

Abstract
Objective
In a previously reported phase II randomized, placebo‐controlled, interventional trial, we demonstrated that treatment with ustekinumab, an anti–interleukin‐12 (IL‐12)/IL‐23 p40 neutralizing monoclonal antibody, improved global and organ‐specific measures of disease activity in patients with active systemic lupus erythematosus (SLE). Utilizing the biomarker data from this phase II clinical study, we sought to determine whether modulation of the expression of IL‐12, IL‐23, or both cytokines by ustekinumab is associated with clinical efficacy in patients with SLE.
Methods
This phase II randomized, placebo‐controlled study enrolled 102 patients with autoantibody‐positive SLE whose disease remained active despite standard‐of‐care therapy. Patients were randomized at a 3:2 ratio to receive ~6 mg/kg ustekinumab intravenously or placebo at week 0, followed by subcutaneous injections of 90 mg ustekinumab or placebo every 8 weeks, with placebo crossover to 90 mg ustekinumab every 8 weeks. The SLE Responder Index 4 (SRI‐4) at week 24 was used to determine which patients could be classified as ustekinumab responders and which could be classified as nonresponders. In addition to measurements of p40 and IL‐23, serum levels of interferon‐γ (IFNγ), IL‐17A, IL‐17F, and IL‐22, as a proxy for the IL‐12 and IL‐23 pathways, were quantified by immunoassay.
Results
Changes in the serum levels of IL‐17A, IL‐17F, and IL‐22 at different time points after treatment were not consistently significantly associated with an SRI‐4 clinical response to ustekinumab in patients with SLE. In contrast, an SRI‐4 response to ustekinumab was significantly associated (P < 0.01) with durable reductions in the serum IFNγ protein levels at several time points relative to baseline, which was not observed in ustekinumab nonresponders or patients who received placebo.
Conclusion
While not diminishing a potential role of IL‐23, these serum biomarker assessments indicate that IL‐12 blockade has an important role in the mechanism of action of ustekinumab treatment in patients with SLE.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a heterogeneous, multiorgan autoimmune disease that is associated with a variety of morbidities and increased risk of death. Ustekinumab, a monoclonal antibody targeting the p40 subunit shared by interleukin‐12 (IL‐12) and IL‐23, demonstrated significant improvement across multiple disease features in patients with moderate‐to‐severe SLE in a phase II study (1). The results of this interventional trial support preclinical and clinical case report data implicating a potential role of IL‐12 and/or IL‐23 in the pathogenesis of SLE. Notably, internal analysis and an independent comprehensive drug repositioning analysis identified ustekinumab as having the highest potential for the treatment of SLE (2).
IL‐12 is a heterodimeric cytokine composed of the subunits p35 and p40, and is a critical cytokine in immunity that is primarily produced by antigen‐presenting cells. IL‐12 promotes the differentiation of Th1 cells, which release the prototypical Th1 cytokine interferon‐γ (IFNγ). Th1‐derived cytokines stimulate innate and adaptive immune cell functions and are important for defense against certain intracellular pathogens. IL‐12 also drives the development and function of human T follicular helper cells, which stimulate B cells to produce immunoglobulins, including pathogenic autoantibodies (3). In addition, IL‐12 promotes the activation and function of cytotoxic cells, such as natural killer cells, γδ T cells, and cytotoxic CD4+ and CD8+ T lymphocytes.
Both IL‐12 and IL‐23 have been implicated in the pathogenesis of SLE. For example, genetic deletion of p35, an IL‐12–specific subunit, resulted in the reduction of both antinuclear antibodies and glomerulonephritis in a mouse model of lupus‐like disease (4). IL‐23 is composed of the p40 subunit, which it shares with IL‐12, and a unique p19 subunit. IL‐23 is also an important factor in the survival and expansion of cells, including those that produce the proinflammatory cytokine IL‐17, such as Th17 cells. The IL‐23/Th17 pathway has been implicated in several immune‐mediated inflammatory diseases, including SLE. Observations from previous research in mice have suggested that the IL‐23/Th17 pathway contributes to lupus pathogenesis and may be particularly relevant to renal disease (5), a finding that has been corroborated in human profiling studies (5, 6). Thus, ustekinumab may modulate SLE by neutralizing the function of 2 proinflammatory cytokines that signal through their shared p40 subunit.
Despite evidence of a potential role of multiple cytokines in SLE, the relative importance of IL‐12 and IL‐23 in SLE pathogenesis is currently unclear. To address this question, we assessed serum biomarkers for both the IL‐12 (IFNγ) pathway and the IL‐23 (IL‐17A, IL‐17F, IL‐22) pathway in order to understand how these distinct mediators are affected following treatment with ustekinumab. For this purpose, we utilized a targeted analytic approach to better understand the mechanism of action of ustekinumab in SLE. Our findings suggest that blockade of the IL‐12 pathway represents an important component of the mechanism of action of ustekinumab in patients with SLE.
PATIENTS AND METHODS
Study design and patients
The present study assessed biomarker data from a phase II multicenter, randomized, placebo‐controlled trial of ustekinumab in adult patients (ages 18–75 years) with active SLE, the details of which have been previously reported (1). Eligible patients were those who had a diagnosis of SLE (in accordance with the Systemic Lupus International Collaborating Clinics classification criteria [7]) for at least 3 months before the first study drug administration. Patients were randomized to receive an intravenous infusion of ustekinumab (~6 mg/kg) or placebo at week 0, followed by subcutaneous injections of 90 mg ustekinumab or placebo every 8 weeks, with placebo crossover to 90 mg ustekinumab every 8 weeks. These treatments were administered in addition to standard‐of‐care therapy.
In the current analysis, efficacy was assessed using the SLE Responder Index 4 (SRI‐4). The SRI‐4 is a composite measure that requires at least a 4‐point improvement in the SLE Disease Activity Index 2000 score, no worsening (<10‐mm increase) in the physician global assessment of disease activity score from baseline, no new British Isles Lupus Assessment Group (BILAG) 2004 domain A score, and no more than 1 new BILAG domain B score (8).
Proteomics analysis
Serum for targeted proteomics analysis was collected from both placebo‐ and ustekinumab‐treated patients at baseline and weeks 4, 8, 12, 24, 28, 40, and 48 (see the time course outlined in Supplementary Figure 1, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41547/abstract). For comparative purposes, serum was also obtained from age‐, sex‐, and race‐matched healthy control subjects (provided by external vendors). Serum levels of IFNγ and p40 were quantified using the Meso Scale Discovery platform. Serum levels of IL‐23, IL‐17A, IL‐17F, and IL‐22 were profiled using the high‐sensitivity Single Molecule Counting Erenna Immunoassay. Serum samples were also collected from ustekinumab‐treated patients with psoriasis from a previous phase III clinical trial, NAVIGATE (A Phase 3, Multicenter, Randomized, Double‐blind Study to Evaluate the Efficacy and Safety of Guselkumab for the Treatment of Subjects With Moderate to Severe Plaque‐type Psoriasis and an Inadequate Response to Ustekinumab; ClinicalTrials.gov identifier: NCT02203032) (9). Patients with psoriasis were evaluated for serum levels of IL‐17A, IL‐17F, IL‐22, and IFNγ using the same methods as described above.
RESULTS
Characteristics of the patients and outcomes
A total of 102 patients with active SLE were randomized in a 3:2 ratio to receive either ustekinumab (n = 60) or placebo (n = 42); patient demographic and disease characteristics have been previously described (1). The primary end point was achieved, with 62% of patients in the ustekinumab group and 33% of patients in the placebo group achieving an SRI‐4 treatment response at week 24 (P = 0.006) (1).
Association of IFNγ suppression with response to ustekinumab
To understand the mechanism of action of ustekinumab treatment in patients with SLE in greater detail, we examined downstream cytokines associated with IL‐12 and IL‐23 signaling. Serum levels of p40 (shared by IL‐12 and IL‐23), p19 (IL‐23), IFNγ, IL‐17A, IL‐17F, and IL‐22 were quantified in the serum samples collected from SLE patients at baseline and longitudinally after ustekinumab administration. Levels of the p40 subunit, the target of ustekinumab, was elevated at baseline in the SLE trial population compared to healthy controls, but were not significantly different between ustekinumab responders and nonresponders (Figure 1A). Over time, the levels of p40 accumulated in only the ustekinumab‐treated patients (Figure 1C), which was indicative of the expected target engagement response and is similar to findings in previous trials with this agent in which binding of ustekinumab to p40 prolongs the half‐life of the antigen. Levels of p40 accumulation after ustekinumab administration were similar between responders and nonresponders (Figure 1C). Unlike what was observed with p40, the baseline levels of IL‐23 were not elevated in the serum of patients with SLE compared to healthy controls, and accumulation of IL‐23 in ustekinumab‐treated patients was not observed (Figures 1B and D).
Figure 1.
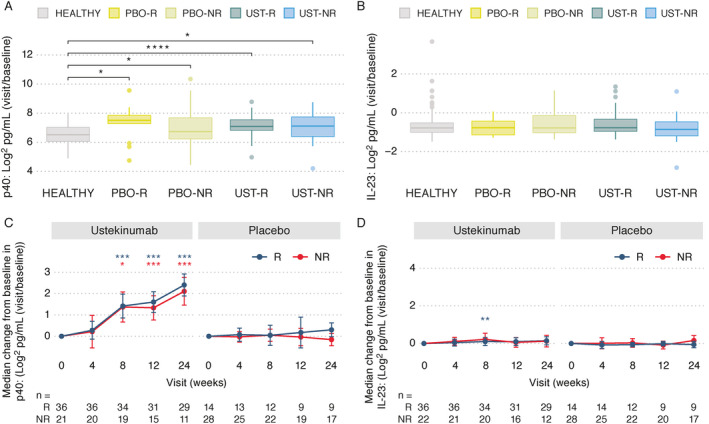
Assessment of serum levels of p40 and interleukin‐23 (IL‐23). A and B, Serum levels of p40 (A) and IL‐23 (B) were determined in healthy controls (n = 60) and at baseline in patients with systemic lupus erythematosus (SLE) in the placebo (PBO) or ustekinumab (UST) treatment groups (subject numbers at week 0 shown at bottom of C and D) by response status at week 24 (responder [R] versus nonresponder [NR]). Data are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the 10th and 90th percentiles. Circles indicate outliers. C and D, Longitudinal serum concentrations of p40 (C) and IL‐23 (D) were compared between the ustekinumab and placebo groups of SLE patients by response status. Results are shown as the median change from baseline with median absolute deviation of the change. Numbers of subjects with available samples are indicated at each time point. * = P < 0.05; ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001 versus baseline within group in C and D or as indicated in A. Asterisk color in C and D matches the treatment response group in which a significant difference was achieved. P values were computed using t‐tests.
Serum IFNγ levels were elevated at baseline in the SLE trial population compared to healthy controls (P < 0.0001). No remarkable differences in the baseline levels of IFNγ were noted between responders and nonresponders in either the ustekinumab or placebo treatment groups (Figure 2A). Following treatment with ustekinumab, levels of IFNγ were significantly down‐modulated over time relative to baseline levels in the responder population only, at week 4 (P < 0.001), week 8 (P < 0.01), and week 12 (P < 0.01), and were significantly lower than those in nonresponders at weeks 4 and 8 (each P < 0.05) (Figure 2B). Serum IFNγ levels remained decreased at the 24‐week time point in ustekinumab responders when compared to nonresponders and patients who received placebo.
Figure 2.
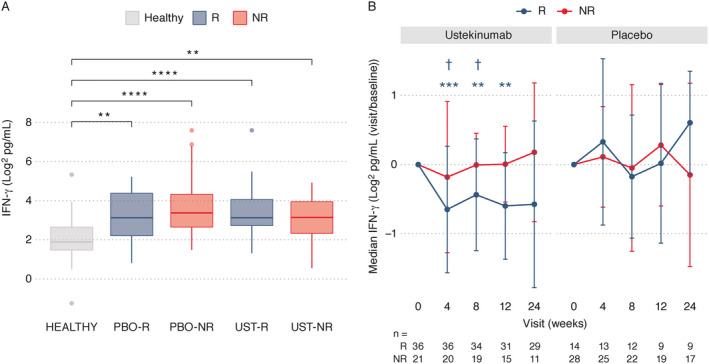
Assessment of serum levels of interferon‐γ (IFNγ). A, Serum levels of IFNγ were determined in healthy controls (n = 60) and at baseline in patients with SLE in the placebo or ustekinumab treatment groups (subject numbers at week 0 shown at bottom of B) by response status at week 24. Data are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the 10th and 90th percentiles. Circles indicate outliers. B, Longitudinal serum concentrations of IFNγ were compared between the ustekinumab and placebo groups of SLE patients by response status. Results are shown as the median change from baseline with median absolute deviation of the change. Numbers of subjects with available samples are indicated at each time point. ** = P < 0.01; *** = P < 0.001; **** = P < 0.0001 versus baseline within group in B or as indicated in A. † = P < 0.05 for responders versus nonresponders. Footnote symbol color matches the treatment response group in which a significant difference was achieved. P values were computed using t‐tests. See Figure 1 for other definitions.
Changes in serum levels of IL‐17A, IL‐17F, and IL‐22 were not significantly associated with an SRI‐4 clinical response to ustekinumab in patients with SLE, but did reach significance at a single time point after baseline for each analyte (week 4 for IL‐17A, week 8 for IL‐17F, and week 12 for IL‐22) (Figure 3). Of note, decreases in the median serum levels of IL‐17A, IL‐17F, and IL‐22 following ustekinumab treatment have previously been observed in a phase III trial of ustekinumab in patients with psoriasis (9) (see Supplementary Figure 2, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41547/abstract). In the current SLE trial, no significant differences in the serum levels of IL‐17A, IL‐17F, and IL‐22 were observed between the responders and nonresponders in either the ustekinumab or placebo treatment groups at any time point. Serum IFNγ and IL‐17A biomarker findings were durable up to week 48 in SLE patients in the ustekinumab group who continued to receive ustekinumab treatment after week 24 (see Supplementary Figure 3, available on the Arthritis & Rheumatology website at http://onlinelibrary.wiley.com/doi/10.1002/art.41547/abstract).
Figure 3.
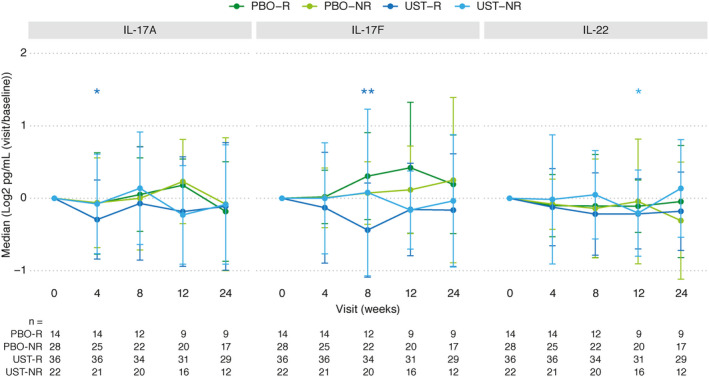
Assessment of serum levels of IL‐17A, IL‐17F, and IL‐22 through week 24 in the placebo and ustekinumab treatment groups of SLE patients according to response status. Results are shown as the median change from baseline with median absolute deviation of the change. Numbers of subjects with available samples at each time point are indicated below the graphs. * = P < 0.05; ** = P < 0.01 versus baseline within group. Asterisk color indicates the treatment response group in which a significant difference was achieved. P values were computed using t‐tests. See Figure 1 for definitions.
DISCUSSION
In this study, targeted proteomics analyses were performed on serum samples from patients with SLE in a phase II trial of ustekinumab to elucidate the mechanism of action of ustekinumab in this cohort. Despite a lack of modulation of type I IFN responses following treatment with ustekinumab in patients with SLE, as was also reported previously (1), we observed significant reductions in the type II IFN (IFNγ) responses in the ustekinumab‐treated responders only. Interestingly, ustekinumab‐treated responders and nonresponders both exhibited similarly elevated serum levels of IFNγ at baseline, suggesting that serum IFNγ expression in ustekinumab responders may be dependent on p40 signaling, whereas in nonresponders, serum IFNγ expression may be perpetuated by additional p40‐independent factors. The reduction in serum IFNγ levels in ustekinumab‐treated responders was observed to be greatest at 4 weeks postdosing, although these reductions were durable up to 48 weeks. It can be speculated that the ustekinumab induction dose may be a factor in the robustness of the reduction in IFNγ expression at 4 weeks. While early reductions in circulating IFNγ levels were associated with an SRI‐4 clinical response to ustekinumab, this and other biomarker findings from our study need to be confirmed with larger studies in SLE. Of note, the phase III LOTUS trial (Multicenter, Randomized, Double‐blind, Placebo‐controlled, Parallel‐group Study of Ustekinumab in Subjects with Active Systemic Lupus Erythematosus; ClinicalTrials.gov identifier: NCT03517722) was recently stopped due to a preplanned interim efficacy analysis, but additional analysis is needed because the participants in this study were not limited to those experiencing abnormalities in the IL‐12/IL‐23 target pathway. The biologic insights derived from the placebo‐controlled ustekinumab phase II study described herein are valuable and may promote additional hypotheses to test in this heterogeneous disease.
It is important to note that IFNγ antagonism has consistently been associated with improvements in disease features in multiple mouse models of lupus (10). IFNγ has also been reported to be one of the earliest dysregulated cytokines preceding SLE classification, even prior to IFNα elevation and autoantibody accumulation, suggesting a potentially more central role of IFNγ in establishing a diagnosis of lupus (11). Despite these data, no substantial benefits in disease activity were observed with a monoclonal antibody targeting IFNγ (AMG‐811) after evaluation in several small trials in patients with lupus (12, 13). It is unclear whether the lack of clear efficacy in those trials was related to the molecule, the dosage studied, or the target itself.
Analysis of cytokines downstream of IL‐23 (IL‐17A, IL‐17F, and IL‐22) failed to demonstrate consistent reductions in cytokine levels or associations with ustekinumab treatment or response in this SLE trial. Indeed, serum IL‐23 levels were neither elevated at baseline nor modulated by ustekinumab treatment in the present study, despite clear target engagement, as demonstrated by accumulation of p40 in ustekinumab‐treated patients. We were unable to reliably quantify IL‐12/p70 in a sufficient number of patients to include in this study, presumably because of limitations in the assay sensitivity. It is important to note that reductions in the serum levels of IL‐17A, IL‐17F, and IL‐22 have been consistently observed with ustekinumab in diseases in which the IL‐23/IL‐17 pathway has been implicated in disease pathogenesis, such as in psoriasis (14) and psoriatic arthritis (15), and baseline levels of these cytokines were elevated in patients relative to healthy controls. However, it should be noted that the small sample size in this phase II SLE trial may be a limitation in detecting a consistent reduction in the levels of IL‐17A, IL‐17F, and IL‐22. Examination of several available Th17 gene signatures indicated that they were not modulated after ustekinumab treatment in this phase II SLE trial (data not shown).
One important limitation of this study is the lack of data on tissue‐based biomarkers, which may be important in attaining robust measurements of IL‐23 pathway biomarkers. Indeed, recent data have suggested that IL‐23 signaling has an important role, particularly in the kidney, among patients with lupus nephritis (5). Because of the small number of patients in the ustekinumab trial who had lupus nephritis (as determined by kidney biopsy), the effect of ustekinumab treatment on kidney manifestations could not be confidently assessed.
Other than the association with the SRI‐4 response, analyses of other measures of disease activity did not reveal any associations with clinical biomarkers or serum IFNγ levels. While not diminishing a potential role of IL‐23, these serum data suggest that blockade of the IL‐12 pathway represents an important component of the mechanism of action of ustekinumab in patients with SLE.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Jordan had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Cesaroni, Seridi, Loza, Sweet, Campbell, Lipsky, van Vollenhoven, Hahn, Tsokos, Chevrier, Rose, Baribaud, Jordan.
Acquisition of data
Franks, Ma, Gordon, Branigan.
Analysis and interpretation of data
Cesaroni, Seridi, Loza, Schreiter, Sweet, Franks, Ma, Orillion, Campbell, Gordon, Branigan, Lipsky, van Vollenhoven, Hahn, Tsokos, Chevrier, Rose, Baribaud, Jordan.
ROLE OF THE STUDY SPONSOR
Employees of Janssen Research & Development participated in the study design, data collection, analysis and interpretation of the data, and writing of the manuscript. All authors approved the manuscript for submission. Writing assistance was provided by Janssen Scientific Affairs. Publication of this article was not contingent on approval by Janssen Research & Development.
ADDITIONAL DISCLOSURE
Author Lipsky is an employee of Ampel BioSolutions.
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Acknowledgment
The authors thank Rebecca Clemente, PhD for providing editorial and submission support.
Clinicaltrials.gov identifier: NCT02349061.
Supported by Janssen Research & Development, LLC.
Drs. Cesaroni and Seridi contributed equally to this work.
Drs. Cesaroni, Seridi, Loza, Sweet, Ma, Orillion, Campbell, Chevrier, Rose, Baribaud, and Jordan, Ms Schreiter, Ms Franks, Mr. Gordon, and Mr. Branigan own stock or stock options in Johnson & Johnson, of which Janssen Research & Development, is a wholly owned subsidiary. Dr. Lipsky has received consulting fees from Janssen (less than $10,000). Dr. van Vollenhoven has received consulting fees, speaking fees, and/or honoraria from AbbVie, AstraZeneca, Biotest, Bristol Myers Squibb, Celgene, Eli Lilly, GlaxoSmithKline, Janssen, Medac, Merck, Novartis, Pfizer, Roche, and UCB (less than $10,000 each) and research support from AbbVie, Arthrogen, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Pfizer, and UCB. Dr. Hahn has received consulting fees, speaking fees, and/or honoraria from Bristol Myers Squibb, Eli Lilly, and Janssen (less than $10,000 each) and research support from Bristol Myers Squibb. Dr. Tsokos has received consulting fees, speaking fees, and/or honoraria from A2 Therapeutics, Abpro, and Silicon Therapeutics (more than $10,000 each) and research support from Janssen and Pfizer.
References
- 1. Van Vollenhoven RF, Hahn BH, Tsokos GC, Wagner CL, Lipsky P, Touma Z, et al. Efficacy and safety of ustekinumab, an IL‐12 and IL‐23 inhibitor, in patients with active systemic lupus erythematosus: results of a multicentre, double‐blind, phase 2, randomised, controlled study. Lancet 2018;392:1330–9. [DOI] [PubMed] [Google Scholar]
- 2. Grammer AC, Ryals MM, Heuer SE, Robl RD, Madamanchi S, Davis LS, et al. Drug repositioning in SLE: crowd‐sourcing, literature‐mining and Big Data analysis. Lupus 2016;25:1150–70. [DOI] [PubMed] [Google Scholar]
- 3. Powell MD, Read KA, Sreekumar BK, Jones DM, Oestreich KJ. IL‐12 signaling drives the differentiation and function of a TH1‐derived TFH1‐like cell population. Sci Rep 2019;9:13991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dai J, Liu B, Cua DJ, Li Z. Essential roles of IL‐12 and dendritic cells but not IL‐23 and macrophages in lupus‐like diseases initiated by cell surface HSP gp96. Eur J Immunol 2007;37:706–15. [DOI] [PubMed] [Google Scholar]
- 5. Dai H, He F, Tsokos GC, Kyttaris VC. IL‐23 limits the production of IL‐2 and promotes autoimmunity in lupus. J Immunol 2017;199:903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zickert A, Amoudruz P, Sundstrom Y, Ronnelid J, Malmstrom V, Gunnarsson I. IL‐17 and IL‐23 in lupus nephritis: association to histopathology and response to treatment. BMC Immunol 2015;16:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petri M, Orbai AM, Alarcón GS, Gordon C, Merrill JT, Fortin PR, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Furie RA, Petri MA, Wallace DJ, Ginzler EM, Merrill JT, Stohl W, et al. Novel evidence‐based systemic lupus erythematosus responder index. Arthritis Rheum 2009;61:1143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Langley RG, Tsai TF, Flavin S, Song M, Randazzo B, Wasfi Y, et al. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double‐blind, phase III NAVIGATE trial. Br J Dermatol 2018;178:114–23. [DOI] [PubMed] [Google Scholar]
- 10. Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR. The role of IFN‐γ in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res 2001;3:136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munroe ME, Lu R, Zhao YD, Fife DA, Robertson JM, Guthridge JM, et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann Rheum Dis 2016;75:2014–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boedigheimer MJ, Martin DA, Amoura Z, Sanchez‐Guerrero J, Romero‐Diaz J, Kivitz A, et al. Safety, pharmacokinetics and pharmacodynamics of AMG 811, an anti‐interferon‐γ monoclonal antibody, in SLE subjects without or with lupus nephritis. Lupus Sci Med 2017;4:e000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Werth VP, Fiorentino D, Sullivan BA, Boedigheimer MJ, Chiu K, Wang C, et al. Pharmacodynamics, safety, and clinical efficacy of AMG 811, a human anti–interferon‐γ antibody, in patients with discoid lupus erythematosus. Arthritis Rheumatol 2017;69:1028–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Campbell K, Branigan P, Yang F, Chen Y, Orlovsky Y, Elloso MM, et al. Selective IL‐23 blockade with guselkumab (GUS) neutralizes Th17‐ and psoriasis‐associated serum, cellular, and transcriptomic measures more potently than dual IL‐12/23 blockade with ustekinumab (UST) [abstract]. Exp Dermatol 2018;27:43–4.28677206 [Google Scholar]
- 15. Siebert S, Loza MJ, Song Q, McInnes I, Sweet K. Ustekinumab and guselkumab treatment results in differences in serum IL17A, IL17F and CRP levels in psoriatic arthritis patients: a comparison from ustekinumab PH3 and guselkumab PH2 programs [abstract]. Ann Rheum Dis 2019;78:A293. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material