

Abstract
Background
Secukinumab has demonstrated sustained long‐term efficacy with a favourable safety profile in various psoriatic disease manifestations in adults.
Objectives
Here, the efficacy and safety of two secukinumab dosing regimens [low dose (LD) and high dose (HD)] in paediatric patients with severe chronic plaque psoriasis over one year are reported.
Methods
In this multicentre, double‐blind study (NCT02471144), patients aged 6 to <18 years with severe chronic plaque psoriasis were stratified and randomized by weight (<25 kg, 25 to <50 kg, ≥50 kg) and age (6 to <12 years, 12 to <18 years) to receive low‐dose (LD: 75/75/150 mg) or high‐dose (HD: 75/150/300 mg) subcutaneous secukinumab or placebo or etanercept 0.8 mg/kg (up to a max of 50 mg).
Results
Overall, 162 patients were randomized to receive secukinumab LD (n = 40) or HD (n = 40), etanercept (n = 41) or placebo (n = 41). The co‐primary objectives of the study were met with both secukinumab doses (LD and HD) showing superior efficacy compared to placebo (P < 0.0001) with respect to PASI 75 response (80.0%, 77.5% vs. 14.6%) and IGA mod 2011, 0 or 1 response (70%, 60% vs. 4.9%) at Week 12. Both secukinumab doses were superior to placebo (P < 0.0001) with respect to PASI 90 response at Week 12 (72.5%, 67.5% vs. 2.4%). The efficacy of both doses was sustained to Week 52 with secukinumab achieving higher responses vs. etanercept (PASI 75/90/100: LD, 87.5%/75.0%/40.0% and HD, 87.5%/80.0%/47.5.% vs. etanercept, 68.3%/51.2%/22.0% and IGA 0 or 1: LD, 72.5% and HD, 75.0% vs. etanercept, 56.1%). The safety profile of secukinumab was consistent with the adult Phase 3 studies, with no new safety signals identified.
Conclusions
Both doses of secukinumab demonstrated high and sustained efficacy up to Week 52 with a favourable safety profile in paediatric patients with severe chronic plaque psoriasis.
Introduction
Psoriasis is a chronic inflammatory condition characterized by well‐defined red plaques with silver or white scales that are itchy and vary in severity. 1 Psoriasis affects 2–4% of the adult population in the western countries with a worldwide prevalence ranging from 0.91% to 8.5%. 1 , 2 , 3 The prevalence of childhood psoriasis varies depending on the study population and is reported to be 1.3–2.1% in Europe and 0.13% in the USA. 4 , 5 Paediatric psoriasis is more common after puberty (0.6–1.3%) than before puberty (0.1–0.5%). 4 Although psoriasis can arise at any age, the median age of onset of paediatric psoriasis is between 7 and 10 years. 6 Paediatric patients with psoriasis may also have co‐morbidities seen in adult patients such as diabetes mellitus, Crohn’s disease, obesity, ischaemic heart disease, hyperlipidaemia, dyslipidaemia, hypertension and metabolic syndrome. 7 , 8 , 9 , 10 Large‐scale epidemiological studies have confirmed the increased risk for psychosocial co‐morbidities like psychiatric disorder, depression and anxiety in children. 11 , 12 , 13 Children suffering from even mild psoriasis have a poorer quality of life (QoL) than that of their peers because of itching, fatigue and feelings of stigmatization, thereby affecting their emotional well‐being and school functioning. 14 Treatment guidelines and therapeutic protocols for adult psoriasis are well established with several approved systemic drugs; however, approved treatment options are scarce for paediatric psoriasis. 15 , 16 , 17 , 18 , 19 There has been a significant gap in the number of ongoing clinical studies in adults versus children and adolescents resulting in a wide use of off‐label systemic drugs (both conventional systemics as well as biologic options) in children and adolescents. 7 , 8 , 20 , 21 Treatment for paediatric psoriasis has expanded in recent years with approval of tumour necrosis factor (TNF)‐α inhibitors, IL‐12/23 inhibitor and IL‐17A inhibitor for varying disease severity (Table S1, Supporting Information). 22 , 23 , 24 , 25 , 26 , 27
Secukinumab is a fully human monoclonal antibody that selectively neutralizes IL‐17A, a cornerstone cytokine involved in the pathogenesis of psoriatic disease. In adults, it has demonstrated sustained long‐term efficacy with a favourable safety profile in various psoriatic disease manifestations. 28 , 29 , 30 , 31 , 32 , 33 , 34 Secukinumab has been recently approved by the European Commission (July, 2020) for the treatment of moderate‐to‐severe plaque psoriasis in children and adolescents from the age of 6 years who are candidates for systemic therapy. This study was designed to evaluate the efficacy and safety of secukinumab in children and adolescents (aged 6 to <18 years). Here, the efficacy and safety of two secukinumab dosing regimens [low (LD) and high dose (HD)] in paediatric patients with severe chronic plaque psoriasis over one year are reported.
Materials and methods
Study design
This was a global, multicentre, randomized, double‐blind placebo‐ and active‐controlled (etanercept in single‐blinded arm) study in paediatric patients aged 6 years to <18 years with severe chronic plaque psoriasis. Patients were randomized using a 1 : 1 : 1 : 1 ratio into one of the treatment arms: secukinumab low dose (LD), secukinumab high dose (HD), etanercept or placebo (Fig. 1). Patients randomized to secukinumab treatment arms (HD and LD) received a dose based on their weight category (<25 kg, 25 to <50 kg, ≥50 kg): patients weighing ≥50 kg received 150 mg (LD group) or 300 mg (HD group), those weighing 25 to <50 kg received 75 mg (LD group) or 150 mg (HD group), and patients weighing <25 kg received 75 mg for both dose groups. If the patient moved into a higher or lower weight group at two consecutive visits with weight measurements during the maintenance or extension treatment period, then the dose was administered according to the new (higher or lower) weight group. Patients in the etanercept arm received weekly subcutaneous dose of 0.8 mg/kg (up to a maximum of 50 mg) of etanercept. Etanercept was open‐label to safety evaluator. Efficacy assessor was blinded to all study treatments.
Figure 1.
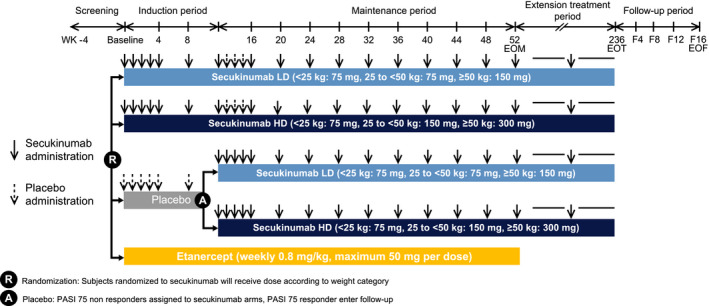
Study design. A, assigned; EOF, end of follow‐up; EOM, end of maintenance period; EOT, end of treatment; HD, high dose; LD, low dose; PASI, Psoriasis Area and Severity Index; R, randomized; SEC, secukinumab; WK, week.
The study consisted of five periods:
Screening period (of up to 4 weeks): to assess the eligibility of the patients and to allow a drug‐free washout period
Induction period (randomization to Week 12): During this time, the study was both active‐ and placebo‐controlled and the co‐primary endpoints (PASI 75 and IGA mod 2011 0 or 1 response) were assessed at Week 12. At the randomization visit, patients randomized to the placebo group were pre‐assigned to either LD or HD secukinumab, in case they did not achieve a Psoriasis Area Severity Index (PASI) 75 response at Week 12. Placebo PASI 75 responders at Week 12, terminated their treatment and entered the post‐treatment follow‐up period.
The maintenance period (Week 12 to Week 52): During this time, the study was active‐controlled and the objectives focused on the maintenance of the response observed at Week 12. Patients who received secukinumab or etanercept during induction continued with the same treatment. Etanercept patients terminated their treatment at Week 52 and entered the treatment‐free follow‐up period. Patients who were on placebo treatment during induction period and were PASI 75 non‐responders at Week 12, were switched to either secukinumab low dose or secukinumab high dose treatment group starting at Week 12 and for the remainder of the study according to their baseline randomization.
Extension treatment period (Week 52 until Week 236): At the end of the maintenance period, all patients on secukinumab entered the extension treatment period and continued to receive the same previous allocated dose of secukinumab. In the extension treatment period, between site visits, secukinumab was administered every 4 weeks at home, either by the patient (self‐injection only for adolescents of at least 12 years of age and under supervision) or by the caregiver. In case a patient or caregiver did not feel confident in performing home administrations, the patient was allowed to receive administration at site.
Follow‐up period (16 weeks): Patients entered the treatment‐free follow‐up period of 16 weeks if they: discontinued treatment at any point; were placebo patients who completed Week 12 and were PASI 75 responders; were etanercept patients who completed Week 52; were secukinumab patients who completed the extension treatment period. This period’s duration was only 8 weeks for placebo PASI 75 responders. Exceptionally, patients who had to start other systemic therapies after stopping study treatment were permitted to terminate the treatment‐free follow‐up earlier.
Enrolment for the study (ClinicalTrials.gov: NCT02471144) occurred between 29 Sep, 2015 and 24 Aug, 2018. At the time of publication of this report, the study is ongoing. The institutional review board of each participating centre approved the study protocol. The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice (GCP) and in compliance with all federal, local, or regional requirements.
Inclusion and key exclusion criteria
Eligible patients were aged 6 to ≤18 years at the time of randomization—patients aged 12 to ≤18 years were enrolled from beginning of trial, and patients aged 6 to <12 years were enrolled after positive DMC recommendation following review of data from approximately first 80 adolescents. Other inclusion criteria were as follows: severe plaque psoriasis at randomization [PASI ≥ 20, Investigator’s Global Assessment modified 2011 (IGA mod 2011) score of 4 and body surface area (BSA) involvement of ≥10%], history of plaque psoriasis ≥3 months and patients regarded by the investigator to be candidates for systemic therapy. Key exclusion criteria were forms of active psoriasis other than plaque‐type psoriasis, and active ongoing inflammatory diseases other than psoriasis that might confound evaluation of benefit of secukinumab and/or etanercept.
Study objectives
The primary objective of the study was to demonstrate the superiority of secukinumab (LD and HD) over placebo at Week 12 as measured by proportion of patients achieving PASI 75 and IGA mod 2011 0 or 1 response (co‐primary endpoints). The key secondary objective was to demonstrate superiority of secukinumab (LD and HD) over placebo at Week 12 as measured by proportion of patients achieving PASI 90. Other secondary objectives: to evaluate efficacy of secukinumab vs. placebo with respect to PASI 100 at Week 12, PASI 75/90/100 and IGA mod 2011 0/1 over time up to Week 52, changes in PASI score and IGA mod 2011 score at Week 12 and over time up to Week 52, changes in Children’s Dermatology Life Quality Index (CDLQI) and achievement of CDLQI 0/1 at Week 12 and over time up to Week 52. Safety outcomes included clinical safety and tolerability of secukinumab assessed by adverse events (AEs). Exploratory objective was to evaluate efficacy of secukinumab vs. etanercept with respect to PASI 75/90/100 and IGA mod 2011 over time up to Week 52.
Statistical methods
The following analysis sets were used for data analysis:
Randomized set: all randomized patients.
Full analysis set (FAS): all patients from randomized set who had been assigned study treatment.
Safety set: all patients who took at least one dose of study treatment during the treatment period.
The analysis of co‐primary and key secondary variables was based on FAS and was performed using the logistic regression analysis with non‐responder imputations (NRI). Week 12 analysis was performed by NRI with extended analysis visit window. Summary statistics were provided for number of patients achieving CDLQI score of 0 or 1, secukinumab groups were compared for controls by Fisher’s exact test. For CDLQI, missing values were replaced by last observation carried forward (LOCF) approach. All safety evaluations were performed on the safety set and were based on data collected till Week 52 visit (or early termination visit) for individual subjects. In this study, an Interactive Response Technology (IRT) error led to additional dosing of some patients after the primary endpoint (Week 12) assessment. Since the error occurred after Week 12, primary endpoint analysis (as well as secondary endpoints at Week 12) were not affected. Additional summary analysis was performed for PASI and IGA response by (IRT dosing error and by bodyweight and dose group to evaluate potential impact of this incident on safety and efficacy beyond Week 12 (refer to Supporting information). The number of patients in the groups that were affected and not affected by the IRT error is small, for any of these differences to be considered as clinically relevant.
Results
Patient disposition
In total, 187 patients were screened, 162 of whom completed the screening phase and were randomized to the four treatment groups: secukinumab LD (n = 40), secukinumab HD (n = 40), placebo (n = 41) and etanercept (n = 41) (Fig. 2). Overall, 156 (96.3%) patients completed the induction period, 151 (97.4%) of whom entered the maintenance period; 140 (90.3%) patients completed the maintenance period.
Figure 2.
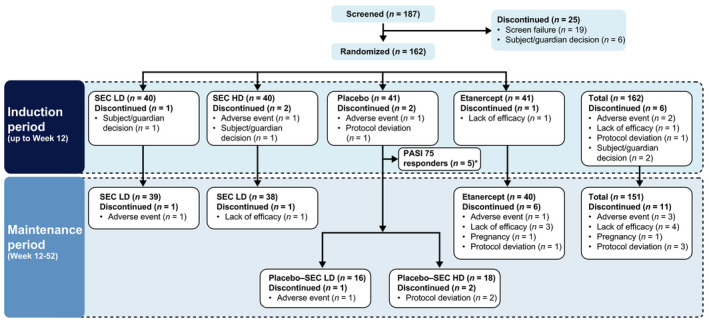
Patient disposition. *Five patients in the placebo group who were PASI 75 responders at Week 12 did not proceed into the maintenance period as defined in the protocol. HD, high dose; LD, low dose; SEC, secukinumab.
Demographics and baseline disease characteristics
The demographics and baseline disease characteristics of the treatment groups were generally balanced and comparable between all treatment groups (Table 1). The mean age of patients was 13.5 years, predominantly female (59.9%) and 77.2% were aged ≥12 years. The mean weight of the overall population was 53.47 kg and was similar in all treatment groups; 92.6% of the patients weighed ≥25 kg. At baseline, the mean total BSA affected by plaque‐type psoriasis was 40.0% and mean PASI score was 28 for overall patients. All patients had a PASI score of >20 and an IGA mod 2011 score of 4 (severe disease) except 2 patients in the secukinumab HD group, one with PASI score <20 and other with IGA mod 2011 score of 3 (moderate disease). Overall, the mean time since diagnosis of psoriasis was 5.22 years (Table 1).
Table 1.
Demographic and baseline disease characteristics (randomized set)
|
SEC LD (N = 40) |
SEC HD (N = 40) |
Placebo (N = 41) |
Etanercept (N = 41) |
Total (N = 162) |
|
|---|---|---|---|---|---|
| Age group (years), n (%) | |||||
| <12 | 8 (20.0) | 9 (22.5) | 10 (24.4) | 10 (24.4) | 37 (22.8) |
| ≥12 | 32 (80.0) | 31 (77.5) | 31 (75.6) | 31 (75.6) | 125 (77.2) |
| Age (years), mean (SD) | 13.7 (2.92) | 13.2 (3.21) | 13.7 (3.27) | 13.5 (2.94) | 13.5 (3.06) |
| Sex, n (%) | |||||
| Male | 13 (32.5) | 17 (42.5) | 19 (46.3) | 16 (39.0) | 65 (40.1) |
| Female | 27 (67.5) | 23 (57.5) | 22 (53.7) | 25 (61.0) | 97 (59.9) |
| Race, Caucasian, n (%) | 34 (85.0) | 34 (85.0) | 36 (87.8) | 30 (73.2) | 134 (82.7) |
| Weight (kg), mean (SD) | 52.60 (15.26) | 53.61 (20.18) | 55.68 (22.28) | 51.96 (19.43) | 53.47 (19.35) |
| Weight strata (kg), n (%) | |||||
| <25 | 2 (5.0) | 3 (7.5) | 3 (7.3) | 4 (9.8) | 12 (7.4) |
| 25 to <50 | 17 (42.5) | 15 (37.5) | 17 (41.5) | 16 (39.0) | 65 (40.1) |
| ≥50 | 21 (52.5) | 22 (55.0) | 21 (51.2) | 21 (51.2) | 85 (52.5) |
| BMI (kg/m2), mean (SD) | 20.32 (3.60) | 21.16 (4.37) | 22.19 (6.20) | 21.00 (4.80) | 21.17 (4.85) |
| Baseline PASI score, mean (SD) | 27.6 (6.89) | 28.0 (8.67) | 28.0 (8.09) | 28.4 (9.05) | 28.0 (8.15) |
| Baseline PASI, n (%) | |||||
| ≤20 | 0 (0.0) | 1 (2.5) | 0 (0.0) | 0 (0.0) | 1 (0.6) |
| >20 | 40 (100.0) | 39 (97.5) | 41 (100.0) | 41 (100.0) | 161 (99.4) |
| Baseline total BSA, mean (SD) | 37.59 (13.86) | 40.26 (17.56) | 38.99 (17.65) | 43.13 (19.56) | 40.01 (17.26) |
| Baseline IGA Mod 2011 score, n (%) | |||||
| 3 = moderate disease | 0 (0.0) | 1 (2.5) | 0 (0.0) | 0 (0.0) | 1 (0.6) |
| 4 = severe disease | 40 (100.0) | 39 (97.5) | 41 (100.0) | 41 (100.0) | 161 (99.4) |
| Time since diagnosis of plaque psoriasis (years), mean (SD) | 4.85 (4.29) | 5.44 (4.67) | 6.03 (5.09) | 4.55 (3.73) | 5.22 (4.47) |
| Diagnosis of psoriatic arthritis, n (%) | 5 (12.5) | 3 (7.5) | 3 (7.3) | 3 (7.3) | 14 (8.6) |
| Previous psoriasis therapies, n (%) | |||||
| Systemic | 26 (65.0) | 21 (52.5) | 20 (48.8) | 19 (46.3) | 86 (53.1) |
| Biologic | 3 (7.5) | 0 (0.0) | 0 (0.0) | 1 (2.4) | 4 (2.5) |
| Non‐biologic systemic | 26 (65.0) | 21 (52.5) | 20 (48.8) | 18 (43.9) | 85 (52.5) |
| Topical | 32 (80.0) | 36 (90.0) | 38 (92.7) | 38 (92.7) | 144 (88.9) |
| Phototherapy | 16 (40.0) | 16 (40.0) | 21 (51.2) | 17 (41.5) | 70 (43.2) |
| Photochemotherapy | 3 (7.5) | 11 (27.5) | 1 (2.4) | 5 (12.2) | 20 (12.3) |
BMI, body mass index; BSA, body surface area; HD, high dose; IGA mod 2011, Investigator’s Global Assessment Modified 2011; LD, low dose; PASI, Psoriasis Area and Severity Index; SD, standard deviation; SEC, secukinumab.
Efficacy
The study met the co‐primary endpoints; both secukinumab doses (LD and HD) were superior (P < 0.0001) to placebo with respect to PASI 75 response (80.0% and 77.5% vs. 14.6%) and IGA mod 2011 0 or 1 response (70.0% and 60.0% vs. 4.9%) at Week 12 (Fig. 3). The study met the key secondary endpoint; both secukinumab doses (LD and HD) were superior (P < 0.0001) to placebo with respect to PASI 90 response (72.5% and 67.5% vs. 2.4%) at Week 12 (Fig. 3). At Week 12, PASI 100 response was achieved by 30.0% and 27.5% of patients in secukinumab LD and HD groups, respectively, and none of the patients in the placebo group (Fig. 3). At Week 12, both secukinumab dose groups (LD and HD) achieved significantly higher (P < 0.05) response vs. etanercept with respect to IGA mod 2011 0/1 (70.0% and 60.0% vs. 34.1%) and PASI 90 (72.5% and 67.5% vs. 29.3%), respectively. Both secukinumab dose groups (LD and HD) demonstrated efficacy as early as Week 4 (PASI 75/90/100: LD, 32.5%/12.5%/7.5% and HD, 55.0%/22.5%/7.5.% vs. etanercept, 12.2%/2.4%/0% and IGA 0 or 1: LD, 15.0% and HD, 32.5% vs. etanercept, 2.4%). Both secukinumab dose groups (LD and HD) achieved numerically higher PASI 75 (80.0% and 77.5% vs. 63.4%) and PASI 100 (30.0% and 27.5% vs. 17.1%) response vs. etanercept at Week 12. Both secukinumab dose groups (LD and HD), achieved comparable PASI 75/90/100 and IGA mod 2011 0/1 responses at Week 12 (Fig. 4). IGA mod 2011 0/1 response increased continuously thereafter reaching maximum at Week 24 in both secukinumab dose groups (LD: 87.5% and HD: 75.0%) and was then sustained to Week 52. The IGA mod 2011 0/1 response was slightly higher in the secukinumab LD group than the secukinumab HD group from Week 12 to Week 40 but similar thereafter and up to Week 52 (LD: 72.5% and HD: 75.0%).
Figure 3.
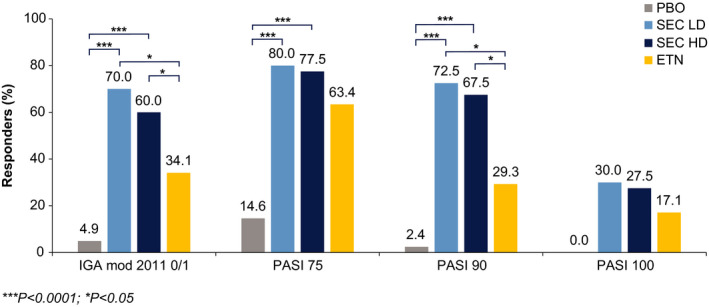
IGA mod 2011 0/1 and PASI responses at Week 12 (NRI with extended analysis visit window). ETN, etanercept; HD, high dose; IGA mod 2011, Investigator’s Global Assessment Modified 2011; LD, low dose; NRI, non‐responder imputation; PASI, Psoriasis Area and Severity Index; PBO, placebo; SEC, secukinumab.
Figure 4.
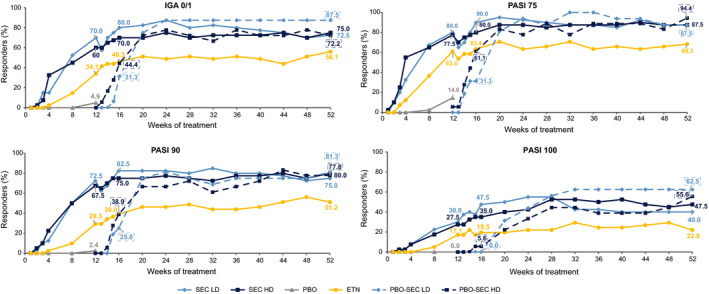
IGA mod 2011 0/1 and PASI responses up to Week 52 (NRI with extended analysis visit window at Week 12). ETN, etanercept; HD, high dose; IGA mod 2011, Investigator’s Global Assessment Modified 2011; LD, low dose; NRI, non‐responder imputation; PASI, Psoriasis Area and Severity Index; PBO, placebo; SEC, secukinumab.
At Week 16, PASI and IGA responses were higher than at Week 12 in all treatment groups (Fig. 4). From Week 16 onwards, PASI response rates increased and reached their maximum between Week 20 and Week 28. Thereafter, responses were sustained to Week 52 in both secukinumab dose groups (LD and HD). Similar to IGA response, PASI 75 and PASI 90 responses were slightly higher in the secukinumab LD than in the HD group midway through the maintenance period and were then comparable at Week 52.
Both secukinumab dose groups (LD and HD) continued to show higher PASI 75/90/100 and IGA mod 2011 0 or 1 responses compared to the etanercept group at each visit up to Week 52 (Fig. 4). At Weeks 24 and 52, it was observed that PASI 75 response rates in secukinumab LD (92.5% and 87.5%, respectively) and HD groups (87.5% for both visits) were higher than in the etanercept group (63.4% and 68.3%, respectively). Similar high response in secukinumab treatment groups than etanercept group were also evident in the PASI 90 and IGA mod 2011 0/1 responses. The placebo non‐responders who were assigned to secukinumab (LD and HD) at Week 12 showed a rapid increase in the responses following the switch to active treatment (Fig. 4), and reached levels comparable to those of the originally randomized secukinumab groups. Analyses by IRT dosing error (Figure S1, Supporting Information) and by bodyweight and dose groups (Table S2, Supporting Information) were also performed. Considering only the subset of patients not affected by the IRT dosing error in both secukinumab dose groups (LD and HD), the responses in both groups were higher compared to both the placebo and the etanercept groups throughout the induction period, and continued to be higher than the etanercept group up to Week 52 (Figure S2, Supporting Information). In the ‘≥25 to <50 kg’ weight category, patients receiving HD [150 mg (N = 15)] had higher PASI 75/90/100 and IGA mod 2011 0 or 1 responses than LD [75 mg (N = 17)] at Week 12 (Table S2, Supporting Information). At Week 52, patients on secukinumab LD showed higher responses than those receiving secukinumab HD. In the ‘≥50 kg’ weight category, throughout the maintenance period, patients on HD [300 mg (N = 21)] showed higher PASI and IGA responses than those receiving LD [150 mg (N = 22)]. At Week 52, the PASI 75/90/100 and IGA mod 2011 0/1 response rates were 100%, 95.2%, 61.9% and 85.7%, respectively, for the 300 mg group and 86.4%, 72.7%, 27.3% and 68.2%, respectively, for the 150 mg group. All rates were numerically higher with 300 mg vs. 150 mg secukinumab. Interpretation of the ‘secukinumab 75 mg and <25 kg’ group is limited due to the low number of patients (N = 5).
The mean baseline PASI score was ~28 in all the treatment groups (Table 1). At Week 12, the mean PASI scores were decreased (improved) from baseline by 82.9% in the secukinumab LD group (reaching 5.12) and by 79.9 % in the secukinumab HD group (reaching 5.56) compared to 29.3% in the placebo group (reaching 19.89) and 74.2 % in the etanercept group (reaching 7.50). At Week 52, the mean PASI scores decreased further from baseline by 92.6% in the secukinumab LD group (reaching 2.13) and by 91.8% in the secukinumab HD group (reaching 2.49), while the etanercept group showed a decrease in PASI score by 77.7% (reaching 7.24).
Health‐related quality of life
At Week 12, the proportion of patients achieving CDLQI score of 0 or 1 in both secukinumab dose groups (LD: 44.7% and HD: 50%) was significantly higher compared with the placebo group (15%) (P < 0.05 for both comparisons) and were numerically higher than the etanercept group (36.6%) (Fig. 5). At Week 52, they were numerically higher (LD: 60.6% and HD: 66.7%) compared to etanercept (44.4%).
Figure 5.
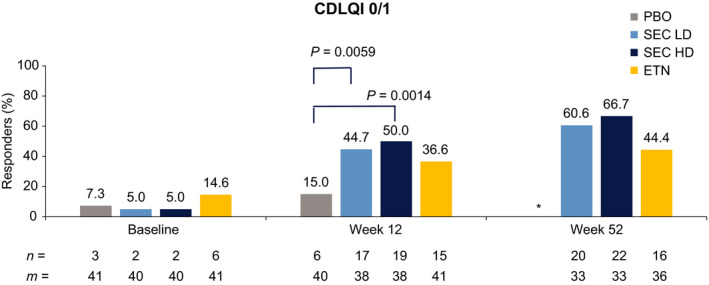
Proportion of patients achieving Children’s Dermatology Life Quality Index 0 or 1 score (LOCF analysis). *Patients on PBO completed treatment at Week 12. CDLQI, Children’s Dermatology Life Quality Index; ETN, etanercept; HD, high dose; LD, low dose; LOCF, last observation carried forward; m, number of patients available for evaluation; n, number of patients; SEC, secukinumab.
Safety
The safety profile of secukinumab for this Week‐52 analysis was based on 228.1 patient‐years of exposure to any secukinumab dose. The incidence of AEs was similar between the secukinumab LD and HD groups (85.0% each). The overall incidence of treatment‐emergent AEs up to Week 52 was similar in the ‘any secukinumab LD’ group (80.4%), the ‘any secukinumab HD’ group (81%) and the etanercept group (82.9%) (Table 2). Overall, the most commonly affected system organ class (SOC) was ‘Infections and infestations’. The incidence of AEs in this SOC was numerically higher in the ‘any secukinumab LD’ group (69.6%) compared to ‘any secukinumab HD’ group (62.1%) and the etanercept group (65.9%). The higher incidence of AEs in this SOC was mainly driven by non‐serious nasopharyngitis, pharyngitis, rhinitis and upper respiratory tract infections. The most commonly reported AEs by preferred term were nasopharyngitis, headache and pharyngitis. The reported level of Candida infections was low (1.8%) and no IBD cases were reported during the 52 week treatment period. The vast majority of the AEs reported up to Week 52 were of mild‐to‐moderate severity. The overall incidence of AEs possibly related to the study medication reported up to Week 52 was higher in the etanercept group (14 patients, 34.1%) compared to ‘any secukinumab LD’ group (13 patients, 23.2%) and comparable with the ‘any secukinumab HD’ group (19 patients, 32.8%). No deaths were reported up to the Week 52 data cut‐off. Overall, non‐fatal SAEs and AEs leading to treatment discontinuation up to Week 52 were infrequent and occurred at similar frequency across groups (Table S3, Supporting Information). During the 12‐week induction period, 7 patients presented with injection site reactions. Two (2.5%) patients in ‘any secukinumab’ dose group [2 (5%) patients in the secukinumab LD group, none in the secukinumab HD group], 3 (7.3%) patients in the etanercept group and 2 (4.9%) patients in the placebo group. All patients reported one unique event, except one etanercept patient reporting injection site pain at five visits (Week 1, 2, 3, 4 and 8). All AEs were mild in intensity, had a duration between 1 and 12 days and required no treatment except injection site hematoma in the placebo group that was treated with arnica cream applications (3 days). Up to Week 52, overall 7 patients (6.1%) in ‘any secukinumab’ dose group and 4 patients (9.8%) in etanercept group experienced injection site reactions up to Week 52. All events were mild in intensity and required no treatment except one case (application site erythema) in the etanercept group treated with loratadine (1 day) (Table 2). Other AEs of interest are discussed in the Supporting information.
Table 2.
Overall safety profile (up to Week 52)
|
SEC LD, N = 40 n (%) |
SEC HD, N = 40 n (%) |
ETN, N = 41 n (%) |
Any SEC LD, N = 56 n (%) |
Any SEC HD, N = 58 n (%) |
Any SEC dose, N = 114 n (%) |
|
|---|---|---|---|---|---|---|
| Patients with AEs | 34 (85.0) | 34 (85.0) | 34 (82.9) | 45 (80.4) | 47 (81.0) | 92 (80.7) |
| Patients with serious or other significant events | ||||||
| Death | 0 | 0 | 0 | 0 | 0 | 0 |
| Non‐fatal SAEs | 3 (7.5) | 4 (10.0) | 5 (12.2) | 5 (8.9) | 5 (8.6) | 10 (8.8) |
| Discontinued study treatment due to any AEs | 1 (2.5) | 1 (2.5) | 1 (2.4) | 2 (3.6) | 1 (1.7) | 3 (2.6) |
| Most frequent AEs (by SOC) | ||||||
| Infections and infestations | 30 (75.0) | 27 (67.5) | 27 (65.9) | 39 (69.6) | 36 (62.1) | 75 (65.8) |
| Gastrointestinal disorders | 12 (30.0) | 13 (32.5) | 14 (34.1) | 14 (25.0) | 17 (29.3) | 31 (27.2) |
| Skin and subcutaneous tissue disorders | 12 (30.0) | 12 (30.0) | 10 (24.4) | 14 (25.0) | 17 (29.3) | 31 (27.2) |
| Most frequent AEs (by PT) | ||||||
| Nasopharyngitis | 12 (30.0) | 16 (40.0) | 11 (26.8) | 14 (25.0) | 20 (34.5) | 34 (29.8) |
| Headache | 5 (12.5) | 6 (15.0) | 4 (9.8) | 8 (14.3) | 9 (15.5) | 17 (14.9) |
| Pharyngitis | 4 (10.0) | 4 (10.0) | 3 (7.3) | 6 (10.7) | 6 (10.3) | 12 (10.5) |
| Rhinitis | 2 (5.0) | 3 (7.5) | 1 (2.4) | 3 (5.4) | 6 (10.3) | 9 (7.9) |
| AEs of interest | ||||||
| Hypersensitivity (SMQ) (narrow) | 3 (7.5) | 9 (22.5) | 5 (12.2) | 4 (7.1) | 11 (19.0) | 15 (13.2) |
| Neutropenia (NMQ) | 2 (5.0) | 1 (2.5) | 1 (2.4) | 2 (3.6) | 1 (1.7) | 3 (2.6) |
| Neutropenia (PT) | 2 (5.0) | 1 (2.5) | 1 (2.4) | 2 (3.6) | 1 (1.7) | 3 (2.6) |
| Candida infections | 1 (2.5) | 1 (2.5) | 0 | 1 (1.8) | 1 (1.7) | 2 (1.8) |
| Nail candida (PT) | 0 | 1 (2.5) | 0 | 0 | 1 (1.7) | 1 (0.9) |
| Vulvovaginal candidiasis (PT) | 1 (2.5) | 0 | 0 | 1 (1.8) | 0 | 1 (0.9) |
| Hepatitis viral infections (HLT) | 0 | 0 | 0 | 0 | 0 | 0 |
| Inflammatory bowel disease (NMQ) | 0 | 0 | 0 | 0 | 0 | 0 |
| Vaccination related complications (HLT) | 0 | 0 | 0 | 0 | 0 | 0 |
| MACE (NMQ) | 0 | 0 | 0 | 0 | 0 | 0 |
| Malignant or unspecified tumours (SMQ) | 0 | 0 | 0 | 0 | 0 | 0 |
| Suicide/self‐injury (SMQ) | 1 (2.5) | 0 | 0 | 1 (1.8) | 0 | 1 (0.9) |
| Suicidal ideation (PT) | 1 (2.5) | 0 | 0 | 1 (1.8) | 0 | 1 (0.9) |
| Injection site reactions † | 3 (7.5) | 4 (10.0) | 4 (9.8) | 3 (5.4) | 4 (6.9) | 7 (6.1) |
AE, adverse event; ETN, etanercept; HD, high dose; HLT, high level term; LD, low dose; NMQ, Novartis customized MedDRA Query; PT, preferred term; SAE, serious adverse event; SEC, secukinumab; SMQ, Standardised MedDRA Query; SOC, system organ class; ‘Any secukinumab’ for both the treatment groups (LD and HD) includes all patients treated with secukinumab from the start of the study and also those who switched from placebo treatment to secukinumab at Week 12.
Injection site reactions corresponding to event coded with HLT ‘Injection site reaction’ and some with HLT ‘Administration site reaction’ or ‘Application and instillation site reactions’.
Discussion
The treatment options for psoriasis have expanded over the past decade, although in children, the options are limited and many therapies (topicals and conventional systemics) are used off‐label. 7 , 8 , 20 , 21 Despite the mostly recent approvals of biologics for use in paediatric psoriasis, there is still a medical need to expand efficacious and safe first‐line systemic treatment options in the paediatric population. In adult patients, secukinumab has been proven to achieve rapid and sustained long‐term efficacy in skin clearance, with onset of relief as early as Week 4, 35 ~80% of patients achieving PASI 90 at Week 16, 33 and long‐lasting clear skin through 5 years. 36 Secukinumab paediatric programme includes 2 studies in patients (aged 6 to <18 years) with severe (NCT02471144) and moderate‐to‐severe (NCT03668613) plaque psoriasis with an active treatment period of 236 and 208 weeks, respectively. In the present study, 99.4% patients had severe disease (baseline PASI > 20 and IGA mod 2011 score of 4) and all patients had received psoriasis therapies prior to study entry. The study achieved the primary objective and the key secondary objective. Both secukinumab doses (LD and HD) were superior to placebo with respect to the co‐primary endpoints of PASI 75 and IGA mod 2011 0 or 1 response and the key secondary endpoint of PASI 90 at Week 12 (P < 0.0001 for all comparisons). Secukinumab treatment demonstrated further improvement in the PASI 75/90/100 and IGA mod 2011 0/1 responses peaking around Week 24 and maintained up to Week 52. Both secukinumab dose groups (LD and HD) achieved comparable PASI 75/90/100 and IGA mod 2011 0/1 responses at Week 12, and efficacy was sustained to Week 52. However, higher response rates were observed with secukinumab HD compared to LD in the bodyweight category ‘≥50 kg’. Thus for patients ≥50 kg, should the LD show insufficient efficacy the HD may be considered for treatment. Both secukinumab doses (LD and HD) demonstrated greater clinical improvement compared to etanercept throughout the study up to Week 52. Results of the patient‐reported outcome of CDLQI indicated continuous improvements in the health‐related quality of life and functional ability of the secukinumab‐treated patients (to a larger extent in the HD group) compared to etanercept patients at Week 52. Importantly, the majority of patients reported no impact on QoL (CDLQI 0 or 1) due to psoriasis symptoms at Week 52. Freeing children of the burden of psoriasis is especially important in this vulnerable patient population to ensure an optimal chance for physical and psychosocial development during childhood.
Secukinumab was well tolerated at both doses (LD and HD). The overall safety profile of secukinumab in this study was consistent with that in adults in pivotal Phase 3 psoriasis trials, 31 , 33 , 35 , 36 and there were no new or unexpected safety signals. Further long‐term efficacy, safety, growth and development data are being collected up to 252 weeks (including 16‐week follow‐up‐period).
In conclusion, both doses of secukinumab demonstrated high and sustained efficacy up to Week 52 in clearing skin and improving health‐related quality of life with a favourable safety profile in paediatric patients with severe chronic plaque psoriasis.
Supporting information
Table S1. Biologics approved for treatment of moderate to severe plaque psoriasis (as of 14 July, 2020)
Table S2. IGA mod 2011 and PASI responses at Weeks 12 and 52 (analyses by body weight and dose groups) (NRI)
Table S3. Treatment emergent serious adverse events, by preferred term (PT) (up to Week 52)
Figure S1. IGA mod 2011 and PASI response up to Week 52 in patients affected and non‐affected by IRT dosing error (NRI)
Figure S2. IGA mod 2011 and PASI response up to Week 52 in non‐affected patients by IRT dosing error in comparison with placebo and etanercept (NRI)
Figure S3. Mean change from baseline in CDLQI total score (LOCF analysis)
Figure S4. Mean change from baseline in individual domain scores of the CDLQI (LOCF analysis)
Acknowledgements
The authors thank Sumeet Sood, PhD, and Avishek Anant, PhD (Novartis Healthcare Pvt. Ltd, Hyderabad), for editorial and medical writing support, which was funded by Novartis Pharma AG, Basel, Switzerland, in accordance with the Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Conflicts of interest
C. Bodemer has received research grants and/or served as speaker/consultant/investigator for Novartis, Bioderma, Roche Posay, Pierre Fabre, Expanscience, Sanofi, Pfizer, Janssen, Amryt, Castel Creek Pharma and Eli Lilly. A. Kaszuba received honoraria for lectures from Novartis, AbbVie and Janssen. K. Kingo has received fees for serving as an investigator in studies sponsored by Celgene, Merck, Mitsubishi Tanabe Pharma, Novartis, Regeneron Pharmaceuticals and Sandoz. A. Tsianakas is an investigator for several Novartis clinical trials including the published one. A. Morita has received research grants, consulting fees, and/or speaker’s fees from AbbVie, Eli Lilly, Esai, Janssen, Kyowa Hakko Kirin, Leo Pharma, Maruho, Mitsubishi Tanabe, Novartis and Taiho Pharmaceutical. E. Rivas is a principal investigator several Novartis clinical trials, also lecture fees, consulting fees and/ or speaker´s fees from Novartis and Sanofi. P. Papanastasiou, D. Keefe, M. Patekar, P. Charef, L. Zhang, S. Cafoncelli and C. Papavassilis are employed by Novartis.
Funding sources
The study was sponsored by Novartis Pharma AG, Basel, Switzerland.
Data availability statement
The datasets generated and/or analysed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.
References
- 1. Johnson‐Huang LM, Lowes MA, Krueger JG. Putting together the psoriasis puzzle: an update on developing targeted therapies. Dis Model Mech 2012; 5: 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parisi R, Symmons DP, Griffiths CE, Ashcroft DM. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 2013; 133: 377–385. [DOI] [PubMed] [Google Scholar]
- 3. Springate DA, Parisi R, Kontopantelis E, Reeves D, Griffiths CEM, Ashcroft DM. Incidence, prevalence and mortality of patients with psoriasis: a U.K. population‐based cohort study. Br J Dermatol 2017; 176: 650–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burden‐Teh E, Thomas KS, Ratib S, Grindlay D, Adaji E, Murphy R. The epidemiology of childhood psoriasis: a scoping review. Br J Dermatol 2016; 174: 1242–1257. [DOI] [PubMed] [Google Scholar]
- 5. Paller AS, Singh R, Cloutier M et al. Prevalence of psoriasis in children and adolescents in the United States: a claims‐based analysis. J Drugs Dermatol 2018; 17: 187–194. [PubMed] [Google Scholar]
- 6. Eichenfield LF, Paller AS, Tom WL et al. Pediatric psoriasis: evolving perspectives. Pediatr Dermatol 2018; 35: 170–181. [DOI] [PubMed] [Google Scholar]
- 7. Menter A, Cordoro KM, Davis DMR et al. Joint American Academy of Dermatology‐National Psoriasis Foundation guidelines of care for the management and treatment of psoriasis in pediatric patients. J Am Acad Dermatol 2020; 82: 161–201. [DOI] [PubMed] [Google Scholar]
- 8. Eisert L, Augustin M, Bach S et al. S2k guidelines for the treatment of psoriasis in children and adolescents – Short version part 2. J Deutsch Dermatol Ges 2019; 17: 959–973. [DOI] [PubMed] [Google Scholar]
- 9. Augustin M, Glaeske G, Radtke MA, Christophers E, Reich K, Schäfer I. Epidemiology and comorbidity of psoriasis in children. Br J Dermatol 2010; 162: 633–636. [DOI] [PubMed] [Google Scholar]
- 10. Tollefson MM, Van Houten HK, Asante D, Yao X, Maradit Kremers H. Association of psoriasis with comorbidity development in children with psoriasis. JAMA Dermatol 2018; 154: 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beattie PE, Lewis‐Jones MS. A comparative study of impairment of quality of life in children with skin disease and children with other chronic childhood diseases. Br J Dermatol 2006; 155: 145–151. [DOI] [PubMed] [Google Scholar]
- 12. Kara T, Topkarcı Z, Yılmaz S, Akaltun İ, Erdoğan B. Pediatric patients with psoriasis and psychiatric disorders: premorbidity and comorbidity in a case‐control study. J Dermatolog Treat 2019; 30: 129–134. [DOI] [PubMed] [Google Scholar]
- 13. Kimball AB, Wu EQ, Guérin A et al. Risks of developing psychiatric disorders in pediatric patients with psoriasis. J Am Acad Dermatol 2012; 67: 651–657.e651‐652. [DOI] [PubMed] [Google Scholar]
- 14. Fortina AB, Bardazzi F, Berti S et al. Treatment of severe psoriasis in children: recommendations of an Italian expert group. Eur J Pediatr 2017; 176: 1339–1354. [DOI] [PubMed] [Google Scholar]
- 15. Menter A, Strober BE, Kaplan DH et al. Joint AAD‐NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol 2019; 80: 1029–1072. [DOI] [PubMed] [Google Scholar]
- 16. Papp K, Gulliver W, Lynde C, Poulin Y, Ashkenas J. Canadian Psoriasis Guidelines Committee. Canadian guidelines for the management of plaque psoriasis: overview. J Cutan Med Surg 2011; 15: 210–219. [DOI] [PubMed] [Google Scholar]
- 17. Canadian Psoriasis Guidelines Addendum Committee . Addendum to the Canadian Guidelines for the Management of Plaque Psoriasis. J Cutan Med Surg 2009; 20: 375–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nast A, Spuls PI, van der Kraaij G et al. European S3‐Guideline on the systemic treatment of psoriasis vulgaris – Update Apremilast and Secukinumab – EDF in cooperation with EADV and IPC. J. Eur. Acad. Dermatol. Venereol. 2017; 31: 1951–1963. [DOI] [PubMed] [Google Scholar]
- 19. Smith CH, Jabbar‐Lopez ZK, Yiu ZZ et al. British Association of Dermatologists guidelines for biologic therapy for psoriasis 2017. Br J Dermatol 2017; 177: 628–636. [DOI] [PubMed] [Google Scholar]
- 20. Eisert L, Augustin M, Bach S et al. S2k guidelines for the treatment of psoriasis in children and adolescents – short version part 2. J Dtsch Dermatol Ges. 2019; 17: 959–973. [DOI] [PubMed] [Google Scholar]
- 21. Lansang P, Bergman JN, Fiorillo L et al. Management of pediatric plaque psoriasis using biologics. J Am Acad Dermatol 2020; 82: 213–221. [DOI] [PubMed] [Google Scholar]
- 22. Stelara [prescribing information]. Horsham, PA: Janssen Biotech, Inc., November 2019. [Google Scholar]
- 23. Stelara [summary of product characteristics]. High Wycombe, Bucks: Janssen‐Cilag Ltd, January 2020. [Google Scholar]
- 24. Enbrel [prescribing information]. Thousand Oaks, CA: Amgen, March 2020. [Google Scholar]
- 25. Enbrel [summary of product characteristics]. Bruxelles, Belgium: Pfizer Europe MA EEIG, November 2019. [Google Scholar]
- 26. Humira [product of summary characteristics]. Berkshire, UK: AbbVie Ltd, November 2019. [Google Scholar]
- 27. Taltz [prescribing information]. Indianapolis, IN: Eli Lilly and Company, March 2020. [Google Scholar]
- 28. Cosentyx [prescribing information]. East Hanover, NJ: Novartis Pharmaceutical Corporation, January 2020. [Google Scholar]
- 29. Cosentyx [summary of product characteristics]. Camberley, UK: Novartis Europharm Limited, November 2019. [Google Scholar]
- 30. Baeten D, Sieper J, Braun J et al. Secukinumab, an interleukin‐17A inhibitor, in ankylosing spondylitis. N Engl J Med 2015; 373: 2534–2548. [DOI] [PubMed] [Google Scholar]
- 31. Langley RG, Elewski BE, Lebwohl M et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med 2014; 371: 326–338. [DOI] [PubMed] [Google Scholar]
- 32. McInnes IB, Mease PJ, Kirkham B et al. Secukinumab, a human anti‐interleukin‐17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2015; 386: 1137–1146. [DOI] [PubMed] [Google Scholar]
- 33. Thaci D, Blauvelt A, Reich K et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol 2015; 73: 400–409. [DOI] [PubMed] [Google Scholar]
- 34. Zeichner JA, Armstrong A. The role of IL‐17 in the pathogenesis and treatment of psoriasis. J Clin Aesthet Dermatol 2016; 9: S3–S6. [PMC free article] [PubMed] [Google Scholar]
- 35. Mrowietz U, Leonardi CL, Girolomoni G et al. Secukinumab retreatment‐as‐needed versus fixed‐interval maintenance regimen for moderate to severe plaque psoriasis: a randomized, double‐blind, noninferiority trial (SCULPTURE). J Am Acad Dermatol 2015; 73: 27–36.e21. [DOI] [PubMed] [Google Scholar]
- 36. Bissonnette R, Luger T, Thaçi D et al. Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate‐to‐severe psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol 2018; 32: 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Biologics approved for treatment of moderate to severe plaque psoriasis (as of 14 July, 2020)
Table S2. IGA mod 2011 and PASI responses at Weeks 12 and 52 (analyses by body weight and dose groups) (NRI)
Table S3. Treatment emergent serious adverse events, by preferred term (PT) (up to Week 52)
Figure S1. IGA mod 2011 and PASI response up to Week 52 in patients affected and non‐affected by IRT dosing error (NRI)
Figure S2. IGA mod 2011 and PASI response up to Week 52 in non‐affected patients by IRT dosing error in comparison with placebo and etanercept (NRI)
Figure S3. Mean change from baseline in CDLQI total score (LOCF analysis)
Figure S4. Mean change from baseline in individual domain scores of the CDLQI (LOCF analysis)
Data Availability Statement
The datasets generated and/or analysed during the current study are not publicly available. Novartis is committed to sharing with qualified external researchers access to patient‐level data and supporting clinical documents from eligible studies. These requests are reviewed and approved on the basis of scientific merit. All data provided are anonymized to respect the privacy of patients who have participated in the trial in line with applicable laws and regulations. The data may be requested from the corresponding author of the manuscript.