

Abstract
For over a century, investigation of Treponema pallidum subsp. pallidum, the spiral‐shaped bacterium that causes syphilis, was hindered by an inability to culture the organism in vitro. A recent breakthrough has enabled continuous in vitro growth of this organism in co‐culture with mammalian tissue culture cells. This article contains the protocols needed to culture T. pallidum in the standard laboratory environment. In addition, protocols for growing and maintaining the required tissue culture cells, for generating isogenic strains by limiting dilution, and for quantitating T. pallidum by darkfield microscopy are included. © 2021 The Authors. Current Protocols published by Wiley Periodicals LLC.
Basic Protocol 1: In vitro cultivation of Treponema pallidum
Basic Protocol 2: Generation of isogenic strains
Support Protocol 1: Alternate harvest procedure
Support Protocol 2: Culture of Sf1Ep cells
Support Protocol 3: Assessment of T. pallidum number and viability
Keywords: cell culture, in vitro cultivation, laboratory growth, spirochetes, syphilis, Treponema pallidum
INTRODUCTION
The spirochete Treponema pallidum subsp. pallidum was identified as the causative agent of syphilis over 100 years ago (Schaudinn & Hoffman, 1905). Closely related organisms cause yaws (T. pallidum subsp. pertenue), bejel (T. pallidum subsp. endemicum), and venereal spirochetosis of rabbits (T. paraluiscuniculi) (Giacani & Lukehart, 2014; Norris, Paster, Moter, & Göbel, 2003). Despite many efforts over the years, this group of bacteria has remained refractory to long‐term in vitro culture until recently. Isolation of new strains and routine maintenance of stocks has typically required infection of rabbits or other laboratory animals. In addition, an inability to isolate clonal populations in vitro, perform mutational analysis, and implement other standard bacteriologic assays has greatly hampered research on these organisms.
Breakthroughs in understanding the biology of T. pallidum, most notably elucidation of its microaerophilic nature and its improved survival in the presence of mammalian cells, led to the ability to keep the organisms alive for several days in vitro (reviewed by Norris, Cox, & Weinstock, 2001). In 1981, limited replication in a tissue culture system was first reported and quickly confirmed (Fieldsteel, Cox, & Moeckli, 1981; Norris, 1982), but little progress was made in continuous culture (Cox, 1994; Norris & Edmondson, 1987; Norris et al., 2001). The T. pallidum genome sequence (Fraser et al., 1998) coupled with limited available biological information confirmed the organism's minimal biosynthetic capabilities but did not provide further clues regarding the in vitro requirements of the spirochetes.
Long‐term in vitro cultivation of T. pallidum was first achieved in 2017 (Edmondson, Hu, & Norris, 2018) using a modification of the system originally described by Fieldsteel et al. (1981). In the current system, continuous growth in vitro is dependent upon co‐culture with Sf1Ep cottontail rabbit epithelial cells in a specialized tissue culture medium (T. pallidum culture medium 2, or TpCM‐2) under microaerobic conditions (1.5% O2 with 5% CO2). In vitro cultures of T. pallidum Nichols strain can apparently be grown indefinitely (currently >3 years) with full retention of multiplication rate, motility, structural integrity, and infectivity in a rabbit model. Thus far, this culture system has been successfully applied to five T. pallidum subsp. pallidum strains (Nichols, SS14, Mexico A, UW231B, and UW249B) and an isolate of T. pallidum subsp. endemicum (Bosnia A) (Edmondson et al., 2018; Edmondson, DeLay, Kowis, & Norris, 2021). In addition, the method is readily scalable, allowing growth in a variety of formats.
This article provides protocols to culture T. pallidum in vitro from organisms extracted from rabbits or from frozen stocks (Basic Protocol 1) and to isolate isogenic clones in vitro (Basic Protocol 2 and Support Protocol 1). Methods for quantification of organisms (Support Protocol 3) and for maintenance of the mammalian cells used for T. pallidum co‐culture (Support Protocol 2) are also described.
CAUTION: T. pallidum is classified as a Biosafety Level 2 (BSL‐2) organism and requires appropriate containment facilities and institutional approvals.
NOTE: T. pallidum is a microaerobe and is very sensitive to atmospheric levels of oxygen. Care should be taken to minimize the amount of time that the spirochetes spend at high oxygen levels. In addition, T. pallidum is physically fragile and easily damaged. Pipet cultures gently and avoid aeration of the cultures.
NOTE: All procedures should be performed under aseptic conditions using sterile supplies and reagents.
STRATEGIC PLANNING
Biosafety
T. pallidum is a BSL‐2 pathogen and poses moderate health risks to personnel. The primary hazards are accidental contact between virulent cultures and mucous membranes or broken skin or contact with an aerosol spray. Appropriate personal protective equipment (PPE) should be worn when handling T. pallidum cultures, including a lab coat, gloves, and eye protection.
Although laboratory‐acquired syphilis is rare (Fitzgerald, Johnson, & Smith, 1976), the infectious dose for humans is less than 100 organisms (Magnuson et al., 1956). Therefore, laboratory personnel should be evaluated for possible prophylactic treatment in the event of a suspected exposure. In addition, periodic monitoring of personnel for possible inadvertent infection through the use of standard serologic tests for syphilis is prudent.
All culture manipulations are performed in a biological safety cabinet, which has the added advantage of protecting the cultures from contamination. Special care should be taken while transporting culture vessels between the biosafety hood and the incubator. Treatment of surfaces with 70% ethanol, 10% bleach, or other disinfectants is adequate for routine disinfection. All culture materials must be autoclaved or disinfected prior to disposal.
Equipment
As a microaerophile, T. pallidum requires small quantities oxygen for survival and growth yet is highly sensitive to the toxic effects of reactive oxygen species (ROS). Therefore, T. pallidum must be maintained in a low‐oxygen environment (Basic Protocols 1 and 2). T. pallidum is most conveniently cultured in a humidified tissue culture incubator with variable oxygen control (a “tri‐gas” incubator). The tri‐gas incubator is connected to a CO2 tank and to an N2 tank. The incubator is set to maintain CO2 at 5% and O2 at 1.5% and a temperature of 34°C. The nitrogen replaces air until the oxygen concentration falls to the set level. A tank switcher is a valuable accessory that helps to ensure that low oxygen is maintained. The incubator uses nitrogen at a rate of 1 to 2 tanks per week depending on how frequently the incubator is opened. Another option is to use an airtight chamber that can be filled with the appropriate gas mixture and placed in a regular tissue culture incubator maintained at 34°C. A GasPak™ 150 vented anaerobic jar (Brewer jar) can be used with equivalent results. The vent is necessary for filling of the jar with low‐oxygen gas mixture.
The Brewer jar is also used during pre‐equilibration of the TpCM‐2 and the Sf1Ep cultures before and after inoculation with T. pallidum (Basic Protocols 1 and 2). Airtight tubing is connected to the vent of the jar, and a vacuum is drawn in the jar using house vacuum or another source (about 12 to 18 in. Hg). The jar is then slowly refilled with 95% N2 and 5% CO2 from a tank containing the gas mixture (see Fig. 1). The procedure is repeated three times for a total of four refills before a final vacuum and refill using a custom gas mixture of 93.5% N2, 5% CO2, and 1.5% O2. Cultures or medium is then removed from the jar and quickly transferred to the low‐oxygen incubator. If incubation is to be carried out in the Brewer jar, the hose on the vent is clamped tightly, the jar is disconnected from the vacuum/gas system, and the entire jar is transferred to the 34°C incubator. It is critical that the hose provides an airtight seal. Thick‐walled Tygon® tubing does not provide a seal that adequately excludes atmospheric oxygen over the entire culture period, so new latex tubing or thin‐walled Tygon® tubing should be used with two clamps.
Figure 1.
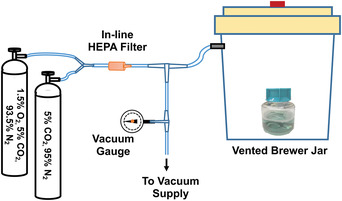
Diagram of the arrangement to evacuate and fill a Brewer jar with a low ‐oxygen gas mixture. Airtight tubing is connected to the vent of a Brewer jar. The tubing connects via a T‐joint to a vacuum supply (usually house vacuum) and to gas cylinders containing custom gas mixtures (5% CO2, balance nitrogen and 1.5% O2, 5% CO2, balance nitrogen). The vacuum is drawn in the jar (about 12 to 18 in. Hg) and is measured using an in‐line vacuum gauge. The evacuated jar is then slowly refilled with the gas mixtures: the Brewer jar is refilled four times with 5% CO2, balance nitrogen before a final vacuum and refill with 1.5% O2, 5% CO2, balance nitrogen. Culture or medium is then removed from the jar and quickly transferred to a low‐oxygen incubator. Alternatively, the tubing between the Brewer jar and the first T‐joint can be clamped tightly, the tubing disconnected from the T‐joint, and the entire Brewer jar transferred to a 34°C incubator.
A microscope with a darkfield condenser is needed for visualization and quantification of T. pallidum (Support Protocol 3). The spirochetes are quite thin (0.18 μm in diameter) and cannot be visualized using standard brightfield microscopy. Although they can be seen by phase‐contrast microscopy, darkfield microscopy is simpler and provides better contrast. A 40× objective lens and 15× ocular lenses are recommended. Quantitation of T. pallidum can be performed using standard glass slides and coverslips (see Current Protocols article; Lukehart & Marra, 2007), but we find that Helber counting chambers with Thoma rulings (Hawksley brand) provide more accurate and reproducible results. An inverted microscope is needed for monitoring and quantifying Sf1Ep cells (using a hemocytometer; Support Protocol 2).
Reagents
T. pallidum is very sensitive to the quality of reagents used for culture, and especially heat‐inactivated fetal bovine serum (FBS). We routinely pre‐screen all FBS lots and find that FBS from different suppliers varies considerably in its ability to support T. pallidum survival and replication (Cox, 1994; Fieldsteel, Stout, & Becker, 1981; Norris & Edmondson, 1986). Different catalog numbers from the same supplier and even different lot numbers can affect in vitro culture of T. pallidum. A particular heat‐inactivated FBS product from Sigma‐Aldrich (cat. no. F4135) has proven to be consistently capable of supporting T. pallidum multiplication and is recommended for in vitro culture. In our studies, newborn calf serum or bovine serum has not been capable of supporting in vitro multiplication.
All reagents should be cell culture grade. Tracking lot numbers of all reagents allows identification of problem medium components in the event of culture failure.
Basic Protocol 1. IN VITRO CULTIVATION OF Treponema pallidum
In vitro cultures of T. pallidum can be initiated from fresh or frozen samples isolated from rabbits or from frozen in vitro cultures. The T. pallidum stock is inoculated into a previously prepared culture of Sf1Ep cells (Support Protocol 2). The cultures are incubated for 1 week, during which the T. pallidum will undergo 4 to 5 successive cell divisions. The spirochetes can be observed attached to the mammalian cells and free in the medium. Approximately 50% to 60% of the organisms are associated with the Sf1Ep cells in actively growing cultures and can be released from the cells by treatment with a trypsin‐EDTA solution. A small aliquot of the trypsinized sample is used to inoculate a new culture. The remaining culture can be used for other experimental purposes.
Materials
T. pallidum extracted from infected rabbit tissue (see Current Protocols article; Lukehart & Marra, 2007) or in vitro cultures, fresh or frozen
Trypsin‐EDTA (e.g., Sigma, T4049)
50% (w/v) glycerol in phosphate‐buffered saline (PBS), filter‐sterilized
Tissue culture incubator maintained at 37°C, 5% CO2
Sterile 50‐ml conical centrifuge tubes or sterile polyethylene medium receiver flasks
Filtration units with polyethersulfone (PES) filters with 0.22‐μm pore size (e.g., Millipore SteriCupTM or SteriFlipTM units)
Vented Brewer jar: GasPak™ 150 vented anaerobic jar (BD BBL™, 260629) or equivalent chamber
Vacuum source (e.g., house vacuum or vacuum pump), gauge, tubing, and connectors
-
Gas cylinders containing custom gas mixtures:
5% CO2 and 95% N2
1.5% O2, 5% CO2, and 93.5% N2
Humidified tri‐gas incubator maintained at 34°C, 1.5% O2, 5% CO2, and 93.5% N2 (e.g., Thermo Scientific Heracell VIOS Tri‐Gas CO2 Incubator)
Incubator tank switch (e.g., NuAire NU‐1550 TankGuard Automatic CO2 Incubator Tank Switch; optional)
Standard tabletop centrifuge (optional)
Inverted microscope
Sterile 15‐ml conical centrifuge tubes (e.g., Falcon, 353046)
Cryovials
Additional reagents and equipment for preparing Sf1Ep cells for T. pallidum culture (see Support Protocol 2, step 18b), preparing TpCM‐2 medium (see Table 1), and quantifying T. pallidum (see Support Protocol 3)
Table 1.
T. pallidum Cultivation Medium 2 (TpCM‐2)
| Component | Amount for 50 ml | Final concentration | Recommended manufacturer/catalog no. |
|---|---|---|---|
| 1× CMRL 1066 without l‐glutamine or phenol red | 37 ml | 0.8× | US Biological/C5900‐03A |
| Sodium pyruvate | 364 µl | 0.73 mM | Sigma/S8636 |
| 0.1% (w/v) resazurin | 50 µl | 0.001% | Sigma/R7017 |
| 1 M MOPS, pH 7.5 | 1 ml | 20 mM | Sigma/M3183 |
| 7.5% (w/v) sodium bicarbonate | 1.08 ml | 19.2 mM | Sigma/S8761 |
| 200 mM l‐glutamine | 500 µl | 2 mM | Sigma/G7513 |
| 100× d‐glucose (15% in water) | 500 µl | To 17.6 mM | Sigma/G6152 |
| 10 g/dl d‐mannitol (10% in water) | 80 µl | 0.88 mM | Sigma /M1902 |
| 5 g/dl l‐histidine (5% in water) | 80 µl | 0.52 mM | Sigma/H6034 |
| dl‐Dithiothreitol | 4 mg | 0.52 mM | Sigma/D9779 |
| Fetal bovine serum, heat inactivated | 10 ml | 20% | Sigma/F4135 |
© American Society for Microbiology (Edmondson et al., 2018). Catalog no. for sodium bicarbonate updated.
Seed Sf1Ep cells
-
1
One day prior to T. pallidum culture initiation or passage, seed Sf1Ep cells into 6‐well tissue culture plates with low‐evaporation lids at 0.5–1.0 × 105 cells per well in Sf1Ep medium (see Support Protocol 2, step 18b).
Six‐well plates are a convenient size for general experiments: they are easy to handle, require small amounts of materials, and allow for several biologic replicates and conditions in each plate. Each well typically produces between 4 × 107 and 2 × 108 T. pallidum, enough for rabbit inoculation or for DNA or protein extraction.
The cultivation procedure is readily scalable. Smaller wells (e.g., in 12‐, 24‐, or 96‐well culture dishes) are convenient for testing many conditions in a smaller format, whereas 75‐cm2 tissue culture flasks can be used for experiments requiring large numbers of organisms. The number of organisms that can be harvested from differently sized culture vessels is roughly proportional to the surface area of the vessel and the volume of medium. Table 2 gives cell densities and medium volumes for culture vessels of various sizes.
Table 2.
Recommended Parameters for Different T. pallidum Co‐Culture Formats
| Culture vessel | Surface area (cm2) | No. of Sf1Ep cells seeded per well | TpCM‐2 volume (ml) | T. pallidum inoculum |
|---|---|---|---|---|
| 75‐cm2 | 75.0 | 8.0 × 105 | 20 | ∼4 × 107 |
| 6‐well | 9.6 | 1.0 × 105 | 4 | 1.25–5.0 × 106 |
| 12‐well | 3.5 | 4.0 × 104 | 2 | 0.25–1 × 106 |
| 24‐well | 1.9 | 2.0 × 104 | 1.5 | 0.1–0.5 × 106 |
| 96‐well | 0.32 | 3.5 × 103 | 250 | <1 × 105 |
-
2
Incubate overnight in a tissue culture incubator maintained at 37°C, 5% CO2, to allow cells to attach and spread.
The cells should be ∼10% confluent.
Prepare TpCM‐2
-
3
One day prior to T. pallidum culture initiation or passage, prepare TpCM‐2 medium (see Table 1) in sterile 50‐ml conical tubes or sterile polyethylene medium receiver flasks. Filter‐sterilize medium after preparation using filtration units with PES filters with 0.22‐μm pore size.
Each 6‐well tissue culture dish requires 30 ml TpCM‐2. We have found that TpCM‐2 frozen at −20°C can be stored for up to 6 months prior to use with no loss in efficacy. The cap of the tube containing the frozen medium is loosened, and the tube is pre‐equilibrated in a low‐oxygen atmosphere and allowed to thaw overnight in the tri‐gas incubator prior to use. A non‐“frost‐free” freezer (with no defrosting cycle) should be used for storage.
-
4
Pre‐gas TpCM‐2 by placing the container (with a loosened lid) in a vented Brewer jar and exchanging atmosphere in the jar five times. Connect Brewer jar to a vacuum source and gauge with vacuum tubing and connectors (see Fig. 1). Draw a vacuum (12 to 18 in. Hg) in jar using vacuum source. Refill jar with 5% CO2 and 95% N2. Repeat vacuum‐and‐refilling procedure four additional times, using 1.5% O2, 5% CO2, and 93.5% N2 for the final refill.
-
5
Quickly transfer TpCM‐2 to a humidified tri‐gas incubator maintained at 34°C, 1.5% O2, 5% CO2, and 93.5% N2 (hereafter referred to as the low‐oxygen incubator) and continue pre‐equilibration overnight. Leave lid of the medium container loose to facilitate pre‐equilibration. Use an incubator tank switch if preferred.
Alternatively, after gas exchange in the Brewer jar, seal the gas inlet of the jar with a hose clamp and transfer the entire jar to a 34°C incubator.
Initiate culture
-
6
Three to four hours prior to the start of an experiment or passage, remove Sf1Ep medium from the 6‐well plates (see step 2) by aspiration.
-
7
Rinse each well with 1 ml pre‐equilibrated TpCM‐2 (prepared the previous day; see step 5) to remove traces of Sf1Ep medium.
-
8
Aspirate and discard TpCM‐2 rinse.
-
9
Add 4 ml pre‐equilibrated TpCM‐2 to each well.
-
10
Place 6‐well plates in the Brewer jar and replace air in the Brewer jar with an atmosphere containing 1.5% O2, 5% CO2, and 93.5% N2 as described in step 4. Quickly transfer plates to the low‐oxygen incubator and allow to equilibrate for at least 3 to 4 hr.
-
11
Centrifuge fresh T. pallidum extracted from infected rabbit tissue (see Current Protocols article; Lukehart & Marra, 2007) in pre‐equilibrated TpCM‐2 for 5 min at 125 × g at room temperature to remove host cells and collect supernatant. Alternatively, thaw a frozen aliquot of T. pallidum extracted from infected rabbit tissue or from in vitro cultures at room temperature or 37°C.
-
12
Dilute fresh or frozen T. pallidum stock in TpCM‐2, such that the inoculum contains 2–5 × 106 motile T. pallidum.
The inoculum volume is typically 200 to 750 µl, although lower or higher volumes may be used.
-
13
Remove Sf1Ep cultures (see step 10) from the low‐oxygen incubator and inoculate them with prepared T. pallidum.
-
14
Place 6‐well plates in the Brewer jar and replace air in the jar with the low‐oxygen mixture described in step 4. Quickly transfer plates to the low‐oxygen incubator.
Be sure that the base of the culture vessel is well supported during transport to avoid accidental spillage.
-
15
Incubate T. pallidum cultures for 7 days in the low‐oxygen incubator.
T. pallidum has a very slow doubling time of 32 to 40 hr. Seven days provides for 4 to 5 generations and is a convenient interval between subcultures.
Culture time can be extended a few days by feeding T. pallidum cultures at 3‐ to 5‐day intervals. Feed the cultures by removing half the TpCM‐2 and replacing it with fresh medium. Despite this procedure, the Sf1Ep cultures may overgrow and fail to support T. pallidum growth after 10 to 14 days of culture.
Subculture and harvest cells
-
16
One day prior to harvest or passage, prepare fresh Sf1Ep cultures and TpCM‐2 as in steps 1 to 5.
-
17
On the day of passage, change medium of the Sf1Ep cultures and pre‐equilibrate as in steps 6 to 10.
-
18
Immediately prior to harvest, remove T. pallidum cultures from the low‐oxygen incubator and examine them with an inverted microscope to assess confluency and overall appearance of the Sf1Ep cells.
-
19
Remove TpCM‐2 growth medium from each well and reserve it in separate sterile 15‐ml conical centrifuge tubes.
Measure the volume of medium to ensure that there has not been significant evaporation.
When the use of trypsin might be deleterious to an experiment, perform Support Protocol 1 as an alternative to steps 19 to 24.
-
20
Rinse each well with 0.35 ml trypsin‐EDTA and transfer rinse to the reserved medium.
-
21
Add an additional 0.35 ml trypsin‐EDTA to each well and incubate plates in the 37°C, 5% CO2 tissue culture incubator for 5 min.
-
22
View plates with the inverted microscope to ensure that the Sf1Ep cells are rounded or floating. Rap edge of each plate to dislodge the Sf1Ep cells from the plate and the T. pallidum from the cells.
Complete trypsinization is important to maximize T. pallidum recovery. Typically, ∼50% of the T. pallidum are attached to the Sf1Ep monolayer in a healthy culture, and these are released by trypsinization. Hold the 6‐well plate flat on the stage of the inverted microscope and rap the side of the plate with a polypropylene rack for microcentrifuge tubes to dislodge the cells from the plate. If the cells do not dislodge, add additional trypsin‐EDTA and incubate at 37°C for 5 min more.
-
23
When trypsinization is complete and all the Sf1Ep cells are detached, add reserved medium (see step 20) back to the corresponding well to stop the trypsinization.
-
24
Rinse well with the reserved medium and return combined trypsin and medium to the sterile 15‐ml conical centrifuge tube.
-
25
Optional: Centrifuge 5 min at 125 × g to pellet Sf1Ep cells.
This step reduces variability due to Sf1EP cell “carryover.”
-
26
Use 125 to 750 µl (typically 250 µl) trypsinized, optionally centrifuged culture to inoculate new 6‐well Sf1Ep cultures prepared in steps 16 to 17.
Three replicate cultures per condition are recommended.
The volume should be adjusted to yield an inoculum of 2–6 × 106 T. pallidum. Slow‐growing strains may routinely require higher inoculation volumes.
-
27
Gas inoculated cultures in the Brewer jar and return new cultures to the low‐oxygen incubator as in step 4.
-
28
Quantify harvested T. pallidum by darkfield microscopy (see Support Protocol 3).
Freeze in vitro cultures
-
29
Place 1 ml trypsinized culture into a cryovial and add 250 µl sterile 50% glycerol (final glycerol concentration of 10%).
-
30
Mix gently by pipetting or inversion and freeze culture at −80°C.
Frozen T. pallidum is thawed at room temperature or 37°C and rapidly inoculated into fresh cultures as described in steps 26 to 27. The cultures may exhibit sluggish motility following thawing.
Basic Protocol 2. GENERATION OF ISOGENIC STRAINS
Clonal populations of T. pallidum can be generated using limiting dilution. An actively growing culture of T. pallidum (Basic Protocol 1) is diluted to a concentration of 0.3 to 2 T. pallidum per well in 96‐well plates containing pre‐seeded Sf1Ep cultures and 200 µl TpCM‐2. The cultures are “fed” once after 1 week of incubation. After 2 weeks, a portion of the culture is used to inoculate a freshly prepared 96‐well plate. After another 2 weeks of incubation, each well is assessed for the presence of viable T. pallidum by darkfield microscopy (Support Protocol 3). Wells positive for spirochetes are slowly expanded into larger dishes. Due to the organism's slow generation time, it may require 2 months to generate a sufficient quantity of a clonal population for characterization, storage by freezing, and use in experiments.
Additional Materials (also see Basic Protocol 1)
In vitro culture of T. pallidum (see Basic Protocol 1, step 15)
96‐well plates (low‐evaporation, clear, flat‐bottom, tissue culture–treated microplates; e.g., Corning Falcon®, 353072)
24‐well plates (low‐evaporation, clear, flat‐bottom, tissue culture‐treated microplates)
12‐well plates (optional)
Additional reagents and equipment for assessing clonality by sequencing tprK locus (see Current Protocols article; Lukehart & Marra, 2007)
-
1
One day prior to the start of the experiment, seed 96‐well plates with 1000 to 3000 Sf1Ep cells per well in 150 µl Sf1Ep medium. Incubate in a tissue culture incubator at 37°C, 5% CO2 overnight to allow cells to attach and begin to grow.
Typically, two plates are prepared for each cloning procedure.
-
2
Prepare TpCM‐2 medium (see Table 1), pre‐gas, and pre‐equilibrate overnight in low‐oxygen incubator (see Basic Protocol 1, steps 3 to 5).
-
3
On the day of the experiment, aspirate medium from each well of the 96‐well plates.
Aspiration of medium from a 96‐well plate with a single‐channel aspirator is quite time consuming. A multi‐channel adapter for the aspirator (e.g., Integra, 155520) is helpful although not essential.
-
4
Rinse each well with 50 µl pre‐equilibrated TpCM‐2 to remove traces of Sf1Ep medium.
Rinsing and filling of the wells are best accomplished by adding the TpCM‐2 to a sterile 50‐ or 100‐ml reagent reservoir and using a 200‐µl multichannel pipet to fill the wells of the 96‐well plate.
-
5
Aspirate TpCM‐2 rinse.
-
6
Add 200 µl TpCM‐2 to each well.
-
7
Pre‐gas 96‐well plates in the Brewer jar and incubate 3 hr in low‐oxygen incubator as described in step 10 of Basic Protocol 1.
-
8
Trypsinize an in vitro culture of T. pallidum (see Basic Protocol 1, steps 19 to 24).
-
9
Count number of T. pallidum by darkfield microscopy and calculate T. pallidum concentration (see Support Protocol 3).
-
10
Dilute culture with pre‐equilibrated TpCM‐2 (see step 2) to a concentration of 10 motile organisms/ml.
-
11
Add 50 µl diluted culture to each well of prepared 96‐well plates (see step 7).
This dilution of T. pallidum results in a plating density of 0.5 organisms per well. We have used concentrations between 0.3 and 2 organisms per well with good results. It is recommended that control wells with higher concentrations of T. pallidum (e.g., 100 per well) be included to ascertain whether the experimental conditions are conducive to T. pallidum survival and multiplication.
-
12
Place 96‐well plates in the Brewer jar and replace atmosphere in the jar as described in step 4 of Basic Protocol 1.
-
13
Quickly transfer plates back to the low‐oxygen incubator.
-
14
After 6 days of incubation, prepare fresh TpCM‐2 and pre‐equilibrate as in step 2.
-
15
On day 7, feed cultures by removing 100 µl medium from each well and then adding 100 µl fresh TpCM‐2.
IMPORTANT NOTE: Use a different pipet tip for each well.
If significant evaporation is observed, add extra TpCM‐2 to bring the volume back to 200 to 250 µl per well.
-
16
After 13 days of incubation, prepare new Sf1Ep cultures in 96‐well plates and new TpCM‐2 (see steps 1 to 2).
-
17
On day 14, exchange medium in the 96‐well plates so that each well contains 200 µl fresh TpCM‐2 (see steps 3 to 7).
-
18
Using supernatant from the 14‐day 96‐well plates as inoculum, seed new plates from step 16 with 50 µl culture from step 17.
After 2 weeks of incubation, the Sf1Ep monolayer will begin to fail, so the potential clones need to be transferred to a fresh Sf1Ep monolayer. Using a larger inoculum adds more “spent” medium to the fresh plates and leads to earlier failure of the new 96‐well Sf1Ep cultures. After the initial 2‐week culture period, a 50‐µl inoculum from a well initially containing a single viable organism is estimated to have expanded to 120 to 140 organisms. (This estimate assumes about five T. pallidum generations per week, attachment of ∼50% of the treponemes to cells, and dilution by feeding at 1 week.)
-
19
Prepare fresh TpCM‐2 on day 20, pre‐equilibrate overnight, and feed cultures on day 21 (as in steps 14 to 15).
-
20
On day 27 of culture, assess each well for T. pallidum growth by darkfield microscopy (see Support Protocol 3) or quantitative PCR. To prevent extended exposure to oxygen, move 50 µl supernatant to a new 96‐well plate and return cloning plate to the incubator.
For slow‐growing strains, feed or passage to a third 96‐well plate if no positive wells are observed (see step 15 or steps 16 to 18).
-
21
If positive wells are observed, prepare fresh TpCM‐2 and pre‐equilibrate overnight in low‐oxygen incubator (see step 2). Seed 24‐well plates with 4 × 104 Sf1Ep cells per well and incubate overnight.
-
22
On day 28, rinse wells with 0.5 ml TpCM‐2 and add 1.5 ml TpCM‐2 per well to 24‐well Sf1Ep cultures. Pre‐equilibrate in low‐oxygen incubator for ≥3 hr (see Basic Protocol 1, step 10).
-
23
Expand wells that contain viable T. pallidum (see Support Protocol 3) by trypsinizing the positive wells and transferring trypsinized culture to the freshly prepared 24‐well plate.
Trypsinization is carried out in a similar manner as trypsinization of 6‐well dishes (see Basic Protocol 1, steps 19 to 24), but on a smaller scale. A 96‐well plate is convenient to hold the reserved medium instead of individual tubes, and 30 µl trypsin‐EDTA is sufficient for rinsing and trypsinizing each well. The entire contents of a trypsinized well can be used to inoculate one well of the 24‐well plate. The 24‐well plate is pre‐gassed as usual (see Basic Protocol 1, step 4) and returned to the low‐oxygen incubator.
-
24
After a 1‐week incubation, examine an aliquot of supernatant of each expanding clone for spirochetes by darkfield microscopy (see Support Protocol 3).
-
25a.
If >2 T. pallidum/field are observed: Slowly expand clones by passage into 12‐well and then 6‐well plates.
-
25b.
If <2 T. pallidum/field are observed: Feed cultures by replacing half the medium with fresh TpCM‐2 and incubating another 3 to 5 days.
It is usually better to passage the culture after 10 to 12 days, even if the organisms are at very low density, as the Sf1Ep cultures will begin to fail.
-
26
Assess clonality by sequencing tprK locus (see Current Protocols article; Lukehart & Marra, 2007).
Support Protocol 1. ALTERNATE HARVEST PROCEDURE
When the use of trypsin might be deleterious to an experiment (e.g., for T. pallidum protein analysis), T. pallidum can also be removed from Sf1Ep cells using dissociation medium. Dissociation medium consists of FBS that has been dialyzed against PBS or Earle's basic salt solution (EBSS) without calcium chloride and magnesium chloride to remove divalent cations to generate a simplified T. pallidum cultivation medium.
Additional Materials (also see Basic Protocol 1)
-
1
Remove TpCM‐2 growth medium from each well of an in vitro culture of T. pallidum and reserve it in separate sterile 15‐ml conical centrifuge tubes.
-
2
Rinse each well with 0.35 ml dissociation medium and combine rinse with the reserved medium.
-
3
Add an additional 0.65 ml dissociation medium to each well and incubate plates in the low‐oxygen incubator for 30 min.
-
4
View plates with the inverted microscope. Rap plates sharply to dislodge the Sf1Ep cells from the plates and the T. pallidum from the cells.
See the annotation to Basic Protocol 1, step 22. Complete removal of Sf1Ep cells from the culture vessel is important to maximize T. pallidum recovery from the cells. If the cells do not dislodge, continue incubation for an additional 10 to 15 min. Make sure the cell monolayer does not dry out.
-
5
Add reserved culture medium back to the cognate wells.
-
6
Rinse wells with the reserved medium and return combined dissociation medium and culture medium to the sterile 15‐ml conical centrifuge tube.
Support Protocol 2. CULTURE OF Sf1Ep CELLS
Sf1Ep cells obtained directly from ATCC® are derived from primary cells and have a finite lifespan of about 27 to 30 passages. Thus, it is important to make a large frozen seed stock of Sf1Ep cells. These cells have an epithelial‐like morphology and take on a polygonal or cobblestone appearance when they reach confluence. Low‐passage Sf1Ep cells grow slowly and are subcultured 1:5 every 1 to 2 weeks.
We have an apparently immortal cell line of Sf1Ep cells that arose spontaneously from the ATCC® cells. These cells grow much faster than the original cells, grow to higher densities, and take on a spindle‐shaped cell morphology when they reach high densities. Initially, these support T. pallidum growth (Basic Protocol 1) as well as the original Sf1Ep cells and are easier to produce in large quantities. However, after reaching passage 60 to 70, T. pallidum growth becomes reduced; thus, Sf1Ep cells above passage 60 should be discarded, and a lower‐passage culture of cells should be thawed. We can supply this cell line to anyone wishing to initiate in vitro cultivation. These are subcultured at 1:15 every week.
Materials
Sf1Ep medium (see recipe)
Frozen stock of Sf1Ep (NBL‐11) cottontail rabbit epithelial cells (Sf1Ep cells; ATCC® CCL‐68™ or equivalent)
70% (v/v) ethanol
Sterile PBS (without calcium chloride and magnesium chloride; e.g., Sigma, D8537)
Trypsin‐EDTA (e.g., Sigma, T4049)
Freezing medium: Sf1Ep medium (see recipe) with 10% (v/v) dimethylsulfoxide (DMSO; sterile cell culture grade; e.g., Sigma, D2650)
Humidified tissue culture incubator maintained at 37°C, 5% CO2
37°C water bath
Sterile 15‐ and/or 50‐ml conical centrifuge tubes (e.g., Falcon, 353046, and Nunc, 339653)
Sterile 5‐ml disposable pipet
Low‐speed centrifuge
75‐cm2 tissue culture flasks (T75 flasks; e.g., Corning, 43061U)
Inverted microscope
Hemocytometer (optional)
6‐well plates with low‐evaporation lids (e.g., Falcon, 353046)
2‐ml cryogenic vials (e.g., Corning, 430659)
Cell‐freezing container (e.g., Thermo Fisher Scientific, 5001‐0001)
Cryogenic liquid nitrogen cell culture storage tank
Thaw Sf1Ep stocks
-
1
Pre‐warm Sf1Ep medium in a humidified tissue culture incubator maintained at 37°C, 5% CO2.
-
2
Quickly thaw a frozen stock of Sf1Ep cells in a water bath at 37°C.
-
3
Wipe outside of vial with 70% ethanol.
-
4
Slowly add 1 ml warm Sf1Ep medium (see step 1) to vial and gently mix with thawed cell stock by pipetting up and down.
The newly thawed cells are fragile and should be handled gently.
-
5
Transfer cell stock to a sterile 15‐ or 50‐ml conical centrifuge tube, add an additional 5 ml warm Sf1Ep medium, and gently mix with a sterile 5‐ml disposable pipet.
-
6
Centrifuge Sf1Ep cells in a low‐speed centrifuge for 7 min at 125 × g.
Steps 6 to 8 are optional but improve recovery of the newly thawed cells. If omitting steps 6 to 8, place the 7 ml cells from step 5 directly into a T75 flask containing 8 ml Sf1Ep medium. The following day, remove one‐half of the medium and replace with fresh warm Sf1Ep medium to dilute the residual DMSO used for freezing.
-
7
Remove and discard supernatant from cell pellet.
-
8
Gently resuspend cell pellet in 5 ml warm Sf1Ep medium and transfer entire amount to a T75 flask containing 10 ml warm Sf1Ep medium.
-
9
Incubate in humidified tissue culture incubator maintained at 37°C, 5% CO2. If using a non‐vented flask, make sure cap is loosened.
Growth of Sf1Ep cell cultures is monitored by observation with an inverted microscope. When the cells become ∼90% confluent, they are ready to be used for T. pallidum culture (see Basic Protocol 1) or to be subcultured (see steps 10 to 19). Cell life can be extended by feeding the cell culture once a week by replacement of one‐half the medium with fresh medium. When the cultures grow to very high density, the cells become quiescent and take longer to recover after subculture.
Maintenance of Sf1Ep stocks and preparation of Sf1Ep cells for T. pallidum culture
-
10
Aspirate cell culture medium and discard.
-
11
Add 5 ml sterile PBS to flask, close flask, and rinse cell monolayer to remove the Sf1Ep medium from the monolayer.
Serum in the medium will inhibit trypsinization.
-
12
Aspirate and discard PBS.
-
13
Add 2.5 ml trypsin‐EDTA to flask.
-
14
Tighten cap, spread trypsin‐EDTA over the cell monolayer, and place flask in the 37°C, 5% CO2 incubator for 5 min.
-
15
Observe cell monolayer under an inverted microscope to confirm complete removal of the cells from the flask. Rap flask sharply to dislodge the Sf1Ep cells from the growth surface.
-
16
Add 5 ml warm Sf1Ep medium to trypsin in the flask and rinse surface that once hosted the monolayer.
-
17
Transfer trypsin‐medium mixture to a sterile 15‐ml conical centrifuge tube. Gently mix with a pipet to disperse the cells.
-
18a
To maintain working stocks of Sf1Ep cells: Transfer an aliquot of trypsin‐medium mixture to a new T75 flask. Split cells about 1:30 for transformed Sf1Ep cells (500 µl per 15 ml Sf1Ep medium) or 1:10 for low‐passage, untransformed Sf1Ep cells (1.5 ml per 15 ml Sf1Ep medium).
-
18b
For T. pallidum culture: Quantitate cells with a hemocytometer. Calculate required number of Sf1Ep cells and dilute cells in warm Sf1Ep medium such that each 2‐ml aliquot will contain 0.5–1.0 × 105 cells. Distribute diluted cells into 6‐well plates with low‐evaporation lids.
Gently pipet up and down before removing a sample to load the hemocytometer or distributing the diluted sample into the 6‐well dishes. The cells settle to the bottom of the tube very quickly.
-
19
Incubate 7 days in a 37°C, 5% CO2 incubator before routine Sf1Ep passage (see step 18a). For T. pallidum culture, use within 1 to 2 days (see Basic Protocol 1).
Sf1Ep cells can be conveniently frozen at the time of passage (see steps 20 to 26).
Freezing Sf1Ep cells
-
20
Trypsinize Sf1Ep cultures as described in steps 10 to 17.
-
21
Pellet trypsinized Sf1Ep cells in a low‐speed centrifuge for 7 min at 125 × g.
-
22
Remove and discard supernatant, being careful not to disturb the small whitish pellet.
-
23
Resuspend cells gently in Sf1Ep freezing medium at a concentration of 1–4 × 106 cells/ml.
-
24
Aliquot 1 ml cell suspension into each 2‐ml cryovial.
-
25
Place cells in a cell‐freezing container and freeze overnight at −80°C.
Mammalian cell viability is improved by slow freezing, typically at about −1°C/min. Specialized freezing racks and programmable coolers are available. A less expensive, low‐tech alternative is to use a styrofoam test tube rack. The styrofoam rack should be ∼1 in. deep to slow cooling of the cryovials when placed in the freezer.
-
26
Move to a cryogenic liquid nitrogen cell culture storage tank after freezing.
Frozen stocks of mammalian cells should be stored in liquid nitrogen or liquid nitrogen vapor in a cryogenic cell storage canister. The cells retain viability for at least several weeks at −80°C but should be maintained at liquid nitrogen temperatures for longer storage.
Support Protocol 3. ASSESSMENT OF T. pallidum NUMBER AND VIABILITY
The most common methods for counting bacteria are the standard plate count method and spectrophotometric (turbidimetric) analysis. Neither of these methods is practical for T. pallidum because the spirochete cannot be plated on solid agar and does not grow to a high enough density to assess turbidity. Turbidity measurement is also complicated by the presence of the tissue culture cells used in T. pallidum in vitro cultivation (Basic Protocols 1 and 2). This protocol describes the use of a Helber chamber for enumeration of T. pallidum.
The Helber chamber is a glass slide with an etched grid surrounded by a circular well. The grid consists of 16 large squares, and each of these large squares is further subdivided into 25 squares (Fig. 2). The top row and the left column of each large square are bisected by another etched line to aid in orientation. Each large square has an area of 1/16 mm2. When a drop of culture is sandwiched between the surface of the slide and the Helber chamber cover glass, a space with a depth of 0.02 mm is formed. Thus, the volume of liquid contained within a single large square is 1.25 × 10‐3 mm3.
Figure 2.

Helber chamber grid. The Helber chamber grid consists of 16 large squares, and each of these large squares is further subdivided into 25 squares. One large square is outlined in red. The top row and the left column of each large square are bisected by another etched line to aid in orientation.
T. pallidum viability is closely related to motility, so the motility of each organism counted should be assessed to determine the viability of the culture. Some movement of organisms occurs due to Brownian motion. Motile organisms rotate on their axis or flex longitudinally.
NOTE: In the absence of a Helber chamber, T. pallidum can be quantitated by the method of Miller (1971) using a standard microscope slide and coverslip (see Current Protocols article; Lukehart & Marra, 2007).
Materials
Helber counting chamber (with cover glass and Thoma ruling; Hawksley)
Lint‐free paper tissues (e.g., Kimwipes)
Darkfield microscope with 40× objective and (preferably) 15× ocular lens
-
1
Clean Helber counting chamber by gently wiping the slide chamber and the cover glass with a lint‐free paper tissue wetted with 70% ethanol. Polish chamber and cover glass with a dry tissue.
Dust and debris refract light under darkfield illumination and make enumeration of T. pallidum more difficult.
-
2
Place 4.5 μl T. pallidum sample within engraved circle on the slide.
The manufacturer's instructions for the Helber chamber suggest using 10 µl, but >4.5 µl frequently results in overfilling of the well and seepage of sample under the cover glass outside of the sample chamber.
-
3
Place Helber chamber cover glass onto the slide and press firmly but gently on edges of the cover glass.
Rainbow‐like rings of color should appear between the cover glass and the slide (Newton's refraction rings).
-
4
Examine slide under a darkfield microscope with a 40× objective and 15× ocular lens.
One large square containing 25 small squares is inscribed within the field created by a 40× objective and 15× ocular lens. If using a 10× ocular lens, the field is larger, so one large square will not fill most of the field. Viewing and enumerating the spirochetes with a 10× ocular lens is more challenging.
-
5
Count number of T. pallidum lying within the inscribed square. Focus up and down to include spirochetes at different depths of field.
Count bacteria crossing the upper horizontal or left‐hand vertical borders as being within the square, whereas those crossing the lower or right‐hand boundaries are ignored.
-
6
Count enough squares to have a count of greater than 60 T. pallidum or, if the T. pallidum numbers are low, count all 16 large squares.
-
7
Perform duplicate counts on each sample.
-
8
Calculate concentration of organisms/ml by multiplying the average number of T. pallidum in one large square by 800,000.
For example, if 62 T. pallidum are counted in four large squares, then # of T. pallidum/ml = 62/4 × 800,000 = 1.24 × 107 T. pallidum/ml.
REAGENTS AND SOLUTIONS
Dissociation medium
32 ml cell culture–grade H2O
5 ml 10× modified EBSS (see recipe)
0.5 ml non‐essential amino acids (Gibco, 11140‐050)
1 ml sodium bicarbonate (Sigma, S8761)
1 ml 1 M MOPS buffer, pH 7.5
364 µl sodium pyruvate (Sigma, S8636)
68 µl 0.5 M EDTA, pH 8.0
4 mg dithiothreitol (DTT)
10 ml dialyzed FBS [dialyzed overnight against two changes of 1× PBS or modified EBSS (diluted from 10×; see recipe)]
Filter sterilize
Aliquot into 15‐ml polypropylene tubes
Store ≤2 years at −20°C
Modified Earle's balanced salt solution (EBSS), 10×
Dissolve 4.0 g KCl, 68.0 g NaCl, 1.4 g NaH2PO4 .H2O, and 10 g d‐glucose in 700 ml cell culture–grade distilled water. Add water to 1000 ml. Adjust pH to 7.6 using 1 N NaOH or 1 N HCl and filter sterilize. Store ≤1 year at room temperature. For 1× solution, dilute 1:10 in sterile cell culture–grade distilled water.
Sf1Ep medium
500 ml Eagle's MEM (Sigma, M4655)
5 ml MEM non‐essential amino acids (Gibco, 11140‐050)
5 ml l‐glutamine (Sigma, G7513)
5 ml sodium pyruvate (Sigma, S8636)
50 ml FBS, heat inactivated (any high‐quality FBS)
Filter sterilize
-
Store ≤3 months at 4°C
It is not necessary to use pre‐screened T. pallidum‐permissive FBS for routine culture of Sf1Ep cells.
COMMENTARY
Background Information
T. pallidum subsp. pallidum is an important human pathogen, causing an estimated 6 to 7 million new cases of syphilis globally each year (Rowley et al., 2019; Newman et al., 2015). The developing world bears an especially high burden, and congenital syphilis remains the second leading infectious cause of stillbirth worldwide (Lawn et al., 2016). In industrialized countries, syphilis is common in communities of men who have sex with men (MSM).
T. pallidum causes invasive and persistent disease. The initial symptom is a painless chancre at the site of spirochete entry that occurs 10 to 90 days after exposure. Motile treponemes can often be recovered from the exudate of primary chancres. These lesions spontaneously resolve in 3 to 6 weeks. The second stage of syphilis is characterized by a widespread rash and a flu‐like illness of fever and myalgia occurring 4 to 10 weeks after initial infection. Both primary and secondary syphilis symptoms resolve regardless of whether treatment is administered. The organisms may remain asymptomatic for years, after which they can manifest in a variety of symptoms, including neurologic and cardiac symptoms and destructive lesions of the skin, bones, and organs. During pregnancy, the spirochete can cross the placenta, resulting in infection of the fetus.
The other subspecies of T. pallidum, T. pallidum subsp. pertenue and subsp. endemicum, also cause chronic disease and destructive lesions. These spirochetes are typically not transmitted sexually, but rather through direct skin contact. The yaws spirochete, T. pallidum subsp. pertenue, is endemic to tropical areas (Pacific, Southeast Asia, and West and Central Africa), has an unknown worldwide incidence, and largely infects children (Giacani & Lukehart, 2014). T. pallidum subsp. endemicum causes bejel [also called endemic (non‐venereal) syphilis] and is endemic to the Middle East and North Africa. These spirochetes are closely related genetically to the syphilis spirochete, with >99.8% identity (Smajs, Strouhal, & Knauf, 2018). Serologically, syphilis, yaws, and bejel are indistinguishable.
Despite their abilities to cause varied disease manifestations and infection over a period of years to decades, the group of T. pallidum–related organisms are obligate pathogens that have not been found in nature except in humans, other primates, rabbits, and hares. Other spirochetes are also obligate parasites of animals, including oral, intestinal, and dermal Treponema species; intestinal Brachyspira organisms; the tick‐transmitted Borrelia species; and a wide variety of termite gut spirochetes. However, these organisms can all be cultivated axenically (without tissue culture cells). In addition, most of these host‐associated spirochetes are obligate anaerobes except Borrelia and Leptospira, which both appear to require oxygen.
The T. pallidum group has apparently undergone an extreme degree of adaptation through genome reduction, so that genes required for survival in mammalian hosts have been retained but most genes required for synthesis of fatty acids, amino acids and nucleic acid precursors have been lost. The reason for the current requirement for mammalian cells in the T. pallidum culture system is unknown but is thought to be related to acquisition of lipids or short‐lived compounds originating from the Sf1Ep cells. Our eventual goal is to develop conditions that support long‐term culture of T. pallidum in the absence of mammalian cells, which would greatly simplify in vitro propagation and both experimental and clinical studies.
The ability to culture T. pallidum in vitro opens many avenues of research that have previously been impossible. Isogenic strains can now be generated by in vitro culture. Systematic evaluation of growth requirements and regulation of T. pallidum gene expression can also be performed independently of a host animal. Development of techniques to genetically manipulate T. pallidum will help to identify genes required for infection. In addition, it may be possible to isolate new strains directly from patients (rather than through rabbit inoculation).
Critical Parameters and Troubleshooting
Strain variations
The Nichols strain of T. pallidum subsp. pallidum grows vigorously and apparently indefinitely in vitro. We have multiple cultures that have been subcultured weekly for >3 years. In addition, infectivity appears to be well maintained in vitro, with as few as 10 organisms able to cause lesions in rabbits after 1.5 years of culture. However, other strains of T. pallidum subsp. pallidum grow less robustly in vitro. In general, we have noted that strains that grow more slowly in rabbits also multiply more slowly in vitro. In our hands, strains of the SS14 cluster grow more slowly in vitro and require a larger inoculum to thrive. T. pallidum subsp. endemicum can grow in the in vitro cultivation system, with existing cultures growing >19 weeks with an average doubling time of ∼50 hr. Despite the close genetic relationship to the syphilis and yaws treponemes, attempts to culture T. pallidum subsp. pertenue have not yet been successful.
Evaporation of medium
Due to the long generation time of T. pallidum (Basic Protocols 1 and 2), culture times are quite long, and evaporation of medium can be a problem. Culture is unsuccessful when evaporation is excessive, likely due to change in the osmolarity of the medium. Use of a humidified incubator is essential, as is use of low‐evaporation cell culture vessels. The volume of medium that is recoverable from a culture after 1 week of incubation can help to gauge evaporation. Typically, it should be ≥95% of the initial culture volume (medium plus inoculum).
In addition, high levels of condensation on culture vessel lids generally indicate poor culture conditions (likely uneven incubator temperatures) that will not support T. pallidum cultivation in vitro.
Sf1Ep cells
High‐passage Sf1Ep cells grow faster as the passage number increases, and eventually, the transformed cells fail to support vigorous T. pallidum growth (Basic Protocols 1 and 2). In our hands, when the passage number increases to 65 to 70, multiplication begins to decline slowly over three or four passages, although motility remains high. If T. pallidum cultures begin to perform poorly, new stocks of low‐passage Sf1Ep cells should be prepared (Support Protocol 2) and utilized in subsequent cultures.
The number of Sf1Ep cells seeded for the co‐culture (Basic Protocols 1 and 2) can be varied from 0.5–2 × 105 cells per well (in 6‐well plates) without a significant effect on T. pallidum growth. However, higher numbers of Sf1Ep cells seeded result in lower T. pallidum viability and multiplication, likely due to consumption of critical nutrients, decreases in pH, or accumulation of toxic products. Likewise, lower concentrations of Sf1Ep cells may result in lower yields and delayed growth.
Understanding Results
The average doubling time of the Nichols strain of T. pallidum is about 38 to 40 hr when the organisms are growing robustly in vitro, similar to the experimentally determined 30 to 33 hr in rabbits (Cumberland & Turner, 1949; Magnuson & Eagle, 1948). Thus, over a 7‐day culture period (Basic Protocols 1 and 2), T. pallidum organisms increase about 20 to 30 fold. If the calculated generation time rises above 45 hr, this is an indication that the culture conditions may be suboptimal. Other strains of T. pallidum subsp. pallidum (SS14, Mexico A, UW231B, and UW249B) and T. pallidum subsp. endemicum (Bosnia A) that we have examined have a somewhat higher average doubling time (ranging from 45 to 56 hr) (Edmondson et al., 2021).
The yield of the Nichols strain of T. pallidum reaches a limit of about 1–2 × 108 organisms per culture for a standard culture in a 6‐well plate with 4 ml TpCM‐2. Increasing the inoculum does not increase yield; rather, the organisms seem to reach a density ceiling. Increasing the medium volume results in higher total culture yield, indicating that growth may be limited by depletion of required nutrients (Edmondson et al., 2018; Norris & Edmondson, 1987).
Time Considerations
The estimation of required time in this example experiment assumes that all necessary solutions have been made or are on hand and that all equipment is ready to use (Fig. 3).
Figure 3.
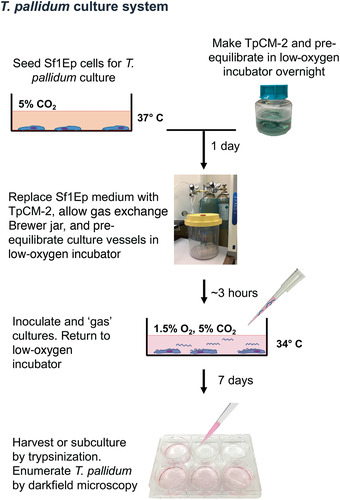
Overview of the T. pallidum in vitro cultivation procedure.
Day before experiment initiation (Basic Protocols 1 and 2):
Make TpCM‐2 and pre‐equilibrate overnight (∼1.5 hr).
Seed Sf1Ep cells (∼1 hr). Sf1Ep cultures can be conveniently passaged at this time (Support Protocol 2).
Day of experiment initiation (Basic Protocols 1 and 2):
Replace Sf1Ep medium with TpCM‐2 and pre‐equilibrate culture vessel (∼30 min for one plate, pre‐incubate for >3 hr). Thaw trypsin at room temperature if passaging an existing T. pallidum culture.
If starting from a frozen T. pallidum stock, thaw sample and dilute in TpCM‐2. Quantitate inoculum using a Helber chamber (Support Protocol 3). Inoculate and “gas” cultures. Return to low‐oxygen incubator. This takes ∼1 hr in total.
If passaging an existing T. pallidum culture, trypsinize existing culture and use an aliquot to seed newly prepared plates (∼30 min/plate). If using Support Protocol 1, allow ∼1 hr/plate.
Incubation period (Basic Protocols 1 and 2):
Incubate 6 to 10 days.
Day of harvest (Basic Protocols 1 and 2 and Support Protocol 3):
Trypsinize and enumerate T. pallidum cultures (∼20 min per culture or 1 hr for triplicate samples).
Author Contributions
Diane G. Edmondson: Funding acquisition; Investigation; Methodology; Project administration; writing‐original draft. Steven J. Norris: Funding acquisition; Investigation; Methodology; Project administration; writing‐review & editing.
Acknowledgments
We thank Bridget D. DeLay for her careful review of the manuscript. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI141958. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Edmondson, D. G. , & Norris, S. J. (2021). In vitro cultivation of the syphilis spirochete Treponema pallidum . Current Protocols, 1, e44. doi: 10.1002/cpz1.44
Literature Cited
- Cox, D. L. (1994). Culture of Treponema pallidum . Methods in Enzymology, 236, 390–405. doi: 10.1016/0076-6879(94)36029-4. [DOI] [PubMed] [Google Scholar]
- Cumberland, M. C. , & Turner, T. B. (1949). The rate of multiplication of Treponema pallidum in normal and immune rabbits. American Journal of Syphilis, 33, 201–211. [PubMed] [Google Scholar]
- Edmondson, D. G. , DeLay, B. D. , Kowis, L. E. , & Norris, S. J. (2021) (in press). Parameters affecting continuous in vitro culture of Treponema pallidum subsp. pallidum strains. mBio, 12e03536–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson, D. G. , Hu, B. , & Norris, S. J. (2018). Long‐term in vitro culture of the syphilis spirochete Treponema pallidum subsp. pallidum . mBio, 9(3), e01153‐18. doi: 10.1128/mBio.01153-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieldsteel, A. H. , Cox, D. L. , & Moeckli, R. A. (1981). Cultivation of virulent Treponema pallidum in tissue culture. Infection and Immunity, 32, 908–915. doi: 10.1128/IAI.32.2.908-915.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fieldsteel, A. H. , Stout, J. G. , & Becker, F. A. (1981). Role of serum in survival of Treponema pallidum in tissue culture. In Vitro, 17, 28–32. doi: 10.1007/BF02618027. [DOI] [PubMed] [Google Scholar]
- Fitzgerald, T. J. , Johnson, R. C. , & Smith, M. (1976). Accidental laboratory infection with Treponema pallidum, Nichols strain. Journal of the American Venereal Disease Association, 3, 76–78. [PubMed] [Google Scholar]
- Fraser, C. M. , Norris, S. J. , Weinstock, G. M. , White, O. , Sutton, G. G. , Dodson, R. , … Venter, J. C. (1998). Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science, 281(5375), 375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- Giacani, L. , & Lukehart, S. A. (2014). The endemic treponematoses. Clinical Microbiology Reviews, 27(1), 89–115. doi: 10.1128/CMR.00070-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawn, J. E. , Blencowe, H. , Waiswa, P. , Amouzou, A. , Mathers, C. , Hogan, D. , … Lancet Stillbirth Epidemiology investigator group (2016). Stillbirths: Rates, risk factors, and acceleration towards 2030. Lancet, 387(10018), 587–603. doi: 10.1016/S0140-6736(15)00837-5. [DOI] [PubMed] [Google Scholar]
- Lukehart, S. A. , & Marra, C. M. (2007). Isolation and laboratory maintenance of Treponema pallidum . Current Protocols in Microbiology, 7, 12A.1.1–12A.1.18. doi: 10.1002/9780471729259.mc12a01s7. [DOI] [PubMed] [Google Scholar]
- Magnuson, H. J. , & Eagle, H. (1948). The minimal infectious inoculum of Spirochaeta pallida (Nichols strain), and a consideration of its rate of multiplication in vivo. American Journal of Syphilis, 32, 1–18. [PubMed] [Google Scholar]
- Magnuson, H. J. , Thomas, E. W. , Olansky, S. , Kaplan, B. I. , DeMello, L. , & Cutler, J. C. (1956). Inoculation syphilis in human volunteers. Medicine, 35, 33–82. doi: 10.1097/00005792-195602000-00002. [DOI] [PubMed] [Google Scholar]
- Miller, J. N. (Ed.) (1971). Spirochetes in body fluids and tissues: Manual of investigative methods. Springfield, IL: Charles C. Thomas. [Google Scholar]
- Newman, L. , Rowley, J. , Vander Hoorn, S. , Wijesooriya, N. S. , Unemo, M. , Low, N. , … Temmerman, M. (2015). Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLOS One, 10(12), e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, S. J. (1982). In vitro cultivation of Treponema pallidum: Independent confirmation. Infection and Immunity, 36(1), 437–439. doi: 10.1128/IAI.36.1.437-439.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, S. J. , & Edmondson, D. G. (1986). Serum requirement for the multiplication of Treponema pallidum in a tissue‐culture system: Association of growth‐promoting activity with the protein fraction. Sexually Transmitted Diseases, 13(4), 207–213. doi: 10.1097/00007435-198610000-00001. [DOI] [PubMed] [Google Scholar]
- Norris, S. J. , & Edmondson, D. G. (1987). Factors affecting the multiplication and subculture of Treponema pallidum subsp. pallidum in a tissue culture system. Infection and Immunity, 53, 534–539. doi: 10.1128/IAI.53.3.534-539.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris, S. J. , Cox, D. L. , & Weinstock, G. M. (2001). Biology of Treponema pallidum: Correlation of functional activities with genome sequence data. Journal of Molecular Microbiology and Biotechnology, 3(1), 37–62. [PubMed] [Google Scholar]
- Norris, S. J. , Paster, B. J. , Moter, A. , & Göbel, U. B. (2003). The genus Treponema. In Dworkin M., Falkow S., Rosenberg E., Schleifer K.‐H., & Stackebrandt E. (Eds.), The prokaryotes: An evolving electronic resource for the microbiological community (1st ed.). New York: Springer Verlag, Inc. [Google Scholar]
- Rowley, J. , Vander Hoorn, S. , Korenromp, E. , Low, N. , Unemo, M. , Abu‐Raddad, L. J. , … Taylor, M. M. (2019). Chlamydia, gonorrhoea, trichomoniasis and syphilis: Global prevalence and incidence estimates, 2016. Bulletin of the World Health Organization, 97(8), 548–562P. doi: 10.2471/BLT.18.228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaudinn, F. , & Hoffman, E. (1905). Vorläufiger bericht über das Vorkommen für Spirochaeten in syphilitischen Krankheitsprodukten und bei Papillomen. Arb. Gesundh. Amt. Berlin, 22, 528–534. [Google Scholar]
- Smajs, D. , Strouhal, M. , & Knauf, S. (2018). Genetics of human and animal uncultivable treponemal pathogens. Infection , Genetics and Evolution, 61, 92–107. doi: 10.1016/j.meegid.2018.03.015. [DOI] [PubMed] [Google Scholar]