

Summary
The coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) and variants has led to significant mortality. We recently reported that an RNA-targeting CRISPR-Cas13 system, called prophylactic antiviral CRISPR in human cells (PAC-MAN), offered an antiviral strategy against SARS-CoV-2 and influenza A virus. Here, we expand in silico analysis to use PAC-MAN to target a broad spectrum of human- or livestock-infectious RNA viruses with high specificity, coverage, and predicted efficiency. Our analysis reveals that a minimal set of 14 CRISPR RNAs (crRNAs) is able to target >90% of human-infectious viruses across 10 RNA virus families. We predict that a set of 5 experimentally validated crRNAs can target new SARS-CoV-2 variant sequences with zero mismatches. We also build an online resource (crispr-pacman.stanford.edu) to support community use of CRISPR-Cas13 for broad-spectrum RNA virus targeting. Our work provides a new bioinformatic resource for using CRISPR-Cas13 to target diverse RNA viruses to facilitate the development of CRISPR-based antivirals.
Keywords: CRISPR, Cas13, SARS-CoV-2, COVID-19, antiviral, RNA virus, infectious disease, PAC-MAN
Graphical abstract
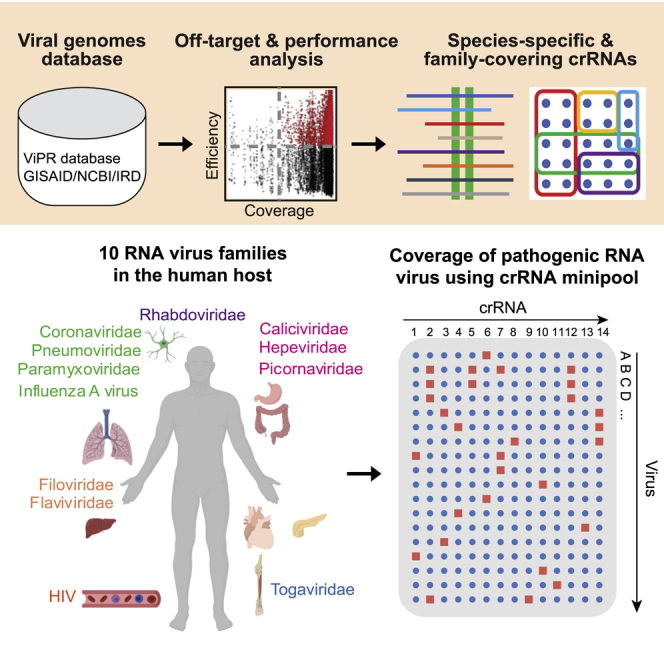
Highlights
A web resource for using CRISPR-Cas13 to target broad RNA virus families
A bioinformatic tool to analyze efficiency and specificity of virus-targeting crRNAs
A minipool of crRNAs can target 90% of human-infectious RNA viruses
Validated crRNAs are predicted to target newly emerging SARS-CoV-2 variants
Lin et al. develop a bioinformatic tool and an online resource for using CRISPR-Cas13 to target diverse human-infecting RNA viruses (10 families and 24,096 strains) with high efficiency and specificity.
Introduction
Since December 2019, the world has been devastated by the coronavirus disease 2019 (COVID-19) pandemic caused by the RNA virus severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2). In addition to SARS-CoV-2, the outbreaks of RNA viruses in recent years, including Zika virus, Ebola virus, Marburg virus, and MERS (Middle East respiratory syndrome) virus, have led the World Health Organization (WHO) to declare public health emergencies. While vaccines and therapeutic options against SARS-CoV-2 have advanced into clinical trials, most RNA viruses lack an approved therapy.1
In search of potential antivirals, we recently developed a CRISPR-Cas13d-based strategy, PAC-MAN (prophylactic antiviral CRISPR in human cells) to target and inhibit SARS-CoV-2 and influenza A virus (IAV) sequences.2 A few researchers also reported that CRISPR-Cas13a/b systems can be used to inhibit RNA viruses, including lymphocytic choriomeningitis virus (LCMV), vesicular stomatitis virus (VSV), human immunodeficiency virus type 1 (HIV-1), human respiratory syncytial virus (HRSV), Dengue virus, Turnip mosaic virus, and tobacco turnip mosaic RNA virus (TuMV).3, 4, 5, 6, 7, 8, 9 The CRISPR-Cas13 systems (subtypes a, b, and d) offer attractive prophylactic or therapeutic options due to 2 key features: (1) CRISPR-Cas13 is highly programmable, which allows it to be rapidly adapted against new RNA viruses by designing the CRISPR RNA (crRNA) sequence5,9 and (2) crRNAs can be designed to target highly conserved viral sequences to cover multiple strains and prevent viral mutational escape.10 One remaining question is whether the system can be expanded to target a broad spectrum of RNA viruses.
Here, we perform a comprehensive in silico analysis using PAC-MAN to target notable pathogenic viral species and human-infectious RNA viruses in the Virus Pathogen Database and Analysis Resource (ViPR).11 We generated an integrated crRNA analysis pipeline by incorporating existing algorithms to compute the coverage, efficiency, and specificity of crRNAs for targeting RNA viruses. Using this pipeline, we identified crRNAs for targeting 16 distinct RNA virus families. We also observed that a minimal set of 14 crRNAs can collectively target >90% of human-infectious strains across 10 non-segmented RNA virus families. For several families of viruses, >90% of sequenced human-infectious strains in the family can be targeted by as few as 1 or 2 crRNAs. In contrast, families of viruses with segmented RNA genomes generally required more crRNAs for full targeting. We built an online resource (http://crispr-pacman.stanford.edu/) to disseminate Cas13d crRNAs that target a broad spectrum of RNA viruses. We experimentally tested a group of predicted crRNAs targeting SARS-CoV-2 genomes and found a high correlation between predicted and validated efficiency. We found that five highly efficient crRNAs can target all of the known SARS-CoV-2 strains, including new variants. This work provides a comprehensive analysis and useful resource for using PAC-MAN to target a broad spectrum of pathogenic RNA viruses and can facilitate the development of CRISPR-based antiviral technology.
Results
An integrated pipeline for Cas13 crRNA analysis for virus-targeting coverage, efficiency, and specificity
We retrieved RNA virus genomes from public databases, including 16 RNA virus families from the ViPR, HIV from NCBI,12 and IAV from the Influenza Research Database (IRD)13 (Table S1). Our pipeline analyzed conserved genome sequences within a designated group of viruses, which generated the primary regions for crRNA targeting (Figure 1A). We generated all possible crRNA candidates with perfect matches to the conserved sequence. The pipeline eliminated crRNAs with putative off-target binding in the human transcriptome (≤2 mismatches, a threshold that was previously defined to distinguish specific versus non-specific crRNAs14). We incorporated a recently reported algorithm to predict crRNA efficiency (range 0–1) and also computed for each crRNA the percentage of viral strains in the group that it targets. We used our pipeline to analyze two types of crRNAs with different features. The first type is species-specific crRNAs, which are comprehensive sets of crRNAs that can target conserved regions in closely related branches of viruses with high coverage, specificity, and predicted efficiency. The second type is family-covering crRNAs, which are minimal pools of crRNAs (“minipools”) that collectively target every virus in a given family.
Figure 1.

Analysis of PAC-MAN for targeting broad-spectrum RNA viruses
(A) An integrated pipeline for Cas13 crRNA analysis for virus-targeting coverage, efficiency, and specificity.
(B) Targeting pandemic/epidemic-causing pathogenic RNA virus species using PAC-MAN.
(C) Schematic of 10 RNA virus families that target different organs and systems in the human host.
(D) The minimal number of crRNAs needed to target all sequenced human-infectious strains in each of 10 RNA virus families and HIV and IAV.
(E) The minimal number of crRNAs needed to target 90% of sequenced human-infectious strains in each of 10 RNA virus families and HIV and IAV.
(F) A cumulative curve showing the predicted minimum number of crRNAs to target all human-infectious strains in 10 RNA virus families in (C).
(G) Coverage of pathogenic RNA viruses by the first 14 crRNAs in the minipool from (F).
(H) A cumulative curve showing the predicted minimum number of crRNAs to target livestock-infectious strains in 10 RNA virus families in (C). Bovine, ovine, swine, feline, and canine viruses are included in the analysis.
Targeting pandemic/epidemic-causing pathogenic RNA virus species using PAC-MAN
We identified species-specific crRNA sets targeting each pandemic/epidemic-causing pathogenic RNA virus species per the WHO; these viruses include SARS, SARS-CoV-2, MERS, Ebola virus, Marburg virus, Zika virus, Chikungunya virus, and Nipah virus. For most pathogenic virus species, we were able to obtain a collection of crRNAs with predicted high specificity (≥3 mismatches in the human transcriptome), high efficiency (score ≥ 0.5), and high coverage (targets >90% of viruses in the group with zero mismatches) (Figure 1B). One exception was Nipah virus, for which we found that no crRNA candidate was able to cover 90% of sequenced strains. Nevertheless, lowering the coverage requirement to 80% virus strain coverage produced a set of crRNAs that can target the Nipah virus.
Analysis of PAC-MAN for targeting a broad spectrum of RNA viruses
We next identified crRNAs that can be used to target a broad spectrum of RNA virus families. We analyzed 10 RNA virus families (calicividirae, coronaviridae, filoviridae, hepeviridae, paramyxoviridae, phenuiviridae, picornaviridae, pneumoviridae, rhabdoviridae, and togaviridae) from the ViPR database that infect various human organs and systems, including 6 positive-sense single-stranded RNA (ssRNA) virus families, 4 negative-sense ssRNA virus families, as well as HIV and IAV species (Figure 1C). For each family or HIV/IAV species, we used an approximation algorithm to obtain a minipool of crRNAs that can collectively target every strain in the family.
Interestingly, we observed that all human-infectious sequences for each of the 10 RNA virus families and HIV can be comprehensively targeted by a small minipool of crRNAs (3–23 crRNAs). One exception is IAV, which requires 78 crRNAs for comprehensive coverage, likely due to its high diversity and its segmented genome, which is split into 8 segments15 (Figure 1D). Since many families contain rare strains with highly variant genomes, if we aim to target at least 90% of viral strains, then the human-infectious members of each family can be targeted by a very small number (1–6) of crRNAs (Figure 1E).
Crucially, we found that a minipool of 14 crRNAs is able to target >90% of human-infectious viruses in the 10 RNA virus families (Figures 1F and S1A). A complete minipool of 90 crRNAs can target all human-infectious strains (24,096) in the 10 families with zero mismatches. We observed that the 14 minipool crRNAs target notable pathogenic viral species with zero mismatches (Figure 1G). This suggests that Cas13-mediated PAC-MAN may offer a potential broad-spectrum antiviral (BSA) strategy for broadly targeting human infectious RNA viruses.
We also analyzed crRNAs that can target animal viruses. For this purpose, we categorized viruses according to their host, including common domestic livestock (cattle, sheep, pig, cat, and dog) that have close interactions with humans. For each category of virus, we observed that a small minipool of crRNAs was able to target the majority of viruses (Figures 1H and S1B). This analysis suggests that beyond human-infectious viruses, PAC-MAN may be used as a BSA strategy in animals, a field that is underdeveloped in the biotechnology sector.
Determinants required of crRNAs targeting a broad spectrum of RNA virus families
We further analyzed whether families with more virus strains required more minipool crRNAs to achieve comprehensive targeting. For the 10 RNA virus families analyzed, we observed a linear relationship between the number of human-infectious strains and the number of crRNAs in the minipool targeting these strains (R = 0.70, p = 0.023; Figure 2A). When we examined all of the strains in each family and the corresponding minipools targeting these strains, we observed a similar linear relationship (R = 0.75. p = 0.012; Figure 2B). This suggests that, as expected, the number of minipool crRNAs required to comprehensively target all of the strains in an RNA virus family is strongly influenced by the number of strains in that family.
Figure 2.

Determinants of required crRNAs targeting broad-spectrum RNA virus families
(A) Correlation between the number of human-infectious strains in each RNA virus family and the number of crRNAs needed for targeting those strains.
(B) Correlation between the number of strains in each RNA virus family and the number of crRNAs needed for targeting all of the strains in that family.
(C) The number of crRNAs needed to target all of the sequenced human-infectious strains in each of 6 RNA virus families with segmented genomes.
(D) The number of crRNAs needed to target 90% of sequenced human-infectious strains in each of 6 RNA virus families with segmented genomes.
(E) Correlation between the number of strains in the RNA virus family and the number of crRNAs needed for targeting all human-infectious viruses in 6 segmented RNA virus families.
The observation that IAV requires more crRNAs for coverage compared to the other 10 families and HIV inspired us to analyze more RNA virus families with segmented genomes. To do this, we included 6 segmented RNA virus families, including 5 negative-sense ssRNA virus families and 1 double-stranded RNA (dsRNA) virus family. Interestingly, we observed that segmented RNA virus families generally require larger minipools to comprehensively target all of the human-infectious strains in that family ( 10-57 crRNAs for full coverage and 3-7 crRNAs for 90% coverage; Figures 2C and 2D). This is likely due to the reassortment process, a shared feature in segmented RNA viruses that have the capacity to exchange genome segments during co-infection.16 Reassortment may greatly increase the diversity of segmented viral genome sequences, making it more challenging to define very few crRNAs to broadly cover strains. Despite the nature of these segmented genomes, we still observed that the number of required minipool crRNAs for comprehensive coverage of human-infectious strains is strongly correlated with the number of human-infectious strains in that family (R = 0.973 , p = 0.001 ; Figure 2E). Overall, our data suggest that PAC-MAN is most effective against non-segmented RNA virus families for broad-spectrum targeting.
An online resource to use CRISPR-Cas13d for BSA targeting
We built an online resource (http://crispr-pacman.stanford.edu) to facilitate the dissemination of analyzed Cas13d crRNAs for 16 RNA virus families, as well as for HIV, IAV, and influenza B virus (IBV). For species-specific crRNAs in each family, we defined a library of crRNAs with predicted high specificity (≥3 mismatches in the human transcriptome and no target in other species in the same family) and high efficiency (score ≥0.5). We provided an interactive tool to facilitate the choice of crRNAs targeting specific genes on a virus. We also implemented a graphical user interface (GUI) for SARS-CoV-2, which integrates with the University of California, Santa Cruz (UCSC) Genome Browser17 and creates a visualization for the mapping of crRNAs to sequence position along the annotated viral genome. An example is provided to visualize where predicted crRNAs target the SARS-CoV-2 genome (Figures 3A–3C). For family-covering crRNAs, we included a phylogenetic tree generated using the Interactive Tree Of Life (iTOL) tool to visualize the evolutionary relationship of the strains targeted by each crRNA.18 We annotated each viral strain in the phylogenetic tree with the genus that the strain belongs to, whether the strain is a human host, and which (if any) of the first 5 crRNAs in the family-covering minipool targets the strain. We provided an example of this phylogenetic tree visualizing family-covering crRNAs for the Filoviridae family, a viral family that includes the pandemic Ebola virus (Figure 3D). We created a video to provide an overview of this online resource (Video S1).
Figure 3.

CRIPSR-PACMAN as an online resource of using CRISPR-Cas13d for broad-spectrum antiviral targeting
(A–C) An exemplar diagram showing the crRNAs designed to target SARS-CoV-2 and its alignment to the genes of SARS-CoV-2. These crRNAs are used for testing in Figure 4. The intensity of colors for each crRNA represents its predicted crRNA score.
(D) An exemplar diagram showing the analysis of family-covering crRNAs targeting the Filoviridae family, including Ebola virus.
See also Table S2.
Validation of predicted crRNAs for targeting SARS-CoV-2 sequences
We next experimentally validated whether the predicted species-specific crRNAs enabled effective Cas13d-mediated inhibition of the SARS-CoV-2 sequence. We used our recently reported SARS-CoV-2 reporters, F1 and F2, which encode a GFP gene fused to SARS-CoV-2 sequences encoding RNA-dependent RNA polymerase (RdRP), which maintains virus proliferation, or nucleocapsid (N), which controls viral packaging19 (Figure S2A). The Cas13d and crRNA were expressed using 2 plasmids, as reported before (Figure S2B). To measure the inhibitory effects of crRNAs, we co-transfected individual crRNA plasmids and SARS-CoV-2 reporter plasmids into A549 lung epithelial cells that expressed Cas13d, and quantified GFP expression using flow cytometry and target RNA abundance using quantitative PCR (qPCR) 48 h post transfection (Figure S2C).
We designed a list of crRNAs targeting RdRP or N without efficiency prediction (Figures 3B and 3C; Table S2). We observed varying repressive effects on the SARS-CoV-2 reporters of individual crRNAs using flow cytometry (Figure S3A; Table S3). Interestingly, there was a statistically significant high correlation between the percentage of GFP repression and the predicted efficiency scores, which were obtained by using a recently reported algorithm14 (R = 0.834, p = 1.667 × 10−5; Figure S3B). These results were consistent with the repressive effects on RNA abundance as measured by qPCR (R = 0.812, p = 4.302 × 10−5; Figures S3C and S3D; Table S4). These data together suggest that the reported algorithm was able to faithfully predict efficient crRNAs for viral genome targeting, despite it being developed based on data from crRNAs targeting human transcripts.
To further validate the performance of the predicted crRNAs, we tested more predicted crRNAs targeting the SARS-CoV-2 sequence with varying efficiency scores (Figures 3B and 3C). Again, we observed a strong correlation between the predicted efficiency and the measured reporter repression (R = 0.875, p = 0.0009; Figures S3E and S3F). We noticed a binary trend: most crRNAs with an efficiency score >0.5 showed efficient repression, whereas crRNAs with an efficiency score <0.5 showed almost no repression. This justified our choice of 0.5 as the threshold to determine efficient crRNAs. These independent experiments confirmed the efficacy of the predicted crRNAs using the algorithm, which also suggests that our computational pipeline can be used to suggest crRNAs that will efficiently inhibit other RNA virus sequences.
Optimized crRNAs targeting SARS-CoV-2 sequences using CRISPR-PAC-MAN
Finally, we defined a minipool of 5 highly efficient crRNAs (crRNA-MP5) targeting sequenced SARS-CoV-2 genomes. Using SARS-CoV-2 reporters, we confirmed that individual crRNAs could repress GFP up to 90% (Figure 4A), with an 85% repression of RNA abundance (Figure 4B). Pooling these crRNAs further enhanced repression up to 95% for GFP expression and 93% for RNA abundance. We further retrieved sequences of 351,670 SARS-CoV-2 isolates from the Global Initiative on Sharing All Influenza Data (GISAID)20 database (as of January 16, 2021). Each crRNA in crRNA-MP5 was able to target almost 99% of SARS-CoV-2 isolates, and the crRNA-MP5 together can target all of the known SARS-CoV-2 strains regardless of the variations in different SARS-CoV-2 clades as identified in Nextstrain21 (Figure 4C). Three new SARS-CoV-2 variants, including the UK variant (B.1.1.7),22 the South Africa variant (B.1.351),23 and the Brazil variant (P.1)24 have emerged since the autumn of 2020. Although multiple mutations have been detected in these new SARS-CoV-2 variants, most reside in the Spike gene and fewer are in the regions targeted by our chosen crRNAs (e.g., RdRP, N). Each crRNA in crRNA-MP5 could still target ∼99% of isolates from each variant (Figure 4D), which were downloaded from GISAID on February 3, 2021. Of note, the coverage of each crRNA-MP5 for the Brazil strain was 100%. The crRNA-MP5 together can target all of the sequenced strains in these 3 variants (Figure 4E). In summary, the optimized minipool of crRNA-MP5 provides a useful resource to be tested against live SARS-CoV-2 virus in vitro and in vivo in the future.
Figure 4.

Optimized crRNAs targeting SARS-CoV-2 sequences using CRISPR-PAC-MAN
(A and B) Repression effects of the optimized minipool of crRNAs (crRNA-MP5) on SARS-CoV-2 GFP reporter (A) and RNA abundance (B).
(A) GFP expression as measured by flow cytometry. p values for each group are included in Table S3. Lines indicate means; error bars indicate SDs (n = 3 independent biological replicates).
(B) mRNA abundance as measured by quantitative real-time PCR. Relative RNA abundance is calculated by normalizing to the reporter-only sample. p values for each group are included in Table S3. Lines indicate means; error bars indicate SDs. The data represent 3 independent biological experiments performed in technical replicates (n = 9).
(C) Bar graphs showing the percentage of genomes of SARS-CoV-2 isolates targeted by individual or multiple crRNAs in the optimized crRNA minipool.
(D and E) Bar graphs showing the percentage of genomes of 3 new SARS-CoV-2 variants targeted by individual (D) or multiple crRNAs (E) in the optimized crRNA minipool.
See also Figures S2 and S3 and Tables S3 and S4.
Discussion
The rapid development of CRISPR-Cas13-based antivirals has raised the question of whether the system can be effectively used to broadly target many families of RNA viruses. Here, we perform a comprehensive in silico analysis using CRISPR-Cas13 to target notable pathogenic viral species and 16 families of human- or animal-infectious RNA viruses in the ViPR database. Our integrated crRNA analysis pipeline incorporates existing algorithms and computes the coverage, efficiency, and specificity of crRNAs for RNA virus targeting. We observed that 10 families of human-infectious viruses can be flexibly targeted by a minipool of 14 crRNAs with >90% coverage. We determined that strain count, diversity, and genome segmentation are potential determinant factors for the number of crRNAs required to comprehensively target a family of viruses. We also observed that, similar to human-infectious viruses, PAC-MAN may also provide a BSA strategy for viruses that infect domestic livestock, thus expanding its potential applications. Importantly, the crRNAs designed here can target all sequenced new SARS-CoV-2 strains to date. Given the rapid development of CRISPR-Cas13 as antivirals against SARS-CoV-2 and other RNA viruses,25 this work provides a resource to the community for using CRISPR-Cas13 PAC-MAN for broad-spectrum targeting with potential use in the development of antivirals.
One major advantage of the PAC-MAN system is its potential as a BSA due to easy multiplexed targeting ability of the CRISPR-Cas13 system. The Cas13 system can use a crRNA minipool or process multiple crRNAs from a crRNA array to target different genes or strains simultaneously. One study demonstrated that 4 crRNAs can be simultaneously delivered in a simple arrayed vector and efficiently knock down 4 target transcripts.26 Using more crRNAs to target many viruses simultaneously will likely require several technical issues to be addressed, including the efficient expression of long crRNA arrays and efficient Cas13d processing of the crRNA arrays. For example, higher expression of the Cas13d protein may help each crRNA pair with Cas13d, thus mitigating the dilution of crRNAs and increasing the knockdown efficiency.2
We further provided an online resource to disseminate specific, efficient, broad-coverage crRNAs targeting RNA viruses. We built a website (http://crispr-pacman.stanford.edu/) with a useful graphical user interface and interactive functionality to facilitate easy access to CRISPR PAC-MAN crRNAs for the broad research community. We experimentally tested different sets of predicted crRNAs targeting the SARS-CoV-2 genomes and found a strong correlation between predicted and validated efficiency. The target site nucleotide context and the secondary structure of mRNA are the major factors determining crRNA targeting efficiency.14 The mRNAs produced from the SARS-CoV-2 reporter plasmid and RNAs from live SARS-CoV-2 virus have the same sequence and likely similar local secondary structure. Although additional experiments examining the inhibitory efficiency of crRNAs in vivo with live viruses are required to confirm this strong correlation, the observed high efficiency of these guides in our study provides a proof-of-concept of the utility of the designed crRNAs against diverse RNA viruses. In summary, this work provides a bioinformatic tool and an online resource for using CRISPR-Cas13-mediated PAC-MAN for BSA targeting with potential future applications in the development of antiviral therapies.
Limitations of study
Although CRISPR-PAC-MAN provides a potential antiviral strategy to combat RNA viruses, there are several challenges that need to be addressed before it enters practical clinical use. For BSA targeting, it remains unknown whether the current CRISPR-Cas13d system can effectively handle the scale of 14 crRNAs simultaneously for the efficient knockdown of each target. Further technical investigation and optimization are needed. Furthermore, delivery is key to successful applications of PAC-MAN in the human body. Conceptually, a sufficiently high delivery threshold is required for efficient use of PAC-MAN against viral infection. Toward this goal, various strategies are being investigated. These strategies include the delivery of mRNA encoding Cas13d and crRNAs using liposomes or lipid nanoparticles27, 28, 29, 30, 31 or delivery of the ribonucleoprotein (RNP) complex containing the Cas13d protein and crRNA to airway epithelia.32, 33, 34 Cas13d and crRNAs can also be delivered using adenovirus or adeno-associated virus (AAV).35, 36, 37 Such delivery modalities could be administered to populations via a nebulizer system or nasal spray, which may present a convenient and cost-effective form of antivirals in pandemics. In the present study, we used A549 human lung epithelial carcinoma cells to validate the targeting efficiency of SARS-CoV-2 sequences by predicted crRNAs (Figure S3). A549 cells have been engineered to stably express Cas13d, and crRNAs were introduced via lentiviral transduction to quantify knockdown efficiency. In future studies, the use of primary cells or an air-liquid interface (ALI) culture system38 will better mimic the tracheobronchial epithelium of the human respiratory tract to study the inhibition in live SARS-CoV-2 virus infection.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Stellar chemically competent cells | TaKaRa | Cat# 636766 |
| Chemicals, peptides, and recombinant proteins | ||
| Puromycin | Gold Biotech | Cat# P-600-1 |
| Fetal Bovine Serum (FBS) | Clontech | Cat# 631367 |
| Trypsin-EDTA solution | Sigma | Cat# T4049-100ML |
| DMEM | ThermoFisher Scientific | Cat# 10569-044 |
| Mirus TranslT-LT1 | Mirus Bio | Cat# MIR 2306 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| A549 | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| A549 + Cas13d (pSLQ5428) | Abbott et al.2 | N/A |
| Oligonucleotides | ||
| Primer used in qPCR: SARS-COV-F1_F: 5′-AACGGGTTTGCGGTGTAAGT-3′ | Abbott et al.2 | N/A |
| Primer used in qPCR: SARS-COV-F1_R: 5′-AA TTTAGCAAAACCAGCTACTTTATCATTGTAG-3′ | Abbott et al.2 | N/A |
| Primer used in qPCR: SARS-COV-F2_F: 5′-CAGGTGGAACCTCATCAGGAG-3′ | Abbott et al.2 | N/A |
| Primer used in qPCR: SARS-COV-F2_R: 5′-GTT ACCATCAGTAGATAAAAGTGCATTAACATTG-3′ | Abbott et al.2 | N/A |
| Primer used in qPCR: GAPDH_F: 5′-TGCACCACCAACTGCTTAGC-3′ | Abbott et al.2 | N/A |
| Primer used in qPCR: GAPDH_R: 5’-GGCATGGACTGTGGTCATGAG-3′ |
Abbott et al.2 | N/A |
| Recombinant DNA | ||
| pHR-PGK-sfGFP-SARS-COV-F1 | Abbott et al.2 | N/A |
| pHR-PGK-sfGFP-SARS-COV-F2 | Abbott et al.2 | N/A |
| pSLQ5428_pHR_EF1a-mCherry-P2A-Rfx_ Cas13d-2xNLS-3xFLAG | Abbott et al.2 | N/A |
| ∗pSLQ5429_pUC_hU6-crScaffold_EF1a-BFP | Abbott et al.2 | N/A |
| ∗pSLQ5465_pHR_hU6-crScaffold_EF1a-PuroRT2A-BFP | Abbott et al.2 | N/A |
| ∗Denotes generic backbone plasmid for crRNAs. Specific target sequences for crRNAs can be found in Table S2. | Abbott et al.2 | N/A |
| Software and algorithms | ||
| Cytobank | Beckman Coulter | N/A |
| FlowJo v10 | https://www.flowjo.com/ | N/A |
| Geneious 2020.0.4 | https://www.geneious.com | N/A |
| MAFFT | Nakamura et al.39 | https://mafft.cbrc.jp/alignment/software/ |
| Bowtie | Langmead et al.40 | http://bowtie-bio.sourceforge.net/index.shtml |
| Bowtie 2 | Langmead et al.41 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| iTOL v5.5 | Letunic and Bork18 | https://itol.embl.de/ |
| Cas13 Guide RNA Design | Wessels et al.14 | https://gitlab.com/sanjanalab/cas13 |
| CRISPR-PACMAN crRNA design tool | This paper | https://github.com/QilabGitHub/CRISPR-PACMAN-pool-generation |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lei S. Qi (stanley.qi@stanford.edu).
Materials availability
All materials and reagents will be made available upon installment of a material transfer agreement (MTA). All plasmids will be available from Addgene (https://www.addgene.org/Stanley_Qi/).
Data and code availability
The codes for CRISPR-PACMAN computational pipeline are deposited in GitHub (https://github.com/QilabGitHub/CRISPR-PACMAN-pool-generation). Additional Supplemental Items are available from Mendeley Data at https://dx.doi.org/10.17632/bgxfwsg3sb.1
Experimental model and subject details
Cell culture
All cell lines were incubated at 37°C and at 5% CO2. Human embryonic kidney (HEK293T) cells and A549 cells (ATCC CCL-185) were cultured in 10% fetal bovine serum (Thermo Fisher Scientific) in DMEM (Thermo Fisher Scientific).
Method details
A general pipeline for crRNA analysis
All RNA virus genomes were retrieved from public databases, including 16 RNA virus families from the Virus Pathogen Database and Analysis Resource (ViPR), human immunodeficiency virus (HIV) from NCBI, and IAV from the Influenza Research Database (IRD) (Table S1).
The viral genomes are aligned by Multiple Alignment using Fast Fourier Transform39 (MAFFT) and a 22-nucleotide (nt) sliding window is passed over the alignment. In each window, if there is a 22-nt sequence with no gap characters that appears in > 50% of genomes then that sequence is added to the crRNA set. In order to eliminate crRNAs with off-targets in the human genome, Bowtie40 is used to identify crRNAs that align to the human transcriptome with ≤ 2 mismatches; these crRNAs as well as crRNAs with the poly-U (> = 4U’s) sequence that may prevent crRNA expression are removed from the guide set. All crRNAs are assigned a predicted efficiency score based on Wessels et al.14 Cas13 guide design method as described in the paragraph below.
To assign each crRNA with a predicted efficiency score, we utilized a recently published computational tool that generates Cas13 crRNAs and predicted efficiency scores given a single input sequence.14 First, for each viral group we choose a representative sequence, which is a NCBI reference sequence if one exists in the viral group, or a random sequence from the viral group. We use the Cas13 Guide Design Tool14 to generate crRNAs against this reference sequence. Because all CRISPR-PACMAN crRNAs are 22nt, in contrast to the 23nt crRNAs generated by the tool, we assign each crRNA the average score of all output crRNAs of which it is a substring.
Species-specific crRNAs refer to those crRNAs that target conserved regions in a group of closely related viral genomes. The viruses in a group can be members of the same viral genus (e.g., betacoronaviruses) and pathogenic species (e.g., SARS-CoV-2 patient isolates). In species-specific crRNAs, we also identify and label crRNAs which target other viral genus or other pathogenic species in the same family.
The minipool of family-covering crRNAs is determined through an iterative process. The crRNAs are ranked by the number of unique genomes they appear in and the crRNA that targets the most genomes is added to the list. The process is repeated, and the crRNAs are ranked by the number of unique genomes that are not already targeted. If multiple crRNAs are tied in a step, the crRNA with the fewest 3-mer repeats (e.g., “AAA” or “GGG”) is selected. The process ends when every virus strain in the family is targeted by at least one crRNA in the list. Note that the ordering of crRNAs in the list is significant; necessarily, the first crRNA in the list will target the largest number of virus strains.
Format of dataset in CRISPR-PAC-MAN server
The downloadable file format of crRNAs is a TSV file, in which each crRNA is annotated with its genomic sequence, spacer DNA for synthesis, % GC content, and additional features. The TSV file for the species-specific crRNAs has an additional column, “Specific to group,” to demonstrate whether the guide is specific to the group (i.e., no alignments with zero mismatches to any sequence in other groups in the same viral family, as determined by Bowtie241). Minipool crRNAs are annotated by the cumulative percentage of sequences in the virus family that can be targeted by this crRNA and preceding crRNAs in the list. The cumulative percentage of sequences targeted by the last crRNA in the list will be 100%. Users can click on coding sequences of interest to selectively download guides targeting those regions. We include a link to the iTOL website for each phylogenetic tree to enable users to interact with the tree. A tutorial video is available at the permanent link: https://www.youtube.com/watch?v=S2sPLE7i_6k.
SARS-CoV-2 targeting in CRISPR-PACMAN
A focused webpage was developed for users to access crRNAs targeting SARS-CoV-2 and the coronavirus family (Figures 3A–3C; http://crispr-pacman.stanford.edu/sarscov2). This webpage hosts: 1) species-specific crRNAs targeting conserved regions of SARS-CoV-2, 2) a coronavirus-covering crRNA minipool that collectively target all sequenced coronaviruses, and 3) custom crRNAs that can target both SARS-CoV-2 and another virus species, such as SARS-Cov or MERS-CoV. Because new sequences of SARS-CoV-2 patient isolates are frequently released, these crRNA sets are frequently updated. This page also highlights experimental validation data for SARS-CoV-2 crRNAs.
Our previous study developed a set of crRNAs to target highly conserved regions among the SARS-CoV-2, SARS-CoV and MERS-CoV genomes.2 These crRNAs are featured as a track on the UCSC Genome Browser,17 which is linked to the SARS-CoV-2 resource page. We characterized each crRNA on the UCSC Genome Browser with relevant features, including predicted efficiency score, reported off-target loci, percentage of SARS-CoV-2 targeted, and percentage of coronaviruses targeted.
Design and cloning of RdRP- and N-targeting crRNAs
The crRNA plasmids were cloned using standard restriction-ligation cloning. Forward and reverse oligos for each spacer (IDT) were annealed and inserted into the backbone using T4 DNA Ligase (NEB). Assembled oligos were either inserted into a pHR backbone, obtained and modified from Addgene (#121514 and #109054, respectively). Spacer sequences for all crRNAs can be found in Table S2.
Design and cloning of SARS-CoV-2 reporters
To clone SARS-CoV-2 reporters, pHR-PGK-scFv GCN4-sfGFP (pSLQ1711) was digested with EcoR1 and Sbf1 and gel purified. The PGK promoter and sfGFP were PCR amplified from pSLQ1711 and gel purified. The SARS-CoV-2-F1 and SARS-CoV-2-F2 fragments were synthesized from Integrated DNA Technologies (IDT). At last, the PGK, sfGFP, and SARS-CoV-2-F1/F2 fragments were inserted into the linearized pSLQ1711 vector using In-Fusion cloning (Takara Bio).
Lentiviral packaging and cell line generation
On day 1, HEK293T cells were seeded into 100mm dishes (Corning). On day 2, cells were ∼50%–70% confluent at the time of transfection. For each dish, 6 μg of pHR vector containing the construct of interest, 4 μg of dR8.91 and 1 μg of pMD2.G (Addgene) were mixed in 1 mL of Opti-MEM reduced serum media (GIBCO) with 30 μL of Mirus TransIT-LT1 reagent and incubated at room temperature for 30 minutes. The transfection complex solution was distributed evenly to HEK293T cultures dropwise. On day 5, lentiviruses are harvested from the supernatant with a sterile syringe and filtered through a 0.22-μm polyvinylidene fluoride filter (Millipore).
For A549 cells, lentivirus precipitation solution (Alstem) was added and precipitated as per the manufacturer’s protocol. One well of a 6-well plate (Corning) of A549 cells at ∼50% confluency was transduced with the precipitated lentivirus. After 2-3 days of growth, the cell supernatant containing the virus was removed and the cells were expanded. Cells were then sorted for mCherry+ cells using a Sony SH800S cell sorter.
SARS-CoV-2 reporter challenge experiments
On day 1, A549 cells stably expressing Cas13d (hereafter referred to as Cas13d A549) were seeded at a density of 80,000 cells per well in a 12-well plate. On day 2, Cas13d A549 cells were transfected with crRNAs targeting the two SARS-CoV-2 reporters or a non-targeting crRNA along with the respective SARS-CoV-2 reporters in an equimolar ratio using Mirus LT1 reagent (100 μL Opti-MEM reduced serum media, 0.5 μg crRNA, 1 μg SARS-CoV-2 reporter, 4 μL Mirus LT1 per well). Wild-type cells were transfected with either the crRNA or SARS-CoV-2 reporter plasmids as compensation controls. GFP expression was measured on day 4 by flow cytometry on a Beckman-Coulter CytoFLEX S instrument. Data was analyzed using FlowJo v10. All p values are available in Table S3.
Quantitative RT-PCR (qRT-PCR)
Real-time qRT-PCR was performed to quantify RNA abundance. For each sample, total RNA was isolated by using the RNeasy Plus Mini Kit (QIAGEN Cat# 74134), followed by cDNA synthesis using the iScript cDNA Synthesis Kit (BioRad, Cat# 1708890). qRT-PCR primers were ordered from Integrated DNA Technologies (IDT). Quantitative PCR was performed using the PrimePCR assay with the SYBR Green Master Mix (BioRad) and run on a Biorad CFX384 real-time system (C1000 Touch Thermal Cycler), according to manufacturers’ instructions. Cq values were used to quantify RNA abundance. The relative abundance of the SARS-CoV-2 fragments was normalized to a GAPDH internal control. Primers used in qPCR are available in Table S4.
Quantification and statistical analysis
Flow cytometry
For quantification of repression efficiency by flow cytometry (Figure 4A), values represent mean of fluorescence signals in GFP positive cells. Statistical analyses were performed using a two-sided t test with unequal variance in Excel to calculate p values (Table S3).
Quantitative RT-PCR (qRT-PCR)
For quantification of RNA abundance by qRT-PCR (Figures 4B and S3C), the relative RNA expression of the SARS-CoV-2 fragments was normalized to a GAPDH internal control. To calculate the relative mRNA abundance, the relative RNA expression of each treatment was normalized by setting the average value in reporter only samples as 1. Statistical analyses were performed using a two-sided t test with unequal variance in Excel to calculate p values (Table S3).
Acknowledgments
The authors thank all of the members from the Stanley Qi lab for useful comments and help on experiments and manuscript preparation. The authors thank David Lewis for helpful insights. The authors thank the researchers who generated and contributed the SARS-CoV-2 sequence data to GISAID (https://www.gisaid.org/). The project is supported by a contract grant from the Defense Advanced Research Projects Agency (DARPA; grant no. HR001119C0060), a COVID-19 Research and Assistance Fund from the School of Engineering at Stanford University, and gift funds from the Li Ka Shing Foundation and the Lucile Packard Foundation for Children’s Health.
Author contributions
X.L., Y.L., A.C., and L.S.Q. conceived the study. X.L., A.C., and M.L.R. designed and developed the platform. Y.L. designed and performed the experiments. T.P. prepared the tutorial video. X.L., A.C., Y.L., and L.S.Q. wrote the manuscript. All of the authors read and commented on the manuscript.
Declaration of interests
Provisional patents have been filed via Stanford University related to the work.
Published: March 23, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100245.
Supplemental information
References
- 1.De Clercq E., Li G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbott T.R., Dhamdhere G., Liu Y., Lin X., Goudy L., Zeng L., Chemparathy A., Chmura S., Heaton N.S., Debs R. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell. 2020;181:865–876.e12. doi: 10.1016/j.cell.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singsuksawat E., Onnome S., Posiri P., Suphatrakul A., Srisuk N., Nantachokchawapan R., Praneechit H., Sae-kow C., Chidpratum P., Hongeng S. Potent Programmable Antiviral Against Dengue Virus in Primary Human Cells by Cas13b Rnp with Short Spacer and Delivery by Virus-Like Particle. bioRxiv. 2020 doi: 10.1101/2020.05.17.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li H., Wang S., Dong X., Li Q., Li M., Li J., Guo Y., Jin X., Zhou Y., Song H., Kou Z. CRISPR-Cas13a Cleavage of Dengue Virus NS3 Gene Efficiently Inhibits Viral Replication. Mol. Ther. Nucleic Acids. 2020;19:1460–1469. doi: 10.1016/j.omtn.2020.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freije C.A., Myhrvold C., Boehm C.K., Lin A.E., Welch N.L., Carter A., Metsky H.C., Luo C.Y., Abudayyeh O.O., Gootenberg J.S. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell. 2019;76:826–837.e11. doi: 10.1016/j.molcel.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yin L., Zhao F., Sun H., Wang Z., Huang Y., Zhu W., Xu F., Mei S., Liu X., Zhang D. CRISPR-Cas13a Inhibits HIV-1 Infection. Mol. Ther. Nucleic Acids. 2020;21:147–155. doi: 10.1016/j.omtn.2020.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahas A., Aman R., Mahfouz M. CRISPR-Cas13d mediates robust RNA virus interference in plants. Genome Biol. 2019;20:263. doi: 10.1186/s13059-019-1881-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aman R., Mahas A., Butt H., Aljedaani F., Mahfouz M. Engineering RNA Virus Interference via the CRISPR/Cas13 Machinery in Arabidopsis. Viruses. 2018;10:732. doi: 10.3390/v10120732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bawage S.S., Tiwari P.M., Santangelo P.J. Synthetic mRNA expressed Cas13a mitigates RNA virus infections. bioRxiv. 2018 doi: 10.1101/370460. [DOI] [Google Scholar]
- 10.Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012;40:D593–D598. doi: 10.1093/nar/gkr859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sayers E.W., Barrett T., Benson D.A., Bolton E., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Federhen S. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2011;39:D38–D51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Squires R.B., Noronha J., Hunt V., García-Sastre A., Macken C., Baumgarth N., Suarez D., Pickett B.E., Zhang Y., Larsen C.N. Influenza research database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respir. Viruses. 2012;6:404–416. doi: 10.1111/j.1750-2659.2011.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wessels H.-H., Méndez-Mancilla A., Guo X., Legut M., Daniloski Z., Sanjana N.E. Massively parallel Cas13 screens reveal principles for guide RNA design. Nat. Biotechnol. 2020;38:722–727. doi: 10.1038/s41587-020-0456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGeoch D., Fellner P., Newton C. Influenza virus genome consists of eight distinct RNA species. Proc. Natl. Acad. Sci. USA. 1976;73:3045–3049. doi: 10.1073/pnas.73.9.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonald S.M., Nelson M.I., Turner P.E., Patton J.T. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat. Rev. Microbiol. 2016;14:448–460. doi: 10.1038/nrmicro.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fernandes J.D., Hinrichs A.S., Clawson H., Navarro Gonzalez J., Lee B.T., Nassar L.R., Raney B.J., Rosenbloom K.R., Nerli S., Rao A.A. The UCSC SARS-CoV-2 Genome Browser. Nat. Genet. 2020;52:991–998. doi: 10.1038/s41588-020-0700-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Letunic I., Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shu Y., McCauley J. GISAID: global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018;34:4121–4123. doi: 10.1093/bioinformatics/bty407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horby P., Huntley C., Davies N., Edmunds J., Ferguson N., Medley G., Semple C. 2021. NERVTAG note on B. 1.1. 7 severity.https://www.gov.uk/government/publications/nervtag-paper-on-covid-19-variant-of-concern-b117 [Google Scholar]
- 23.Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., du Plessis L., Pybus O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faria N.R., Claro I.M., Candido D., Moyses Franco L.A., Andrade P.S., Coletti T.M., Silva C.A.M., Sales F.C., Manuli E.R., Aguiar R.S., CADDE Genomic Network . 2021. Genomic characterisation of an emergent SARS-CoV-2 lineage in Manaus: preliminary findings.https://virological.org/t/genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-manaus-preliminary-findings/586 [Google Scholar]
- 25.Blanchard E.L., Vanover D., Bawage S.S., Tiwari P.M., Rotolo L., Beyersdorf J., Peck H.E., Bruno N.C., Hincapie R., Michel F. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol. 2021 doi: 10.1038/s41587-021-00822-w. Published online February 3, 2021. [DOI] [PubMed] [Google Scholar]
- 26.Konermann S., Lotfy P., Brideau N.J., Oki J., Shokhirev M.N., Hsu P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell. 2018;173:665–676.e14. doi: 10.1016/j.cell.2018.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang C.-Y., Uno T., Murphy J.E., Lee S., Hamer J.D., Escobedo J.A., Cohen F.E., Radhakrishnan R., Dwarki V., Zuckermann R.N. Lipitoids--novel cationic lipids for cellular delivery of plasmid DNA in vitro. Chem. Biol. 1998;5:345–354. doi: 10.1016/s1074-5521(98)90173-9. [DOI] [PubMed] [Google Scholar]
- 28.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKinlay C.J., Vargas J.R., Blake T.R., Hardy J.W., Kanada M., Contag C.H., Wender P.A., Waymouth R.M. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl. Acad. Sci. USA. 2017;114:E448–E456. doi: 10.1073/pnas.1614193114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sago C.D., Lokugamage M.P., Paunovska K., Vanover D.A., Monaco C.M., Shah N.N., Gamboa Castro M., Anderson S.E., Rudoltz T.G., Lando G.N. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. USA. 2018;115:E9944–E9952. doi: 10.1073/pnas.1811276115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Handumrongkul C., Ye A.L., Chmura S.A., Soroceanu L., Mack M., Ice R.J., Thistle R., Myers M., Ursu S.J., Liu Y. Durable multitransgene expression in vivo using systemic, nonviral DNA delivery. Sci. Adv. 2019;5:eaax0217. doi: 10.1126/sciadv.aax0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amirkhanov R.N., Stepanov G.A. Systems of delivery of CRISPR/Cas9 ribonucleoprotein complexes for genome editing. Russ. J. Bioorganic Chem. 2019;45:431–437. [Google Scholar]
- 33.Krishnamurthy S., Wohlford-Lenane C., Kandimalla S., Sartre G., Meyerholz D.K., Théberge V., Hallée S., Duperré A.M., Del’Guidice T., Lepetit-Stoffaes J.P. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019;10:4906. doi: 10.1038/s41467-019-12922-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu C.-F., Chen G.-J., Luo Y.-L., Zhang Y., Zhao G., Lu Z.D., Czarna A., Gu Z., Wang J. Rational designs of in vivo CRISPR-Cas delivery systems. Adv. Drug Deliv. Rev. 2021;168:3–29. doi: 10.1016/j.addr.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 35.Halbert C.L., Allen J.M., Miller A.D. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 2001;75:6615–6624. doi: 10.1128/JVI.75.14.6615-6624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santry L.A., Ingrao J.C., Yu D.L., de Jong J.G., van Lieshout L.P., Wood G.A., Wootton S.K. AAV vector distribution in the mouse respiratory tract following four different methods of administration. BMC Biotechnol. 2017;17:43. doi: 10.1186/s12896-017-0365-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Lieshout L.P., Domm J.M., Wootton S.K. AAV-Mediated Gene Delivery to the Lung. Methods Mol. Biol. 2019;1950:361–372. doi: 10.1007/978-1-4939-9139-6_21. [DOI] [PubMed] [Google Scholar]
- 38.Sungnak W., Huang N., Bécavin C., Berg M., Queen R., Litvinukova M., Talavera-López C., Maatz H., Reichart D., Sampaziotis F., HCA Lung Biological Network SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020;26:681–687. doi: 10.1038/s41591-020-0868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura T., Yamada K.D., Tomii K., Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34:2490–2492. doi: 10.1093/bioinformatics/bty121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The codes for CRISPR-PACMAN computational pipeline are deposited in GitHub (https://github.com/QilabGitHub/CRISPR-PACMAN-pool-generation). Additional Supplemental Items are available from Mendeley Data at https://dx.doi.org/10.17632/bgxfwsg3sb.1