

Abstract
S=2 oxoiron(IV) species act as reactive intermediates in the catalytic cycle of nonheme iron oxygenases. The few available synthetic S=2 FeIV=O complexes known to date are often limited to trigonal bipyramidal and very rarely to octahedral geometries. Herein we describe the generation and characterization of an S=2 pseudotetrahedral FeIV=O complex 2 supported by the sterically demanding 1,4,7‐tri‐tert‐butyl‐1,4,7‐triazacyclononane ligand. Complex 2 is a very potent oxidant in hydrogen atom abstraction (HAA) reactions with large non‐classical deuterium kinetic isotope effects, suggesting hydrogen tunneling contributions. For sterically encumbered substrates, direct HAA is impeded and an alternative oxidative asynchronous proton‐coupled electron transfer mechanism prevails, which is unique within the nonheme oxoiron community. The high reactivity and the similar spectroscopic parameters make 2 one of the best electronic and functional models for a biological oxoiron(IV) intermediate of taurine dioxygenase (TauD‐J).
Keywords: bioinorganic chemistry, enzyme models, high-valent iron, hydrogen atom abstraction, electron transfer
A highly reactive S=2 pseudotetrahedral oxoiron(IV) complex 2 supported by a sterically demanding 1,4,7‐tri‐tert‐butyl‐1,4,7‐triazacyclononane ligand has been synthesized and spectroscopically characterized as one of the best electronic and functional models for a biological oxoiron(IV) intermediate of taurine dioxygenase (TauD‐J).
Introduction
High‐valent oxoiron(IV) intermediates act as the active oxidants in the catalytic cycles of a variety of mononuclear non‐heme iron oxygenases. [1] These high‐valent species have been characterized by rapid freeze quench methods in few cases [1] and were unambiguously shown by UV/Vis, Mössbauer, and X‐ray absorption spectroscopic methods to contain high‐spin (S=2) iron(IV) centres. However, the available experimental data could not reveal other important structural features, such as the number, identity, and disposition of ligands in the FeIV coordination sphere. Density functional theoretical (DFT) studies [2] on the taurine:αKG dioxygenase (TauD) system have shown that the spectroscopic properties of the hydrogen‐abstracting oxoiron(IV) key intermediate (TauD‐J) are consistent with both suggested structural models (Scheme 1), that is, with trigonal bipyramidal (TBP) as well as distorted octahedral (Oh) coordinations. Significant synthetic efforts in the past decade have led to the generation of oxoiron(IV) cores in both TBP and Oh geometries (Scheme 1). Although the majority of the synthetic complexes exhibit S=1 ground states in Oh geometry, [3] DFT‐studies predicted stabilization of the more reactive [4] S=2 oxoiron(IV) units [5] either by enforcing a TBP geometry at the iron(IV) centre[ 5a , 5b , 5c , 5d ] or by weakening the equatorial donation in Oh geometry. [5e]
Scheme 1.
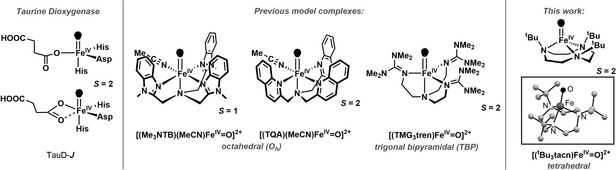
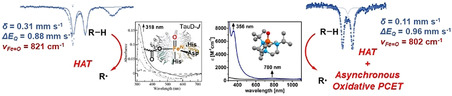
Left: Proposed structures of S=2 TauD‐J based on DFT studies; [2] middle: selected examples of S=1 and S=2 oxoiron(IV) cores in TBP and Oh geometries; right: A pseudotetrahedral S=2 oxoiron(IV) complex 2 reported in this work; in the inset is shown the DFT calculated structure of 2 in the S=2 state.
Results and Discussion
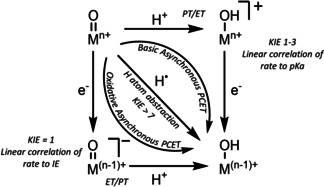
In the context of the existing ambiguity related to the coordination number of iron in biological oxoiron(IV) intermediates, [2] and the limitation of the synthetic S=2 oxoiron(IV) cores to mainly TBP and in rare cases to Oh geometries, we have now sought to identify a tripodal ligand that allows for trapping an FeIV=O core in a geometry different from the known TBP or Oh geometries. Herein we report the synthesis and characterization of the S=2 pseudotetrahedral [FeIV(O)(tBu3tacn)]2+ (2, tBu3tacn [6] =1,4,7‐tri‐tert‐butyl‐1,4,7‐triazacyclononane) complex, which exhibits spectroscopic and reactivity properties distinct from the oxoiron(IV) cores in TBP or Oh geometries. In particular, in direct contrast to the vast majority of previous oxoiron(IV) cores,[ 3a , 3b , 3c , 3d , 3e , 3f , 3g , 5a , 5b , 5c , 5d , 5e ] where the reactivity with substrates containing C−H bonds is controlled by the C−H bond dissociation energies (BDEC‐H), complex 2 demonstrates a mechanistic promiscuity in its C−H oxidation reactions. Sterically less hindered C−H bonds are oxidized via a conventional direct hydrogen atom abstraction (HAA; Scheme 2) mechanism that is characterized by large deuterium kinetic isotope effects (KIEs), which are greater than the semi‐classical limit of 7, implying a significant contribution of hydrogen tunnelling. [7] In contrast, for sterically encumbered substrates, where the direct access to the FeIV=O core is blocked, the C−H oxidation reaction proceeds with a significantly lower KIE and presumably involves a proton‐coupled electron transfer (PCET) mechanism along a spectrum of “asynchronicity” [8] in which the transition state for the net H‐atom transfer contains more electron transfer character (Scheme 2; Oxidative asynchronous PCET).
Scheme 2.
Mechanisms of net hydrogen atom transfer.
Combination of equimolar amounts of the previously reported tBu3tacn ligand[ 6 , 9 ] and FeII(OTf)2(CH3CN)2 in CH2Cl2 afforded [FeII(tBu3tacn)(OTf)](OTf) (1), whose crystal structure (Figure S1; Tables S1,S2) exhibited a distorted tetrahedral geometry (N‐Fe‐N angles of 86.5–88.3°) with an Fe‐O distance of 1.935(2) Å and three Fe‐N distances of 2.105(2)–2.124(2) Å. The zero‐field Mössbauer spectrum of 1 (Figure S2) revealed a single doublet with an isomer shift (δ) of 0.97 mm s−1 and a large quadrupole splitting (ΔE Q=1.98 mm s−1), consistent with an S=2 spin state, which is also supported by DFT [11] (Table S3). Reaction of 1 in pure CH2Cl2 or butyronitrile (PrCN) at −90 °C with 2‐(tert‐butylsulfonyl)‐iodosobenzene (sPhIO) [12] yielded a transient species 2 (Figure 1 A; half‐life at −70 °C=20 min) with electronic absorption features centered at λ max=356 nm (ϵ=7500 M−1 cm−1) and 780 nm (ϵ=150 M−1 cm−1). Notably, the presence of a well‐defined strong absorption band in the near‐UV region is typical of S=2 oxoiron(IV) cores (Table S4);[ 5a , 5b , 5c , 5d ] in 2 this band at λ max=356 nm is slightly red‐shifted (Table 1) relative to that of TauD‐J (λ max=318 nm). [1a] The S=2 spin state of 2 was additionally corroborated by the Evans [13] NMR method (Figure S3) at −90 °C which yielded the magnetic moment μ eff=4.50 μB (theoretical value for S=2: 4.90 μB). An electron spray ionization mass spectrum (Figure S4) of 2 exhibited a signal at m/z=518.7, consistent with its formulation as [FeIV(O)(tBu3tacn)(OTf)]+ (m/z calc=518.2). However, the 19F‐NMR spectrum (Figure S5) of 2 displayed a single resonance at −77.0 ppm, which confirmed that the triflate (OTf) anion is not bound to the Fe‐centre in 2. This observation together with the same UV‐vis spectrum (Figure S6) of 2 in both coordinating (PrCN) and non‐coordinating (CH2Cl2) solvents corroborates the absence of any exogeneous ligands binding to the iron centre. Thus, the 4‐coordinate geometry found in 1 is retained in 2, leading us to formulate the latter as [FeIV(O)(tBu3tacn)]2+.
Figure 1.
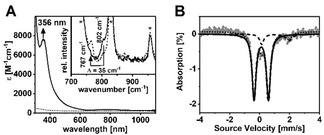
A) UV/Vis spectra of 1 (dashed line) and 2 (solid line) in CH2Cl2 at −90 °C; inset shows the rRaman spectra of 16O‐ (solid line) and 18O‐labelled (dashed line) 2 (4 mM solution) in CH2Cl2 upon 406 nm irradiation at −90 °C; solvent signals are indicated by an asterisk; B) Zero‐field Mössbauer spectrum (grey) of a frozen sample of 2 in PrCN/CH2Cl2 (10:1) and simulation with δ=0.11 mm s−1 and ΔE Q=0.96 mm s−1 for the main species (solid line, 87 %). The minor species (dashed line) with δ=0.97 mm s−1 and ΔE Q=1.98 mm s−1 corresponds to unreacted 1.
Table 1.
Comparison of the spectroscopic properties of TauD‐J and 2.
|
|
2 |
|
|---|---|---|
|
λ max [nm] |
318 |
356 |
|
R (Fe‐O) [Å] |
1.62 |
1.66 |
|
ν Fe=O [cm−1] |
821 |
802 |
|
δ [mm−1 s−1] |
0.31 |
0.11 |
|
ΔE Q [mm−1 s−1] |
0.88 |
0.96 |
|
Axx, Ayy, Azz [T] |
S=2: −18.4, −17.6, −31 |
S=2: −10.1, −3.3, −36.1 |
|
E o [eV] |
7123.8 |
7123.2 |
The 4‐coordinate geometry of 2 was also supported by Extended X‐ray Absorption Fine Structure (EXAFS) analysis (Figure S7A, Table S5), which yielded a good fit with an oxygen ligand at 1.66 Å, assigned to the Fe=O bond, and a further shell of three nitrogen ligands at 2.06 Å, corresponding to the N donors of tBu3tacn. The Fe K‐edge X‐ray absorption spectrum (Figure S7B) of 2 reveals an edge energy of 7123.2 eV (vs. 7119.7 eV for 1), which is within the range of values found for synthetic FeIV=O complexes.[ 3b , 5a , 5b , 5c ] Furthermore, in contrast to the pre‐edge features of existing S=1 complexes that can be modelled with a single Gaussian, [13] in the pre‐edge region of 2 two spectral features at ≈7115 and ≈7117 eV are tentatively discernible (Figure S7A,C), which may be rationalized in terms of a splitting of the α and β orbitals by spin polarization in the S=2 oxoiron(IV) cores.[ 5a , 5b ]
Resonance Raman spectroscopy revealed a ν(Fe=O) stretching mode at 802 cm−1 in 2 (Figure 1 A, inset) that shifted to 767 cm−1 upon 18O‐labelling. The observed ν(Fe=O) mode has one of the lowest energies reported to date for oxoiron(IV) cores. This may be attributed to the high spin (S=2) ground state of 2 as this would (in a simplified pseudotetrahedral ligand field) require a d(x 2−y 2)1d(xy)1d(xz,yz)2d(z 2)0 electronic configuration with an Fe−O bond order (BO) of 2.0. [15] Notably, the high‐spin ground state of 2 is unique for a pseudotetrahedral geometry; previously reported pseudotetrahedral M‐X (X=O2−, NR2−, or N3−) complexes, [16] including the recent CoIII≡O complex,[ 8 , 17 ] all possess a low‐spin ground state with a M‐X BO of 3. The zero field Mössbauer spectrum of 2 exhibits a doublet (87 % yield) with a quadrupole splitting, ΔE Q=0.96 mm s−1, and an isomer shift, δ=0.11 mm s−1 (Figure 1 B). Although, the ΔE Q value is very close to the value reported for TauD‐J (Table 1), [1b] the δ‐value is significantly lower, which may reflect the nitrogen‐rich character in 2 in contrast to the harder oxygen‐containing ligand sphere in TauD‐J. In applied magnetic fields, the spectra of 2 exhibit paramagnetic hyperfine structures, which were analysed by assuming an S=2 center yielding a non‐axial A‐tensor with Axx/gnβn=−10.1 T, Ayy/gnβn=−3.3 T and Azz/gnβn=−36.1 T (Figure S8). The structure of 2 as obtained by DFT calculations (Scheme 1, inset) reveals an off‐axis tilt of the oxo ligand resulting in a deviation from the C3 symmetry, which may account for the non‐axial A‐tensor determined from magnetic Mössbauer studies. The quintet state was calculated to be more stable than the triplet and the singlet states by 0.8 and 6.6 kcal mol−1, respectively (Table S3). Furthermore, among all spin states, the calculated spectroscopic properties of the S=2 state provide the best description of the experimental data. The calculated Fe=O and Fe−N bond distances (1.63 and 2.06 Å, respectively), Fe=O stretching mode frequency (893 cm−1, 18O isotope shift −36 cm−1), and Mössbauer δ‐value (0.06 mm s−1), on the ground S=2 state are in satisfactory agreement with experiments (Table S3). Notably, the calculated data for the S=1 and S=0 states deviate significantly from the experiments, such that we take the calculations as a further support for the S=2 ground state in 2.
The oxidative reactivity of 2 (Figures S9–S18; Table S6) has been investigated with several substrates in oxygen atom transfer (OAT) and HAA reactions and the second order rate constants derived from these studies in CH2Cl2 are compared with three of the most reactive high‐valent Fe‐oxo intermediates reported to date (namely the [(TQA)FeIV(O)(CH3CN)]2+ (TQA=tris(2‐quinolylmethyl)amine), [5e] [(Me3NTB)FeIV(O)]2+ (Me3NTB=tris((N‐methyl‐benzimidazol‐2‐yl)methyl)amine) [3c] and [(TMCO)FeIV(O)(CH3CN)]2+ (TMCO=4,8,12‐trimethyl‐1‐oxa‐4,8,12‐triazacyclotetradecane) [3f] complexes (Table 2). In reactions with ethylbenzene, 1,4‐cyclohexadiene (1,4‐CHD), and toluene, 2 is a stronger oxidant than [(TMCO)FeIV(O)(CH3CN)]2+, but comparable to [(TQA)FeIV(O)(CH3CN)]2+ and [(Me3NTB)FeIV(O)]2+. Interestingly, the reactivity trend is reversed in reactions with 9,10‐dihydroanthracene (DHA), where 2 exhibits the least reactivity. Furthermore, when the logarithms of the statistically corrected second order rate constants (k 2’) were plotted vs. the BDEC‐H values of the substrates (Figure 2 A, Figure S20A), the linear correlation typically observed for oxoiron(IV) cores[ 3a , 3b , 3c , 3d , 3e , 3f , 3g , 3h , 3i , 5 ] is found to be not valid for 2. While the respective log (k 2) values associated with 2 for the oxidation of 1,4‐CHD, 1,3‐cyclohexadiene (1,3‐CHD), ethylbenzene, cyclohexene and toluene fall on a line (Figure 2 A, black points), xanthene, DHA, indene and fluorene substrates (Figure 2 A, inset) deviate from this pattern and exhibit significantly lower rates than predicted by the linear relationship. Particularly interesting is the large rate difference of two orders of magnitude for DHA and 1,4‐CHD, which are known to have small difference in BDEC‐H values. [18] Furthermore, large deuterium KIEs of 7 (Figure S9), 12 (Figure S10), and 53 (Figure 2 C, Figure S11) were recorded for toluene, 1,4‐CHD, and ethylbenzene reactions, respectively, suggesting a HAA mechanism with significant contribution of hydrogen‐tunnelling, as is frequently proposed in C−H bond activation reactions of FeIV=O species. [7] In contrast, significantly reduced KIEs of 1.2 (Figure 2 D, Figure S12) and 2.1 (Figure S13) were determined for DHA and xanthene, respectively, thereby pointing to a change of mechanism. Further mechanistic insights were obtained by plotting the rate constants against the pK a and the ionization energies (IE) of the substrates. The log (k2) vs. IE plot (Figure 2 B, Figure S20B) revealed that for reactions of 2 with xanthene, DHA, indene and fluorene the rate decreased linearly with increasing IE, whereas the rates for 1,4‐CHD, 1,3‐CHD, ethylbenzene, cyclohexene and toluene scatter irregularly. Furthermore, no linear trend was observed in the log (k 2) vs. pK a plot (Figure S20C) for all the investigated substrates. Thus, the tBu3tacn ligand blocks the HAA pathway by presumably impeding access of the bulkier polycyclic hydrocarbons to the Fe=O unit in 2. An alternative oxidative asynchronous PCET mechanism (Scheme 2) prevails in such cases, which are typically characterized by low KIEs and a linear correlation of the reaction rates to IEs.
Table 2.
Comparison of the reaction rate constants k 2′ (normalized to the number of equivalent H atoms) at −40 °C for the C−H activation reaction of 2 and the highly reactive intermediates (TMCO)FeIV=O, (Me3NTB)FeIV=O and (TQA)FeIV=O towards a selection of substrates.
|
Substrate (BDEC‐H, kcal/mol) |
k 2′ [M−1 s−1] |
|||
|---|---|---|---|---|
|
|
2 |
(TMCO) FeIV=O |
(Me3NTB) FeIV=O |
(TQA) FeIV=O |
|
1,4‐CHD (76.0) |
1.0×102[a] |
nd |
7.8×102 |
nd |
|
DHA (76.3) |
1.6[b] |
Too fast (−90 °C) |
2.4×102 |
nd |
|
Ethylbenzene (85.4) |
3.3[b] |
0.10[c] |
0.75 |
1.1 |
|
Toluene (89.7) |
0.43[b] |
0.0044[c] |
0.16 |
0.21 |
nd=rate not determined; k 2′ values at −40 °C were calculated from the values measured at [a] −90 °C; [b] −70 °C; [c] −60 °C; [d] −50 °C and corrected for the temperature difference by doubling the rate for every 10 degrees rise in temperature.
Figure 2.
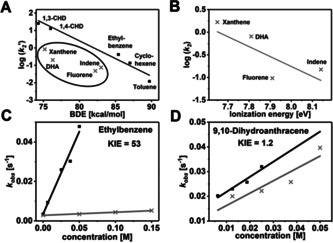
A) Plot of the logarithm of the second order rate constants k 2′ (normalized to the number of equivalent H atoms) of the reactions of 2 with different substrates vs. the BDEC‐H of the respective substrates; the inset shows the substrates that deviate from a linear correlation; B) Plot of the logarithm of the second order rate constants k 2 of the reactions of 2 with different polycyclic substrates vs. the ionization energy of the respective substrates; C) plot of the first‐order rate constants k obs vs. the concentration of ethylbenzene (black) and d 10‐ethylbenzene (grey) for determination of the second‐order rate constants k 2 and the deuterium KIE; D) plot of the first‐order rate constants k obs vs. the concentration of DHA (black) and d 4‐DHA (grey) for determination of the second‐order rate constant k 2 and the deuterium KIE.
Conclusion
Taken together the results presented herein unequivocally validate the formation of a terminal oxoiron(IV) complex 2 in a pseudotetrahedral geometry. The computational and experimental analyses are consistent with the presence of an S=2 FeIV=O core in 2. Complex 2 represents the only example of a high‐spin complex with metal‐ligand multiple bond character in a pseudotetrahedral geometry; notably, a pseudotetrahedral oxoiron(IV) complex has been very recently demonstrated to possess an S=0 state in the gas‐phase. [19] The absorption spectrum, Mössbauer ΔE Q, Fe K‐edge energy, and the ν(Fe=O) mode of 2 (Table 1) bear very close resemblance to the corresponding spectroscopic properties of TauD‐J. 2 also exhibits the distinct high‐reactivity features known from the strongly oxidizing iron‐oxo cores in biology and accordingly possesses one of the most reactive oxoiron(IV) cores that have been synthesized to date. Furthermore, a large KIE of 53 has been determined for the reaction of 2 with ethylbenzene, which compares well with the KIE of 57 [1] determined for the oxidation of taurine by TauD‐J. The uniqueness of 2 within the non‐heme oxoiron family is, however, emphasized in its ability to oxidize sterically hindered C−H bonds by an IE‐driven asynchronous PCET mechanism. Although limited examples of C−H oxidation by a basicity controlled PCET mechanism (Scheme 2) are known,[ 8 , 20 ] evidence of oxidative PCET mechanism has stayed elusive prior to this study. In conclusion, the high reactivity and the similar spectroscopic parameters of 2 and TauD‐J make 2 one of the best structural, electronic and functional models for TauD‐J.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy—EXC 2008–390540038—UniSysCat to K.R., P.H., and H.D., and the Heisenberg‐Professorship to K.R., and MINECO (CTQ2017‐87392‐P) and FEDER (UNGI10‐4E‐801) to M.S. K.W. also thanks Einstein Foundation Berlin (ESB)—Einstein Center of Catalysis (EC2) for its support. Open access funding enabled and organized by Projekt DEAL.
K. Warm, A. Paskin, U. Kuhlmann, E. Bill, M. Swart, M. Haumann, H. Dau, P. Hildebrandt, K. Ray, Angew. Chem. Int. Ed. 2021, 60, 6752.
Dedicated to Professor Wolfgang Kaim on the occasion of his 70th birthday
References
- 1.
- 1a. Price J. C., Barr E. W., Tirupati B., Bollinger J. M., Krebs C., Biochemistry 2003, 42, 7497–7508; [DOI] [PubMed] [Google Scholar]
- 1b. J. M. Bollinger, Jr. , Price J. C., Hoffart L. M., Barr E. W., Krebs C., Eur. J. Inorg. Chem. 2005, 4245–4254; [Google Scholar]
- 1c. Krebs C., Galonić Fujimori D., Walsh C. T., Bollinger J. M., Acc. Chem. Res. 2007, 40, 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sinnecker S., Svensen N., Barr E. W., Ye S., Bollinger J. M., Neese F., Krebs C., J. Am. Chem. Soc. 2007, 129, 6168–6179. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Grapperhaus C. A., Mienert B., Bill E., Weyhermüller T., Wieghardt K., Inorg. Chem. 2000, 39, 5306–5317; [DOI] [PubMed] [Google Scholar]
- 3b. Rohde J.-U., In J.-H., Lim M. H., Brennessel W. W., Bukowski M. R., Stubna A., Münck E., Nam W., Que L., Science 2003, 299, 1037; [DOI] [PubMed] [Google Scholar]
- 3c. Seo M. S., Kim N. H., Cho K.-B., So J. E., Park S. K., Clémancey M., Garcia-Serres R., Latour J.-M., Shaik S., Nam W., Chem. Sci. 2011, 2, 1039–1045; [Google Scholar]
- 3d. Meyer S., Klawitter I., Demeshko S., Bill E., Meyer F., Angew. Chem. Int. Ed. 2013, 52, 901–905; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 935–939; [Google Scholar]
- 3e. Wang D., Ray K., Collins M. J., Farquhar E. R., Frisch J. R., Gómez L., Jackson T. A., Kerscher M., Waleska A., Comba P., Costas M., Que L., Chem. Sci. 2013, 4, 282–291; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3f. Monte Pérez I., Engelmann X., Lee Y.-M., Yoo M., Kumaran E., Farquhar E. R., Bill E., England J., Nam W., Swart M., Ray K., Angew. Chem. Int. Ed. 2017, 56, 14384–14388; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 14576–14580; [Google Scholar]
- 3g. Gordon J. B., Vilbert A. C., DiMucci I. M., MacMillan S. N., Lancaster K. M., Moënne-Loccoz P., Goldberg D. P., J. Am. Chem. Soc. 2019, 141, 17533–17547; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3h. Nam W., Acc. Chem. Res. 2015, 48, 2415–2423; [DOI] [PubMed] [Google Scholar]
- 3i. Larson V. A., Battistella B., Ray K., Lehnert N., Nam W., Nat. Rev. Chem. 2020, 4, 404–419; [DOI] [PubMed] [Google Scholar]
- 3j. Engelmann X., Monte-Pérez I., Ray K., Angew. Chem. Int. Ed. 2016, 55, 7632–7649; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7760–7778; [Google Scholar]
- 3k. Sacramento J. J. D., Goldberg D. P., Acc. Chem. Res. 2018, 51, 2641–2652; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3l. Chen J., Jiang Z., Fukuzumi S., Nam W., Wang B., Coord. Chem. Rev. 2020, 421, 213443; [Google Scholar]
- 3m. Guo M., Corona T., Ray K., Nam W., ACS Cent. Sci. 2019, 5, 13–28; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3n. Que L., Acc. Chem. Res. 2007, 40, 493–500; [DOI] [PubMed] [Google Scholar]
- 3o. McDonald A. R., L. Que, Jr. , Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar]
- 4.
- 4a. Shaik S., Hirao H., Kumar D., Acc. Chem. Res. 2007, 40, 532–542; [DOI] [PubMed] [Google Scholar]
- 4b. Bernasconi L., Louwerse M. J., Baerends E. J., Eur. J. Inorg. Chem. 2007, 3023–3033; [Google Scholar]
- 4c. Decker A., Rohde J.-U., Klinker E. J., Wong S. D., Que L., Solomon E. I., J. Am. Chem. Soc. 2007, 129, 15983–15996; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Geng C., Ye S., Neese F., Angew. Chem. Int. Ed. 2010, 49, 5717–5720; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5853–5856. [Google Scholar]
- 5.
- 5a. England J., Guo Y., Van Heuvelen K. M., Cranswick M. A., Rohde G. T., Bominaar E. L., Münck E., Que L., J. Am. Chem. Soc. 2011, 133, 11880–11883; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. England J., Martinho M., Farquhar E. R., Frisch J. R., Bominaar E. L., Münck E., L. Que, Jr. , Angew. Chem. Int. Ed. 2009, 48, 3622–3626; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3676–3680; [Google Scholar]
- 5c. Lacy D. C., Gupta R., Stone K. L., Greaves J., Ziller J. W., Hendrich M. P., Borovik A. S., J. Am. Chem. Soc. 2010, 132, 12188–12190; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5d. Bigi J. P., Harman W. H., Lassalle-Kaiser B., Robles D. M., Stich T. A., Yano J., Britt R. D., Chang C. J., J. Am. Chem. Soc. 2012, 134, 1536–1542; [DOI] [PubMed] [Google Scholar]
- 5e. Biswas A. N., Puri M., Meier K. K., Oloo W. N., Rohde G. T., Bominaar E. L., Münck E., Que L., J. Am. Chem. Soc. 2015, 137, 2428–2431; [DOI] [PubMed] [Google Scholar]
- 5f. Puri M., Que L., Acc. Chem. Res. 2015, 48, 2443–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Thangavel A., Wieliczko M., Bacsa J., Scarborough C. C., Inorg. Chem. 2013, 52, 13282–13287. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Mandal D., Shaik S., J. Am. Chem. Soc. 2016, 138, 2094–2097; [DOI] [PubMed] [Google Scholar]
- 7b. Mandal D., Mallick D., Shaik S., Acc. Chem. Res. 2018, 51, 107–117; [DOI] [PubMed] [Google Scholar]
- 7c. Klinker E. J., Shaik S., Hirao H., L. Que, Jr. , Angew. Chem. Int. Ed. 2009, 48, 1291–1295; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1317–1321. [Google Scholar]
- 8. Goetz M. K., Anderson J. S., J. Am. Chem. Soc. 2019, 141, 4051–4062. [DOI] [PubMed] [Google Scholar]
- 9. Karahalis G. J., Thangavel A., Chica B., Bacsa J., Dyer R. B., Scarborough C. C., Inorg. Chem. 2016, 55, 1102–1107. [DOI] [PubMed] [Google Scholar]
- 10. Riggs-Gelasco P. J., Price J. C., Guyer R. B., Brehm J. H., Barr E. W., Bollinger J. M., Krebs C., J. Am. Chem. Soc. 2004, 126, 8108–8109. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Swart M., Gruden M., Acc. Chem. Res. 2016, 49, 2690–2697; [DOI] [PubMed] [Google Scholar]
- 11b. Swart M., Chem. Phys. Lett. 2013, 580, 166–171. [Google Scholar]
- 12. Macikenas D., Skrzypczak-Jankun E., Protasiewicz J. D., J. Am. Chem. Soc. 1999, 121, 7164–7165. [Google Scholar]
- 13. Evans D. F., J. Chem. Soc. (Resumed) 1959, 2003–2005. [Google Scholar]
- 14. Jackson T. A., Rohde J.-U., Seo M. S., Sastri C. V., DeHont R., Stubna A., Ohta T., Kitagawa T., Münck E., Nam W., Que L., J. Am. Chem. Soc. 2008, 130, 12394–12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Berry J. F., DeBeer George S., Neese F., Phys. Chem. Chem. Phys. 2008, 10, 4361–4374. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Betley T. A., Peters J. C., J. Am. Chem. Soc. 2003, 125, 10782–10783; [DOI] [PubMed] [Google Scholar]
- 16b. Hu X., Meyer K., J. Am. Chem. Soc. 2004, 126, 16322–16323; [DOI] [PubMed] [Google Scholar]
- 16c. Cowley R. E., Bontchev R. P., Sorrell J., Sarracino O., Feng Y., Wang H., Smith J. M., J. Am. Chem. Soc. 2007, 129, 2424–2425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16d. Saouma C. T., Peters J. C., Coord. Chem. Rev. 2011, 255, 920–937; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16e. Wu B., Hernández Sánchez R., Bezpalko M. W., Foxman B. M., Thomas C. M., Inorg. Chem. 2014, 53, 10021–10023; [DOI] [PubMed] [Google Scholar]
- 16f. Shay D. T., Yap G. P. A., Zakharov L. N., Rheingold A. L., Theopold K. H., Angew. Chem. Int. Ed. 2005, 44, 1508–1510; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 1532–1534; [Google Scholar]
- 16g. Scepaniak J. J., Vogel C. S., Khusniyarov M. M., Heinemann F. W., Meyer K., Smith J. M., Science 2011, 331, 1049–1052; [DOI] [PubMed] [Google Scholar]
- 16h. Ray K., Heims F., Pfaff F. F., Eur. J. Inorg. Chem. 2013, 3784–3807. [Google Scholar]
- 17. Goetz M. K., Hill E. A., Filatov A. S., Anderson J. S., J. Am. Chem. Soc. 2018, 140, 13176–13180. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Luo Y.-R., Comprehensive Handbook of Chemical Bond Energies, CRC, Boca Raton, 2007; [Google Scholar]
- 18b. Klein J. E. M. N., Dereli B., L. Que, Jr. , Cramer C. J., Chem. Commun. 2016, 52, 10509–10512. [DOI] [PubMed] [Google Scholar]
- 19. Andris E., Segers K., Mehara J., Rulíšek L., Roithová J., Angew. Chem. Int. Ed. 2020, 59, 23137–23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Usharani D., Lacy D. C., Borovik A. S., Shaik S., J. Am. Chem. Soc. 2013, 135, 17090–17104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Parsell T. H., Yang M.-Y., Borovik A. S., J. Am. Chem. Soc. 2009, 131, 2762–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary