

Abstract
Intravascular large B-cell lymphoma (IVLBCL) is a rare form of diffuse LBCL. The patient was a 71-year-old female admitted to our hospital with hypoxia. On admission, chest computed tomography revealed a ground-glass opacity. Interstitial pneumonia associated with systemic scleroderma was suspected because of positive anti-centromere antibody. Thereafter, steroid pulse therapy and plasma exchange were performed. Although ground-glass opacity improved, bilateral pleural effusion appeared, so we performed a random skin biopsy because of her elevated serum lactate dehydrogenase and soluble interleukin-2 receptor levels. The patient was diagnosed with IVLBCL with symptoms improving after 6 cycles of rituximab plus chemotherapy treatment.
Keywords: anti-centromere antibody, ground-glass opacity, intravascular diffuse large B-cell lymphoma, serum lactate dehydrogenase, random skin biopsy
Introduction
The World Health Organization classification defines intravascular diffuse large B-cell lymphoma (IVLBCL) as a rare subtype of non-Hodgkin’s lymphoma, where neoplastic cells grow mainly in the lumina of small vessels.1 The first case of IVLBCL was reported by Pfleger et al, as “angioendotheliomatosis proliferans systemisata” with an endothelial origin.2 In 1982, Ansel et al suggested a lymphoid origin,3 and in 1986, the lymphoid nature of this entity was confirmed by Wick et al.4 Its clinical variants and strange characteristics, including absence of lymphadenopathy or mass formation and its aggressive clinical behavior, often lead to the delay of accurate and timely diagnosis and to fatal complications.5 Thus, its prognosis is extremely poor. Clinical manifestations of IVLBCL are extremely variable since multiple organs are involved in its signs and symptoms. Like other aggressive forms of lymphoma, symptoms associated with specific organs should be examined and investigated with specific medical procedures, including biopsy.
We report an IVLBCL case with ground-glass opacity (GGO) of the bilateral lungs and anti-centromere antibody positivity in blood tests, which required distinction from interstitial pneumonia related to collagen disease. If a dyspneic patient has elevated serum levels of lactate dehydrogenase (LDH) and soluble interleukin 2 receptor (sIL-2R), it is important to suspect IVLBCL regardless of imaging findings in the pulmonary field for an early diagnosis due to the rapid progression of IVLBCL and the associated fatalities.
Case Report
A 71-year-old female patient was admitted to our hospital with a slight fever, night sweats, and dyspnea. Chest computed tomography (CT) and X-ray showed GGO on the bilateral lung (Figure 1A). Brain magnetic resonance imaging revealed subacute cerebral infarction in the right corona radiata. Her vital signs were as follows: body temperature of 37.1 °C; blood pressure 126/71 mm Hg; respiratory rate 24 breaths/minute; oxygen saturation 91% with 5 L/min oxygen inhalator. Slight crackles were present at the end of expiration and slight pansystolic murmur was located at the apex. The finger-nose test was slightly poor on her left side. An initial laboratory examination results obtained within 2 weeks after the admission. It showed remarkably high levels of LDH (1027 U/I; normal range = 120-220), anti-nuclear antibody (×1280; <40), centromere antibody (×1280; <40), soluble interleukin-2 receptor (1180; normal range = 145-519 U/I), and C-reactive protein (3.49; normal range = 0-0.35 g/dL; Table 1). Community-acquired pneumonia was suspected on admission, but her hypoxia worsened.
Figure 1.

Chest computed tomography (CT) in the course of treatment. (A) Ground-glass opacity (GGO) was revealed on the chest CT at bilateral lung fields. (B) Bilateral pleural effusion appeared after GGO was improved. (C) The bilateral pleural effusion disappeared after 6 cycles of R-chemotherapy treatment for diagnosis with IVL.
Table 1.
Laboratory Data on Admission.
| Hematology | Serology | KL-6 190 (<500) U/mL |
| WBC 6900 (3.5 to 8.5)/µL | CRP 3.49 (0 to 0.35) mg/dL | SP-D 161 (<110) ng/mL |
| SEG 71.1% (40% to 70%) | β-2MG 2.9 (0.9 to 2.0) mg/L | HIV antigen Ab (−) |
| LY 14.9% (20% to 50%) | BNP 19.2 (0 to 20) pg/mL | T-SPOT (−) |
| MO 12.7% (2% to 9%) | IgG 816 (870 to 1700) mg/dL | β-D glucan <6.0 (<11.0) pg/mL |
| EO 0.6% (1% to 6%) | IgA 292 (110 to 410) mg/dL | Mycoplasma pneumoniae Ab (−) |
| BA 0.7% (0% to 2%) | IgM 59 (35 to 220) mg/dL | CMV antigenemia (−) |
| RBC 374 × 104 (370 to 490)/µL | C3 109 (65-135) mg/dL | Anti-trichosporon asahii Ab (−) |
| Hb 1.7 (11.5 to 15) g/dL | C4 16 (13-35) mg/dL | CEA <0.5 (<0.5) ng/mL |
| PLT 15.7 × 104 (150 to 350)/µL | RF <15 (<15) IU/mL | Coagulation |
| Biochemistry | Antinuclear Ab ×1280 (<40) | PT(I) 1.11 (0.8 to 1.2) |
| TP 5.9 (6.7 to 8.2) g/dL | DiscreteSp ×1280 (<40) | APTT 26.4 (24 to 38) s |
| Alb 3.1 (4.3 to 5.4) g/dL | Centromere Ab 134 (12) IU/mL | D-dimer 1.3 (0 to 1.0) µg/mL |
| Na 134 (6.7 to 8.2) mEq/L | ds-DNA Ab <10 (<12.0) U/mL | Arterial blood gasa |
| K 4.2 (3.6 to 4.8) mEq/L | RNP Ab <2.0 (<10.0) U/mL | pH 7.401 (7.35 to 7.40) |
| Cl 100 (99 to 107) mEq/L | Sm Ab <1.0 (<10.0) U/mL | pCO2 36.7 (35.0 to 45.0) mm Hg |
| BUN 19 (8 to 20) mg/dL | SS-A Ab <1.0 (<10.0) U/mL | pO2 67.6 (80 to 100) mm Hg |
| Cr 0.6 (0.4 to 0.8) mg/dL | SS-B Ab <1.0 (<10.0) U/mL | HCO3− 22.3 (20.0 to 26.0) mmol/L |
| UA 5.8 (3 to 7) mg/dL | Scl-70 Ab <1.0 (<10.0) U/mL | BE 2.1(−3.0 to 3.0) mmol/L |
| T-Bil 0.7 (0.4 to 1.3) mg/dL | Jo-1 Ab <1.0 (<10.0) U/mL | AnGap 11.1 (10.0 to 18.0) mmol/L |
| AST 91 (10 to 35) U/L | PR3-ANCA <1.0 (<3.5) U/mL | Lac 0.93 (0.37 to 1.67) mmol/L |
| ALT 17 (5 to 40) U/L | MPO-ANCA <1.0 (<3.5) U/mL | Urinalysis |
| LD 1027 (120 to 220) U/L | ARS Ab <5.0 (<25.0) U/mL | Sterptococcus pneumoniae Ag (−) |
| ALP 200 (100 to 320) U/L | GBM Ab <2.0 (<3.0) U/mL | Legionella pneumophila Ag (−) |
| γ-GTP 25 (5 to 40) U/L | sIL-2R 1180 (145 to 519) U/mL | Blood culture: negative |
Abbreviations: WBC, white blood cell; SEG, segmented bands; LY, lymphocyte; MO, monocyte; EO, eosinophil; BA, basophil; RBC, red blood cell; Hb, hemoglobin; PLT, platelets; Tp, total protein; Alb, albumin; Na, sodium; K, potassium; Cl, chloride; BUN, blood urea nitrogen; Cr, creatinine; UA, uric acid; T-Bil, total bilirubin; AST, aspartate aminotransferase; ALT, alanine aminotransferase; ALP, alkaline phosphatase; γ-GTP, γ-glutamyl transpeptidase; CRP, C-reactive protein; BNP, B-type natriuretic peptide; Ig, immunoglobulin; Ab, antibody; CMV, cytomegalovirus; PT, prothrombin time; APTT, activated partial thromboplastin time; pCO2, partial pressure of carbon dioxide; pO2, partial pressure of oxygen; HCO3−, bicarbonate; Ag, antigen.
Oxygen 5 L/min inhalation.
Although she did not have a history of collagen disease, the presence of symptoms of hand stiffness and GGO on imaging necessitated screening for autoimmune disease.
Then, interstitial pneumonia associated with systemic scleroderma was suspected due to the high titer of anti-centromere antibody, rapid progress in renal dysfunction with hematuria and proteinuria. She received steroid pulse therapy (mPSL 1 g/day for 3 days) and plasma exchange. Although her GGO improved, bilateral pleural effusion on chest X-ray was detected (Figure 1B). Thereafter, random skin biopsies were performed from multiple locations including the thigh and forearm; although there were no skin findings, examination revealed large cells with big dysmorphic nuclei in capillaries (Figure 2), on hematoxylin-eosin staining. Moreover, bone marrow biopsy was performed; it revealed CD20, CD79a, and CD5 positive lymphoma cells on immunohistochemistry (Figure 3A-C). Finally, the patient was diagnosed with IVLBCL, and after 6 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) treatment she experienced a stark improvement in laboratory data and clinical findings (Figure 1C).
Figure 2.
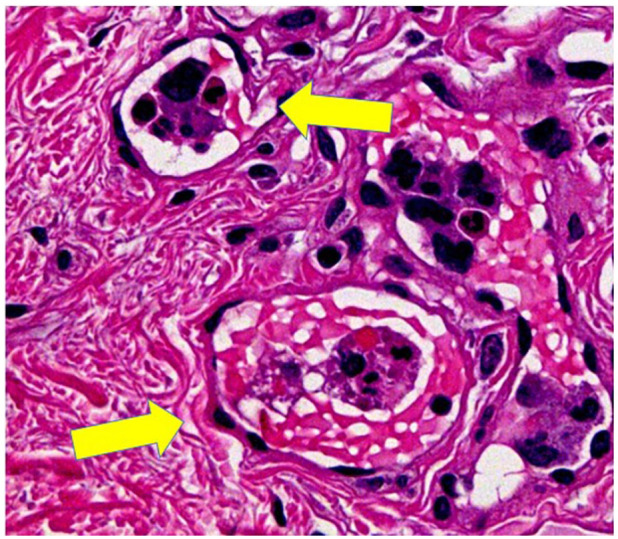
Large lymphoma cells with irregular dysmorphic nuclei with distant rims and abundant endoplasm within capillaries (hematoxylin-eosin stain) in the skin (yellow arrows).
Figure 3.

CD 20 (A), CD 79a (B), and CD5 (C) were positive in lymphoma by immunohistochemistry in bone marrow.
Discussion
Our IVLBCL case needed to be differentiated from interstitial pneumonia related to systemic scleroderma with positive anti-centromere antibody because their clinical courses are quite similar and due to some reports on malignant lymphoma associated autoimmune disease.6 Although she received steroid pulse therapy and plasma exchange for interstitial pneumonia related to systemic scleroderma, her condition did not improve, while other symptoms started to appear, which required a careful review of the case itself and skin and bone marrow biopsies for the final diagnosis. We were finally able to give IVLBCL as a diagnosis and she improved after a 6-cycle of R-CHOP treatment.
The case fulfilled the criteria of IVLBCL, which are (1) presence of lymphoma cells in the blood vessels, (2) no swelling on lymph nodes, and (3) quickly worsening symptoms before diagnosis.7,8
In IVLBCL, laboratory tests often suggest anomalies such as anemia, thrombocytopenia, elevated LDH, and sIL-2R.9 This case did not present anemia or thrombocytopenia, but the highly elevated LDH and sIL-2R and LDH values decreased after R-CHOP therapy.
There are various reports on the imaging characteristics of lung lesions in IVLBCL patients; these include the presence of GGO, granular shadows, nodules, and the absence of any abnormalities. It is therefore essential to identify IVLBCL when LDH and sIL-2R levels are elevated. Sometimes IVLBCL pulmonary lesions can be diagnosed by TBLB, but otherwise, bone marrow biopsy and random skin biopsy might be useful. In this case, bronchoscopy could not be performed due to rapid deterioration of respiratory failure with IVLBCL finally being diagnosed by random skin and bone marrow biopsy. Most cases with pulmonary lesions present with respiratory symptoms such as cough, dyspnea, and hypoxemia, which are known to improve quickly in response to treatment for IVLBCL.10
Furthermore, our case was CD5 positive with bone marrow infiltration. It is reported that CD5 is expressed on some diffuse LBCLs, and that CD5+ are older than the negative group, have more advanced stages and poor prognosis.11 Brunet et al reported no difference in prognosis between positive and negative CD5 in IVLBCL.8
Moreover, differences in clinical features between Western countries and Asia have been reported. In Western countries, patients present most commonly with symptoms related to involvement of the central nervous system and skin.7,8 In Asia, patients frequently present with the involvement of the bone marrow, spleen, liver, and lung, while neurological and cutaneous symptoms are less common.12,13 In this case, she had combined both Western and Asian characteristics such as subacute cerebral infarction with involvement of the bone marrow and lungs.
Although IVLBCL is a rare and difficult disease to diagnose, especially in suspected interstitial pneumoniae related to collagen disease, IVLBCL should be included among the possible differential diagnoses.
Because IVL often presents with fever and dyspnea, patients are more likely to be examined by respiratory physicians. If the patient is dyspneic and has elevated LDH and sIL-2R, regardless of imaging findings in the pulmonary field, it is important to suspect IVL for early diagnosis because of its rapid progression and associated fatalities.
Acknowledgments
We would like to thank Masami Yoshihara, the medical interpreter and the medical coordinator at Tokyo Saiseikai Central Hospital, and Editage (www.editage.com) for English language editing. We thank Keisuke Kamata, MD, PhD, at Hakkaido University Hospital, for helping us with the medical treatment.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethics Approval: Ethical approval to report this case was obtained from Saiseikai Central Hospital of Institutional Review Board (CR-112).
Informed Consent: Verbal informed consent was obtained from the patient(s) for their anonymized information to be published in this article.
ORCID iD: Tomoyo Oguri  https://orcid.org/0000-0002-7655-1517
https://orcid.org/0000-0002-7655-1517
References
- 1. Ponzoni M, Ferreri AJ, Campo E, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. J Clin Oncol. 2007;25:3168-3173. [DOI] [PubMed] [Google Scholar]
- 2. Pfleger L, Tappeiner J. On the recognition of systematized endotheliomatosis of the cutaneous blood vessels (reticuloendotheliosis?) [in German]. Hautarzt. 1959;10:359-363. [PubMed] [Google Scholar]
- 3. Ansell J, Bhawan J, Cohen S, Sullivan J, Sherman D. Histocytic lymphoma and malignant angioendotheliomatosis. One disease or two? Cancer. 1982;50:1506-1152. [DOI] [PubMed] [Google Scholar]
- 4. Wick MR, Mills SE, Scheithauer BW, Cooper PH, Davitz MA, Parkinson K. Reassessment of malignant “angioendotheliomatosis.” Evidence in favor of its reclassification as “intravascular lymphomatosis.” Am J Surg Pathol. 1986;10:112-123. [PubMed] [Google Scholar]
- 5. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. Lancet Oncol. 2009;10:895-902. [DOI] [PubMed] [Google Scholar]
- 6. Váróczy L, Gergely L, Zeher M, Szegedi G, Illés A. Malignant lymphoma-associated autoimmune diseases—a descriptive epidemiological study. Rheumatol Int. 2002;22:233-237. [DOI] [PubMed] [Google Scholar]
- 7. Ferreri AJ, Campo E, Seymour JF, et al. ; International Extranodal Lymphoma Study Group (IELSG). Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the “cutaneous variant.” Br J Haematol. 2004;127:173-183. [DOI] [PubMed] [Google Scholar]
- 8. Brunet V, Marouan S, Routy JP, et al. Retrospective study of intravascular large B-cell lymphoma cases diagnosed in Quebec: a retrospective study of 29 case reports. Medicine (Baltimore). 2017;96:e5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimada K, Matsue K, Yamamoto K, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. J Clin Oncol. 2008;26:3189-3195. [DOI] [PubMed] [Google Scholar]
- 10. Sakurai A, Tomii K, Haruna A, et al. Two cases of successfully treated intravascular lymphoma presenting with fever and dyspnea [in Japanese]. Nihon Kokyuki Gakkai Zasshi. 2011;49:743-749. [PubMed] [Google Scholar]
- 11. Yamaguchi M, Seto M, Okamoto M, et al. De novo CD5+ diffuse large B-cell lymphoma: a clinicopathologic study of 109 patients. Blood. 2002;99:815-821. [DOI] [PubMed] [Google Scholar]
- 12. Shimada K, Murase T, Matsue K, et al. Central nervous system involvement in intravascular large B-cell lymphoma: a retrospective analysis of 109 patients. Cancer Sci. 2010;101:1480-1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoon SE, Kim WS, Kim SJ. Asian variant of intravascular large B-cell lymphoma: a comparison of clinical features based on involvement of the central nervous system. Korean J Intern Med. 2020;35:946-956. [DOI] [PMC free article] [PubMed] [Google Scholar]