]
- Sekella P. T.; Rueda D.; Walter N. G. (2002) A biosensor for theophylline based on fluorescence detection of ligand-induced hammerhead ribozyme cleavage. RNA 8, 1242–1252. 10.1017/S1355838202028066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausländer S.; Ketzer P.; Hartig J. S. (2010) A ligand-dependent hammerhead ribozyme switch for controlling mammalian gene expression. Mol. BioSyst. 6, 807–814. 10.1039/b923076a. [DOI] [PubMed] [Google Scholar]
- Jaffrey S. R.; Pasternak G. W.; Coyle J. T. (2018) RNA-Based Fluorescent Biosensors for Detecting Metabolites in vitro and in Living Cells. Adv. Pharmacol. 82, 187–203. 10.1016/bs.apha.2017.09.005. [DOI] [PubMed] [Google Scholar]
- Su Y.; Hammond M. C. (2020) RNA-based fluorescent biosensors for live cell imaging of small molecules and RNAs. Curr. Opin. Biotechnol. 63, 157–166. 10.1016/j.copbio.2020.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige J. S.; Nguyen-Duc T.; Song W.; Jaffrey S. R. (2012) Fluorescence imaging of cellular metabolites with RNA. Science 335, 1194–1194. 10.1126/science.1218298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.; Hickey S. F.; Keyser S. G. L.; Hammond M. C. (2016) In Vitro and In Vivo Enzyme Activity Screening via RNA-Based Fluorescent Biosensors for S-Adenosyl-l-homocysteine (SAH). J. Am. Chem. Soc. 138, 7040–7047. 10.1021/jacs.6b01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M.; Litke J. L.; Jaffrey S. R. (2015) Imaging metabolite dynamics in living cells using a Spinach-based riboswitch. Proc. Natl. Acad. Sci. U. S. A. 112, E2756. 10.1073/pnas.1504354112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepsen M. D. E.; Sparvath S. M.; Nielsen T. B.; Langvad A. H.; Grossi G.; Gothelf K. V.; Andersen E. S. (2018) Development of a genetically encodable FRET system using fluorescent RNA aptamers. Nat. Commun. 9, 18–18. 10.1038/s41467-017-02435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger C. A.; Wilson S. C.; Sales-Lee J.; Hammond M. C. (2013) RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messengers Cyclic di-GMP and Cyclic AMP-GMP. J. Am. Chem. Soc. 135, 4906–4909. 10.1021/ja311960g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger C. A.; Wilson S. C.; Hickey S. F.; Gonzalez T. L.; Su Y.; Hallberg Z. F.; Brewer T. F.; Iavarone A. T.; Carlson H. K.; Hsieh Y.-F.; Hammond M. C. (2015) GEMM-I riboswitches from Geobacter sense the bacterial second messenger cyclic AMP-GMP. Proc. Natl. Acad. Sci. U. S. A. 112, 5383. 10.1073/pnas.1419328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger C. A.; Chen C.; Whiteley A. T.; Portnoy D. A.; Hammond M. C. (2015) RNA-Based Fluorescent Biosensors for Live Cell Imaging of Second Messenger Cyclic di-AMP. J. Am. Chem. Soc. 137, 6432–6435. 10.1021/jacs.5b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. C.; Wilson S. C.; Hammond M. C. (2016) Next-generation RNA-based fluorescent biosensors enable anaerobic detection of cyclic di-GMP. Nucleic Acids Res. 44, e139 10.1093/nar/gkw580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter E. B.; Polaski J. T.; Morck M. M.; Batey R. T. (2017) Recurrent RNA motifs as scaffolds for genetically encodable small-molecule biosensors. Nat. Chem. Biol. 13, 295–301. 10.1038/nchembio.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo J.; Dippel A. B.; Wang X. C.; Hammond M. C. (2018) In Vivo Biochemistry: Single-Cell Dynamics of Cyclic Di-GMP in Escherichia coli in Response to Zinc Overload. Biochemistry 57, 108–116. 10.1021/acs.biochem.7b00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You M.; Litke J. L.; Wu R.; Jaffrey S. R. (2019) Detection of Low-Abundance Metabolites in Live Cells Using an RNA Integrator. Cell Chem. Biol. 26, 471–481. 10.1016/j.chembiol.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J. W.; Atilho R. M.; Sherlock M. E.; Stockbridge R. B.; Breaker R. R. (2017) Metabolism of Free Guanidine in Bacteria Is Regulated by a Widespread Riboswitch Class. Mol. Cell 65, 220–230. 10.1016/j.molcel.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahi T.; Weaver D. J.; Lamb J. D.; Harrison R. G. (2016) A new approach for trace analysis of guanidine compounds in surface water with resorcinarene-based ion chromatography columns. Analyst 141, 939–946. 10.1039/C5AN01076D. [DOI] [PubMed] [Google Scholar]
- Price D.; Clairmont A. R. (1969) Explosive behavior of nitroguanidine. Symp. (Int.) Combust., [Proc.] 12, 761–770. 10.1016/S0082-0784(69)80457-1. [DOI] [Google Scholar]
- Qiu J.; Lee H.; Zhou C. (2005) Analysis of guanidine in high salt and protein matrices by cation-exchange chromatography and UV detection. Journal of Chromatography A 1073, 263–267. 10.1016/j.chroma.2004.10.045. [DOI] [PubMed] [Google Scholar]
- Sherlock M. E.; Malkowski S. N.; Breaker R. R. (2017) Biochemical Validation of a Second Guanidine Riboswitch Class in Bacteria. Biochemistry 56, 352–358. 10.1021/acs.biochem.6b01270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherlock M. E.; Breaker R. R. (2017) Biochemical Validation of a Third Guanidine Riboswitch Class in Bacteria. Biochemistry 56, 359–363. 10.1021/acs.biochem.6b01271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenkeit F.; Eckert I.; Hartig J. S.; Weinberg Z. (2020) Discovery and characterization of a fourth class of guanidine riboswitches. Nucleic Acids Res. 48, 12889–12899. 10.1093/nar/gkaa1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvail H.; Balaji A.; Yu D.; Roth A.; Breaker R. R. (2020) Biochemical Validation of a Fourth Guanidine Riboswitch Class in Bacteria. Biochemistry 59, 4654–4662. 10.1021/acs.biochem.0c00793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss C. W.; Xiong Y.; Strobel S. A. (2017) Structural Basis for Ligand Binding to the Guanidine-I Riboswitch. Structure 25, 195–202. 10.1016/j.str.2016.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Wang J.; Lilley D. M. J. (2017) The Structure of the Guanidine-II Riboswitch. Cell Chem. Biol. 24, 695–702. 10.1016/j.chembiol.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Wang J.; Wilson T. J.; Lilley D. M. J. (2017) Structure of the Guanidine III Riboswitch. Cell Chem. Biol. 24, 1407–1415. 10.1016/j.chembiol.2017.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger C. A., Hallberg Z. F., and Hammond M. C. (2015) Live Cell Imaging Using Riboswitch-Spinach tRNA Fusions as Metabolite-Sensing Fluorescent Biosensors in RNA Scaffolds: Methods and Protocols (Ponchon L., Ed.) pp 87–103, Springer New York, New York, NY. [DOI] [PubMed] [Google Scholar]
- Truong J.; Hsieh Y.-F.; Truong L.; Jia G.; Hammond M. C. (2018) Designing fluorescent biosensors using circular permutations of riboswitches. Methods 143, 102–109. 10.1016/j.ymeth.2018.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orelle C.; Carlson E. D.; Szal T.; Florin T.; Jewett M. C.; Mankin A. S. (2015) Protein synthesis by ribosomes with tethered subunits. Nature 524, 119–124. 10.1038/nature14862. [DOI] [PubMed] [Google Scholar]
- Batey R. T.; Gilbert S. D.; Montange R. K. (2004) Structure of a natural guanine-responsive riboswitch complexed with the metabolite hypoxanthine. Nature 432, 411–415. 10.1038/nature03037. [DOI] [PubMed] [Google Scholar]
- Smith K. D.; Lipchock S. V.; Ames T. D.; Wang J.; Breaker R. R.; Strobel S. A. (2009) Structural basis of ligand binding by a c-di-GMP riboswitch. Nat. Struct. Mol. Biol. 16, 1218–1223. 10.1038/nsmb.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dippel A. B.; Anderson W. A.; Evans R. S.; Deutsch S.; Hammond M. C. (2018) Chemiluminescent Biosensors for Detection of Second Messenger Cyclic di-GMP. ACS Chem. Biol. 13, 1872–1879. 10.1021/acschembio.7b01019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponchon L.; Dardel F. (2007) Recombinant RNA technology: the tRNA scaffold. Nat. Methods 4, 571–576. 10.1038/nmeth1058. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Bremer H. (1995) Control of the Escherichia coli rrnB P1 promoter strength by ppGpp. J. Biol. Chem. 270, 11181–11189. 10.1074/jbc.270.19.11181. [DOI] [PubMed] [Google Scholar]
- Thavarajah W.; Silverman A. D.; Verosloff M. S.; Kelley-Loughnane N.; Jewett M. C.; Lucks J. B. (2020) Point-of-Use Detection of Environmental Fluoride via a Cell-Free Riboswitch-Based Biosensor. ACS Synth. Biol. 9, 10–18. 10.1021/acssynbio.9b00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige J. S.; Wu K. Y.; Jaffrey S. R. (2011) RNA mimics of green fluorescent protein. Science 333, 642–646. 10.1126/science.1207339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.; Strack R. L.; Svensen N.; Jaffrey S. R. (2014) Plug-and-Play Fluorophores Extend the Spectral Properties of Spinach. J. Am. Chem. Soc. 136, 1198–1201. 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. T.; Niemela S. L.; Miller R. H. (1989) One-step preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc. Natl. Acad. Sci. U. S. A. 86, 2172–2175. 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S. C.; Cohen D. T.; Wang X. C.; Hammond M. C. (2014) A neutral pH thermal hydrolysis method for quantification of structured RNAs. RNA 20, 1153–1160. 10.1261/rna.045856.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R.-Y.; Venter J. C.; Hutchison C. A.; Smith H. O. (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Sudarsan N.; Wickiser J. K.; Nakamura S.; Ebert M. S.; Breaker R. R. (2003) An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 17, 2688–2697. 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.