

Abstract
The reactivity of the geminal frustrated Lewis pair (FLP) (F5C2)3SnCH2P(tBu)2 (1) was explored by reacting it with a variety of small molecules (PhOCN, PhNCS, PhCCH, tBuCCH, H3CC(O)CH=CH2, Ph[C(O)]2Ph, PhN=NPh and Me3SiCHN2), featuring polar or non‐polar multiple bonds and/or represent α,β‐unsaturated systems. While most adducts are formed readily, the binding of azobenzene requires UV‐induced photoisomerization, which results in the highly selective complexation of cis‐azobenzene. In the case of benzil, the reaction does not lead to the expected 1,2‐ or 1,4‐addition products, but to the non‐stereoselective (tBu)2PCH2‐transfer to a prochiral keto function of benzil. All adducts of 1 were characterised by means of multinuclear NMR spectroscopy, elemental analyses and X‐ray diffraction experiments.
Keywords: activation, fluoroalkyl groups, frustrated Lewis pairs, small molecules, tin
A highly selective complexation of cis‐azobenzene was found when reacting the isomeric compound with the intramolecular frustrated Lewis pair (F5C2)3SnCH2P(tBu)2 under UV‐induced photoisomerization conditions.

Introduction
With the discovery of metal‐free reversible heterolytic hydrogen splitting in 2006, Stephan et al. laid the foundation for frustrated Lewis pair (FLP) chemistry. [1] Just over a decade later, the concept of frustrated Lewis pairs has emerged as an important tool in main group chemistry for describing, predicting and exploring new pathways of reactions. [2] Whereas Lewis’ definition of acids and bases included the mutual neutralization of both by forming a stable adduct, [3] the FLP concept describes the inhibition of adduct formation by steric shielding of acid and base while maintaining their reactivity potential.[ 2a , 2b , 2c , 2d , 2e , 2f ] This can be used for the activation of various unreactive small molecules,[ 2a , 2b , 2c , 2d , 2e , 2f ] but also for capturing unstable species. [4] Recent results show that even classical Lewis acid‐base adducts can exhibit FLP reactivity. [5] Typically, inter‐ or intramolecular combinations of group 13 Lewis acids (B, Al) and group 15 Lewis bases (N, P) serve as active FLP systems.[ 2a , 2b , 2c , 2d , 2e , 2f , 6 ] However, the range of Lewis acidic functions is continuously being expanded to include group 14 (Si, [7] Ge,[ 7a , 8 ] Sn[ 8 , 9 ]) elements and rare‐earth [10] or transition metals. [11] Besides catalytic hydrogenation of unsaturated substrates [12] and conversion of CO2 and CO into chemical feedstock,[ 2a , 2b , 2c , 2d , 2e , 2f , 13 ] the activation of dinitrogen is a major aspect of current FLP research. [14] Due to the high N≡N bond stability in dinitrogen and the associated high global energy consumption while converting dinitrogen to ammonia, the investigation of N2 containing compounds is of interest in order to gain knowledge for a long‐term low‐energy alternative to the Haber–Bosch process. [14a] Thus, Stephan and Melen consider the hydroboration of diazomethane derivatives as a promising step towards metal‐free dinitrogen activation. [14]
Recent results from Stephan et al. show that azobenzene, featuring an N=N double bond, is also suitable for use in FLP chemistry. They reported on the phosphinoboration of azobenzene by Ph2PBcat (cat=catecholate), yielding the PhNNPh‐linked B/P‐FLP Ph2P(NPh)2Bcat. Such hydrazine‐backbone‐type FLPs were found to bind another equivalent of azobenzene via 1,2‐addition, yielding six‐membered heterocycles. [15]
Recently, we reported on the tetrel‐based FLP systems (F5C2)3ECH2P(tBu)2 (E=Si, Ge, Sn (1)). They differ significantly in their reactivities.[ 7a , 7b , 9a ] Of these, Sn/P‐FLP (F5C2)3SnCH2P(tBu)2 (1), is capable of reversibly binding CO2 and turned out to be the most versatile FLP. [9a] Consequently, we set out to extend the scope with regard to binding nitrogen containing small molecules.
Results and Discussion
Reacting the Sn/P‐FLP (F5C2)3SnCH2P(tBu)2 (1) with a mixture of cis‐ and trans‐azobenzene in an NMR test reaction led to nearly no conversion. This is probably due to the steric hindrance for the 1,2‐addition of the trans‐isomer, which is energetically favoured and more abundant under normal conditions. [16] To prove this hypothesis, we attempted to use the known trans‐ into cis‐conversion of azobenzenes via photoisomerization (Scheme 1) to produce the less hindered cis‐isomer. After a total of five hours of UV irradiation (λ=365 nm), a complete conversion of FLP 1 was observed in the 1H NMR spectrum (Figure 1). The immediate formation of the corresponding azobenzene adduct 2 was not only indicated by the disappearing signals of the starting materials, but also by the formation of eight characteristic adduct signals. Apart from the tert‐butyl and methylene proton signals at 0.84 and 1.70 ppm, six resonances for two different phenyl groups were observed in the range of 6.55 to 7.40 ppm. Unexpectedly, a longer irradiation period led to decomposition of excess FLP 1 and adduct 2.
Scheme 1.

UV‐induced photoisomerization of azobenzene and its reaction with FLP 1.
Figure 1.

1H NMR spectra of a mixture of 1 (•) and cis/trans‐azobenzene (+) in C6D6 (*) solution after different times (Δt) of UV irradiation (λ=365 nm). Initially, no formation of adduct 2 (▪) takes place, but it is complete after 5 h.
In a reaction on preparative scale, 1 was reacted with the isomeric mixture of azobenzene under UV irradiation (λ=365 nm) for 5 hours and afforded the corresponding PhNNPh adduct 2 in 53 % yield after work‐up. The molecular structure of 2 (Figure 2) in the crystal exhibits an envelope‐like five‐membered heterocycle with two exocyclic N−Ph units. Compared to the N−N bond length (trans: 1.189 Å, [17] cis: 1.251 Å [18] ) in azobenzene, the N−N bond in adduct 2 is significantly longer with 1.446(3) Å. The sum of angles at N(1) and N(2) are 358.6(6)° and 357.9(6)°, respectively, indicating them to be nearly planar. A sum of angles for the equatorial groups of 360.0(3)° and a τ Sn parameter of 0.78 (Table 1) indicate a slightly distorted trigonal bipyramidal coordination sphere at the tin atom. The latter is calculated by subtracting the two largest bond angles at Sn and dividing the result by 60°.
Figure 2.

Molecular structure of compound 2 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.785(3), P(1)−C(8) 1.879(3), P(1)−N(2) 1.668(3), Sn(1)−N(1) 2.199(2), Sn(1)−C(1) 2.242(3), Sn(1)−C(5) 2.360(3), Sn(1)−C(7) 2.164(3), N(1)−N(2) 1.446(3), C(16)−N(1) 1.403(4), C(22)−N(2) 1.432(4); P(1)‐C(7)‐Sn(1) 112.7(2), C(7)‐P(1)‐N(2) 103.8(1), P(1)‐N(2)‐N(1) 116.2(2), P(1)‐N(2)‐C(22) 126.8(2), C(22)‐N(2)‐N(1) 114.9(2), Sn(1)‐N(1)‐N(2) 115.3(2), C(16)‐N(1)‐N(2) 112.9(2), C(16)‐N(1)‐Sn(1) 130.4(2), N(1)‐Sn(1)‐C(1) 95.4(1), N(1)‐Sn(1)‐C(5) 168.5(1), N(1)‐Sn(1)‐C(7) 83.0(1).
Table 1.
Selected NMR and structural parameters of compounds 2–9.
|
Comp. |
δ(31P) [ppm][a] |
δ(119Sn) [ppm][a] |
2 J Sn,P [Hz][a] |
∢(Sn‐C‐P) [°] |
τ Sn [b] |
|---|---|---|---|---|---|
|
2 |
77.2 |
−348.5 |
70 |
112.7(2) |
0.78 |
|
3 |
58.9 |
−318.8 |
77 |
96.2(1) |
0.56 |
|
4 |
35.3 |
−273.2 |
68 |
110.3(3) |
0.77 |
|
5 |
31.5 |
−274.4 |
64 |
117.2(1) |
0.74 |
|
6 |
49.1 |
−268.5 |
77 |
111.4(1) |
0.79 |
|
7 |
60.5 |
−275.8 |
105 |
110.1(3) |
0.22 |
|
8 |
53.4 |
–[c] |
–[c] |
122.2(1) |
0.76 |
|
9 |
8.5 |
−397.4 |
– |
– |
– |
[a] In C6D6 at ambient temperature. [b] Calculated for pentacoordinated Sn according to Addison et al. [19] [c] Not detectable due to poor solubility.
τ Sn parameters close to 1 indicate a trigonal bipyramidal coordination sphere while those close to 0 indicate a square pyramidal one. [19] Compared to the equatorial Sn−C bonds (2.164(3)–2.257(3) Å) the axial Sn(1)−C(5) bond is longer (2.360(3) Å). The 31P and 119Sn NMR chemical shifts of 2 are 77.2 and −348.5 ppm (Table 1).
In contrast to azobenzene, diazomethane derivatives feature terminal N2 moieties; they have recently been discussed as a pre‐stage towards metal‐free dinitrogen activation. [14] Differences in their reaction behaviour are illustrated when 1 is reacted with trimethylsilyldiazomethane (Me3SiCHN2). In analogy to the related (F5C2)3SiCH2P(tBu)2,[ 7a , 7b ] conversion of FLP 1 with Me3SiCHN2 provides 1,1‐adduct 3 in quantitative yields. The molecular structure of 3 in the solid contains a four‐membered NSnCP‐heterocycle (Figure 3) involving only the terminal nitrogen atom of Me3SiCHN2. Thus, binding Me3SiCHN2 results in a narrowed angle Sn(1)‐C(7)‐P(1) of 96.2(1)° compared to 2 (112.7(1)°). The angles C(5)‐Sn(1)‐N(1) (161.6(1)°) and Ceq‐Sn(1)‐N(1) (69.6(1)°‐96.6(1)°) as well as a τ Sn parameter of 0.56 indicate a clearly distorted trigonal bipyramidal coordination sphere at the tin atom (Table 1). The N−N bond in 3 is slightly shorter than a typical N−N single bond and than that in 2; it is probably due to the hydrazine‐type structure, that is, one nitrogen atom bonded to a three‐coordinate carbon atom.
Figure 3.

Molecular structure of compound 3 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.780(2), P(1)−C(8) 1.852(2), P(1)−N(1) 1.653(2), Sn(1)−N(1) 2.251(2), Sn(1)−C(1) 2.243(2), Sn(1)−C(5) 2.294(2), Sn(1)−C(7) 2.196(2), N(1)−N(2) 1.393(2), C(16)−N(2) 1.279(2), C(16)−Si(1) 1.875(2); P(1)‐C(7)‐Sn(1) 96.2(1), C(7)‐P(1)‐N(1) 95.3(1), P(1)‐N(1)‐N(2) 115.4(1), P(1)‐N(1)‐Sn(1) 98.0(1), Sn(1)‐N(1)‐N(2) 146.6(1), N(1)‐Sn(1)‐C(1) 90.9(1), N(1)‐Sn(1)‐C(5) 161.6(1), N(1)‐Sn(1)‐C(7) 69.6(1).
In addition to the investigation of the N2‐functional azo compounds PhNNPh and Me3SiCHN2, the examination of C≡X triple bond systems (X=C, N) is of particular interest, as they are isosteric to dinitrogen, although of course more polar and easier to dissociate than N2. The reaction of 1 with nitrile derivatives, such as benzonitrile and pivalonitrile does neither result in adduct formation nor in R−CN⋅⋅⋅Sn coordination (R=Ph, tBu), as was concluded from the absence of changes in the NMR resonances of 1. In contrast, the reaction of 1 with phenyl cyanate PhOCN (R=OPh; Scheme 2) surprisingly proceeded under 1,2‐addition to the C−N bond affording adduct 4 in 44 % yield. Since crystals of 4 show a phase transition at 100 K, data for X‐ray diffraction were recorded at 200 K. The molecular structure of 4 (Figure 4) in the crystal displays a five‐membered heterocycle with a N(1)−C(16) double bond of 1.227(6) Å and an exocyclic C‐O‐Ph unit. The Sn(1)‐C(7)‐P(1) angle of 110.3(3)° matches the angle of the corresponding constitutional isomeric phenyl isocyanate adduct (110.7(1)° [9a] ). Compound 4 shows a slightly distorted trigonal bipyramidal coordination at the tin atom, indicated by a τ Sn parameter of 0.77 (Table 1).
Scheme 2.

Reactions of FLP 1 with selected substrates at ambient temperature.
Figure 4.

Molecular structure of compound 4 in the solid state. Ellipsoids are set at 30 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.772(5), P(1)−C(8) 1.862(6), P(1)−C(16) 1.869(5), Sn(1)−N(1) 2.155(4), Sn(1)−C(1) 2.233(7), Sn(1)−C(5) 2.313(8), Sn(1)−C(7) 2.202(5), O(1)−C(16) 1.375(6), N(1)−C(16) 1.227(7); P(1)‐C(7)‐Sn(1) 110.3(3), C(7)‐P(1)‐C(16) 101.4(2), O(1)‐C(16)‐P(1) 107.2(3), N(1)‐C(16)‐P(1) 124.0(4), N(1)‐C(16)‐O(1) 128.8(4), C(16)‐N(1)‐Sn(1) 117.0(3), N(1)‐Sn(1)‐C(1) 91.2(2), N(1)‐Sn(1)‐C(5) 171.7(2), N(1)‐Sn(1)‐C(7) 86.9(2).
Less surprisingly, no addition to the C−N but to the C−S bond was found, when reacting 1 with the heterocumulene homologue phenyl isothiocyanate PhNCS by forming adduct 5 in 79 % yield. The molecular structure of 5 reveals an envelope‐like CSSnCP‐heterocycle (Figure 5) and a C−S bond, which are well comparable to the related CS2 adduct, and agrees well with expectations from the HSAB concept.[ 9a , 20 ] Compared to the corresponding PhOCN adduct 4, compound 5 has a slightly widened Sn(1)‐C(7)‐P(1) angle of 117.2(1)°, close to that of the related CS2 adduct (118.4(1)° [9a] ). In addition, the molecular structure of 5 shows a slightly more distorted trigonal bipyramidally coordinate tin atom (τ Sn=0.74, Table 1). With a chemical shift of 164.3 ppm, the 13C NMR signal of the SC(NPh) unit is between those of the C=X moieties (X=S, NPh) in the corresponding CS2 and PhOCN adducts with 226.8 and 150.0 ppm, respectively. [9a]
Figure 5.

Molecular structure of compound 5 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.782(2), P(1)−C(8) 1.858(2), P(1)−C(16) 1.845(2), Sn(1)−S(1) 2.618(1), Sn(1)−C(1) 2.263(2), Sn(1)−C(5) 2.294(2), Sn(1)−C(7) 2.177(2), S(1)−C(16) 1.730(2), N(1)−C(16) 1.276(3); P(1)‐C(7)‐Sn(1) 117.2(1), C(7)‐P(1)‐C(16) 108.5(1), S(1)‐C(16)‐P(1) 120.7(1), N(1)‐C(16)‐P(1) 111.2(2), N(1)‐C(16)‐S(1) 128.0(2), C(16)‐S(1)‐Sn(1) 101.8(1), S(1)‐Sn(1)‐C(1) 87.1(1), S(1)‐Sn(1)‐C(5) 173.8(1), S(1)‐Sn(1)‐C(7) 86.4(1).
In addition, selective addition to less polar triple bond systems was observed when reacting 1 with terminal alkynes of the type HC≡CR (R=Ph, tBu; Scheme 2). In both cases, five‐membered SnCPCC heterocycles, with the incorporated C−R unit (R=Ph, tBu) each being bound by the phosphorus atom (Figure 6/Figure 7), were obtained as adducts. This proves that FLP 1 is capable of reacting with substrates of relatively low polarity. X‐ray diffraction analysis provided nearly similar values for the Sn(1)‐C(7)‐P(1) bond angles (111.4(1)°/ 110.1(3)°) and the C(16)−C(17) bond lengths (1.339(4) Å/1.336(7) Å) of both. These C−C bond lengths are typical lengths of C=C double bonds. However, a significant structural difference is observed in the coordination geometry of the tin atom. While a τ Sn parameter of 0.79 indicates a slightly distorted trigonal bipyramidal coordination sphere in HCCPh adduct 6, a τ Sn parameter of 0.22 reveals a square pyramidal one in HCCtBu adduct 7 (Table 1).
Figure 6.
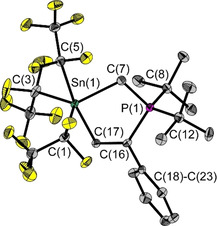
Molecular structure of compound 6 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.779(3), P(1)−C(8) 1.861(3), P(1)−C(12) 1.865(3), P(1)−C(16) 1.833(2), Sn(1)−C(1) 2.256(3), Sn(1)−C(3) 2.226(5), Sn(1)−C(5) 2.346(3), Sn(1)−C(7) 2.198(3), Sn(1)−C(17) 2.207(3), C(16)−C(17) 1.339(4); P(1)–C(7)‐Sn(1) 111.4(1), C(7)‐P(1)‐C(16) 105.8(1), C(17)‐C(16)‐P(1) 116.4(2), C(17)‐C(16)‐C(18) 121.1(2), C(18)‐C(16)‐P(1) 122.4(2), C(16)‐C(17)‐Sn(1) 120.7(2), C(17)‐Sn(1)‐C(1) 90.7(1), C(17)‐Sn(1)‐C(3) 97.7(1), C(17)‐Sn(1)‐C(5) 170.2(1), C(17)‐Sn(1)‐C(7) 84.5(1).
Figure 7.

Molecular structure of compound 7 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.776(5), P(1)−C(8) 1.884(5), P(1)−C(12) 1.871(5), P(1)−C(16) 1.859(5), Sn(1)−C(1) 2.305(6), Sn(1)−C(3) 2.234(5), Sn(1)−C(5) 2.307(5), Sn(1)−C(7) 2.226(5), Sn(1)−C(17) 2.163(5), C(16)−C(17) 1.336(7); P(1)‐C(7)‐Sn(1) 110.1(3), C(7)‐P(1)‐C(16) 104.3(2), C(17)‐C(16)‐P(1) 112.3(4), C(17)‐C(16)‐C(18) 117.9(5), C(18)‐C(16)‐P(1) 129.7(4), C(16)‐C(17)‐Sn(1) 125.1(4), C(17)‐Sn(1)‐C(1) 90.2(2), C(17)‐Sn(1)‐C(3) 104.1(2), C(17)‐Sn(1)‐C(5) 149.2(2), C(17)‐Sn(1)‐C(7) 82.3(2).
Furthermore, in order to investigate whether adduct formation proceeds via 1,2‐ or 1,4‐addition when reacting 1 with α,β‐unsaturated carbonyl compounds, 1 was reacted with methyl vinyl ketone and benzil. In the case of conversion with methyl vinyl ketone the corresponding 1,4‐addition product was obtained in 77 % yield in the absence of light (in order to prevent methyl vinyl ketone from light‐induced polymerization). The molecular structure of 8 (Figure 8) reveals a seven‐membered heterocycle with exocyclic methyl group, comparable to those described by Uhl and co‐workers. [21] They assume that the favoured 1,4‐adduct formation of their geminal Al/P FLP with acrolein derivatives is due to the initiating Al⋅⋅⋅O‐interaction under activation of the Cβ atom of the vinyl‐carbonyl moiety. [21] Compared to the previously described four‐ and five‐membered ring systems 2–7, the Sn(1)‐C(1)‐P(1) angle in 8 is significantly expanded with 122.2(1)°.
Figure 8.

Molecular structure of compound 8 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(1) 1.801(2), P(1)−C(2) 1.820(2), P(1)−C(12) 1.866(2), P(1)−C(16) 1.878(2), Sn(1)−C(1) 2.185(2), Sn(1)−C(6) 2.331(2), Sn(1)−C(8) 2.248(2), Sn(1)−C(10) 2.252(2), Sn(1)−O(1) 2.086(2), O(1)−C(4) 1.345(2), C(3)−C(4) 1.347(3); P(1)‐C(1)‐Sn(1) 122.2(1), C(1)‐P(1)‐C(2) 107.8(1), O(1)‐Sn(1)‐C(1) 96.0(1), O(1)‐Sn(1)‐C(6) 173.6(1), O(1)‐Sn(1)‐C(8) 80.6(1), O(1)‐Sn(1)‐C(10) 87.6(1).
In contrast to that, the reaction of FLP 1 with benzil, PhC(O)C(O)Ph, did neither lead to the expected 1,2‐ nor 1,4‐addition product, but to the formation of the racemic compound 9. Racemate 9 crystallizes as a two‐component perfect inversion twin with two different molecules in the asymmetric unit. The molecular structure of 9 (Figure 9) reveals a non‐stereoselective transfer of the (tBu)2PCH2 unit to one prochiral keto function of the prior benzil. This results in coordination of the tin atom through the formed alcoholate and the intact keto function. While the Sn(1)−O(2) and Sn(2)−O(4) bond lengths are given as 2.005(4) and 2.027(4) Å, the Sn(1)−O(1) and Sn(2)−O(3) bonds are elongated correspondingly with 2.254(4) and 2.248(4) Å, respectively. In addition, weak intramolecular Sn⋅⋅⋅P interactions are indicated by Sn⋅⋅⋅P distances of 3.058(1) and 2.992(1) Å. This results in an almost octahedral coordination sphere for the tin atom. In solution, the 119Sn NMR signal is observed at −397.4 ppm, while the 31P NMR resonance is shifted to high field at 8.5 ppm. Furthermore, a chemical inequivalence of the methylene and tert‐butyl protons ensues from the newly formed stereo centre. Thus, the diastereotopic protons of the methylene unit are found at 2.98 and 2.36 ppm with a geminal coupling constant of 15 Hz.
Figure 9.

Molecular structure of the (S)‐isomer of compound 9 in the solid state. Ellipsoids are set at 50 % probability; hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: P(1)−C(7) 1.850(5), P(1)−C(8) 1.895(5), P(1)−C(12) 1.896(5), O(1)−C(16) 1.238(6), O(2)−C(17) 1.415(6), Sn(1)−O(1) 2.254(4), Sn(1)−O(2) 2.005(4), Sn(1)−C(1) 2.294(6), Sn(1)−C(3) 2.270(6), Sn(1)−C(5) 2.263(6); O(1)‐Sn(1)‐O(2) 75.2(1), O(1)‐Sn(1)‐C(1) 170.4(2), O(1)‐Sn(1)‐C(3) 85.4(2), O(1)‐Sn(1)‐C(5) 86.1(2), O(2)‐Sn(1)‐C(1) 96.2(2), O(2)‐Sn(1)‐C(3) 152.2(2), O(2)‐Sn(1)‐C(5) 93.3(2), C(1)‐Sn(1)‐C(3) 104.1(2), C(1)‐Sn(1)‐C(5) 90.2(2), C(3)‐Sn(1)‐C(5) 105.1(2).
Conclusions
In summary, we have extended the scope of the recently presented Sn/P FLP 1 regarding its ability of binding small molecules (PhN=NPh, Me3SiCHN2, PhOCN and PhNCS) containing N≡C and N=X (X=C, S, N) functions. In case of azobenzene UV‐induced photoisomerization of the isomeric species enabled the highly selective complexation of cis‐azobenzene. In addition, adduct formation towards less polar C≡C triple bonds (HCCPh, HCCtBu) and α,β‐unsaturated carbonyl compounds (H3CC(O)CH=CH2, Ph[C(O)]2Ph) was verified, wherein conversion of 1 with benzil resulted in the non‐stereoselective transfer of the (tBu)2PCH2 unit to benzil. The obtained results underline the diversity and reactivity of the presented Sn/P FLP 1, assign its value and encourage us to further investigate this class of compounds in future work.
Experimental Section
Crystallographic data
Deposition numbers 2025718, 2025719, 2025720, 2025721, 2025722, 2025723, 2025724, and 2025725 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Felix Friedrich and Erik Stratmann for lab assistance, Marco Wissbrock for recording NMR spectra and Barbara Teichner for performing elemental analyses. We gratefully acknowledge financial support from Deutsche Forschungsgemeinschaft (DFG, grant Mi477/31‐1 project no. 320753677). Open access funding enabled and organized by Projekt DEAL.
P. Holtkamp, D. Poier, B. Neumann, H.-G. Stammler, N. W. Mitzel, Chem. Eur. J. 2021, 27, 3793.
References
- 1. Welch G. C., San Juan R. R., Masuda J. D., Stephan D. W., Science 2006, 314, 1124–1126. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Stephan D. W., Science 2016, 354, aaf7229;27940818 [Google Scholar]
- 2b. Stephan D. W., Acc. Chem. Res. 2015, 48, 306–316; [DOI] [PubMed] [Google Scholar]
- 2c. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2015, 54, 6400–6441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6498–6541; [Google Scholar]
- 2d. Stephan D. W., Erker G., Chem. Sci. 2014, 5, 2625–2641; [Google Scholar]
- 2e. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2010, 49, 46–76; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 50–81; [Google Scholar]
- 2f. Stephan D. W., Dalton Trans. 2009, 3129–3136; [DOI] [PubMed] [Google Scholar]
- 2g. Mömming C. M., Otten E., Kehr G., Fröhlich R., Grimme S., Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2009, 48, 6643–6646; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6770–6773; [Google Scholar]
- 2h. Spies P., Erker G., Kehr G., Bergander K., Fröhlich R., Grimme S., Stephan D. W., Chem. Commun. 2007, 5072–5074; [DOI] [PubMed] [Google Scholar]
- 2i. Welch G. C., Stephan D. W., J. Am. Chem. Soc. 2007, 129, 1880–1881. [DOI] [PubMed] [Google Scholar]
- 3. Lewis G. N., Valence and the Structure of Atoms and Molecules, The Chemical Catalog Company, Inc., New York, 1923. [Google Scholar]
- 4.
- 4a. Longobardi L. E., Wolter V., Stephan D. W., Angew. Chem. Int. Ed. 2015, 54, 809–812; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 823–826; [Google Scholar]
- 4b. Bebbington M. W. P., Bontemps S., Bouhadir G., Bourissou D., Angew. Chem. Int. Ed. 2007, 46, 3333–3336; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3397–3400; [Google Scholar]
- 4c. Stute A., Heletta L., Fröhlich R., Daniliuc C. G., Kehr G., Erker G., Chem. Commun. 2012, 48, 11739–11741; [DOI] [PubMed] [Google Scholar]
- 4d. Pleschka D., Layh M., Rogel F., Uhl W., Philos. Trans. R. Soc. A 2017, 375, 20170011; [DOI] [PubMed] [Google Scholar]
- 4e. Boom D. H. A., Jupp A. R., Nieger M., Ehlers A. W., Slootweg J. C., Chem. Eur. J. 2019, 25, 13299–13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnstone T. C., Wee G. N. J. H., Stephan D. W., Angew. Chem. Int. Ed. 2018, 57, 5881–5884; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5983–5986. [Google Scholar]
- 6.
- 6a. Appelt C., Westenberg H., Bertini F., Ehlers A. W., Slootweg J. C., Lammertsma K., Uhl W., Angew. Chem. Int. Ed. 2011, 50, 3925–3928; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4011–4014; [Google Scholar]
- 6b. Körte L. A., Blomeyer S., Heidemeyer S., Mix A., Neumann B., Mitzel N. W., Chem. Commun. 2016, 52, 9949–9952. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Kinder T. A., Pior R., Blomeyer S., Neumann B., Stammler H.-G., Mitzel N. W., Chem. Eur. J. 2019, 25, 5899–5903; [DOI] [PubMed] [Google Scholar]
- 7b. Waerder B., Pieper M., Körte L. A., Kinder T. A., Mix A., Neumann B., Stammler H.-G., Mitzel N. W., Angew. Chem. Int. Ed. 2015, 54, 13416–13419; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13614–13617; [Google Scholar]
- 7c. Kinder T. A., Blomeyer S., Franke M., Depenbrock F., Neumann B., Stammler H.-G., Mitzel N. W., Eur. J. Inorg. Chem. 2019, 3933–3939; [Google Scholar]
- 7d. Herrington T. J., Ward B. J., Doyle L. R., McDermott J., White A. J. P., Hunt P. A., Ashley A. E., Chem. Commun. 2014, 50, 12753–12756; [DOI] [PubMed] [Google Scholar]
- 7e. Schäfer A., Reißmann M., Schäfer A., Saak W., Haase D., Müller T., Angew. Chem. Int. Ed. 2011, 50, 12636–12638; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12845–12848; [Google Scholar]
- 7f. Weicker S. A., Stephan D. W., Chem. Eur. J. 2015, 21, 13027–13034. [DOI] [PubMed] [Google Scholar]
- 8. Yu Y., Li J., Liu W., Ye Q., Zhu H., Dalton Trans. 2016, 45, 6259–6268. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Holtkamp P., Friedrich F., Stratmann E., Mix A., Neumann B., Stammler H.-G., Mitzel N. W., Angew. Chem. Int. Ed. 2019, 58, 5114–5118; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5168–5172; [Google Scholar]
- 9b. Freitag S., Henning J., Schubert H., Wesemann L., Angew. Chem. Int. Ed. 2013, 52, 5640–5643; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5750–5754; [Google Scholar]
- 9c. Freitag S., Krebs K. M., Henning J., Hirdler J., Schubert H., Wesemann L., Organometallics 2013, 32, 6785–6791; [Google Scholar]
- 9d. Krebs K. M., Freitag S., Schubert H., Gerke B., Pöttgen R., Wesemann L., Chem. Eur. J. 2015, 21, 4628–4638; [DOI] [PubMed] [Google Scholar]
- 9e. Scott D. J., Phillips N. A., Sapsford J. S., Deacy A. C., Fuchter M. J., Ashley A. E., Angew. Chem. Int. Ed. 2016, 55, 14738–14742; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14958–14962; [Google Scholar]
- 9f. Turnell-Ritson R. C., Sapsford J. S., Cooper R. T., Lee S. S., Földes T., Hunt P. A., Pápai I., Ashley A. E., Chem. Sci. 2018, 9, 8716–8722; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9g. Dickie D. A., Coker E. N., Kemp R. A., Inorg. Chem. 2011, 50, 11288–11290. [DOI] [PubMed] [Google Scholar]
- 10. Pieper M., Lamm J.-H., Neumann B., Stammler H.-G., Mitzel N. W., Dalton Trans. 2017, 46, 5326–5336. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Pieper M., Auer P., Schwarzbich S., Kühn F., Neumann B., Stammler H.-G., Mitzel N. W., Z. Anorg. Allg. Chem. 2017, 643, 909–915; [Google Scholar]
- 11b. Xu X., Kehr G., Daniliuc C. G., Erker G., J. Am. Chem. Soc. 2013, 135, 6465–6476; [DOI] [PubMed] [Google Scholar]
- 11c. Chapman A. M., Haddow M. F., Wass D. F., J. Am. Chem. Soc. 2011, 133, 8826–8829; [DOI] [PubMed] [Google Scholar]
- 11d. Chapman A. M., Haddow M. F., Wass D. F., J. Am. Chem. Soc. 2011, 133, 18463–18478; [DOI] [PubMed] [Google Scholar]
- 11e. Dobrovetsky R., Stephan D. W., Isr. J. Chem. 2015, 55, 206–209; [Google Scholar]
- 11f. Dobrovetsky R., Stephan D. W., Angew. Chem. Int. Ed. 2013, 52, 2516–2519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2576–2579; [Google Scholar]
- 11g. Jochmann P., Stephan D. W., Angew. Chem. Int. Ed. 2013, 52, 9831–9835; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10014–10018; [Google Scholar]
- 11h. Chang K., Wang X., Fan Z., Xu X., Inorg. Chem. 2018, 57, 8568–8580. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Chase P. A., Welch G. C., Jurca T., Stephan D. W., Angew. Chem. Int. Ed. 2007, 46, 8050–8053; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8196–8199; [Google Scholar]
- 12b. Wang H., Fröhlich R., Kehr G., Erker G., Chem. Commun. 2008, 5966–5968; [DOI] [PubMed] [Google Scholar]
- 12c. Chernichenko K., Nieger M., Leskelä M., Repo T., Dalton Trans. 2012, 41, 9029–9032; [DOI] [PubMed] [Google Scholar]
- 12d. Lam J., Szkop K. M., Mosaferi E., Stephan D. W., Chem. Soc. Rev. 2019, 48, 3592–3612. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Ashley A. E., O'Hare D., Top. Curr. Chem. 2012, 334, 191–217; [DOI] [PubMed] [Google Scholar]
- 13b. Courtemanche M.-A., Légaré M.-A., Maron L., Fontaine F.-G., J. Am. Chem. Soc. 2014, 136, 10708–10717; [DOI] [PubMed] [Google Scholar]
- 13c. Wang X., Xia C., Wu L., Green Chem. 2018, 20, 5415–5426. [Google Scholar]
- 14.
- 14a. Melen R. L., Angew. Chem. Int. Ed. 2018, 57, 880–882; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 890–892; [Google Scholar]
- 14b. Tang C., Liang Q., Jupp A. R., Johnstone T. C., Neu R. C., Song D., Grimme S., Stephan D. W., Angew. Chem. Int. Ed. 2017, 56, 16588–16592; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16815–16819. [Google Scholar]
- 15. LaFortune J. H. W., Trofimova A., Cummings H., Westcott S. A., Stephan D. W., Chem. Eur. J. 2019, 25, 12521–12525. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Wolf E., Cammenga H. K., Z. Phys. Chem. 1977, 107, 21–38; [Google Scholar]
- 16b. Eckardt N., Flammersheim H. J., Cammenga H. K., J. Therm. Anal. Calorim. 1998, 52, 177–185. [Google Scholar]
- 17. Harada J., Ogawa K., Tomoda S., Acta Crystallogr. Sect. B 1997, 53, 662–672. [Google Scholar]
- 18. Mostad A., Rømming C., Acta Chem. Scand. 1971, 25, 3561–3568. [DOI] [PubMed] [Google Scholar]
- 19. Addison A. W., Rao T. N., Reedjik J., van Rijn J., Verschoor G. C., J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar]
- 20. Pearson R. G., J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- 21. Lange M., Tendyck J. C., Wegener P., Hepp A., Würthwein E.-U., Uhl W., Chem. Eur. J. 2018, 24, 12856–12868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary