

Abstract
Objectives
Pyruvate kinase (PK) deficiency is caused by PKLR gene mutations, leading to defective red blood cell glycolysis and hemolytic anemia. Rates of comorbidities and complications by transfusion history and relative to the general population remain poorly quantified.
Methods
Data for patients aged ≥ 18 years with two confirmed PKLR mutations were obtained from the PK deficiency Natural History Study (NCT02053480). Frequencies of select conditions were compared with an age‐ and sex‐matched cohort from a general insured US population without PK deficiency.
Results
Compared with the matched population (n = 1220), patients with PK deficiency (n = 122) had significantly higher lifetime rates of osteoporosis, liver cirrhosis, and pulmonary hypertension; splenectomy and cholecystectomy rates were also significantly higher in the 8 years before the index date. Sixty‐five (53.3%) patients with PK deficiency were classified as regularly transfused, 30 (24.6%) as occasionally transfused, and 27 (22.1%) as never transfused. Regularly transfused patients were significantly more likely than never transfused patients to have had splenectomy, cholecystectomy, and/or thrombosis. Liver iron overload was reported in 62% of patients and occurred regardless of transfusion cohort.
Conclusions
Even never transfused patients with PK deficiency had higher rates of select comorbidities and complications than individuals without PK deficiency.
Keywords: blood transfusion, comorbidity, pyruvate kinase deficiency
1. INTRODUCTION
Pyruvate kinase (PK) deficiency is a rare congenital disorder caused by autosomal recessive mutations in the liver‐ and red cell‐specific PKLR gene, leading to a defect in the glycolytic pathway in red blood cells and hereditary hemolytic anemia. 1 , 2 , 3 This glycolytic defect leads to a reduction in adenosine triphosphate, and shortened reticulocyte and red cell lifespans due to an inability to maintain the red cell electrochemical gradient and membrane integrity. 4 As a consequence, damaged PK‐defective erythrocytes undergo brisk hemolysis in the spleen. 2 , 5
The diagnosed prevalence of PK deficiency in the general population is estimated to be between 3.2 and 8.5 cases per million individuals. 6 , 7 , 8 , 9 , 10 Higher prevalence is observed in specific populations, such as in the US Amish community, due to the founder effect. 11 , 12
In addition to anemia, PK deficiency is associated with serious complications. Estimates of complication rates observed in 254 pediatric and adult patients in the PK deficiency Natural History Study (NHS) include iron overload (48%), gallstones (45%), and bone fracture (17%). 13 Furthermore, PK deficiency has been shown to negatively affect quality of life due to impacts on physical, emotional, and social functioning. 14
Current management of PK deficiency is supportive and includes red blood cell transfusions, iron chelation therapy, and/or splenectomy. 15 Both the hemolysis and supportive treatments are associated with short‐ and long‐term risks. Splenectomy is associated with the risk of thrombosis and pulmonary hypertension 16 ; transfusions are associated with iron loading. Hemolysis is associated with transfusion‐associated and transfusion‐independent iron loading, as well as extramedullary hematopoiesis, osteopenia, and pigmented gallstones. 1 , 15 There are currently no approved pharmacologic options for altering the disease pathophysiology of PK deficiency.
The aims of this analysis were to (a) compare prevalence rates of comorbidities and complications among adults with PK deficiency with those observed in a general insured US population without PK deficiency and (b) better understand the impact of transfusion history on the prevalence of comorbidities and complications in adults with PK deficiency.
2. METHODS
This was a retrospective analysis of cross‐sectional data from two separate data sources, attempting to estimate the lifetime prevalence of select conditions based on reported medical history.
2.1. Data source and inclusion criteria: PK deficiency population
The PK deficiency NHS is a longitudinal, retrospective, and prospective cohort study that enrolled 278 patients with biochemically or molecularly diagnosed PK deficiency from June 2014 to April 2017 at 31 centers in six countries in North America and Europe (ClinicalTrials.gov NCT02053480). 13 The study protocol was approved by each site's Institutional Review Board and/or ethics committee, and all patients gave informed consent. Patient demographics and clinical, laboratory, transfusion, and radiologic data were collected in a case report form completed by the site, based on medical record review. When medical records were not available, data were either reported as missing or a patient report was used, when possible. This analysis of comorbidities and complications focused only on the retrospective data collected at the time of patients' enrollment in the PK deficiency NHS, and not the prospective data collected over the 2 years following enrollment. The inclusion criteria for this analysis were age ≥ 18 years at the time of enrollment, diagnosis of PK deficiency by molecular testing (two confirmed mutations in the PKLR gene), and sufficient data on transfusion history to enable classification into one of three mutually exclusive cohorts.
2.2. PK deficiency cohort definitions
The regularly transfused cohort included patients who received six or more transfusions over any 12‐month period, the occasionally transfused cohort included patients who had received transfusions in the past but never more than four in a single year, and the never transfused cohort included patients who had never received a transfusion.
Baseline demographic characteristics, clinical characteristics, transfusion history, medical conditions, procedures, and medication use were collected from the PK deficiency NHS enrollment survey. Unless otherwise noted, medical conditions were reported as lifetime history. Diagnosis and procedure dates were collected in the PK deficiency NHS for select conditions and events. Select medical procedures were also reported.
2.3. Data source and inclusion criteria: matched general population
The frequencies of select conditions and procedures observed in the PK deficiency NHS population were compared with age‐ and sex‐matched cohorts of individuals without any hereditary or acquired hemolytic anemia diagnoses (ie, International Classification of Diseases, 9th Revision, Clinical Modification [ICD‐9‐CM] codes 282 and 283; ICD, 10th Revision, CM [ICD‐10‐CM] codes D55‐D59) who had ≥ 5 years of continuous enrollment in one of three US MarketScan® (Truven, IBM Watson Health) Research Databases. Patients with PK deficiency are typically coded in claims data with one of these codes, as there is no specific ICD‐9‐CM or ICD‐10‐CM code for PK deficiency. Excluding patients with any hereditary or acquired hemolytic anemia helped ensure no patients with PK deficiency were included in the comparator population. The MarketScan databases include anonymized inpatient, outpatient, and outpatient prescription drug claims data for ~205 million individuals insured commercially or as part of the national Medicare or Medicaid programs between 1995 and 2017. All patients who met the study eligibility requirements in these databases were included in the sampling frame for this study. For the MarketScan analyses, all database records are de‐identified and fully compliant with US patient confidentiality requirements, including the Health Insurance Portability and Accountability Act of 1996. Because this study used de‐identified patient records and did not involve the collection, use, or transmittal of individually identifiable data, Institutional Review Board approval for this analysis was not necessary.
Individuals in the MarketScan databases were matched at a 1:10 (PK deficiency NHS:MarketScan) ratio based on age (±1 year), sex, and year of enrollment in the PK deficiency NHS.
Baseline demographic characteristics were reported as of the index date, which was designated as the midpoint of the year of the corresponding matched PK deficiency NHS patient's study enrollment year. All matched patients were required to have a minimum of 5 years of continuous enrollment immediately prior to the index date. Medical conditions and procedures were identified using ICD‐9‐CM, ICD‐10‐CM, Current Procedural Terminology, and Healthcare Common Procedure Coding System codes.
2.4. Data analysis and statistical interpretation
Data on comorbidities and complications extracted from the PK deficiency NHS represented the lifetime history of comorbidities. Conversely, claims data did not include entire medical histories but only included diagnosis codes for conditions that were diagnosed and/or treated in the years during which the patient had continuous enrollment in the health plan, previously diagnosed conditions that were medically relevant and coded during their time in the health plan, or conditions for which a “history of” an ICD‐9‐CM or ICD‐10‐CM code exists and the patient's physician chose to include the code in the patient's record. Thus, there was a risk of a relative underreporting of conditions in the general population that occurred prior to the enrollment period (eg, during childhood) and were fully resolved and/or did not warrant any medical follow‐up. To minimize this risk of bias, comparisons were limited to certain conditions, and two different analytic approaches were used: (a) A focus on conditions that were chronic and required ongoing management and thus would be expected to appear in claims data, regardless of how long ago the condition was initially diagnosed (osteoporosis, pulmonary hypertension, and liver cirrhosis); and (b) a narrowing of the observation window for the PK deficiency population to be aligned with the average 8‐year observation window for the general population, with a focus only on conditions for which a diagnosis or procedure date was available in the PK deficiency NHS population, so that events (such as splenectomy, cholecystectomy, gallstones, and congestive heart failure) that occurred prior to the narrowed observation window could be distinguished and excluded from the analysis. As it was not possible to identify conditions that had not yet been diagnosed, prevalence rates reported for both the PK deficiency population and the matched general population represented diagnosed prevalence rather than actual disease prevalence. Prevalence data on comorbidities and complications were tabulated by transfusion cohort and in aggregate, and compared using Fisher's exact two‐tailed tests of significance. Results were deemed significant when the P value was <.05. Adjustments were not made for multiple comparisons given the exploratory nature of this analysis, the danger of missing clinically important findings introduced by penalizing results, and increasing the risk of type II error in a rare disease setting where data are limited. 17 , 18
3. RESULTS
3.1. Study populations
Of the 254 patients with two confirmed mutations of the PKLR gene in the PK deficiency NHS, 131 patients met the inclusion criteria of being aged ≥ 18 years. Of these, nine could not be categorized into a transfusion cohort (two patients had regular transfusions at a frequency of five transfusions over a 12‐month period and seven patients had missing transfusion data that prohibited categorization). Therefore, the final PK deficiency study sample consisted of 122 adults: 65 (53.3%) were included in the regularly transfused cohort, 30 (24.6%) in the occasionally transfused cohort, and 27 (22.1%) in the never transfused cohort.
The 122 adult patients with PK deficiency were matched to 1220 individuals from the general population; individuals from the general population had an average of 8.1 years of continuous enrollment. Baseline demographics and characteristics are shown in Table 1. The transfusion cohorts were not different with regard to age at enrollment, sex, race, or ethnicity. Twenty‐seven Amish patients (homozygous for the R479H splice variant) accounted for 22.1% of the PK deficiency study population and the majority of these patients (n = 20) were in the regularly transfused cohort (30.8% of the regularly transfused cohort). Patients with a non‐missense/non‐missense PKLR genotype were more likely to be in the regularly transfused cohort than in the never transfused cohort.
TABLE 1.
Baseline demographics and characteristics of the Pyruvate Kinase deficiency Natural History Study population
| Regularly transfused cohort (n = 65) | Occasionally transfused cohort (n = 30) | Never transfused cohort (n = 27) | P value | ||
|---|---|---|---|---|---|
| Regularly transfused vs occasionally transfused cohorts | Regularly transfused vs never transfused cohorts | ||||
| Male, n (%) | 30 (46.2) | 17 (56.7) | 16 (59.3) | .383 | .360 |
| Mean (SD) age, y | 34.2 (11.0) | 39.5 (14.7) | 37.2 (16.3) | .083 | .383 |
| White, n (%) | 63 (96.9) | 30 (100) | 27 (100) | >.999 | >.999 |
| Hispanic or Latino, n (%) | 2 (3.1) | 1 (3.3) | 1 (3.7) | >.999 | >.999 |
| Genotype, n (%) | .085 | .003 | |||
| Amish R479H/R479H | 20 (30.8) | 4 (13.3) | 3 (11.1) | ||
| Missense/missense | 21 (32.3) | 14 (46.7) | 19 (70.4) | ||
| Missense/non‐missense | 11 (16.9) | 8 (26.7) | 5 (18.5) | ||
| Non‐missense/non‐missense | 12 (18.5) | 2 (6.7) | 0 | ||
| Other a | 1 (1.5) | 2 (6.7) | 0 | ||
The general population (n = 1220; data not shown) was age and sex matched to the pyruvate kinase deficiency population. The percent male and mean age across each transfusion cohort were identical to the pyruvate kinase deficiency population. Race, ethnicity, Amish status, and genotype were either not available or not applicable. For sex, race, and ethnicity, comparisons are based on a two‐tailed Fisher's exact test. For age, comparisons are based on a two‐sample t test. For genotype, comparisons are based on a chi‐square test.
Three patients could not be classified due to having three variants (n = 1) or due to having a promoter variant of uncertain significance (n = 2).
3.2. Comparisons with matched general population
3.2.1. Comorbidities and complications: lifetime rate
Osteoporosis, pulmonary hypertension, and liver cirrhosis were the only conditions captured in the PK deficiency NHS that are chronic in nature, require ongoing management, and/or would be expected to appear in claims data regardless of how long ago the condition was initially diagnosed. Rates of all of these conditions were significantly higher in patients with PK deficiency than in the matched general population: pulmonary hypertension (4.6% vs 0.3%, respectively; P < .001), osteoporosis (15.6% vs 0%; P < .001), and liver cirrhosis (5.6% vs 0.4%; P < .001) (Figure 1).
FIGURE 1.
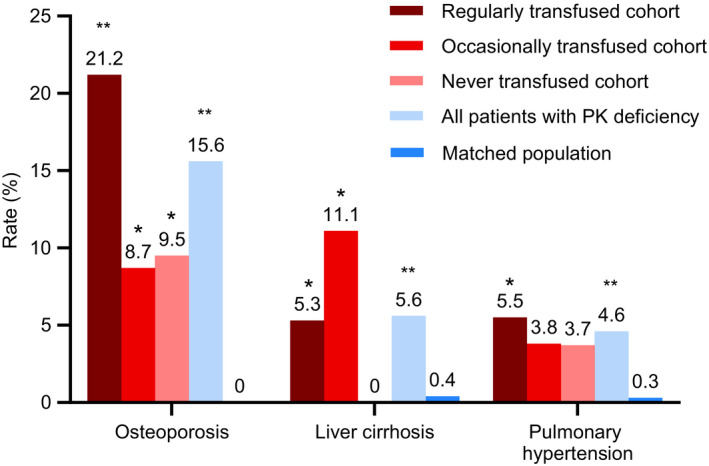
Comparison of lifetime prevalence estimates of select comorbidities and complications between the PK deficiency NHS population and matched patients from MarketScan data. All comparisons are based on a two‐tailed Fisher's exact test. *P < .05 for PK deficiency NHS population vs matched general population. **P < .001 for PK deficiency NHS population vs matched general population. NHS, Natural History Study; PK, pyruvate kinase
3.2.2. Comorbidities and complications: restricted observation window
Of the conditions and procedures that were assessed based on rates over the 8 years leading up to the index date, patients with PK deficiency had significantly higher rates than the general population for splenectomy (4.9% vs 0.2%, respectively; P < .001), cholecystectomy (13.1% vs 3.6%; P < .001) (Figure 2), and gallstones (16.9% vs 4.3%; P < .001). Rates of congestive heart failure over the previous 8 years were not significantly different between the two populations.
FIGURE 2.
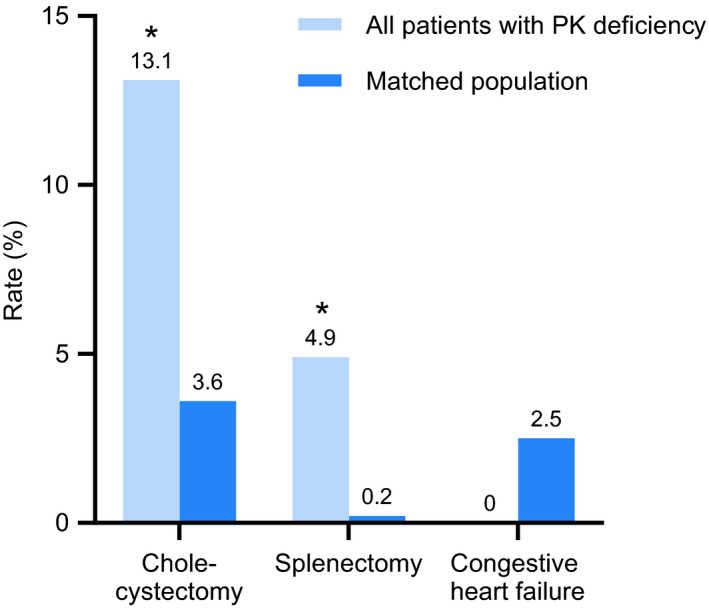
Comparison of prevalence estimates of select comorbidities and complications limited to an average of 8 y prior to enrollment in PK deficiency NHS or index date in MarketScan. *P < .001 for PK deficiency NHS population vs matched general population. NHS, Natural History Study; PK, pyruvate kinase
3.3. Comorbidities and complications in the PK deficiency NHS population
For conditions for which unbiased lifetime comparisons with the general population could not be made, lifetime prevalence rates just for the PK deficiency NHS population, stratified by transfusion cohort, are presented in Table 2. Statistically significant differences between transfusion cohorts were observed for splenectomy, cholecystectomy, aplastic crises, thrombosis, and liver iron overload.
TABLE 2.
General, hematologic, and endocrine comorbid conditions and complications in the Pyruvate Kinase deficiency Natural History Study population
|
Regularly transfused cohort, n/N (%) (n = 65) |
Occasionally transfused cohort, n/N (%) (n = 30) |
Never transfused cohort, n/N (%) (n = 27) |
All patients with PK deficiency, n/N (%) (n = 122) |
P value | |||
|---|---|---|---|---|---|---|---|
| Regularly transfused vs occasionally transfused cohorts | Occasionally transfused vs never transfused cohorts | Regularly transfused vs never transfused cohorts | |||||
| Chronic diseases | |||||||
| Arrhythmia | 5/62 (8.1) | 5/28 (17.9) | 1 (3.7) | 11/117 (9.4) | .275 | .193 | .663 |
| Congestive heart failure | 2/62 (3.2) | 1/28 (3.6) | 0 (0) | 3/117 (2.6) | >.999 | >.999 | >.999 |
| Pulmonary hypertension | 3/55 (5.5) | 1/26 (3.8) | 1 (3.7) | 5/108 (4.6) | >.999 | >.999 | >.999 |
| Osteoporosis | 11/52 (21.2) | 2/23 (8.7) | 2/21 (9.5) | 15/96 (15.6) | .321 | >.999 | .323 |
| Liver cirrhosis | 3/57 (5.3) | 3/27 (11.1) | 0/24 (0) | 6/108 (5.6) | .381 | .238 | .551 |
| Hematological conditions | |||||||
| Bone enlargement/bony expansion | 8/60 (13.3) | 1/24 (4.2) | 0/25 (0) | 9/109 (8.3) | .435 | .490 | .098 |
| Extramedullary hematopoiesis | 11/49 (22.4) | 1/21 (4.8) | 2/22 (9.1) | 14/92 (15.2) | .092 | >.999 | .319 |
| Liver iron overload | 54 (83.1) | 15 (50.0) | 7 (25.9) | 76 (62.3) | .001 | .102 | <.001 |
| Cardiac iron overload | 2/34 (5.9) | 0/4 (0.0) | 0/3 (0.0) | 2/41 (4.9) | >.999 | NC | >.999 |
| History of thrombosis | 13 (20.0) | 3 (10.0) | 0/25 (0) | 16/120 (13.3) | .376 | .242 | .016 |
| Deep vein thrombosis | 7 (10.8) | 2 (6.7) | 0/25 (0) | 9/120 (7.5) | .715 | .495 | .184 |
| Pulmonary embolism | 5 (7.7) | 1 (3.3) | 0/25 (0) | 6/120 (5.0) | .661 | .317 | >.999 |
| Stroke | 1 (1.5) | 1 (3.3) | 0/25 (0) | 2/120 (1.7) | .534 | >.999 | >.999 |
| Other | 3 (4.6) | 1 (3.3) | 0/25 (0) | 4/120 (3.3) | >.999 | >.999 | .557 |
| Central venous line | 2 (3.1) | 1 (3.3) | 0/25 (0) | 3/120 (2.5) | >.999 | >.999 | >.999 |
| Presplenectomy thrombosis | 1 (1.5) | 0 (0) | 0/25 (0) | 1/120 (0.8) | >.999 | >.999 | >.999 |
| Postsplenectomy thrombosis | 11 (16.9) | 3 (10.0) | 0/25 (0) | 14/120 (11.7) | .537 | .242 | .031 |
| Postsplenectomy thrombosis a | 11/65 (16.9) | 3/20 (15.0) | 0/6 (0) | 14/91 (15.4) | >.999 | >.999 | .580 |
| Endocrine conditions | |||||||
| Growth hormone deficiency | 2/59 (3.4) | 1/27 (3.7) | 0/23 (0) | 3/109 (2.8) | >.999 | >.999 | >.999 |
| Hypoparathyroidism | 3/59 (5.1) | 0/21 (0) | 0/23 (0) | 3/103 (2.9) | .563 | NC | .556 |
| Thyroid disease | 6/63 (9.5) | 2/27 (7.4) | 2/24 (8.3) | 10/114 (8.8) | >.999 | >.999 | >.999 |
| Hypothyroidism | 5/63 (7.9) | 1/27 (3.7) | 1/24 (4.2) | 7/114 (6.1) | NC | NC | NC |
| Hyperthyroidism | 0/63 (0) | 0/27 (0) | 1/24 (4.2) | 1/114 (0.9) | NC | NC | NC |
| Unknown | 1/63 (1.6) | 1/27 (3.7) | 0/24 (0) | 2/114 (1.8) | NC | NC | NC |
| Other conditions/events | |||||||
| Splenectomy | 65 (100.0) | 20 (66.7) | 6 (22.2) | 91 (74.6) | <.001 | .001 | <.001 |
| Cholecystectomy | 46 (70.8) | 19 (63.3) | 8 (29.6) | 73 (59.8) | .485 | .017 | <.001 |
| Gallstones | 47/63 (74.6) | 20/28 (71.4) | 13 (48.1) | 80/118 (67.8) | .799 | .102 | .027 |
| Leg ulcers | 3/58 (5.2) | 1/27 (3.7) | 0/23 (0) | 4/108 (3.7) | >.999 | >.999 | .554 |
| Aplastic crises | 13/59 (22.0) | 8/27 (29.6) | 0 (0) | 21/113 (18.6) | .589 | .004 | .007 |
| Bone fracture | 20/61 (32.8) | 6/24 (25.0) | 6/23 (26.1) | 32/108 (29.6) | .604 | >.999 | .608 |
| Sepsis | 3 (4.6) | 1/29 (3.4) | 1/26 (3.8) | 5/120 (4.2) | >.999 | >.999 | >.999 |
Denominators are only specified when data for the full cohort are not available.
Note:Liver iron overload is defined as patients meeting any of the following criteria: (a) Ever received chelation therapy, (b) Ever been prescribed phlebotomy for iron removal, (c) had ferritin > 1000 ng/mL in the 12 mo prior to enrollment, (d) biopsy liver iron concentration > 3 mg Fe/g DW, (e) T2 magnetic resonance imaging liver iron concentration > 3 mg Fe/g DW, or (f) Ferriscan liver iron concentration > 3 mg Fe/g DW. Cardiac iron overload is defined as T2 ≤ 20 ms.
Abbreviations: DW, dry weight; NC, not calculated; PK, pyruvate kinase.
Limited denominator to patients who had a splenectomy. All comparisons are based on a two‐tailed Fisher's exact test.
For many conditions, a gradient could be seen, with the highest rates reported for patients in the regularly transfused cohort and the lowest rates in the never transfused cohort. For liver iron overload, the highest rates were observed in the regularly transfused cohort (83.1%). However, notably, 25.9% of patients in the never transfused cohort were reported to have a history of liver iron overload despite never receiving a transfusion. Rates of extramedullary hematopoiesis were numerically higher in the regularly transfused cohort vs the occasionally transfused cohort (22.4% vs 4.8%, respectively [P = .092], vs 9.1% in the never transfused cohort [P = .319]). A history of thrombosis correlated with splenectomy status and was observed in 16 (13.3%) patients in the PK deficiency population, with the majority being in the regularly transfused cohort (13 [20.0%] patients) compared with 3 (10.0%) in the occasionally transfused cohort (P = .376) and no patients in the never transfused cohort (P = .016).
Because all 65 patients in the regularly transfused cohort had been splenectomized, compared with only 20 of 30 (66.7%) patients in the occasionally transfused cohort and 6 of 27 (22.2%) patients in the never transfused cohort (P < .001 for regularly transfused vs occasionally transfused and regularly transfused vs never transfused; P = .001 for occasionally transfused vs never transfused), additional analyses were performed whereby patients were stratified by splenectomy status rather than transfusion history. Thrombosis occurred in 16 of 90 (17.8%) splenectomized patients vs 0 of 30 non‐splenectomized patients (P = .011). The majority of patients (14 of 16) experienced thrombotic events postsplenectomy (11 from the regularly transfused cohort and 3 from the occasionally transfused cohort).
Gallstones and iron overload were more commonly observed in patients who underwent splenectomy, likely related to the greater severity of hemolysis in patients who underwent splenectomy. Although gallstones were frequently reported among non‐splenectomized patients (15 of 30 [50.0%] patients), they were significantly more common among patients who had a splenectomy (65 of 88 [73.9%]; P = .023). Similarly, iron overload was also significantly more common among splenectomized patients (72 of 91 [79.1%]) than non‐splenectomized patients (four of 31 [12.9%]; P < .001).
The rates of pulmonary hypertension in the splenectomized subgroup were not statistically significantly different from non‐splenectomized patients. However, of the five cases of pulmonary hypertension, four occurred in splenectomized patients (three in the regularly transfused cohort and one in the occasionally transfused cohort) and one case occurred in a non‐splenectomized patient in the never transfused cohort. The prevalence of sepsis was not different between transfusion cohorts or by splenectomy status.
The association between transfusion cohort and liver iron overload was no longer significant when splenectomy was added to a logistic regression model. However, this finding may not be related to the splenectomy, but rather to greater underlying disease severity in patients who were splenectomized.
Given the relatively high proportion of Amish patients in the PK deficiency study adult population (22.1%) and concerns of possible confounding due to potential differences in clinical characteristics or treatment patterns in this population, a subgroup analysis restricted to the non‐Amish patients was conducted. Although limited by small sample sizes in the transfusion cohorts, the results were consistent with findings for the full population (Amish and non‐Amish patients), with the exception that gallstones over the previous 8 years were less common in the regularly transfused cohort of the non‐Amish population (7.0%) than the regularly transfused cohort of the full population (16.9%). This is likely attributable to screening ultrasounds performed as part of the PK deficiency NHS for the Amish cohort as compared with the non‐Amish cohort, and/or with the lower lifetime rates of cholecystectomy among Amish patients (41.4% vs 65.6% for non‐Amish patients; P = .029) that left them at ongoing risk of gallstones in the 8 years prior to enrollment.
4. DISCUSSION
The aim of this analysis was to compare the prevalence of select comorbidities and complications in patients with PK deficiency with age‐ and sex‐matched individuals without PK deficiency, and to determine if the prevalence of conditions in patients with PK deficiency varies by transfusion history. Patients with PK deficiency had higher lifetime rates of pulmonary hypertension, osteoporosis, and liver cirrhosis, and higher 8‐year rates of splenectomy, gallstones, and cholecystectomy than age‐ and sex‐matched individuals from a general insured US population who did not have PK deficiency.
The purpose of comparing with an age‐ and sex‐matched population of patients without PK deficiency was to contextualize the rates of comorbidities and complications previously reported in the PK deficiency literature, and to demonstrate that regardless of transfusion status, patients with PK deficiency have measurable disease burden that exceeds what would be seen in the general population. Comparisons with the general population are particularly helpful for healthcare decision makers who may not be familiar with the disease and prefer a familiar benchmark to help prioritize policies, interventions, and other treatment‐related decisions.
Evaluation of the individual transfusion cohorts in the PK deficiency population revealed significant differences in occurrence of splenectomy, cholecystectomy, aplastic crises, thrombosis, and liver iron overload between those in the never transfused, occasionally transfused, or regularly transfused cohorts, with the latter cohort typically having the highest rates. However, even patients with PK deficiency in the never transfused cohort were shown to have significant complications, including liver iron overload, gallstones, osteoporosis, and extramedullary hematopoiesis.
In order to optimize treatment of patients with PK deficiency, it is important to distinguish the complications that are due to the disease itself compared with those that occur as a result of treatment, such as splenectomy and transfusions. Splenectomy, in general, increases the risk of venous thromboembolism, including in individuals with PK deficiency. Given that the majority of thrombotic events occurred postsplenectomy, this risk is likely to be related to the splenectomy rather than the anemia. 19 In addition to thrombosis, patients who underwent splenectomy in this study had a high rate of liver iron overload. Splenectomy may be a marker of, or correlate with, a more severe hemolytic phenotype with a greater degree of iron absorption, but the rate of splenectomy was also highest in those in the regularly transfused cohort, with a greater risk of transfusion‐related iron loading. In this setting, splenectomy is often performed to minimize transfusional iron burden. Iron overload is a serious consequence of PK deficiency, occurring regardless of transfusion or splenectomy status. 20 Even patients in the never transfused cohort had iron overload, indicating that this complication occurs as a consequence of the disease itself, in addition to its management. Similarly, the occurrence of osteoporosis and pulmonary hypertension in patients who had not undergone transfusion or splenectomy suggests that they may be due to PK deficiency itself. All three of these complications are also reported in patients with β‐thalassemia, where they are considered to be caused, at least in part, by ineffective erythropoiesis. 21 , 22 , 23
Osteoporosis rates were significantly higher in the PK deficiency population than the general population. The lack of osteoporosis in the matched healthy population is likely due to the relatively young age of the cohorts (mean age, 36 years). A high rate of other bone conditions, such as bone enlargement/bony expansion and bone fractures, 13 is seen in patients with PK deficiency, similar to the bone disease encountered in thalassemia. 24
There were a number of limitations to this analysis. As it was not possible to identify conditions that have not yet been diagnosed, prevalence rates estimated from both the PK deficiency NHS and MarketScan databases are based on diagnosed disease only and thus may underestimate the true prevalence of these comorbidities and complications. The data from the PK deficiency population are subject to the limitations typical of an NHS, including bias due to collection of data via clinical care, missing data, and small numbers that prohibited multivariate analyses, which would have allowed for a meaningful exploration of possible differences by genotype, splenectomy status, or other characteristics. The general population data are limited by their retrospective nature, such that data were collected for reimbursement purposes only and not for research. In addition, there may be a risk of a relative underreporting of conditions in the general population as the claims database did not include entire medical histories. The limited 8‐year timeline may also have impacted findings, and data for uninsured individuals, which constitutes < 10% of the US population, were not included. Additionally, the PK deficiency NHS is a multinational registry; however, the general population data are restricted to individuals in the United States and contain very limited information on ethnic background. Accordingly, we were unable to assess any possible impact of ethnicity differences on study outcomes. Despite the international effort, the PK deficiency NHS cohort included a limited number of patients. Therefore, it remains necessary to continue to expand upon the collection of longitudinal data on the natural history of patients affected by PK deficiency, with a view to better understand the disease burden and inform physicians on how to manage this patient population. This longitudinal data will continue to be collected through the PK deficiency Global Longitudinal Registry (Peak Registry; ClinicalTrials.gov NCT03481738).
The rate of comorbidities and complications are significantly higher in the PK deficiency population than a general insured US population. This analysis has also demonstrated the high rate of comorbidities and complications in adults with PK deficiency regardless of transfusion status, and the impact of transfusion frequency on the prevalence of these comorbidities.
CONFLICT OF INTEREST
ANB and YY are employees of and hold stock in Agios. EH received funding for this research from Agios through a contract with IBM Watson Health. EJvB has received research support from Agios, Bayer, Mechatronics, and Novartis; and has served as a consultant for Agios. HA‐S reports research support from Agios, Amgen, and Dova; and has served as a consultant for Agios and Dova. WB has received research support from Alexion and Novartis; has served on advisory boards for Agios, Alexion, Bioverativ, and Incyte; and has served on speaker bureaus for Agios, Alexion, and Novartis. SWE is a consultant for Agios. BG is a research investigator and consultant for Agios. HMY has served as a consultant for Agios, Bayer, Novo Nordisk, Octapharma, and Takeda; and has served on speaker bureaus for Bayer and Takeda. SC has served as a consultant and on advisory boards for Agios and Alexion. KHMK has received honoraria from and is a consultant for Agios, Apellis, bluebird bio, Celgene, and Pfizer; and has served on a data safety monitoring board for Bioverativ. EJN has served as a consultant and on advisory boards for Agios, Celgene, and Genentech; has served as a consultant for Pfizer; has served on advisory boards for Baxalta/Shire (now Takeda) and Novartis; has served as a consultant, on advisory boards, and received honoraria from Octapharma; has served on advisory boards and received honoraria from Novo Nordisk; and has served on data monitoring committees for ApoPharma, Acceleron, Bayer, and Imara. MV has served as a consultant for Vertex. SS has served as a consultant for Acceleron, Agios, bluebird bio, and Celgene/BMS; has served on a clinical trial steering committee for CRISPR/Vertex; and participated in clinical trials with Celgene/BMS, Dispersol, La Jolla, and Terumo. RFG has served on advisory boards for Dova; has served on advisory boards and received research funding from Agios; and has received research funding from Novartis and Pfizer. The remaining authors declare no competing financial interests.
AUTHOR CONTRIBUTION
ANB conceptualized the study; ANB and EH developed the study protocol; RFG provided critical feedback on the study protocol; YY and EH performed the statistical programming and analyses; ANB created tables and figures; ANB, EJvB, HA, WB, SWE, BG, HMY, SC, MS, KHMK, EJN, HW, MV, SS, and RFG provided critical feedback and assisted with interpretation of study results; EJvB, HA, WB, SWE, BG, HMY, SC, MS, KHMK, EJN, HW, MV, SS, and RFG contributed to Pyruvate Kinase deficiency Natural History Study data collection; all co‐authors contributed to editing and provided final approval of the paper.
ACKNOWLEDGEMENTS
The Pyruvate Kinase deficiency Natural History Study was supported by research funding from Agios Pharmaceuticals, Inc. We would like to thank the patients taking part in the Pyruvate Kinase deficiency Natural History Study. Editorial assistance with manuscript preparation was provided by Christine Ingleby, PhD, CMPP, Excel Scientific Solutions, Horsham, UK, and supported by Agios Pharmaceuticals, Inc.
Boscoe AN, Yan Y, Hedgeman E, et al. Comorbidities and complications in adults with pyruvate kinase deficiency. Eur J Haematol. 2021;106:484–492. 10.1111/ejh.13572
Novelty Statements: What is the new aspect of your work?: The rates of key co‐occurring conditions in patients with pyruvate kinase (PK) deficiency are assessed according to blood transfusion history and are compared with the rates in the matched general population. What is the central finding of your work?: Patients with PK deficiency have higher rates of select comorbidities and complications than matched individuals from the general population, and these can occur even in patients who have never been transfused. What is (or could be) the specific clinical relevance of your work?: These findings raise awareness about the excess disease burden experienced by patients with PK deficiency and should alert clinicians to monitor these patients for certain comorbidities and complications, regardless of the perceived severity of their illness as measured by transfusion requirements.
DATA AVAILABILITY STATEMENT
For original data, please contact the corresponding author.
REFERENCES
- 1. Grace RF, Zanella A, Neufeld EJ, et al. Erythrocyte pyruvate kinase deficiency: 2015 status report. Am J Hematol. 2015;90(9):825‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype‐phenotype association. Blood Rev. 2007;21(4):217‐231. [DOI] [PubMed] [Google Scholar]
- 3. Bianchi P, Fermo E, Lezon‐Geyda K, et al. Genotype‐phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am J Hematol. 2020;95(5):472‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Wijk R, van Solinge WW. The energy‐less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood. 2005;106(13):4034‐4042. [DOI] [PubMed] [Google Scholar]
- 5. Nathan DG, Oski FA, Miller DR, Gardner FH. Life‐span and organ sequestration of the red cells in pyruvate kinase deficiency. N Engl J Med. 1968;278(2):73‐81. [DOI] [PubMed] [Google Scholar]
- 6. Glader B. Other hereditary red blood cell disorders. In: Rimoin D, Connor J, Pyeritz R, Korf B, eds. Emery and Rimoin's Principles and Practices of Medical Genetics. 5th edn. Philadelphia, PA: Churchill Livingstone; 2007:1675. [Google Scholar]
- 7. Carey PJ, Chandler J, Hendrick A, et al. Prevalence of pyruvate kinase deficiency in northern European population in the north of England. Northern Region Haematologists Group. Blood. 2000;96(12):4005‐4006. [PubMed] [Google Scholar]
- 8. de Medicis E, Ross P, Friedman R, et al. Hereditary nonspherocytic hemolytic anemia due to pyruvate kinase deficiency: a prevalence study in Quebec (Canada). Hum Hered. 1992;42(3):179‐183. [DOI] [PubMed] [Google Scholar]
- 9. Christensen RD, Eggert LD, Baer VL, Smith KN. Pyruvate kinase deficiency as a cause of extreme hyperbilirubinemia in neonates from a polygamist community. J Perinatol. 2010;30(3):233‐236. [DOI] [PubMed] [Google Scholar]
- 10. Secrest MH, Storm M, Carrington C, et al. Prevalence of pyruvate kinase deficiency: a systematic literature review. Eur J Haematol. 2020;105(2):173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baronciani L, Beutler E. Molecular study of pyruvate kinase deficient patients with hereditary nonspherocytic hemolytic anemia. J Clin Invest. 1995;95(4):1702‐1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rider NL, Strauss KA, Brown K, et al. Erythrocyte pyruvate kinase deficiency in an old‐order Amish cohort: longitudinal risk and disease management. Am J Hematol. 2011;86(10):827‐834. [DOI] [PubMed] [Google Scholar]
- 13. Grace RF, Bianchi P, van Beers EJ, et al. Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood. 2018;131(20):2183‐2192. [DOI] [PubMed] [Google Scholar]
- 14. Grace RF, Cohen J, Egan S, et al. The burden of disease in pyruvate kinase deficiency: patients' perception of the impact on health‐related quality of life. Eur J Haematol. 2018;101(6):758‐765. [DOI] [PubMed] [Google Scholar]
- 15. Grace RF, Mark Layton D, Barcellini W. How we manage patients with pyruvate kinase deficiency. Br J Haematol. 2019;184(5):721‐734. [DOI] [PubMed] [Google Scholar]
- 16. Kimmig LM, Palevsky HI. Review of the association between splenectomy and chronic thromboembolic pulmonary hypertension. Ann Am Thorac Soc. 2016;13(6):945‐954. [DOI] [PubMed] [Google Scholar]
- 17. Li G, Taljaard M, Van den Heuvel ER, et al. An introduction to multiplicity issues in clinical trials: the what, why, when and how. Int J Epidemiol. 2017;46(2):746‐755. [DOI] [PubMed] [Google Scholar]
- 18. Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology. 1990;1(1):43‐46. [PubMed] [Google Scholar]
- 19. Thomsen RW, Schoonen WM, Farkas DK, Riis A, Fryzek JP, Sørensen HT. Risk of venous thromboembolism in splenectomized patients compared with the general population and appendectomized patients: a 10‐year nationwide cohort study. J Thromb Haemost. 2010;8(6):1413‐1416. [DOI] [PubMed] [Google Scholar]
- 20. van Beers EJ, van Straaten S, Morton DH, et al. Prevalence and management of iron overload in pyruvate kinase deficiency: report from the Pyruvate Kinase Deficiency Natural History Study. Haematologica. 2019;104(2):e51‐e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gaudio A, Morabito N, Catalano A, Rapisarda R, Xourafa A, Lasco A. Pathogenesis of thalassemia major‐associated osteoporosis: a review with insights from clinical experience. J Clin Res Pediatr Endocrinol. 2019;11(2):110‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Machado RF, Gladwin MT. Pulmonary hypertension in hemolytic disorders: pulmonary vascular disease: the global perspective. Chest. 2010;137(6):30S‐38S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hershko C. Pathogenesis and management of iron toxicity in thalassemia. Ann N Y Acad Sci. 2010;1202:1‐9. [DOI] [PubMed] [Google Scholar]
- 24. Vogiatzi MG, Macklin EA, Fung EB, et al. Bone disease in thalassemia: a frequent and still unresolved problem. J Bone Miner Res. 2009;24(3):543‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
For original data, please contact the corresponding author.